CN1034503C - 吡唑并嘧啶酮抗心绞痛剂的制备方法 - Google Patents
吡唑并嘧啶酮抗心绞痛剂的制备方法 Download PDFInfo
- Publication number
- CN1034503C CN1034503C CN92105591A CN92105591A CN1034503C CN 1034503 C CN1034503 C CN 1034503C CN 92105591 A CN92105591 A CN 92105591A CN 92105591 A CN92105591 A CN 92105591A CN 1034503 C CN1034503 C CN 1034503C
- Authority
- CN
- China
- Prior art keywords
- methyl
- alkyl
- reaction
- formula
- definition
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/14—Decongestants or antiallergics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/16—Otologicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/08—Vasodilators for multiple indications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
Abstract
本发明有关于制造一系列吡唑并[4,3-d]嘧啶-7-酮类化合物的方法,其中所述的化合物它们是3′,5′-环磷酸鸟苷磷酸二酯酶(cGMP PDE)的强有力的和选择性的抑制剂,可应用于许多医疗领域,包括治疗各种心血管疾病诸如心绞痛,高血压,心力衰竭和动脉粥样硬化。
Description
本发明有关于一系列吡唑并[4,3-d]嘧啶-7-酮类化合物,它们是3′,5′-环磷酸鸟苷磷酸二酯酶(cGMP PDE)的强有力的和选择性的抑制剂,可应用于许多医疗领域,包括治疗各种心血管疾病诸如心绞痛,高血压,心力衰竭和动脉粥样硬化。
本发明的化合物呈现出对于cGMP PDEs抑制的选择性,而没有对于3′,5′-环磷酸腺苷磷酸二酯酶(cAMP PDEs)的抑制性,由于这种选择性PDE抑制的结果,cGMP水平被升高,它转而导致有益的抗血小板,抗中性白细胞,抗血管痉挛和血管舒张的活性,并具有对于内皮衍生的松弛因素(EDRF)和硝基血管舒张剂的药效的增效作用。因此,这类化合物可用来治疗许多疾病,包括稳定,不稳定和变异型(Prinzme-tal)的心绞痛,高血压,肺动脉高压,充血性心力衰竭,动脉粥样硬化,以及在经皮穿刺冠状动脉腔内成形术后(即post-PTCA)出现的降低的血管畅通症状,末梢血管疾病,中风,支气管炎,慢性哮喘,过敏性哮喘,过敏性鼻炎,青光眼,以及由于内脏功能失调导致的疾病,例如肠过敏性综合症(IBS)等。
欧洲专利申请EP-A-0201188公开了一些吡唑并[4,3-d]嘧啶-7-酮可作为腺苷受体拮抗物和PDE抑制剂,用于治疗心血管疾病诸如心力衰竭或心搏乏力。但是这些化合物都不是特别强有力的PDE抑制剂,也未报导它们是cGMP PDE的选择性抑制剂。
本发明的化合物具有下列式(I)以及它们的药物上可接受的盐: 其中R1是H;任意选择地被一个或多个氟取代基取代的C1-C3烷基;或C3-C5环烷基:R2是H,或任意选择地被一个或多个氟取代基取代的,或被C3-C6环烷基取代的C1-C6烷基;R3是任意选择地被一个或多个氟取代基取代的,或被C3-C6环烷基取代的C1-C6烷基;C3-C5环烷基;C3-C6烯基;或C3-C6炔基;R4是任意选择地被OH,NR5R6,CN,CONR5R6或CO2R7取代的C1-C4烷基;任意选择地被CN,CONR5R6或CO2R7取代的C2-C4烯基;任意选择地被NR5R6取代的烷酰基;任意选择地被NR5R6取代的羟基C2-C4烷基;任意选择地被OH或NR5R6取代的(C2-C3烷氧基)C1-C2烷基;CONR5R6;CO2R7;卤素;NR5R6;NHSO2NR5R6;NHSO2R8;或苯基或杂环基,其上可任意地被甲基取代;R5和R6各自独立地是H或C1-C4烷基,或者和它们所连接的氮原子一起,形成吡咯烷基,哌啶子基,吗啉代基,4-(NR9)-哌嗪基或咪唑基,其中所说的基团可任意地被甲基或羟基所取代;R7是H或C1-C4烷基;R8是任意选择地被NR5R6取代的C1-C3烷基;R9是H;任意选择地被苯基取代的C1-C3烷基;羟基C2-C3烷基;或C1-C4烷酰基。
其中R1是H;任意选择地被一个或多个氟取代基取代的C1-C3烷基;或C3-C5环烷基:R2是H,或任意选择地被一个或多个氟取代基取代的,或被C3-C6环烷基取代的C1-C6烷基;R3是任意选择地被一个或多个氟取代基取代的,或被C3-C6环烷基取代的C1-C6烷基;C3-C5环烷基;C3-C6烯基;或C3-C6炔基;R4是任意选择地被OH,NR5R6,CN,CONR5R6或CO2R7取代的C1-C4烷基;任意选择地被CN,CONR5R6或CO2R7取代的C2-C4烯基;任意选择地被NR5R6取代的烷酰基;任意选择地被NR5R6取代的羟基C2-C4烷基;任意选择地被OH或NR5R6取代的(C2-C3烷氧基)C1-C2烷基;CONR5R6;CO2R7;卤素;NR5R6;NHSO2NR5R6;NHSO2R8;或苯基或杂环基,其上可任意地被甲基取代;R5和R6各自独立地是H或C1-C4烷基,或者和它们所连接的氮原子一起,形成吡咯烷基,哌啶子基,吗啉代基,4-(NR9)-哌嗪基或咪唑基,其中所说的基团可任意地被甲基或羟基所取代;R7是H或C1-C4烷基;R8是任意选择地被NR5R6取代的C1-C3烷基;R9是H;任意选择地被苯基取代的C1-C3烷基;羟基C2-C3烷基;或C1-C4烷酰基。
在上面的定义中,除非另外指明,具有三个或更多碳原子的烷基可以是直链的或带支链的。此外,具有四个或更多碳原子的烯基或炔基,或具有三个碳原子的烷氧基,也可以是直链的或带支链的。卤素意指氟,氯,溴或碘,杂环基可选自噻吩基,吡啶基,吡唑基,咪唑基,三唑基,唑基,噻唑基或嘧啶基。
式(I)化合物可以含有一个或多个不对称中心,因此它们可以对映异构体或非对映异构体的形式存在;进一步,一些式(I)化合物含有烯基,可以顺式或反式异构体的形式存在,在每种情况下,本发明既包括混合物,也包括分开的各别异构体。
式(I)化合物也可以互变异构的形式存在,本发明既包括它们的混合物,也包括分开的各别互变异构体。
在本发明中也包括放射性标记的式(I)化合物的衍生物,它适合于生物学方面的研究。
含有碱性中心的式(I)化合物的药物上可接受的盐,是与药物上可接受的酸进行加成而形成的盐。实例包括盐酸盐,氢溴酸盐,硫酸盐或硫酸氢盐,磷酸盐或磷酸氢盐,醋酸盐,苯甲酸盐,丁二酸盐,富马酸盐,马来酸盐,乳酸盐,柠檬酸盐,酒石酸盐,葡糖酸盐,甲烷磺酸盐,苯磺酸盐,和对甲苯磺酸盐。式(I)化合物也可提供药物上可接受的金属盐,特别是与碱形成的碱金属盐,实例包括钠盐和钾盐。
式(I)化合物的经优选的基团是其中的R1是H,甲基或乙基;R2是C1-C3烷基;R3是C2-C3烷基;R4是任意选择地被OH,NR5R6,CONR5R6或CO2R7取代的C1-C2烷基;任意选择地被NR5R6取代了的乙酰基;任意选择地被NR5R6取代的羟乙基;任意选择地被OH或NR5R6取代了的乙氧基甲基;CH=CH-CN;CH=CH-CONR5R6;CH=CHCO2R7;CO2H;CONR5R6;Br;NR5R6;NHSO2NR5R6;NHSO2R8;或吡啶基或咪唑基,其上可任意选择地被甲基取代;R5和R6各自独立的是H,甲基或乙基,或者和它们所连接的氮原子一道,形成一个哌啶子基,吗啉代基,4-(NR9)-1-哌嗪基或咪唑基,其中所说的基团可任意选择地被甲基或羟基取代;R7是H或叔丁基;R8是甲基或CH2CH2CH2NR5R6;R9是H,甲基,苄基,2-羟乙基或乙酰基。
式(I)化合物的特别优选的基团是其中的R1是甲基;R2是正丙基;R3是乙基或正丙基;R4是CH2NR5R6,CH2OCH2CH2NR5R6,CH2OCH2CH3,CH2OCH2CH2OH,COCH2NR5R6,CH(OH)CH2NR5R6,CH=CH-CON(CH3)2,CH=CHCO2R7,CO2H,CONR5R6,Br,NHSO2NR5R6,NHSO2CH2CH2CH2NR5R6,2-吡啶基,1-咪唑基或1-甲基-2-咪唑基;R5和R6和它们所连接的氮原子形成一个哌啶子基,4-羟基哌啶子基,吗啉代基,4-(NR9)-1-哌嗪基或2-甲基-1-咪唑基;R7是H或叔丁基;R9是H,甲基,苄基,2-羟乙基或乙酰基。
本发明特别优选的具体的化合物包括:
5-[2-乙氧基-5-(1-甲基-2-咪唑基)苯基]-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮;
5-[2-乙氧基-5-(4-甲基-1-哌嗪基羰基)苯基]-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮;
5-[5-(4-乙酰基-1-哌嗪基)乙酰基-2-乙氧基苯基]-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮;
5-(2-乙氧基-5-吗啉代基乙酰基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮;和
5-(5-吗啉代基乙酰基-2-正丙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮。
依赖于R4的性质,式(I)化合物可以由下式(II)的化合物通过不同的方法来制备,或(II)中的R1,R2和R3的定义与前相同: 例如,当式(I)化合物中的R4是C2-C4烷酰基时,所需的产物可经过通常的Friedel-Crafts酰基化反应来制备,即在大约0℃至反应介质的回流温度,在一种适当的溶剂例如二氯甲烷中,在大约3倍过量的Lewis酸诸如三氯化铝或三溴化铝存在的条件下,把式(II)化合物与两倍过量的具有通式(C1-C3烷基)COY的酰卤反应,其中Y是卤素,最好是氯或溴。当R4是取代了NR5R6的C2-C4烷酰基,其中R5和R6的定义与前相同时,产物可以由式(II)化合物开始,经过相应的卤代酮中间体,即一种式(I)化合物,其中的R4是CO(C1-C3烯基)X,X是卤素,最好是氯或溴;把这适当的卤代酮在室温,在一种合适的溶剂例如乙腈中,在至少一当量碱存在的条件下(碱是用来清除反应中产生的酸(HX)付产物的)与所需的具有通式R5R6NH的胺进行反应而制得。所用的碱可以是一种无机盐例如无水碳酸钾,一种叔胺例如三乙胺,或者就用过量的作为反应物的胺。在R5或R6二者当中有一个是H的情况下,最好是用具有通式R5NHP或R6NHP的经保护的胺,其中P是适当的保护基例如苄基,它在以后可通过催化氢化除去。当R5和R6二者都是H时,最好是用具有通式R2′NH的氨,其中P′是一种保护基诸如叔丁氧羰基。这种情况下可用非碱性氨化试剂的钾盐来和卤代酮反应;去保护基反应可以用例如氯化氢进行酸解来实现,这样可以盐酸盐的形式方便地析离所需的氨基酮产物。如上所述,中间体卤代酮也可经由Friedel-Crafts反应来制得,即把化合物(II)与具有通式X(C1-C3烯基)COY的适当的卤代酰卤进行反应,其中X和Y的定义与前面相同。
例如,当式(I)化合物中的R4是C2-C4烷酰基时,所需的产物可经过通常的Friedel-Crafts酰基化反应来制备,即在大约0℃至反应介质的回流温度,在一种适当的溶剂例如二氯甲烷中,在大约3倍过量的Lewis酸诸如三氯化铝或三溴化铝存在的条件下,把式(II)化合物与两倍过量的具有通式(C1-C3烷基)COY的酰卤反应,其中Y是卤素,最好是氯或溴。当R4是取代了NR5R6的C2-C4烷酰基,其中R5和R6的定义与前相同时,产物可以由式(II)化合物开始,经过相应的卤代酮中间体,即一种式(I)化合物,其中的R4是CO(C1-C3烯基)X,X是卤素,最好是氯或溴;把这适当的卤代酮在室温,在一种合适的溶剂例如乙腈中,在至少一当量碱存在的条件下(碱是用来清除反应中产生的酸(HX)付产物的)与所需的具有通式R5R6NH的胺进行反应而制得。所用的碱可以是一种无机盐例如无水碳酸钾,一种叔胺例如三乙胺,或者就用过量的作为反应物的胺。在R5或R6二者当中有一个是H的情况下,最好是用具有通式R5NHP或R6NHP的经保护的胺,其中P是适当的保护基例如苄基,它在以后可通过催化氢化除去。当R5和R6二者都是H时,最好是用具有通式R2′NH的氨,其中P′是一种保护基诸如叔丁氧羰基。这种情况下可用非碱性氨化试剂的钾盐来和卤代酮反应;去保护基反应可以用例如氯化氢进行酸解来实现,这样可以盐酸盐的形式方便地析离所需的氨基酮产物。如上所述,中间体卤代酮也可经由Friedel-Crafts反应来制得,即把化合物(II)与具有通式X(C1-C3烯基)COY的适当的卤代酰卤进行反应,其中X和Y的定义与前面相同。
上述具有通式(IA)的酮: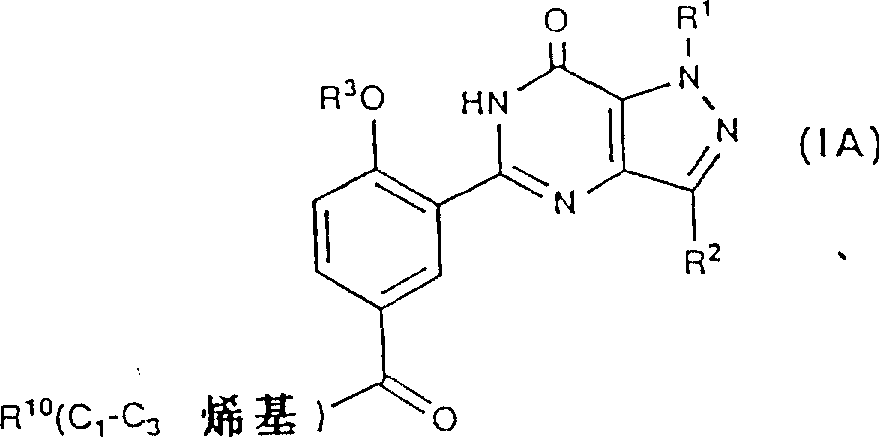 其中R10是H或NR5R6,R1,R2,R3,R5和R6的定义与前面相同,可被还原而形成相应的具有通式(1B)的醇衍生物:
其中R10是H或NR5R6,R1,R2,R3,R5和R6的定义与前面相同,可被还原而形成相应的具有通式(1B)的醇衍生物: 其中的R1,R2,R3和R10的定义与前面相同,所用的还原剂最好是硼氢化钠,反应可在接近室温,在一种合适的溶剂例如乙醇中进行。
其中的R1,R2,R3和R10的定义与前面相同,所用的还原剂最好是硼氢化钠,反应可在接近室温,在一种合适的溶剂例如乙醇中进行。
对于R4是羟甲基,R1,R2和R3的定义与前面相同的式(I)化合物,可以这样来制备,即把式(II)化合物在标准的氯甲基化反应的条件下,例如用多聚甲醛和浓盐酸,在室温至大约120℃范围内的温度下反应,制得具有式(III)的中间体氯甲基衍生物: 然后在大约室温至大约100℃的温度范围内,在一种合适的溶剂诸如乙二醇和二甲亚砜混合物中,用碱金属氢氧化物例如氢氧化钠或氢氧化钾处理而制得。
然后在大约室温至大约100℃的温度范围内,在一种合适的溶剂诸如乙二醇和二甲亚砜混合物中,用碱金属氢氧化物例如氢氧化钠或氢氧化钾处理而制得。
上述具有式(III)的氯甲基衍生物,其中R1,R2和R3的定义与前面相同,是进一步合成其它具有式(I)的化合物的有价值的中间体。例如,在大约室温,在大约一当量碱金属,最好是钠存在的条件下,用C2-或C3-烷醇处理式(III)化合物,即各自得到相应的C2-或C3-烷氧甲基衍生物,相似地,当应用C2-或C3-二醇时,即得到类似的羟基(C2-或C3-烷氧)甲基化合物。后者可通过端基羟基的活化而被进一步转化,即在大约0℃至大约室温的温度范围内,在吡啶溶剂中,用大约10%过量的甲磺酰氯进行常规的甲磺酰化反应,然后把甲磺酸酯与例如具有通式R5R6NH的胺进行反应。反应最好是在反应介质的回流温度,在一种合适的溶剂例如乙腈中,与多达五倍过量的胺存在的条件下进行。像上面讨论过的,当R5或R6中有一个是H,或者二者都是H时,最好是采用保护氨基-去保护基的方案。这样就提供了这样的式(I)化合物,其中的R4是任意取代了OH或NR5R6的(C2-C3)烷氧甲基,其中R1,R2,R3,R5和R6的定义与前面相同。
上述化合物的较高级的同系物,即那些式(I)化合物,其中R4是任意取代了OH或NR5R6的(C2-C3)烷氧乙基,可通过相似的程序,由具有式(III)的2-氯乙基-,2-溴乙基或2-甲磺酰氧乙基类似物来合成,后者又可由相应的2-羟乙基前体通过标准的程序`得。这前体可由R4是溴,R1,R2和R3的定义与前面相同的式(I)化合物(即下式(IV)),通过用正丁基锂的锂-溴交换反应,然后把芳基锂中间体(参看下面内容)与环氧乙烷反应而制得:
具有式(III)的氯甲基中间体也可以被用来制备R4是CH2NR5R6,R1,R2,R3,R5和R6的定义与前面相同的式(I)化合物,即把它与具有通式R5R6NH的适当的胺进行反应(或与带保护基的胺反应,参见上文)。反应最好是在由大约0℃至反应介质的回流温度范围内,在一种合适的溶剂例如2-丁酮中,与3倍过量的胺进行。类似地,R4是(C2-C4烯基)NR5R6的式(I)化合物可以方便地由适当的氯,溴或甲磺酰氧前体来制备,这些前体又可从相应的醇衍生而来(见上面关于2-羟乙基类似物合成的方法)。3-羟丙基和4-羟丁基类似物是通过相应的烯醇的催化氢化反应而得到的,这些烯醇是把上述具有式(IV)的溴化合物各自与烯丙醇或3-丁烯-1-醇置于Heck反应的条件下(参见下文)而制得的。
氯甲基中间体可进一步用来制备相应的甲基衍生物,即R4是CH3,R1,R2和R3与前面定义相同的式(I)化合物。这可以在室温和大约50p.s.i(磅/平方英吋,3.45巴)的氢气压力下,在适当的溶剂诸如乙酸乙酯中,用钯炭催化剂进行催化氢化来完成。类似地,当R4是乙基,正丙基或正丁基时,这类式(I)化合物也可以由相应的氯代烷衍生物制得,而后者又是按上述标准的方法由适当的醇衍生而来的。也可以用别的醇衍生物,例如相应的溴化物或甲磺酸酯。
上述溴衍生物(IV),在合成更进一步的式(I)化合物中是有价值的中间体,它可以在适当的溶剂中由式(II)化合物直接溴化而制得。这可以通过例如在室温,在二甲基甲酰胺中与60%过量的N-溴代丁二酰亚胺反应而完成,也可以在由大约室温至大约100℃的温度范围内,在冰醋酸中与相似过量程度的溴进行反应来完成。另外,(IV)和相应的氟,氯,碘类似物也可以由伯胺(参见下文)经过典型的重氮化-卤化反应系列的程序,例如Schiemann,Sandmeyer和Gatterman反应的程序而制得。
通过应用Heck反应的方法,溴中间体(IV)还可以与丙烯腈或适当的丙烯酰胺或酯衍生物反应而被转化为R4是CH=CH-CN,CH=CHCONR5R6,或CH=CHCO2R7的式(I)化合物,其中R1,R2,R3,R5,R6和R7的定义与前面相同。这反应一般是在反应介质的回流温度,在适当的溶剂诸如乙腈中,在大约0.1当量三芳基膦,最好是三-邻甲苯基膦,和大约0.05当量的醋酸钯(II)存在的条件下,与大约过量50%的烯试剂和叔胺诸如三乙胺这样的反应条件下进行的。得到的丙烯酸酯,如果需要,可用氢氧化钠水溶液,以甲醇为助溶剂,水解成为相应的肉桂酸。还有,所有这些这样合成的烯基产物都可进行催化氢化反应,即在室温和大约15p.s.i.(1.0巴)的氢气压力下,与5%钯炭催化剂一道进行反应而提供R4是CH2CH2CN,CH2CH2CONR5R6或CH2CH2CO2R7的式(I)化合物,其中R1,R2,R3,R5,R6和R7的定义与前面在式(I)化合物中的相同。另一种还原方案,是把丙烯腈衍生物(肉桂腈类似物)用阮内镍在冰醋酸中彻底氢化以提供R4是3-氨基丙基的式(I)化合物,其中R1,R2和R3与前面的定义相同。
较高级的同系物,即那些R4是取代了CN,CONR5R6或CO2R7的C3-C4烷基或C3-C4烯基或者是4-氨基丁基的式(I)化合物,可以由前面所说的烯醇衍生而来,后者是由具有式(IV)的溴化合物与烯丙醇或3-丁烯-1-醇之间发生Heck反应而制得的。为把端基的羟基经过适当的活泼衍生物,例如相应的氯化物,溴化物或甲基磺酸酯,转化为所需的官能团所必需的常规实验程序,对于本专业熟练的技术人员来说是众所周知的,并且也同样适用于2-羟乙基类似物(见前),它们为Heck合成方法提供了另一个选择。R4是CH2CN,CH2CONR5R6,CH2CO2R7或CH2CH2NH2的式(I)化合物,可以通过把具有式(III)的氯甲基中间体,与碱金属氰化物例如氰化钠或氰化钾反应,然后把得到的腈通过标准的转化反应而制得。
作为上述Heck反应方案的一般选择,所需的烯类化合物(和经过催化氢化衍生的烷类化合物)可以用Wittig-Horner方案来制得,其中具有式(I)的醛,即R4是CHO,R1,R2和R3的定义与前相同的式(I)化合物,在适当的碱存在的条件下,与适当的鏻盐或磷酸酯进行反应。醛本身可通过前面叙述的式(IV)的芳基锂衍生物的甲酰基化反应而制得(即用二甲基甲酰胺),并且通过类比,也是R4是C2-C4烯基或C2-C4烷基的式(I)化合物的一种方便的前体,其中R1,R2和R3的定义与前相同。
这种芳基锂中间体在制备R4是CONR5R6或CO2R7的式(I)化合物时也是有用的,其中R1,R2,R3,R5,R6和R7的定义与前相同。例如,把(IV)在-78℃用大约5倍过量的正丁基锂在己烷中的溶液进行锂化,得到的芳基锂在大约-40℃用二氧化碳淬灭,并在大约0℃用水溶液处理,包括小心地酸化到pH值为3,即得到相应的苯甲酸衍生物。酸可在温和的条件下活化,诸如那些用于由氨基酸偶联形成肽键中的程序,并像需要那样转化为酯或酰胺衍生物。例如,用碳化二亚胺/1一羟基苯并三唑活化苯甲酸,并在大约0℃至室温的温度范围内,在适当的溶剂诸如二氯甲烷中,在所需的具有通式R5R6NH的胺或通式R7OH的醇存在的条件下进行化合,即分别产生相应的酰胺或酯。
具有式(IV)的溴中间体也可用来合成R4是苯基或杂环基,其上可任意地被甲基取代的式(I)化合物,其中R1,R2和R3的定义与前相同。当R4是苯基或C-连接的杂环基时,它们可由相应的苯基锂或杂环基锂中间体,经过就地产生的锌酸盐衍生物的钯催化偶联而引入;苯基锂或杂环基锂则又是由杂环或如果必需,由卤代杂环用正丁基锂处理而制得的。例如,苯基锂或杂环基锂(在大约额外1当量正丁基锂存在的条件下反应,以供应吡唑并嘧啶酮底物中的活泼氢原子)在-78℃,在干燥的四氢呋喃中用大约2当量的无水氯化锌处理,然后在室温,用(IV)和钯催化剂,最好是四(三苯基膦)钯(O)处理。反应混合物可被加热到回流温度,并且如果必须,再加入最多大约另外2当量的无水氯化锌。当R4是N连接的杂环基时,反应可以在大约反应介质的回流温度,在适当的溶剂例如二甲基甲酰胺中,用多至5倍过量的适当的杂环化合物,在约10%过量的碱例如无水碳酸钾存在的条件下(用以清除溴化氢付产物),与大约10%过量的铜青铜和大约0.25当量的碘催化剂一起进行反应。
R4是NHSO2NR5R6或NHSO2R8的式(I)化合物,其中R1,R2,R3,R5,R6和R8与前面的定义相同,可以由相应的伯胺合成,后者又是由化合物(II),用例如常规的浓硝酸/浓硫酸混合酸进行硝化,然后把硝基芳烃用常规的实验程序进行催化氢化还原而制得的。反应一般是用等摩尔量的式(I)的伯胺,其中R4是NH2,R1,R2和R3的定义和前面相同,分别和所需的通式为R5R6NSO2卤或R8SO2卤的氨基磺酰卤或烷基磺酰卤(最好是氯化物),在大约0℃至大约室温的温度范围内,在一种合适的溶剂例如二氯甲烷中,在过量叔胺诸如三乙胺或吡啶存在的条件下(用来清除酸付产物)进行的。如果需要,吡啶可方便地既用作碱又用作溶剂,并且反应也可以通过加入大约0.1至0.2当量的4-叔-氨基吡啶诸如4-二甲氨基吡啶进行催化。当R5和R6都是H时,所需的产物也可以通过把伯胺在大约100℃,在一种合适的溶剂例如1,4二噁烷中,与磺酰胺反应而制得。
在把式(II)化合物转化为式(I)化合物时,如果R3是一种在用来引入R4的特定条件下对反应或除去是敏感的基团,则所说的R3基团本身可以在合成的最后一步引入。这样,具有式(II)的酚,其中R3是H,R1和R2的定义与前相同,可以例如通过Pd(O)中介的O-烯丙基类似物的去保护基而制得,即一种式(II)化合物,其中的R3是烯丙基,R1和R2的定义与前相同,这种酚可作为后面引入各种R4取代基的反应的底物。这样,为提供式(I)化合物,最好还必须进行酚基的O-烷基化反应。这可以在反应混合物的回流温度,在一种合适的溶剂例如2-丁酮中,在一种碱诸如无水碳酸钾存在的条件下,用合适的氯代烷,溴代烷或磺酸酯在标准的条件下来完成。另外,烷基化反应也可以在典型的Mitsunobu反应条件下进行。
式(II)化合物可由下面的式(V)化合物其中R1,R2和R3的定义与前面相同,通过应用已知的形成嘧啶环的环化反应而制得。例如,环化反应可以在乙醇-水介质中,在回流温度,也可在过氧化氢存在的条件下,用一种碱诸如氢氧化钠或碳酸钾,处理式(V)化合物来进行。在这些条件下,有关的具有式(VI)的腈,其中R1,R2和R3的定义与前面的定义相同,也可以用作(IV)的前体。
在一种供选择的环化实验程序中,式(II)化合物可在大约1400℃,用多聚磷酸处理(V)而制得。
式(V)和式(VI)化合物可分别由式(VII)和式(VIII)化合物:其中R1和R2的定义与前面相同,通过与下面的式(IX)化合物进行反应而制得,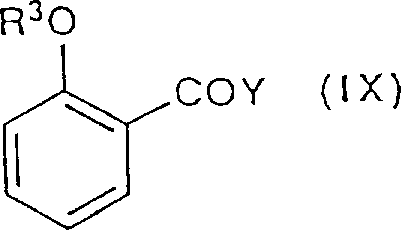 式(IX)中R3和Y的定义与前面相同。
式(IX)中R3和Y的定义与前面相同。
反应一般是在大约0℃至室温的温度范围内,在一种惰性溶剂诸如二氯甲烷中,可在一种催化剂诸如4-二甲氨基吡啶存在的条件下,用过量的(IX),在用过量叔胺诸如三乙胺作为酸付产物(HY)的清除剂的条件下进行的。
式(I)化合物可以更直接地由下面的式(X)化合物:其中的R3,R4和Y的定义与前面相同,当这类酰卤是容易得到的化合物时,通过与(VII)或者(VIII)进行反应,然后像上面叙述过的那样进行关环反应而制得。很清楚,这一供选择的合成途径只有当R4与两步反应的条件相容才是合适的,例如当R4是乙酰基时,由实例17所说明的那样。
具有式(VII)和式(VIII)的氨基吡唑,具有式(IX)和式(X)的酰卤,以及用来把各种R4取代基引入式(II)化合物以便提供式(I)化合物的中间体,如果不能从市场买到,后面又没有叙述制备它们的方法时,可以由容易得到的原料,用合适的试剂和反应条件,用常规的合成实验程序,按照文献中的先例来制得。
一些式(I)化合物,其中的R9的定义与前面相同但不是氢时,可以直接由相应的4-N-未取代的哌嗪类似物,用适当的标准合成实验程序来制备,它是R9是氢的化合物的前体。
所有上面的反应都完全是常规的,并且所必需的试剂和完成这些反应的条件可以容易地通过参考标准的教科书和后面提供的实例而确定下来,对于本专业熟练的人员,虽然还有许多选择和变通办法,使得所有式(I)定义的化合物都可以被制备出来。
本发明化合物的生物活性是由下面的试验方法来测定的。磷酸二酯酶活性
化合物对cGMP和cAMP PDEs的亲合力是通过测定它们的IC50值(即酶的活性有50%被抑制时所需抑制剂的浓度)来评价的。PDE酶基本上是按照W.J.Thompson等人的方法(见Biochem.,1971,10311),由家兔血小板和老鼠的肾中析离的。独立于钙/调钙蛋白(Ca/CAM)的cGMP PDE和抑制了cGMP的cAMP PDE酶是由家免血小板得到的,而与Ca/CAM有关的cGMP PDE(馏份1)则是由老鼠肾中四种主要的PDE酶中析离的。试验是用经修改的,由W.J.Thompson和M.M.Appleman提出的“分批”方法来完成的(参见Biochem.,1979,18,5228)。这些试验的结果表明本发明化合物对两种cGMP PDEs都是强有力的和选择性的抑制剂。
血小板抗凝聚活性
这是通过测定在活体外化合物抑制血小板凝聚作用(由血小板活化因素PAF所诱导)的能力以及增强活体外鸟苷酸环化酶诸如硝普盐和EDRF的激活剂的抗凝聚作用的能力来评价的。洗过的血小板基本是用J.F.Mustard等人(见Methods in Enzymol.,1989,169,3)的方法制备的,凝聚作用是用标准的比浊技术,按G.V.R.Born叙述的方法(见J.Physiol.(Lond),1962,162,67p)测定的。抗高血压活性
这是通过给自生高血压症的老鼠静脉内注射或口服待试验的化合物来评价的,血压是经过插入在清醒的或麻醉的动物颈总动脉中的插管记录的。
为在治疗和预防心绞痛,高血压或充血性心力衰竭时给人类服用,化合物的口服剂量对于一位通常的成年患者(体重70公斤),一般应在每天4-800毫克的范围内。这样,对于一位典型的成年患者,可以每天一次或数次服用一次或多次剂量的,含有2-400毫克活性化合物以及适当的药物上可接受的赋形剂或载体的个别片剂或胶囊剂。对于静脉内,颊内或舌下给药的剂量,按需要典型地应在每一次剂量1-400毫克范围内。在实践中医生将决定对于一位具体患者实际使用的剂量体制,它将随着年龄,体重和特定的患者对药物的反应等因素而变化。上述剂量是在平均情况下的示范值,对一些个别病例使用较高或较低的剂量范围可能有好处,但也应属于本发明范围之内。
为人类服用,式(I)化合物可以单独服用,但一般是与一种药物载体混合服用,后者将按照想用的给药途径和标准的药物实际经验来选择。例如,它们可以含有诸如淀粉或乳糖这类赋形剂的片剂形式或胶囊剂的形式或卵形剂的形式,单独或与赋形剂混合,经口服,颊内,或舌下来用药,也可以含有香料或着色剂的酏剂或悬浮液的形式来用药。这些化合物也可不经肠道注射给药,例如静脉内注射,肌内注射,皮下注射或冠状动脉内注射。对于非肠道用药,最好是使用消毒过的水溶液的形式,其中可包含其它物质,例如盐,诸如甘露糖或葡萄糖这类单糖等,以使得溶液与血液等渗。
这样,本发明提供了一种含有式(I)化合物或它们的药物上可接受的盐,与药物上可接受的稀释剂或载体一起形成的药物构成。
本发明也提供了式(I)化合物或它的药物上可接受的盐,或者包含这两种物质的药物构成在医药方面的应用。
本发明进一步提供了应用式(I)化合物或它的药物上可接受的盐,或包含这两类物质的药物构成,来制造医疗稳定的,不稳定的和变异型(Prinzmetal)的心绞痛,高血压,肺动脉高压,充血性心力衰竭,动脉粥样硬化,中风,末梢血管疾病,以及在经皮穿刺冠状动脉腔内成形术后(即post-PTCA)出现的降低的血管畅通症状,慢性哮喘,支气管炎,过敏性哮喘,过敏性鼻炎,青光眼以及由于内脏功能失调导致的疾病,例如肠过敏性综合症(IBS)这些疾病的药品。
进一步的特点是,本发明提供了医疗或预防哺乳动物(包括人类)发生的稳定的,不稳定的和变异型(Prinzmetal)的心绞痛,高血压,肺动脉高压,充血性心力衰竭,动脉粥样硬化,中风,末梢血管疾病,以及在经皮穿刺冠状动脉腔内成形术后(即post-PTCA)出现的降低的血管畅通症状,慢性哮喘,支气管炎,过敏性哮喘,过敏性鼻炎,青光眼以及由于内脏功能失调导致的疾病,例如肠过敏性综合症(IBS)等疾病的方法,它包括给所说的哺乳动物服用治疗上有效剂量的式(I)化合物或它的药物上可接受的盐,或包含这两类物质的药物构成。
本发明也包括这里公开的,具有式(II),(III)和(IV)的任何一种新的中间体化合物。
本发明化合物及其所用的中间体的合成,可用以下的实例和制备实例来解释,化合物的纯度通常是在Merck Kieselgel 60 F254型号的层析板上进行薄板层析来监测的。1H核磁共振谱是用Nicolet QE-300型或Bruker AC-300型核磁共振仪记录的,并在所有情况下与提出的结构式一致。
实例15-(2-乙氧基-5-哌啶子基乙酰基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
在室温,把哌啶(0.22毫升,0.0022摩尔)加到搅拌着的5-(5-溴乙酰基-2-乙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(制备例8,0.95克,0.0022摩尔)和无水碳酸钾(0.6克,0.0044摩尔)在乙腈中形成的悬浮液中。18小时后混合物在真空下蒸发,残余物溶于水(50毫升)并把溶液用乙酸乙酯提取(3×30毫升)。汇合有机提取液,用食盐水洗涤(3×20毫升),干燥(Na2SO4)并在真空下蒸发,得到的黄色固体在硅胶柱(12克)上层析,用甲醇在二氯甲烷中作梯度洗脱(0-2%甲醇)即得到灰白色的固体,由乙酸乙酯-己烷中重结晶给出灰白色粉末状的标题化合物(0.27克,28%),熔点149-151℃。元素分析实验值:C,66.13;H,6.90;N,15.95。按C24H31N5O5计算应为C,65.88;H,7.14;N,16.01%。
实例2-8
用实例1的实验程序和适当的胺,制备了以下的实例:

 a 0.50 H2O
a 0.50 H2O
实例95-{2-乙氧基-5-[1-羟基-2-(1-哌嗪基)乙基]苯基}-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
把硼氢化钠(0.01克,0.0027摩尔)加到搅拌着的5-[2-乙氧基-5-(1-哌嗪基乙酰基)苯基]-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(0.12克,0.0027摩尔)在乙醇(10毫升)中形成的悬浮液中,得到的溶液在室温搅拌18小时,通过真空下蒸发除去溶剂,残余物悬浮在饱和的碳酸钠水溶液(50毫升)中,并用二氯甲烷提取此混合物(3×20毫升)。汇合有机提取液,干燥(Na2SO4),在真空下蒸发即给出一油状物。与乙醚一起研磨得-白色固体,把它由乙酸乙酯-己烷中重结晶即给出白色粉末状的标题化合物(0.050克,42%),熔点139-140℃。元素分析实验值:C,62.55;H,7.44;N,18.79。按C3H32N6O3计算应为:C,62.71;H,7.32;N,19.08%。
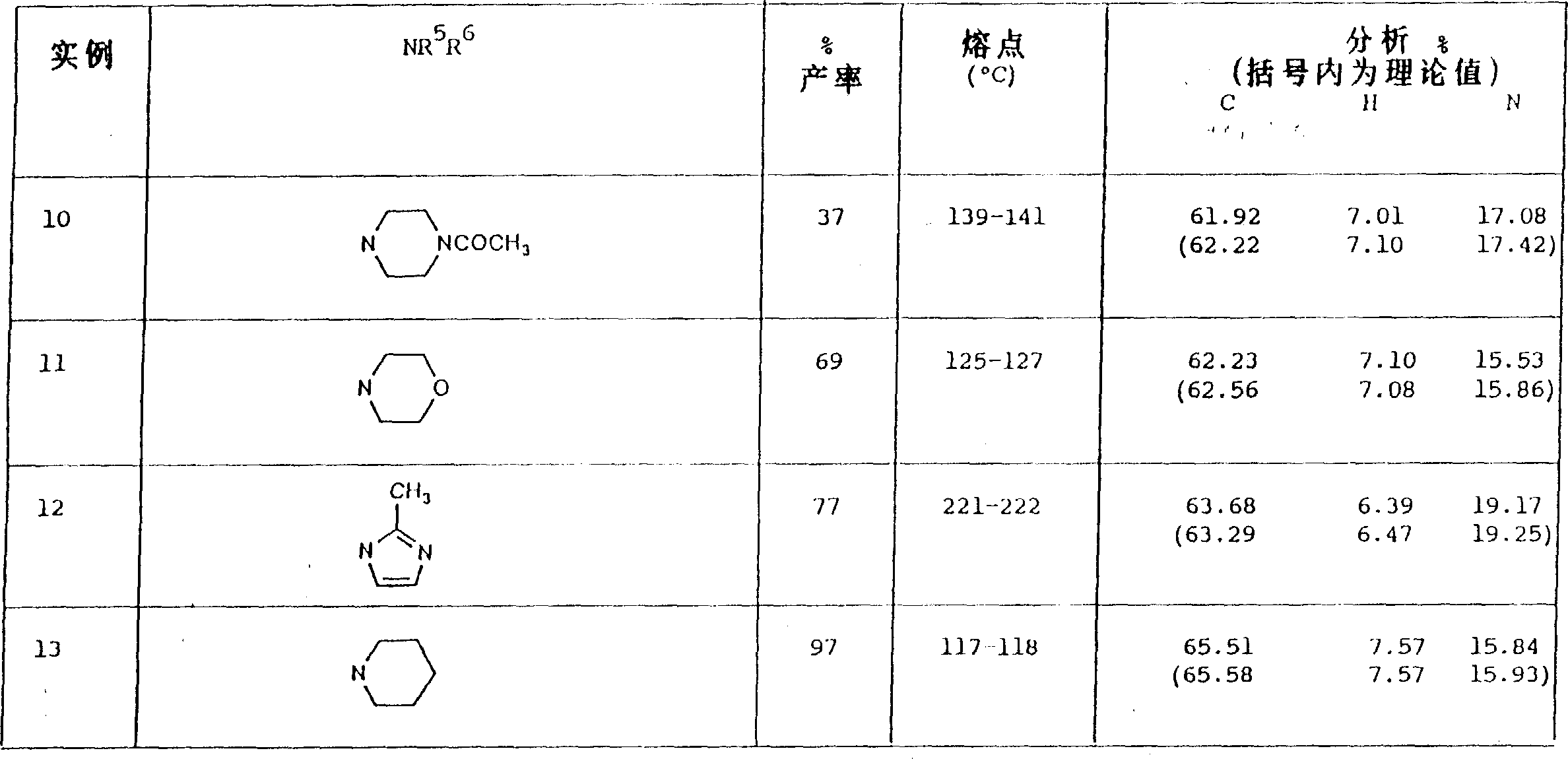
实例10-13
用实例9的实验程序和适当的酮(各自来自实例3,4,5和1),制备了以下的实例: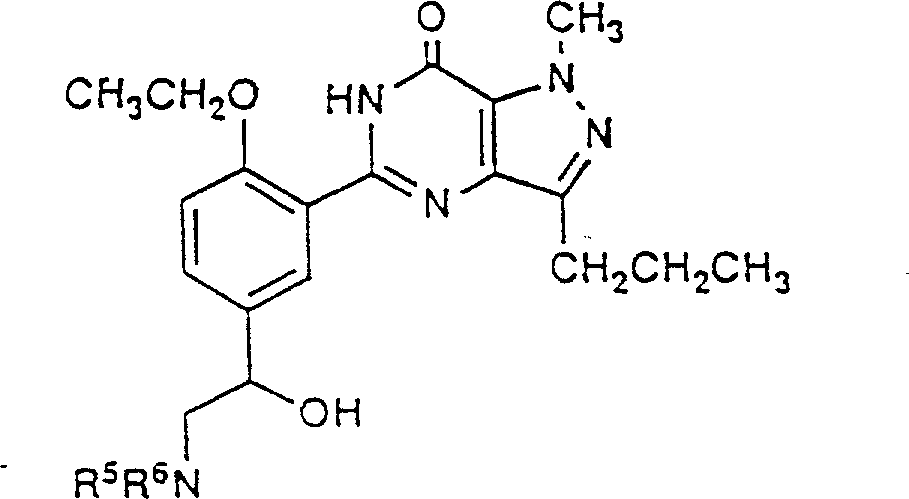
实例141-甲基-5-(5-吗啉代基乙酰基-2-正丙氧基苯基)-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
这化合物是按实例1的实验程序,用吗啉和5-(5-溴乙酰基-2-正丙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(制备例11)制得的,得到的是白色结晶(47%),熔点128-129℃。元素分析实验值C,63.62;H,7.07;N,15.53。按C24H31N5O4计算应为:C,63.56;H,6.89;N,15.44%。实例151-甲基-5-[5-(4-甲基-1-哌嗪基乙酰基)-2-正丙氧基苯基]-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
这化合物是按实例1的实验程序,用4-甲基哌嗪和5-(5-溴乙酰基-2-正丙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(制备例11)制备的,得到的是白色固体(27%)熔点124-125℃。元素分析实验值:C,63.96;H,7.19;N,17.80。按C25H34N6O3计算应为:C,64.36;H,7.34;N,18.01%
实例165-[5-(1-羟基-2-吗啉代基乙基)-2-正丙氧基苯基]-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮。
这化合物是按照实例9的实验程序,由1-甲基-5-(5-吗啉代基乙酰基-2-正丙氧基苯基)-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]-嘧啶-7-酮制备的,得到的是白色固体(28%),熔点1.04-105℃,元素分析实验值:C,62.90;H,7.50;N,15.48。按C24H33N5O4计算应为:C,63.28;H,7.30;N,15.37%。
实例175-(5-乙酰基-2-乙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮。
这标题化合物是按照制备例7的实验程序,由4-(5-乙酰基-2-乙氧基苯甲酰氨基)-1-甲基-3-正丙基吡唑-5-羧酸胺(制备例15)制备的,得到的是白色固体(77%),熔点196-198℃,元素分析实验值:C,64.35;H,6.16;N,15.85。按C19H22N4O3计算应为:C,64.39;H,6.26;N,15.81%。
实例185-(5-溴-2-正丙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮。
把N-溴代丁二酰亚胺(2.6克,0.016摩尔)在二甲基甲酰胺(40毫升)中的溶液在室温滴加到搅拌着的5-(2-正丙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(制备例10,4.0克,0.010摩尔)在二甲基甲酰胺(40毫升)中的溶液中。7小时后在真空下蒸发除去溶剂,残余物悬浮在饱和的碳酸钠水溶液中,并用乙酸乙酯(3×50毫升)提取得到的溶液。汇合有机提取液,干燥(Na2SO4),在真空下蒸发,残余物和醚一起研磨,然后由乙酸乙酯-己烷结晶,给出白色结晶状的标题化合物(3.39克,68%),熔点117-118℃。元素分析实验值:C,53.15;H,5.03;N,13.78。按C18H21BrN4O2计算为:C,53.34;H,5.22;N,13.82%。
实例19(E)-3-(1-甲基-7-氧代-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]-嘧啶-5-基)-4-正丙氧基肉桂酸叔丁酯
往5-(5-溴-2-正丙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(1.0克,0.0025摩尔)和三乙胺(0.38克,0.0038摩尔)在乙腈(2毫升)中的溶液中加入醋酸钯(II)(0.03克,0.00013摩尔),三-邻-甲苯基膦(0.076克,0.00025摩尔)和丙烯酸叔丁酯(0.48克,0.0038摩尔)。把混合物回流加热4小时,然后冷却并在真空下蒸发。残余物被悬浮于水(30毫升)中,用二氯甲烷(3×20毫升)提取。汇合有机提取液,干燥(Na2SO4),在真空下蒸发即给出黄绿色的固体。在硅胶(12克)上层析,用甲醇在二氯甲烷中的溶液进行梯度洗脱(0-2%甲醇),然后由乙酸乙酯-己烷中结晶即给出白色固体状的标题化合物(0.65克,58%)熔点167-168℃。元素分析实验值:C,66,47;H,7.00;N,12.31,按C25H32N4O4计算应为:C,66.35;H,7.13;N,12.38%。
实例20(E)-3-(1-甲基-7-氧代-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-5-基)-4-正丙氧基肉桂酸
往(E)-3-(1-甲基-7-氧代-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-5-基)-4-正丙氧基肉桂酸叔丁酯(0.40克,0.00088摩尔)在甲醇(2.3毫升)中的溶液中加入2N氢氧化钠水溶液(2.28毫升,0.0046摩尔)并把混合物回流加热18小时。在真空下蒸发以除去甲醇,残余物溶于水(25毫升)中,溶液用乙酸乙酯(4×15毫升)提取,分出水相,用盐酸把水溶液酸化到pH值为1,然后用甲醇和乙酸乙酯的混合物提取(3∶97,4×20毫升)。汇合有机提取液,干燥(Na2SO4),在真空下蒸发,然后把残余物由乙酸乙酯中结晶,即给出白色固体状的标题化合物(0.27克,77%)。熔点229-230℃。元素分析实验值:C,63.64;H,5.98;N,14.14。按C21H28N4O4计算应为:C,63.46;H,6.34;N,14.10%。
实例215-(5-溴-2-乙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮。
把溴(0.93克,0.0058摩尔)滴加到搅拌着的5-(2-乙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(制备例7,1.1克,0.00352摩尔)在冰醋酸(20毫升)中的溶液中。反应混合物在100℃搅拌6.5小时并通过真空下蒸发除去溶剂。残余物被溶解在9∶1的甲醇在二氯甲烷中的混合物(50毫升)中,溶液用饱和的碳酸氢钠水溶液(50毫升),水(50毫升)和饱和的食盐水(50毫升)相继洗涤,然后干燥(MgSO4)并在真空下蒸发。残余物在硅胶(15克)上层析,用甲醇和二氯甲烷(1∶99)的混合物洗脱,再由乙腈中结晶,即给出标题化合物(0.62克,45%),熔点157-159℃,元素分析实验值:C,52.41;H,5.25;N,14.01。按C17H19BrN4O2计算应为:C,52.18;H,4.89;N,14.32%。
实例22(E)-4-乙氧基-3-(1-甲基-7-氧代-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-5-基)肉桂酸叔丁酯
这标题化合物是按照实例19的实验程序,由5-(5-溴-2-乙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮制备的,得到白色结晶状固体(31%),熔点179-180℃。元素分析实验值:C,65.83;H,6.90N,12.75。按C24H30N4O4计算应为:C,65.89;H,6.68;N,12.81%。
实例23(E)-4-乙氧基-3-(1-甲基-7-氧代-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-5-基)肉桂酸
这标题化合物是按照实例20的实验程序,由(E)-4-乙氧基-3-(1-甲基-7-氧代-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-5-基)肉桂酸叔丁酯制备的,得到的是白色结晶状物质(66%),熔点234-236℃。元素分析实验值:C,63.01;H,5.59;N,14.62。按照C20H22N4O4计算应为:C,62.82;H,5.80;N,14.65%。
实例243-[4-乙氧基-3-(1-甲基-7-氧代-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-5-基)苯基]丙酸
把(E)-4-乙氧基-3-(1-甲基-7-氧代-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-5-基)肉桂酸(0.426克,0.0011摩尔)在甲醇(28.5毫升),乙酸乙酯(100毫升)和水(1.5毫升)的混合物中形成的溶液,在室温,在氢气压力下与5%钯炭催化剂(0.05克)一起搅拌3小时。过滤除去催化剂,真空下蒸发除去溶剂。残余物由乙酸乙酯-己烷中结晶即给出米色结晶状的标题化合物(0.23克,54%),熔点165-167℃。元素分析实验值:C,62.24;H,6.17;N,14.09。按C20H24N4O4计算应为:C,62.39;H,6.33;N,14.41%。
实例25(E)-4-乙氧基-3-(1-甲基-7-氧代-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-5-基)肉桂酸二甲酰胺
这标题化合物是按照实例19的实验程序,由N,N-二甲基丙烯酰胺和5-(5-溴-2-乙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮制备的,产物由乙酸乙酯-己烷中结晶后为无色结晶(38%),熔点219-221℃。元素分析实验值:C,64.15;H,6.46;N,16.96。按照C22H27N5O3计算应为:C,64.53;H,6.65;N,17.10%。
实例263-[4-乙氧基-3-(1-甲基-7-氧代-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-5-基)苯基]丙酸二甲酰胺
这标题化合物是按照实例24的实验程序,由(E)-4-乙氧基-3-(1-甲基-7-氧代-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-5-基)肉桂酸二甲酰胺制备的,在由乙酸乙酯-己烷中结晶后,得到无色结晶(74%),熔点155-157℃,元素分析实验值:C,64.09;H,7.04;N,16.71。按C22H29N5O3计算应为:C,64.21;H,7.10;N,17.02%。
实例27(E)-4-乙氧基-3-(1-甲基-7-氧代-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-5-基)肉桂腈
这标题化合物是按照实例19的实验程序,由丙烯腈和5-(5-溴-2-乙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H吡唑并[4,3-d]嘧啶-7-酮制备的,得到的是灰白色的结晶(33%)。元素分析实验值:C,65.99;H,5.52;N,19.07。按C20H21N5O2计算应为:C,66.10;H,5.82;N,19.27%。
实例285-[5-(3-氨基丙基)-2-乙氧基苯基]-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
把(E)-4-乙氧基-3-(1-甲基-7-氧代-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-5-基)肉桂腈(0.25克,0.00064摩尔)在冰醋酸(25毫升)中的溶液与阮内镍催化剂(25毫克)在室温,50磅/平方英吋的氢气压下搅拌3小时。得到的混合物过滤并把滤液真空下蒸发,残余物在饱和碳酸钠水溶液(50毫升)和二氯甲烷(30毫升)之间分配,分层后水相进一步用二氯甲烷提取(2×30毫升)。汇合有机相,干燥(Na2OS4),在真空下蒸发,得一棕色固体,把它由己烷-乙酸乙酯中结晶即给出浅黄褐色的晶体(96毫克,38%),熔点115-117℃。元素分析实验值:C,65.29;H,7.35;N,18.66,接C20H27N5O2计算应为:C,65.02;H,7.37;N,18.96%。
实例294-乙氧基-3-(1-甲基-7-氧代-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]-嘧啶-5-基)苯甲酸
在-78℃和干燥氮气的保护下,把正丁基锂(1.53毫升2.5M的在己烷中的溶液,0.0038摩尔)滴加到搅拌着的5-(5-溴-2-乙氧基-苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(0.60克,0.00074摩尔)在干燥的四氢呋喃(25毫升)中形成的溶液中。在-78℃反应3小时后,允许溶液温热到-40℃,并往溶液中通入二氧化碳气泡。得到的溶液允许温热到室温然后倾入水中。用2N盐酸酸化到pH值为3并用9∶1的二氯甲烷和甲醇提取(4×50毫升)。汇合有机提取液,干燥(MgSO4)并在真空下蒸发,给出无色固体。把这固体在硅胶(20克)上进行层析。用甲醇在二氯甲烷中的溶液作梯度洗脱(2-5%甲醇),得到的固体溶解在9∶1的二氯甲烷和甲醇(50毫升)的混合物中;然后把溶液用饱和碳酸钠水溶液(50毫升)洗涤,干燥(MgSO4)并在真空下蒸发,即给出白色粉末状的标题化合物(0.144克,26%),熔点,285-288℃。元素分析实验值:C,60.74;H,5.72;N,15.61。按C18H20N4O4计算应为C,60.66;H,5.66;N15.72%。
实例305-[2-7氧基-5-(4-甲基哌嗪基羰基)苯基]-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
把4-乙氧基-3-(1-甲基-7-氧代-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-5-基)苯甲酸(0.095克,0.00027摩尔),1-甲基哌嗪(0.265克,0.00265摩尔),1-(3-二甲氨基丙基)-3-乙基羰二亚胺盐酸盐(0.077克,0.0004摩尔)和1-羟基苯并三唑(0.054克,0.0004摩尔)在二氯甲烷中(25毫升)所形成的溶液在室温搅拌18小时。反应溶液用水(25毫升)洗涤,干燥(MgSO4)并在真空下蒸发,然后把得到的残余物由乙酸乙酯-己烷中结晶,即给出无色结晶状的标题化合物(0.03克,25%),熔点196-197℃。元素分析实验值:C,63.12;H,6.81;N,18.96。按照C23H30N6O3计算应为:C,62.99;H,6.90;N,19.16%。
实例315-[2-乙氧基-5-(1-咪唑基)苯基]-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
把5-(5-溴-2-乙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(0.20克,0.00051摩尔),咪唑(0.172克,0.0025摩尔),无水碳酸钾(0.077克,0.00056摩尔),铜-青铜(0.036克,0.00057摩尔)和碘(0.015克,0.00012摩尔)在二甲基甲酰胺(10毫升)中所形成的溶液在氮气保护下加热回流4.5小时,冷却后倾入水(50毫升)中,混合物用9∶1的二氯甲烷和甲醇形成的混合溶剂提取(6×50毫升)。汇合提取液,干燥(MgSO4),在真空下蒸发即给出淡棕色的油,把这油状物在硅胶(20克)上进行层析,用二氯甲烷,甲醇和三乙胺(97.8∶2∶0.2)组成的混合溶剂洗脱,得-黄色固体,把它由乙酸乙酯-己烷中重结晶,即得到标题化合物,为一奶油色固体(0.073克,38%),熔点193-194℃,元素分析实验值:C,63.61;H,5.97;N,22.03。按照C20H22N6O2计算应为:C,63.48;H,5.86;N,22.21%。
实例325-[2-乙氧基-5-(1-甲基-2-咪唑基)苯基]-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
在-78℃把正丁基锂(9.6毫升1.6M的在己烷中的溶液,0.0153摩尔)加到搅拌着的1-甲基咪唑(0.628克,0.0077摩尔)在干四氢呋喃(10毫升)中形成的溶液中,并把得到的溶液搅拌0.25小时。加入无水氯化锌(2.08克,0.0153摩尔)在干四氢呋喃(15毫升)中形成的溶液,允许反应混合物温热到室温,然后加入5-(5-溴-2-乙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]-嘧啶-7-酮(1.0克,0.0026摩尔)和四(三苯基膦)钯(O)(0.036克,0.031毫摩尔),反应混合物加热回流18小时。进一步加入无水氯化锌,并把得到的混合物再加热回流60小时,然后冷却;加入甲醇(2毫升);在真空下蒸发除去溶剂,残余物与乙二胺四醋酸二钠二水合物(23.0克,0.0618摩尔)在水(100毫升)中形成的溶液一起加热到100℃反应0.2不时,然后把得到的溶液用饱和碳酸钠水溶液碱化到pH值为8,然后用二氯甲烷提取(6×100毫升)。汇合有机提取液,干燥(Na2SO4)并在真空下蒸发,即给出黄色固体,把它通过在硅胶(13克)上进行层析,用甲醇-二氯甲烷进行梯度洗脱(0-3%甲醇)来提纯然后由乙酸乙酯中结晶,即得到灰白色固体状的标题化合物(0.542克,53%),熔点199-202℃。元素分析实验值:C,64.45;H,6.27;N,21.56。按照C21H24N6O2计算应为C,64.27;H,6.16;N,21.42%。
实例335-[2-乙氧基-5-(2-吡啶基)苯基]-1-甲基-3-正丙基-1,6-5氢-7H-吡唑并[4,3-d]嘧啶-7-酮
这标题化合物是按照实例32中所叙述的实验程序,用5-(5-溴-2-乙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮制备的,得到的是灰白色固体(33%),熔点216-218℃。元素分析实验值:C,67.61;H,5.81;N,17.63。按照C22H23N5O2计算应为:C,67.85;H,5.95;N,17.98%。
实例341-甲基-5-(5-吗啉代基甲基-2-正丙氧基苯基)-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
在0℃,把5-(5-氯甲基-2-正丙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(制备例16,0.60克,0.0016摩尔)在2-丁酮(10毫升)中形成的溶液滴加到搅拌着的吗啉(0.42克,0.0048摩尔)在2-丁酮(40毫升)中的溶液中去,然后把反应溶液回流加热16小时,冷却,在真空下蒸发,残余物悬浮于水(50毫升)中,悬浮液用乙酸乙酯提取(3×20毫升),汇合有机提取液,用食盐水(2×30毫升)洗涤,干燥(Na2SO4),在真空下蒸发,残余物在硅胶(12克)上进行层析,用甲醇在二氯乙烷中的溶液作梯度洗脱(0-2%甲醇),得到一个油状物。它与己烷研磨即固化。由乙酸乙酯-己烷中重结晶,即给出标题化合物,为一无色固体(0.36克,53%),熔点106-107℃。元素分析实验值:C,64.76;H,7.34;N,16.36。按C23H31N5O3计算应为:C,64.92;H,7.34;N,16.46%。
实例351-甲基-5-[5-(4-甲基-1-哌嗪基甲基)-2-正丙氧基苯基)-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
这标题化合物是按照实例34叙述的实验程序,用5-(5-氯甲基-2-正丙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮和1-甲基哌嗪制备的,得到的是一个无色的固体(36%),熔点149-150℃。元素分析实验值:C,65.68;H,7.83;N,19.10。按照C24H34N6O2计算应为:C,65.73;H,7.81;N,19.16%。
实例361-甲基-5-(5-甲基-2-正丙氧基苯基)-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
把5-(5-氯甲基-2-正丙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(0.5范,0.0013摩尔)在乙酸乙醋(50毫升)中形成的溶液与10%钯炭催化剂在50磅/平方英吋的氢气压力和室温的条件下一起搅拌。1小时后,把混合物过滤,把滤液在真空下蒸发即得一淡绿色的固体。在硅胶(4克)上进行层析,用甲醇在二氯甲烷中的溶液作梯度洗脱,得一白色固体,把它由己烷-乙酸乙酯中重结晶,即给出无色针状的标题化合物(0.12克,26%),熔点115-116℃。元素分析实验值:C,66.66;H,7.12;N,16.55。按C19H24N4O2计算应为:C,67.04;H,7.11;N,16.46%。
实例375-(5-羟甲基-2-正丙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
往5-(5-氯甲基-2-正丙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(0.5克,0.0013摩尔)在二甲亚砜(10毫升)中形成的溶液中加入氢氧化钠(0.26克,0.0065摩尔)和乙二醇(0.41克,0.0065摩尔),反应混合物在100℃加热6小时,允许冷却,并倾入水(50毫升)中,然后用乙酸乙醋(3×30毫升)提取水溶液混合物。汇合的提取液过滤,干燥(Na2SO4),并在真空下蒸发,得到的油状物通过在硅胶(6克)上进行层析,用甲醇在二氯甲烷中的溶液作梯度洗脱(0-3%甲醇)来提纯,固体产物由己烷-乙酸乙酯重结晶,即得到标题化合物,为一白色固体(2%)熔点174-175℃。元素分析实验值:C,63.97;H,6.66;N,15.57。按C19H24N4O3计算应为:C,64.03;H,6.79;N,15.72%。
实例385-(5-乙氧甲基-2-正丙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
在1小时内把钠(0.15克,0.0013摩尔)分几部份加入乙醇(40毫升)中,然后往溶液中加入5-(5-氯甲基-2-正丙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(0.5克,0.0013摩尔,在室温反应3天以后,在真空下蒸发以除去溶剂,残余的固体被悬浮于水(50毫升)中,用乙酸乙酯(3×30毫升)提取悬浮液。汇合提取液,干燥(Na2SO4),真空下蒸发,得一绿色固体。在硅胶(6克)上进行层析,用甲醇在二氯甲烷中的溶液作梯度洗脱,得到的产物经己烷-乙酸乙酯混合溶剂结晶后,即得到标题化合物,为一白色固体(0.2克,39%),熔点89-90℃。元素分析实验值:C,65.87;H,7.57;N,14.66。按C21H28N4O3计算应为C,65.60;H,7.34;N,14.57%。
实例395-[5-(2-羟乙氧基甲基)-2-正丙氧基苯基]-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
这化合物是按照实例38的实验程序,由5-(5-氯甲基-2-正丙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮和乙二醇制备的,得到的是一个白色固体(45%),熔点101-102℃。元素分析实验值:C,63.13;H,6.88;N,13.98。按C21H28N4O4计算应为:C,62.98;H,7.05;N,13.99%。
实例401-甲基-5-[5-(2-吗啉代基乙氧甲基)-2-正丙氧基苯基]-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(a)在0℃把甲烷磺酰氯(0.56克,0.0049摩尔)加到搅拌着的5-[5-(2-羟乙氧基甲基)-2-正丙氧基苯基]-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(1.8克,0.0045摩尔)在吡啶(25毫升)中形成的溶液中。在室温反应18小时以后,真空蒸发除去溶剂,残余物在2N盐酸(30毫升)和二氯甲烷(30毫升)之间进行分配。水层分出后再用二氯甲烷(2×30毫升)提取,汇合有机溶液,干燥(Na2SO4),真空下蒸发,得一棕色油状物。在硅胶(12克)上进行层析,用甲醇在二氯甲烷中的溶液作梯度洗脱(0-3%甲醇)得一油状物,把它与己烷一起研磨,然后再由己烷-乙酸乙酯中结晶,即给出所需的甲烷磺酸酯,为一白色结晶物质(0.19克,9%),熔点74-76℃。元素分析实验值:C,55.71;H,6.25;N,11.69。按C22H30N4O6S计算应为:C,55.21;H,6.32;N,11.71%。(b)把吗啉(0.19克,0.0021摩尔)加到上述甲烷磺酸酯,即5-[5-(2-甲烷磺酰氧基乙氧基甲基)-2-正丙氧基苯基]-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(0.20克,0.00042摩尔)在乙腈(25毫升)中形成的溶液中,并把搅拌着的混合物加热回流18小时。在真空下蒸发除去溶剂,残余物溶解于饱和的碳酸钠水溶液中,溶液用乙酸乙酯(3×20毫升)提取。汇合提取液,干燥(Na2SO4),在真空下蒸发,残余物在硅胶(4克)上进行层析,用甲醇在二氯甲烷中的溶液作梯度洗脱(0-2%甲醇),在真空下蒸发适当的馏份,然后由己烷中结晶,即得到标题化合物,为一白色结晶物质(0.098克,48%),熔点65-66℃。元素分析实验值:C,64.17;H,7.09;N,14.96。按C25H35N5O5计算应为:C,63.94;H,7.51;N,14.91%。
实例415-(2-乙氧基-5-甲烷磺酰氨基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
在0℃把甲烷磺酰氯(0.157克,0.00137摩尔)加到搅拌着的5-(5-氨基-2-乙氧基苯基)-1-甲基-3-正丙基-1.6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(0.45克,0.00137摩尔)在干燥吡啶(30毫升)中所形成的溶液中。混合物在室温搅拌18小时然后在真空下蒸发,残余物被悬浮于饱和的碳酸氢钠水溶液(50毫升)中,混合物用二氯甲烷提取(2×30毫升)。汇合有机提取液,用食盐水洗涤(2×30毫升),干燥(Na2SO4)并在真空下蒸发。残余物用醚研磨,在硅胶(12克)上进行层析。用98.5∶1.5的二氯甲烷和甲醇的混合物洗脱,所需产物再由乙酸乙酯-己烷中结晶即得到标题化合物,为一白色粉末状物质(0.32克,58%),熔点205-206℃。元素分析实验值:C,53.63;H,5.66;N,17.24%按C18H23N5O4S计算应为:C,53.32;H,5.72;N,17.27%。
实例425-[2-乙氧基-5-(3-吗啉代基丙基磺酰氨基)苯基]-1-甲基-3-正丙基-1.6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
这标题化合物是按照实例41的实验程序,由5-(5-氨基-2-乙氧基苯基)-1-甲基-3-正丙基-1.6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮和3-吗啉代基丙基磺酰氯制备的。得到的是棕色结晶(14%)。熔点157-159℃。元素分析实验值:C,55.42;H,6.53;N,16.01。按C24H34N6O5S计算应为:C,55.58;H,6.61;N,16.21%。
实例435-[2-乙氧基-5-(4-甲基-1-哌嗪基)磺酰氨基苯基]-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
这标题化合物是按照实例41的实验程序,由4-甲基-1-哌嗪基-磺酰氯和5-(5-氨基-2-乙氧基苯基)-1-甲基-3-正丙基-1.6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮制备的,得到的是一个桔黄色粉末状物质(13%),熔点152-153℃。元素分析实验值:C,54.32;H,6.38;N,19.88。按C22H31N7O4S计算应为:C,53.97;H,6.38;N,20.03%。
实例445-[5-(4-苄基-1-哌嗪基磺酰氨基苯基)-2-乙氧基]-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
把4-苄基-1-哌嗪基-磺酰氯(制备例19,0.9克,0.0029摩尔)加到搅拌着的5-(5-氨基-2-乙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(0.94克,0.0029摩尔),4-二甲氨基吡啶(0.050克,0.00041摩尔)和三乙胺(1.09克,0.0108摩尔)在二氯甲烷(50毫升)中所形成的溶液中。反应溶液在室温搅拌48小时,然后在真空下蒸发。残余物悬浮于饱和的碳酸氢钠水溶液中(50毫升),悬浮液用二氯甲烷(3×30毫升)提取。汇合有机提取液,逐次用饱和碳酸氢钠水溶液(2×20毫升)和食盐水(3×20毫升)洗涤,干燥(Na2SO4),在真空下蒸发。残余物在硅胶(20克)上进行层析,用甲醇在二氯甲烷中的溶液作梯度洗脱(0-4%甲醇),所需的产物由乙酸乙酯-己烷中结晶即得到标题化合物,为一灰白色粉末(0.185克,11%)。元素分析实验值:C,58.30;H,6.20;N,16.80。按C28H35N7O4S;0.5H2O计算应为:C,58.52;H,6.31;N,17.06%。
制备例11-甲基-3-正丙基吡唑-5-羧酸乙酯
把3-正丙基吡唑-5-羧酸乙酯(24.1克,0.132摩尔)(用Chem.Pharm.Bull.,1984,32,1588发表的方法制备)和硫酸二甲酯(16.8克,0.133摩尔)的混合物加热到90℃反应2.5小时。混合物溶解于二氯甲烷中,溶液用碳酸钠水溶液洗涤。分出有机相,干燥(MgSO4),在真空下蒸发即得一固体。在硅胶(300克)上进行层析,用二氯甲烷洗脱,得到产物为一无色油状物(20.4克,79%)。Rf值0.8,(硅胶∶二氯甲烷,甲醇,醋酸;80∶20∶1)。
制备例21-甲基-3-正丙基-吡唑-5-羧酸
把1-甲基-3-正丙基-吡唑-5-羧酸乙酯(20.2克,0.10摩尔)悬浮在6N氢氧化钠水溶液中(50毫升,0.30摩尔)。混合物加热到80℃反应2小时,然后用水(50毫升)稀释并用浓盐酸(25毫升)酸化。过滤即得到淡棕色结晶状的羧酸(12.3克,71%)。熔点150-154℃。元素分析实验值:C,56.99;H,7.25;N,16.90。按C8H12N2O2计算应为:C,57.13;H,7.19;N,16.66%。
制备例31-甲基-4-硝基-3-正丙基吡唑-5-羧酸
维持温度在60℃以下,把1-甲基-3-正丙基吡唑-5-羧酸(12.1克,0.072摩尔)分几部份加到发烟硫酸(13毫升)和发烟硝酸(11毫升)所形成的混合物中。加完后,混合物在60℃加热过夜,然后冷却到室温并倾倒在冰上;过滤即得到硝基吡唑,为白色固体(11.5克,75%),熔点124-127℃。元素分析实验值:C,45.43;H,5.22;N,19.42。按C8H11N3O4计算应为:C,45.57;H,5.20;N,19.71%。
制备例41-甲基-4-硝基-3-正丙基吡唑-5-羧酰胺
1-甲基-4-硝基-3-正丙基吡唑-5-羧酸(11.3克,0.053摩尔)加到亚硫酰氯(50毫升)中。并把得到的混合物加热回流3小时,然后把反应混合物冷却,在真空下蒸发以除去过量的亚硫酰氯。油状残余物被溶解在丙酮(50毫升)中,并把这溶液小心地加到冰(50克)和浓氢氧化铵水溶液(50毫升)组成的混合物中,过滤收集沉淀,即得到吡唑羧酰胺,为一淡黄色固体(8.77克,78%),熔点141-143℃。元素分析实验值:C,45.22;H,5.71;N,26.12。按C8H12N4O3计算应为:C,45.28;H,5.70;N,26.40%。
制备例54-氨基-1-甲基-3-正丙基吡唑-5-羧酰胺
把1-甲基-4-硝基-3-正丙基吡唑-5-羧酰胺(3.45克,16.2毫摩尔)和氯化亚锡二水合物(18.4克,81毫摩尔)一起悬浮于乙醇中,混合物加热回流2小时。得到的溶液冷却到室温,用2N氢氧化钠水溶液碱化到PH值为9,并用二氯甲烷(3×150毫升)提取。汇合有机提取液。干燥(MgSO4)并在真空下蒸发。残余物与醚一起研磨,即给出氨基吡唑,为一灰白色固体(2.77克,94%),熔点98-101℃。元素分析实验值:C,52.84;H,7.81;N,30.38。按C8H14N4O计算应为:C,52.73;H,7.74;N,30.75%。
制备例64-(2-乙氧基苯甲酰氨基)-1-甲基-3-正丙基吡唑-5-羧酰胺
在0℃,把2-乙氧基苯甲酰氯(6.1克,33.0毫摩尔)在二氯甲烷(50毫升)中的溶液加到搅拌着的4-氨基-1-甲基-3-正丙基吡唑-5-羧酰胺(3.0克,16.4毫摩尔),4-二甲氨基吡啶(0.02克,0.164毫摩尔)和三乙胺(3.34克,33.0毫摩尔)在二氯甲烷(50毫升)中所形成的溶液中。得到的反应混合物允许被温热到室温,并继续搅拌2小时。在真空下蒸发除去溶剂。残余物被溶解于19∶1的二氯甲烷和甲醇组成的混合物中(250毫升),然后用1N盐酸(100毫升)洗涤溶液,干燥(MgSO4),在真空下蒸发。粗产物在硅胶(200克)上进行层析,用97∶3的二氯甲烷和甲醇的混合物洗脱,得到粉红色的固体,由乙酸乙酯-己烷中结晶即得到吡唑-5-羧酰胺,为淡粉红色的固体(2.2克,40%),熔点153-155℃。元素分析实验值:C,61.66;H,6.77;N,16.95。按C17H22N4O3计算应为:C,61.80;H,6.71;N,16.96%
制备例75-(2-乙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
把4-(2-乙氧基苯甲酰氨基)-1-甲基-3-正丙基吡唑-5-羧酰胺(223克,0.676摩尔)分部加入氢氧化钠(54克,1.35摩尔)和30%过氧化氢溶液(224毫升)在水(2000毫升)中所形成的溶液中。再加入乙醇(700毫升),把得到的混合物加热回流2.5小时,冷却,然后在真空下蒸发。得到的固体用2N盐酸(380毫升)处理,在外部冷却的条件下,混合物用二氯甲烷(1×700毫升,3×200毫升)提取。汇合的有机提取液逐次用饱和的碳酸钠水溶液(3×400毫升)和食盐水(300毫升)洗涤,然后干燥(Na2SO4)并在真空下蒸发。
残余物在硅胶(1000克)上进行层析,用甲醇在二氯甲烷中的溶液作梯度洗脱(0-1%甲醇),然后把粗产物与乙醚(300毫升)一起研磨,即给出标题化合物,为一无色固体(152.2克,72%),熔点143-146℃。元素分析实验值:C,65.56;H,6.44;N,18.14。按C17H20N4O2计算应为:C,65.36;H,6.45;N,17.94%。
制备例85-(5-溴乙酰基-2-乙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
在0℃,在1小时内把三氯化铝(12.8克,0.096摩尔)分部加入搅拌着的5-(2-乙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(10.0克,0.032摩尔)和溴乙酰溴(5.6毫升,0.064摩尔)在二氯甲烷(150毫升)中所形成的溶液中。在室温反应18小时后,把反应混合物倾入冰和水(400克)中,并把得到的混合物剧烈地搅拌。分出有机相,水相进一步用二氯甲烷(2×100毫升)提取,汇合有机溶液,干燥(Na2SO4),在真空下蒸发,即给出灰白色的固体,把它和醚一道研磨即得到标题化合物。为一白色固体(10.87克,78%)熔点159-160℃。元素分析实验值:C,52.54;H,4.88;N,12.78。按C19H21BrN4O3计算应为:C,52.67;H,4.88;N,12.93%。
制备例91-甲基-4-(2-正丙氧基苯甲酰氨基)-3-正丙基吡唑-5-羧酰胺
这酰胺化合物是按制备例6叙述的实验程序,由2-正丙氧基苯甲酰氯制备的,得到的是一个粉红色固体(63%),熔点148-149℃。元素分析实验值:C,62.97;H,7.00;N,16.29。按C18H24N4O3计算应为:C,2.77;H,7.02;N,16.27%。
制备例101-甲基-5-(2-正丙氧基苯基)-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
把1-甲基-4-(2-正丙氧基苯甲酰氨基)-3-正丙基吡唑-5-羧酰胺(0.34克,0.99摩尔)加到搅拌着的由30%过氧化氢溶液(1毫升),碳酸钾(0.54克,3.92毫摩尔),水(10毫升)和乙醇(5毫升)所组成的混合物中。混合物加热回流38小时然后在真空下蒸发。残余物被悬浮於水(20毫升)中,然后用2N盐酸把悬浮液酸化,并用二氯甲烷(3×20毫升)提取。汇合提取液,干燥(Na2SO4)并在真空下蒸发。得到的残余物在硅胶(6克)上进行层析,用甲醇在二氯甲烷中的溶液进行梯度洗脱(0-1%甲醇),得一油状物,把它用醚逐次研磨,即得到所需的产物,为一白色固体(0.19克,59%),熔点111-114℃。元素分析实验值:C,66.26;H,6.92;N,17.15。按C18H22N4O2计算应为:C,66.23;H,6.80;N,17.17%。
制备例115-(5-溴乙酰基-2-正丙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
在0℃,把三氯化铝(6.0克,0.045摩尔)分部加入搅拌着的1-甲基-5-(2-正丙氧基苯基)-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(5.0克,0.0153摩尔)和2-溴乙酰氯(2.5毫升,0.0303摩尔)在二氯甲烷(100毫升)中所形成的溶液中。允许反应混合物温热到室温并搅拌18小时,加热回流3小时,然后小心地把它加到冰和水(100克)的混合物中。把得到的混合物搅拌1小时后用二氯甲烷(2×50毫升)提取。汇合的有机提取液用食盐水(2×50毫升)洗涤。干燥(Na2SO4),然后在真空下蒸发即给出一灰白色的固体,把它与醚一起研磨即得到标题化合物,为一白色固体(4.1克,60%),取出小量试样由乙酸乙酯-己烷重结晶,可得到纯净产物,熔点136-137℃。元素分析实验值:C,53.82;H,5.24;N,12.57。按C20H23BrN4O3计算应为:C,53.70;H,5.18;N,12.52%。
制备例125-乙酰基-2-乙氧基苯甲酸甲酯
把碘乙烷(16.4克,0.105摩尔)加到搅拌着的5-乙酰基-2-羟基苯甲酸甲酯(10克,51.5毫摩尔)和无水碳酸钾(14.4克,0.104摩尔)在2-丁酮(200毫升)中所形成的混合物中,并把得到的混合物加热回流3天。真空下蒸发除去溶剂,残余物在水(100毫升)和乙酸乙酯(100毫升)之间进行分配。分出水相,进一步用乙酸乙酯提取(4×100毫升)。汇合的有机溶液干燥(Na2SO4)后在真空下蒸发。残余物在硅胶(130克)上进行层析。用甲醇在二氯甲烷中的溶液作梯度洗脱(0-1%甲醇),即给出标题化合物,为一无色结晶(10.15克,89%),熔点50-55℃。元素分析实验值:C,64.88;H,6.38。按C12H14O4计算应为:C,64.85;H,6.35%。
制备例135-乙酰基-2-乙氧基苯甲酸
把由5-乙酰基-2-乙氧基苯甲酸甲酯(9.6克,0.043摩尔),5M氢氧化钠水溶液(44毫升,0.217摩尔),水(80毫升)和1,4-二噁烷(80毫升)所组成的反应混合物在室温搅拌18小时。在真空下蒸发除去溶剂,残余物溶解于水(100毫升)中,并把得到的溶液用浓盐酸酸化到PH值为1。水溶液混合物用乙酸乙酯提取(4×100毫升)。汇合的提取液干燥(Na2SO4)后在真空下蒸发。得到的固体由乙酸乙酯中结晶即得到标题化合物,为一无色固体(5.4克,60%),熔点122-125℃。元素分析实验值:C,63.20;H,5.81。按C11H12O4计算应为:C,63.45;H,5.81%。
制备例145-乙酰基-2-乙氧基苯甲酰氯
把草酰氯(3.66克,0.029摩尔)滴加到搅拌着的5-乙酰基-2-乙氧基苯甲酸(3.0克,0.014摩尔)在二氯甲烷(15毫升)和二甲基甲酰胺(0.1毫升)中所形成的溶液中。在室温反应3小时后在真空下蒸发除去溶剂,残余物用己烷(3×30毫升)进行恒沸蒸馏,即给出标题化合物,它无需进一步提纯即可用于下一步反应中。
制备例154-(5-乙酰基-2-乙氧基苯甲酰氨基)-1-甲基-3-正丙基吡唑-5-羧酰胺
这标题化合物是按照制备例6的实验程序,由5-乙酰基-2-乙氧基苯甲酰氯和4-氨基-1-甲基-3-正丙基吡唑-5-羧酰胺制备的,得到的是一个白色固体(60%),熔点225-227℃。元素分析实验值:C,61.35;H,6.25;N,15.07。按C19H24N4O4计算应为:C,61.28;H,6.50;N,15.04%。
制备例165-(5-氯甲基-2-正丙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
在室温,把1-甲基-5-(2-正丙氧基苯基)-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(0.80克,0.00246摩尔)分部加入搅拌着的浓盐酸(10毫升)中。然后加入多聚甲醛(0.20克,0.00246摩尔),得到的溶液在120℃搅拌22小时。把反应混合物冷却并倾入冰和水(50克)的混合物中,得到的混合物用乙酸乙酯(3×30毫升)提取。汇合有机提取液,干燥(Na2SO4),在真空下蒸发,得到一个白色固体,把它和乙醚一起研磨,然后由乙酸乙酯-己烷中结晶,即给出标题化合物,为一无色结晶状物(0.65克,70%),熔点102-104℃。元素分析实验值:C,60.91;H,6.14;N,14.94。按C19H23N4O2计算应为:C,60.88;H,6.18;N,14.95%。
制备例175-(2-乙氧基-5-硝基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
在0℃,把浓硝酸(0.5毫升)滴加到搅拌着的5-(2-乙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(2.0克,0.0064摩尔)在浓硫酸(10毫升)中形成的溶液中。得到的桔黄色溶液在室温搅拌18小时。然后把反应溶液滴加到搅拌着的冰和水(200克)的混合物中。过滤收集沉淀出来的固体,然后把固体溶解在二氯甲烷(50毫升)中,溶液逐次用食盐水(2×30毫升)和水(30毫升)洗涤,干燥(Na2SO4)并在真空下蒸发。得到一个黄色固体。由乙腈中结晶即得到标题化合物,为黄色针状结晶(1.40克,61%),熔点214-216℃。元素分析实验值:C,57.36;H,5.21;N,19.49。按照C17H19N5O4计算应为:C,57.13;H,5.36;N,19.60%。
制备例185-(5-氨基-2-乙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮
把5-(2-乙氧基-5-硝基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮(0.64克,0.0018摩尔)溶解于乙醇(50毫升)中,把溶液在室温和50磅/平方英吋的氢气压力下与5%钯炭催化剂(0.050克)一起搅拌4小时。过滤反应混合物以除去催化剂。滤液在真空下蒸发,残余物与醚一起研磨,即得到标题化合物,为一灰白色固体(0.56克,95%),熔点147-148℃。元素分析实验值:C,62.63;H,6.60;N,21.57。按C17H21N5O2计算应为:C,62.86;H,6.47;N,21.39%
制备例194-苄基-1-哌嗪基磺酰氯
把1-苄基哌嗪(20.0克,0.114摩尔)在乙腈中(45毫升)形成的溶液加到硫酰氯(28毫升,0.346摩尔)在乙腈中(50毫升)形成的溶液中,混合物加热回流17小时,然后冷却。在真空下蒸发除去溶剂,残余物与醚(2×50毫升)一道研磨,即产生标题化合物(27.8克,89%),它无需进一步提纯即可用于下一步反应中。生物活性下表列出了本发明中的一些化合物的体外活性。
表
体外PDE抑制数据
在钙/钙调节蛋白间的选择性
独立的cGMP PDE和抑制了cGMP的cAMP PDE
安全性质
| 实例号 | IC50(nM) | 选择性比例 | |
| cGMP | cAMP | ||
| 341114151620252930313239 | 2.21.84.91.03.43.73.73.45.53.41.45.3 | 86,00063,00057,00057,00075,00053,00059,00084,00084,00058,00056,00038,00054,000 | 39,09035,00011,63257,00022,05814.32415,94524,70515,27241,42816,47027,14210,188 |
本发明的一些化合物进行过一些毒性试验,对老鼠进行静脉注射的治疗剂量达1毫克/公斤体重,口服剂量达3毫克/公斤体重时,没有观察到不利的急性中毒迹象。对于小鼠,在静脉注射剂量达到100毫克/公斤体重时,仍无死亡现象发生。
Claims (8)
1.一种制备具有下式(1)的化合物及其药物上可接受的盐的方法: 式中:R1是C1-C3烷基;R2是C1-C6烷基;R3是C1-C6烷基;R4是随意选择地被OH,NR5R6,CONR5R6或CO2R7取代的C1-C4烷基;随意选择地被CN,CONR5R6或CO2R7取代的C2-C4烯基;随意选择地被NR5R6取代的C2-C4烷酰基;随意选择地被NR5R6取代的羟基C2-C4烷基;随意选择地被OH或NR5R6取代的(C2-C3烷氧基)C1-C2烷基;CONR5R6;CO2R7;卤素;NHSO2NR5R6;NHSO2R8;吡啶基;或咪唑基其上可随意选择地被甲基取代;R5和R6各自独立地是H或C1-C4烷基,或者和它们所连接的氮原子一起形成哌啶子基,吗啉代基,4-(NR9)-哌嗪基或咪唑基,其中所说的基团可随意选择地被甲基或羟基所取代;R7是H或C1-C4烷基;R8是随意选择地被NR5R6的取代C1-C3烷基;R9是H;随意选择地被苯基取代的C1-C3烷基;羟基C2-C3烷基;或C1-C4烷酰基,其条件是当R1是C1-C3烷基,R2是甲基而R3是C1-C6烷基时,R4不能是C1-C4烷基或卤素,该方法包括把具有式(II)的化合物:其中R1,R2和R3的定义和在本权利要求中前面所限定的相同,对于式(I)化合物
式中:R1是C1-C3烷基;R2是C1-C6烷基;R3是C1-C6烷基;R4是随意选择地被OH,NR5R6,CONR5R6或CO2R7取代的C1-C4烷基;随意选择地被CN,CONR5R6或CO2R7取代的C2-C4烯基;随意选择地被NR5R6取代的C2-C4烷酰基;随意选择地被NR5R6取代的羟基C2-C4烷基;随意选择地被OH或NR5R6取代的(C2-C3烷氧基)C1-C2烷基;CONR5R6;CO2R7;卤素;NHSO2NR5R6;NHSO2R8;吡啶基;或咪唑基其上可随意选择地被甲基取代;R5和R6各自独立地是H或C1-C4烷基,或者和它们所连接的氮原子一起形成哌啶子基,吗啉代基,4-(NR9)-哌嗪基或咪唑基,其中所说的基团可随意选择地被甲基或羟基所取代;R7是H或C1-C4烷基;R8是随意选择地被NR5R6的取代C1-C3烷基;R9是H;随意选择地被苯基取代的C1-C3烷基;羟基C2-C3烷基;或C1-C4烷酰基,其条件是当R1是C1-C3烷基,R2是甲基而R3是C1-C6烷基时,R4不能是C1-C4烷基或卤素,该方法包括把具有式(II)的化合物:其中R1,R2和R3的定义和在本权利要求中前面所限定的相同,对于式(I)化合物
当其中R4为(A):C2-C4烷酰基或羟基C2-C4烷基时,在一种Lewis酸存在的条件下与具有通式(C1-C3烷基)COY而其中Y是卤素的酰卤进行反应,接着随意选择地用还原方法把所得到的酮还原为相应的醇;
当其中R4为(B):C2-C4烷酰基或羟基C2-C4烷基,每个烷基被NR5R6取代,其中R5和R6的定义与本权利要求前面的定义相同时,在一种Lewis酸存在的条件下,与具有通式X(C1-C3烯基)COY而其中X是卤素Y的定义与本权利要求中前面的定义相同的卤代酰卤进行反应,然后把得到的卤代酮与具有通式R5R6NH的胺反应,并接着任意选择地用还原方法将得到的氨基酮还原,
当其中R4为(C):任意选择地被OH,NR5R6,CONR5R6或CO2R7取代的C1-C4烷基,其中R5,R6和R7与本权利要求前面的定义相同或者是溴时:
(i)在氯甲基化反应条件下进行反应,然后把所得到的氯甲基化中间体分别地进行下列反应:
(a)还原反应,或者
(b)与碱金属氢氧化物反应,或者
(c)与具有通式R5R6NH的胺反应;
(ii)在芳香溴化反应条件下进行反应;
当其中R4为(D):2-位被CN,CONR5R6,或CO2R7取代的C2-C4烯基,或2位被CN,CONR5R6,CO2R7或CH2NH2取代的C2-C4烷基,其中R5,R6和R7的定义与本权利要求前面的定义相同时,经上述(C)(ii)的溴衍生物,与适当的α,β-不饱和腈,酰胺或酯分别地进行反应,然后把任何得到的酯水解,把得到的烯基还原并且在腈的情况下进一步或伴随还原腈基成为相应的伯氨基;
当R4为(E):任意选择地被OH或NR5R6取代的(C2-C3烷氧基)C1-C2烷基,其中R5和R6的定义与本权利要求前面的定义相同时,
经上述(C)(i)中所得的氯甲基衍生物,与
(a)C2-或C3-烷醇进行反应,或者与
(b)C2-或C3-二醇进行反应,可随意选择地然后活化羟基以得到甲烷磺酸酯,并且与具有通式R5R6NH的胺进行反应,或者与具有式为R5NHP,R6NHP或P′2NH的被保护的胺进行反应,其中P和P′是适当的氨基保护基;
当R4为(F):CONR5R6或CO2R7,其中R5,R6和R7的定义与本权利要求前面的定义相同时,
可经过上述(C)(ii)中所得的的溴衍生物,与锂-溴交换试剂反应,然后把得到的芳基锂衍生物与二氧化碳反应,并把得到的羧酸以适当活化的形式,分别与具有通式R5R6NH的胺或具有通式R7OH的醇反应而转化为酰胺或酯衍生物;
当R4为(G):NHSO2NR5R6或NHSO2R8,其中R5,R6和R8的定义与本权利要求前面的定义相同时,
可在芳香硝化反应的条件下进行反应,然后把得到的硝基化合物还原为相应的伯胺,并将所说的胺与具有通式R5R6NSO2卤素的氨磺酰卤化物反应,或者与具有通式R8SO2卤素的磺酰卤化物反应,其中卤素最好是氯;或者当R5和R6都是H时,与磺酰胺进行反应;
当R4为(H):吡啶基或咪唑基并可任意选择地被甲基取代时,可经过上述(C)(ii)中所得到的溴衍生物,进行以下的反应:
(i)当R4是吡啶基或任意选择地被取代的C-连接的咪唑基时,在一种钯催化剂存在的条件下,该溴衍生物与适当的吡啶基的或任意选择地被取代的咪唑基的锌酸盐衍生物进行反应,或者
(ii)当R4是N-连接的咪唑基时,在铜-青铜,碘和一种碱存在的条件下该溴衍生物与该咪唑化合物进行反应;
然后在每种情况下,通过可任意选择地析离成或形成产物的药物上可接受的盐。
2.一种制备具有式(I)的化合物或它的药物上可接受的盐的方法,其中R1,R2,R3和R4的定义与前面权利要求1中的定义相同,该方法包括把具有式(II)的化合物,其中R3是H,R1和R2的定义与在权利要求1中所限定的相同,按照权利要求1中所限定的任何一种方法进行反应,然后通过酚基的O-烷基化反应引入R3,并任意选择地进行析离成或形成产物的药物上可接受的盐。
3.一种制备式(I)化合物或它的药物上可接受的盐的方法,其中R1,R2,R3和R4的定义与在权利要求1中所限定的相同,该方法包括把具有式(X)的化合物: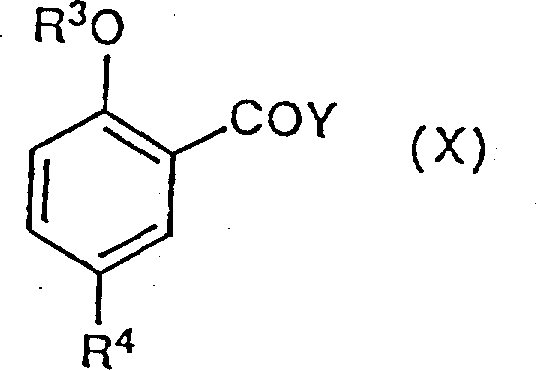
式(X)中Y是氯或溴,R3和R4的定义与在权利要求1中所限定的相同,与一种具有式(VII)或者式(VIII)的氨基吡唑进行反应:
式中R1和R2的定义与在权利要求1中所限定的相同,然后把各自形成的酰胺,可任意选择地在过氧化氢存在的条件下,通过用碱处理而环化,并任意选择地进行析离成或形成该产物的药物上可接受的盐。
4.根据权利要求1的方法,其中:
在(A)中,Y是氯或溴,所应用的Lewis酸是三氯化铝或三溴化铝,所应用的还原剂是硼氢化钠;
在(B)中,X和Y是氧或溴;
在(C)中,
(i)进行氯甲基化是用多聚甲醛和浓盐酸进行反应,以及
(a)进行还原作用是用钯-催化氢化作用进行反应,
(b)碱金属氢氧化物是氢氧化钠,
(c)与R5R6NH的反应用过量所说的胺来进行;
(ii)用溴或N-溴代丁二酰亚胺来进行芳香溴化反应;
在(D)中,各自与适当的α,β-不饱和腈,酰胺或酯的反应是在Heck反应的条件下用三-O-甲苯基膦,醋酸钯(II)和三乙胺来进行,可供选择进行的酯的水解作用是用氢氧化钠水溶液在甲醇中来进行,可供选择进行的烯基的还原作用可用钯-催化的氢化作用来进行,可选择进行的氰基的进一步还原作用或相伴随地还原作用可用阮内镍在冰醋酸中来进行;
在(E)中,
(a)与C2-或C3-烷醇的反应是在1当量金属钠存在的条件下进行
的,或
(b)与C2-或C3-二醇的反应是在1当量金属钠存在的条件下进行
的,羟基转化为它的甲基磺酸酯是用甲磺酰氯在吡啶作为溶
剂的条件下进行的,与R5R6NH的反应是周过量所说的胺来
进行的,
在(F)中,锂-溴交换反应是用正丁基锂进行的,而羧酸的活化是用碳化二亚胺结合1-羟基苯并三唑来进行的;
在(G)中,硝化反应是用浓硝酸结合浓硫酸来完成的,该硝基化合物通过催化氢化来还原,与氨基磺酰氯的反应或与磺酰氯的反应是在过量吡啶或三乙胺存在的条件下进行,可供选择地是在4-二甲氨基吡啶存在的条件下进行的;
在(H)中,
(i)钯催化剂是四个(三苯基膦)-钯(O),而吡啶基或任意选择
地被取代的咪唑基锌酸盐衍生物是由相应的吡啶基的或任
意选择地被取代的咪唑的衍生物和无水氯化锌制得的;
(ii)咪唑化合物是过量存在的,所用的碱是无水碳酸钾。
5.根据权利要求2的方法,其中的O-烷基化反应是用适当的氯代烷,溴代烷或磺酸烷基酯在碳酸钾存在的条件下进行的。
6.根据权利要求1至5中任意一项权利要求所述的方法,其中R1是甲基或乙基,R2是C1-C3烷基;R3是C2-C3烷基;R4是随意选择地被OH,NR5R6,CONR5R6或CO2R7取代的C1-C2烷基;随意选择地被NR5R6取代的乙酰基;被NR5R6取代的羟乙基;随意选择地被OH或NR5R6取代的乙氧基甲基;CH=CHCN;CH=CHCONR5R6;CH=CHCO2R7;CO2H;CONR5R6;Br;NHSO2NR5R6;NHSO2R8;吡啶基;或可任意选择地被甲基取代的咪唑基;R5和R6各自独立地是H,甲基或乙基,或和它们所连接的氮原子一起形成哌啶子基,吗啉代基,4-(NR9)-1-哌嗪基或咪唑基,其中所说的基团可任意选择地被甲基或羟基取代;R7是H或叔丁基;R8是甲基或CH2CH2CH2NR5R6;R9是H,甲基、苄基,2-羟乙基或乙酰基,其条件是当R1是甲基或乙基,R2是甲基以及R3是C2-C3烷基时R4不能是甲基、乙基或Br。
7.根据权利要求6的方法,其中R1是甲基;R2是正丙基;R3是乙基或正丙基;R4是CH2NR5R6,CH2OCH2CH2NR5R6,CH2OCH2CH3,CH2OCH2CH2OH,COCH2NR5R6,CH(OH)CH2NR5R6,CH=CHCON(CH3)2,CH=CHCO2R7,CO2H,CONR5R6,Br,NHSO2NR5R6,NHSO2CH2CH2CH2NR5R6,2-吡啶基,1-咪唑基或1-甲基-2-咪唑基;R5和R6和它们所连接的氮原子一起形成一个哌啶子基,4-羟基哌啶子基,吗啉代基,4-(NR9)-1-哌嗪基或2-甲基-1-咪唑基;R7是H或叔丁基;R9是H,甲基,苄基,2-羟乙基或乙酰基。
8.根据权利要求7的方法,其中生成的式(I)化合物是选自:
5-[2-乙氧基-5-(1-甲基-2-咪唑基)苯基]-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮;
5-[2-乙氧基-5-(4-甲基-1-哌嗪基羰基)苯基]-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮;
5-[5-(4-乙酰基-1-哌嗪基)乙酰基-2-乙氧基苯基]-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]嘧啶-7-酮;
5-(2-乙氧基-5-吗啉代基乙酰基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]-嘧啶-7-酮;以及
5-(5-吗啉代基乙酰基-2-正丙氧基苯基)-1-甲基-3-正丙基-1,6-二氢-7H-吡唑并[4,3-d]-嘧啶-7-酮,
以及它们的药物上可接受的盐。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB9114760.3 | 1991-07-09 | ||
| GB919114760A GB9114760D0 (en) | 1991-07-09 | 1991-07-09 | Therapeutic agents |
| US9114760.3 | 1991-07-09 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1068329A CN1068329A (zh) | 1993-01-27 |
| CN1034503C true CN1034503C (zh) | 1997-04-09 |
Family
ID=10698038
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN92105591A Expired - Fee Related CN1034503C (zh) | 1991-07-09 | 1992-07-09 | 吡唑并嘧啶酮抗心绞痛剂的制备方法 |
Country Status (27)
| Country | Link |
|---|---|
| US (2) | US5272147A (zh) |
| EP (1) | EP0526004B1 (zh) |
| JP (1) | JP2554824B2 (zh) |
| KR (1) | KR950011739B1 (zh) |
| CN (1) | CN1034503C (zh) |
| AT (1) | ATE159019T1 (zh) |
| AU (1) | AU636816B2 (zh) |
| BR (2) | BR9202525A (zh) |
| CA (1) | CA2073226C (zh) |
| CZ (1) | CZ281316B6 (zh) |
| DE (1) | DE69222595T2 (zh) |
| DK (1) | DK0526004T3 (zh) |
| EG (1) | EG19978A (zh) |
| ES (1) | ES2107506T3 (zh) |
| FI (1) | FI103509B (zh) |
| GB (1) | GB9114760D0 (zh) |
| GR (1) | GR3025424T3 (zh) |
| HU (2) | HU220046B (zh) |
| IE (1) | IE922222A1 (zh) |
| IL (1) | IL102368A (zh) |
| MX (1) | MX9204021A (zh) |
| NO (1) | NO180750C (zh) |
| NZ (1) | NZ243472A (zh) |
| PL (2) | PL170615B1 (zh) |
| RU (1) | RU2114113C1 (zh) |
| TW (1) | TW358096B (zh) |
| ZA (1) | ZA925084B (zh) |
Families Citing this family (142)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5250534A (en) * | 1990-06-20 | 1993-10-05 | Pfizer Inc. | Pyrazolopyrimidinone antianginal agents |
| GB9126260D0 (en) * | 1991-12-11 | 1992-02-12 | Pfizer Ltd | Therapeutic agents |
| US5294612A (en) * | 1992-03-30 | 1994-03-15 | Sterling Winthrop Inc. | 6-heterocyclyl pyrazolo [3,4-d]pyrimidin-4-ones and compositions and method of use thereof |
| GB9218322D0 (en) * | 1992-08-28 | 1992-10-14 | Pfizer Ltd | Therapeutic agents |
| GB9301192D0 (en) * | 1993-06-09 | 1993-06-09 | Trott Francis W | Flower shaped mechanised table |
| GB9315017D0 (en) * | 1993-07-20 | 1993-09-01 | Glaxo Lab Sa | Chemical compounds |
| GB9514465D0 (en) * | 1995-07-14 | 1995-09-13 | Glaxo Lab Sa | Chemical compounds |
| US6143746A (en) * | 1994-01-21 | 2000-11-07 | Icos Corporation | Tetracyclic cyclic GMP-specific phosphodiesterase inhibitors, process of preparation and use |
| US5776962A (en) * | 1994-08-03 | 1998-07-07 | Cell Pathways, Inc. | Lactone compounds for treating patient with precancerous lesions |
| US5696159A (en) * | 1994-08-03 | 1997-12-09 | Cell Pathways, Inc. | Lactone compounds for treating patients with precancerous lesions |
| GB9423911D0 (en) | 1994-11-26 | 1995-01-11 | Pfizer Ltd | Therapeutic agents |
| GB9423910D0 (en) | 1994-11-26 | 1995-01-11 | Pfizer Ltd | Therapeutic agents |
| US5656629A (en) * | 1995-03-10 | 1997-08-12 | Sanofi Winthrop, Inc. | 6-substituted pyrazolo (3,4-d)pyrimidin-4-ones and compositions and methods of use thereof |
| US6046206A (en) * | 1995-06-07 | 2000-04-04 | Cell Pathways, Inc. | Method of treating a patient having a precancerous lesions with amide quinazoline derivatives |
| US6232312B1 (en) | 1995-06-07 | 2001-05-15 | Cell Pathways, Inc. | Method for treating patient having precancerous lesions with a combination of pyrimidopyrimidine derivatives and esters and amides of substituted indenyl acetic acides |
| US6060477A (en) * | 1995-06-07 | 2000-05-09 | Cell Pathways, Inc. | Method of treating a patient having precancerous lesions with phenyl cycloamino pyrimidinone derivatives |
| US6200980B1 (en) * | 1995-06-07 | 2001-03-13 | Cell Pathways, Inc. | Method of treating a patient having precancerous lesions with phenyl purinone derivatives |
| US5874440A (en) * | 1995-06-07 | 1999-02-23 | Cell Pathways, Inc. | Method of treating a patient having precancerous lesions with phenyl pyrimidinone derivatives |
| US6262059B1 (en) | 1995-06-07 | 2001-07-17 | Cell Pathways, Inc. | Method of treating a patient having precancerous lesions with quinazoline derivatives |
| US6046216A (en) * | 1995-06-07 | 2000-04-04 | Cell Pathways, Inc. | Method of treating a patient having precancerous lesions with phenyl pyridinone derivatives |
| GB9514473D0 (en) * | 1995-07-14 | 1995-09-13 | Glaxo Lab Sa | Chemical compounds |
| GB9514464D0 (en) | 1995-07-14 | 1995-09-13 | Glaxo Lab Sa | Medicaments |
| EA199800907A1 (ru) | 1996-05-10 | 1999-04-29 | Айкос Корпорейшн | Производные карболина |
| DE69720542D1 (de) * | 1996-05-31 | 2003-05-08 | Mochida Pharm Co Ltd | PYRIDOCARBAZOL DERIVATE DIE EINEN cGMP-PDE INHIBILIERENDEN EFFEKT HABEN |
| GB9612514D0 (en) * | 1996-06-14 | 1996-08-14 | Pfizer Ltd | Novel process |
| HRP970348A2 (en) * | 1996-07-17 | 1998-04-30 | Bayer Ag | New nitropyrazole esters, process for their preparation and their use for the preparation of nytropyrazolamides |
| US6331543B1 (en) | 1996-11-01 | 2001-12-18 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiesterase inhibitors, compositions and methods of use |
| USRE37234E1 (en) * | 1996-11-01 | 2001-06-19 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiestrase inhibitor compounds, compositions and their uses |
| WO1998019672A1 (en) * | 1996-11-01 | 1998-05-14 | Nitromed Inc. | Nitrosated and nitrosylated phosphodiesterase inhibitor compounds, compositions and their uses |
| US5958926A (en) | 1996-11-01 | 1999-09-28 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiesterase inhibitor compounds, compositions and their uses |
| US5874437A (en) * | 1996-11-01 | 1999-02-23 | Nitromed, Inc. | Nitrosated and nitrosylated phosphodiesterase inhibitor compounds, compositions and their uses |
| DE19701277A1 (de) * | 1997-01-16 | 1998-07-23 | Bayer Ag | Verfahren zur Herstellung von 1-Alkyl-pyrazol-5-carbonsäureestern |
| WO1998049166A1 (en) * | 1997-04-25 | 1998-11-05 | Pfizer Limited | PYRAZOLOPYRIMIDINONES WHICH INHIBIT TYPE 5 CYCLIC GUANOSINE 3',5'-MONOPHOSPHATE PHOSPHODIESTERASE (cGMP PDE5) FOR THE TREATMENT OF SEXUAL DYSFUNCTION |
| CA2238283C (en) | 1997-05-30 | 2002-08-20 | Cell Pathways, Inc. | Method for identifying compounds for inhibition of neoplastic lesions, pharmaceutical compositions from such compounds and uses of such compounds and compositions for treating neoplastic lesions |
| US5858694A (en) * | 1997-05-30 | 1999-01-12 | Cell Pathways, Inc. | Method for identifying compounds for inhibition of cancerous lesions |
| GB9722520D0 (en) | 1997-10-24 | 1997-12-24 | Pfizer Ltd | Compounds |
| AU734734B2 (en) * | 1997-10-28 | 2001-06-21 | Vivus, Inc. | Local administration of phosphodiesterase inhibitors for the treatment of erectile dysfunction |
| US5919474A (en) * | 1997-10-28 | 1999-07-06 | Vivus, Inc. | Transurethral administration of vasoactive agents to treat peripheral vascular disease, related vascular diseases, and vascular impotence associated therewith |
| US6037346A (en) * | 1997-10-28 | 2000-03-14 | Vivus, Inc. | Local administration of phosphodiesterase inhibitors for the treatment of erectile dysfunction |
| US6403597B1 (en) | 1997-10-28 | 2002-06-11 | Vivus, Inc. | Administration of phosphodiesterase inhibitors for the treatment of premature ejaculation |
| US6548490B1 (en) | 1997-10-28 | 2003-04-15 | Vivus, Inc. | Transmucosal administration of phosphodiesterase inhibitors for the treatment of erectile dysfunction |
| US6156753A (en) * | 1997-10-28 | 2000-12-05 | Vivus, Inc. | Local administration of type III phosphodiesterase inhibitors for the treatment of erectile dysfunction |
| US6127363A (en) * | 1997-10-28 | 2000-10-03 | Vivus, Inc. | Local administration of Type IV phosphodiesterase inhibitors for the treatment of erectile dysfunction |
| US6472425B1 (en) | 1997-10-31 | 2002-10-29 | Nitromed, Inc. | Methods for treating female sexual dysfunctions |
| IL135462A0 (en) * | 1997-11-12 | 2001-05-20 | Bayer Ag | 2-phenyl substituted imidazotriazinones as phosphodiesterase inhibitors |
| US5852035A (en) * | 1997-12-12 | 1998-12-22 | Cell Pathways, Inc. | Method for inhibiting neoplastic cells and related conditions by exposure to substituted N- arylmethyl and heterocyclmethyl-1H-pyrazolo (3,4-B) quinolin-4-amines |
| US20030040514A1 (en) * | 1999-11-12 | 2003-02-27 | Wyllie Michael G. | Combination effective for the treatment of impotence |
| US6410584B1 (en) | 1998-01-14 | 2002-06-25 | Cell Pathways, Inc. | Method for inhibiting neoplastic cells with indole derivatives |
| US6046199A (en) * | 1998-01-14 | 2000-04-04 | Cell Pathways, Inc. | Method of inhibiting neoplastic cells with tetracyclic pyrido[3,4-B]indole derivatives |
| US5942520A (en) * | 1998-01-27 | 1999-08-24 | Cell Pathways, Inc. | Method for inhibiting neoplastic cells by exposure to substituted N-cycloalkylmethyl-1-H-pyrazolo (3,4-B) quinolone-4 amines |
| TW542719B (en) * | 1998-02-23 | 2003-07-21 | Pfizer Res & Dev | Method of treating impotence due to spinal cord injury |
| US5990117A (en) * | 1998-04-15 | 1999-11-23 | Cell Pathways, Inc. | Method for inhibiting neoplastic cells and related conditions by exposure to quinazoline derivatives |
| EA200000969A1 (ru) * | 1998-04-20 | 2001-06-25 | Пфайзер Инк. | ИНГИБИТОРЫ цГМБ ФДЭ5 НА ОСНОВЕ ПИРАЗОЛПИРИМИДИНОНА ДЛЯ ЛЕЧЕНИЯ ПОЛОВЫХ ДИСФУНКЦИЙ |
| US6087368A (en) * | 1998-06-08 | 2000-07-11 | Bristol-Myers Squibb Company | Quinazolinone inhibitors of cGMP phosphodiesterase |
| DE19829616A1 (de) | 1998-07-02 | 2000-01-05 | Bayer Ag | Verfahren zur Herstellung von 1-Alkyl-pyrazol-5-carbonsäureestern |
| US6180629B1 (en) | 1998-08-14 | 2001-01-30 | Cell Pathways, Inc. | [4,5]-Fused-1,3-disubstituted-1,2-diazine-6-one derivatives with nitrogen containing substitutents in position one for the treatment of neoplasia |
| DE19837067A1 (de) | 1998-08-17 | 2000-02-24 | Bayer Ag | Verfahren zur Herstellung von 1-Alkyl-pyrazol-5-carbonsäureestern |
| US6268372B1 (en) | 1998-09-11 | 2001-07-31 | Cell Pathways, Inc. | Method for inhibiting neoplastic cells and related conditions by exposure to 2,9-disubstituted purin-6-ones |
| US6124303A (en) * | 1998-09-11 | 2000-09-26 | Cell Pathways, Inc. | Method for inhibiting neoplastic cells and related conditions by exposure to 9-substituted 2-(2-N-aloxyphenyl) purin-6-ones |
| US6326379B1 (en) | 1998-09-16 | 2001-12-04 | Bristol-Myers Squibb Co. | Fused pyridine inhibitors of cGMP phosphodiesterase |
| US6200771B1 (en) | 1998-10-15 | 2001-03-13 | Cell Pathways, Inc. | Method of using a novel phosphodiesterase in pharmaceutical screeing to identify compounds for treatment of neoplasia |
| US6130053A (en) * | 1999-08-03 | 2000-10-10 | Cell Pathways, Inc. | Method for selecting compounds for inhibition of neoplastic lesions |
| IL132406A0 (en) * | 1998-10-21 | 2001-03-19 | Pfizer Prod Inc | Treatment of bph with cgmp elevators |
| GB9823102D0 (en) | 1998-10-23 | 1998-12-16 | Pfizer Ltd | Pharmaceutically active compounds |
| GB9823103D0 (en) * | 1998-10-23 | 1998-12-16 | Pfizer Ltd | Pharmaceutically active compounds |
| GB9823101D0 (en) | 1998-10-23 | 1998-12-16 | Pfizer Ltd | Pharmaceutically active compounds |
| UA67802C2 (uk) | 1998-10-23 | 2004-07-15 | Пфайзер Рісьоч Енд Дівелепмент Компані, Н.В./С.А. | Фармацевтична композиція з контрольованим вивільненням інгібітора цгмф фде-5 (варіанти), спосіб її одержання та спосіб лікування еректильної дисфункції |
| KR100353014B1 (ko) * | 1998-11-11 | 2002-09-18 | 동아제약 주식회사 | 발기부전 치료에 효과를 갖는 피라졸로피리미디논 화합물 |
| US6133271A (en) * | 1998-11-19 | 2000-10-17 | Cell Pathways, Inc. | Method for inhibiting neoplastic cells and related conditions by exposure thienopyrimidine derivatives |
| US6187779B1 (en) | 1998-11-20 | 2001-02-13 | Cell Pathways, Inc. | Method for inhibiting neoplastic cells and related conditions by exposure to 2,8-disubstituted quinazoline derivatives |
| US6369092B1 (en) | 1998-11-23 | 2002-04-09 | Cell Pathways, Inc. | Method for treating neoplasia by exposure to substituted benzimidazole derivatives |
| US6034099A (en) * | 1998-11-24 | 2000-03-07 | Cell Pathways, Inc. | Method for inhibiting neoplastic lesions by administering 4-(arylmethylene)- 2, 3- dihydro-pyrazol-3-ones |
| US6486155B1 (en) | 1998-11-24 | 2002-11-26 | Cell Pathways Inc | Method of inhibiting neoplastic cells with isoquinoline derivatives |
| US6077842A (en) * | 1998-11-24 | 2000-06-20 | Cell Pathways, Inc. | Method of inhibiting neoplastic cells with pyrazolopyridylpyridazinone derivatives |
| US6225315B1 (en) | 1998-11-30 | 2001-05-01 | Pfizer Inc | Method of treating nitrate-induced tolerance |
| DK1140941T3 (da) * | 1998-12-23 | 2005-02-14 | Bristol Myers Squibb Pharma Co | Nitrogenholdige heterobicykliske forbindelser som faktor Xa-inhibitorer |
| US6025394A (en) | 1999-01-29 | 2000-02-15 | Cell Pathways, Inc. | Method for treating patients with acne by administering substituted sulfonyl indenyl acetic acids, amides and alcohols |
| US6020379A (en) * | 1999-02-19 | 2000-02-01 | Cell Pathways, Inc. | Position 7 substituted indenyl-3-acetic acid derivatives and amides thereof for the treatment of neoplasia |
| US6428769B1 (en) * | 1999-05-04 | 2002-08-06 | Aradigm Corporation | Acute testosterone administration |
| US7258850B2 (en) * | 1999-05-04 | 2007-08-21 | Aradigm Corporation | Methods and compositions for treating erectile dysfunction |
| US7235625B2 (en) | 1999-06-29 | 2007-06-26 | Palatin Technologies, Inc. | Multiple agent therapy for sexual dysfunction |
| CN1077108C (zh) * | 1999-07-13 | 2002-01-02 | 成都地奥制药集团有限公司 | 用于制备药物昔多芬的前体化合物 |
| IL137429A0 (en) * | 1999-07-28 | 2001-07-24 | Pfizer Prod Inc | Methods and compsitions for treating diseases and conditions of the eye |
| TWI265925B (en) * | 1999-10-11 | 2006-11-11 | Pfizer | Pyrazolo[4,3-d]pyrimidin-7-ones useful in inhibiting type 5 cyclic guanosine 3',5'-monophosphate phosphodiesterases(cGMP PDE5), process and intermediates for their preparation, their uses and composition comprising them |
| CZ20021151A3 (cs) * | 1999-10-11 | 2003-03-12 | Pfizer Inc. | 5-(2-substituovaný-5-heterocyklylsulfonylpyrid-3-yl)-dihydropyrazolo[4,3-d]-pyrimidin-7-ony jako inhibitory fosfodiesterasy |
| US6555547B1 (en) | 2000-02-28 | 2003-04-29 | Cell Pathways, Inc. | Method for treating a patient with neoplasia by treatment with a vinca alkaloid derivative |
| DE10010067A1 (de) * | 2000-03-02 | 2001-09-06 | Bayer Ag | Neue Imidazotriazinone und ihre Verwendung |
| US6569638B1 (en) | 2000-03-03 | 2003-05-27 | Cell Pathways, Inc | Method for screening compounds for the treatment of neoplasia |
| US6271228B1 (en) * | 2000-04-28 | 2001-08-07 | Pfizer Inc. | Blood pressure stabilization during hemodialysis |
| PE20020394A1 (es) | 2000-08-18 | 2002-06-21 | Agouron Pharma | Compuestos de pirazol y composiciones farmaceuticas que los contienen, que modulan y/o inhiben la actividad de erab/hadh2 |
| US6821978B2 (en) | 2000-09-19 | 2004-11-23 | Schering Corporation | Xanthine phosphodiesterase V inhibitors |
| US6548508B2 (en) | 2000-10-20 | 2003-04-15 | Pfizer, Inc. | Use of PDE V inhibitors for improved fecundity in mammals |
| US6784185B2 (en) | 2001-03-16 | 2004-08-31 | Pfizer Inc. | Pharmaceutically active compounds |
| JP2004527510A (ja) * | 2001-03-16 | 2004-09-09 | ファイザー インコーポレイテッド | CGMPPDE阻害剤としてのピラゾロ[4,3−d]ピリミジノン化合物 |
| GB0107751D0 (en) * | 2001-03-28 | 2001-05-16 | Pfizer Ltd | Pharmaceutically active compounds |
| US6794387B2 (en) | 2001-03-28 | 2004-09-21 | Pfizer Inc. | Pharmaceutically active compounds |
| EP1392314B1 (de) * | 2001-05-09 | 2006-12-20 | Bayer HealthCare AG | NEUE VERWENDUNG VON 2-[2-Ethoxy-5-(4-methyl-piperazin-1-sulfonyl)-phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-on |
| HUP0202719A3 (en) * | 2001-08-21 | 2006-01-30 | Pfizer Prod Inc | Pharmaceutical compositions for the treatment of female sexual dysfunctions |
| US6479493B1 (en) | 2001-08-23 | 2002-11-12 | Cell Pathways, Inc. | Methods for treatment of type I diabetes |
| EP1312363A1 (en) * | 2001-09-28 | 2003-05-21 | Pfizer Products Inc. | Methods of treatment and kits comprising a growth hormone secretagogue |
| WO2003042216A1 (en) | 2001-11-09 | 2003-05-22 | Schering Corporation | Polycyclic guanine derivative phosphodiesterase v inhibitors |
| DE60222931T2 (de) | 2001-12-13 | 2008-07-10 | Asubio Pharma Co., Ltd. | Pyrazolopyrimidinonderivate mit pde7-hemmender wirkung |
| GB0202282D0 (en) | 2002-01-31 | 2002-03-20 | Pfizer Ltd | Treatment of male sexual dysfunction |
| US7342884B2 (en) * | 2002-03-13 | 2008-03-11 | Harmonic, Inc. | Method and apparatus for one directional communications in bidirectional communications channel |
| US7276529B2 (en) | 2002-03-20 | 2007-10-02 | Celgene Corporation | Methods of the treatment or prevention of exercise-induced asthma using (+)-2-[1-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4-acetylaminoisoindoline-1,3-dione |
| US7208516B2 (en) | 2002-03-20 | 2007-04-24 | Celgene Corporation | Methods of the treatment of psoriatic arthritis using (+)-2-[1-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4-acetylaminoisoindoline-1,3-dione |
| US7893101B2 (en) | 2002-03-20 | 2011-02-22 | Celgene Corporation | Solid forms comprising (+)-2-[1-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4-acetylaminoisoindoline-1,3-dione, compositions thereof, and uses thereof |
| US6962940B2 (en) | 2002-03-20 | 2005-11-08 | Celgene Corporation | (+)-2-[1-(3-Ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4-acetylaminoisoindoline-1,3-dione: methods of using and compositions thereof |
| DE10232113A1 (de) | 2002-07-16 | 2004-01-29 | Bayer Ag | Vardenafil Hydrochlorid Trihydrat enthaltende Arzneimittel |
| GB0219961D0 (en) | 2002-08-28 | 2002-10-02 | Pfizer Ltd | Oxytocin inhibitors |
| EP1572213A1 (en) * | 2002-11-26 | 2005-09-14 | Pfizer Products Inc. | Method of treatment of transplant rejection |
| US7323462B2 (en) | 2002-12-10 | 2008-01-29 | Pfizer Inc. | Morpholine dopamine agonists |
| KR20050085563A (ko) | 2002-12-13 | 2005-08-29 | 워너-램버트 캄파니 엘엘씨 | 하부요로증상을 치료하기 위한 알파-2-델타 리간드 |
| EP1599468B1 (en) | 2003-01-14 | 2007-10-03 | Arena Pharmaceuticals, Inc. | 1,2,3-trisubstituted aryl and heteroaryl derivatives as modulators of metabolism and the prophylaxis and treatment of disorders related thereto such as diabetes and hyperglycemia |
| US20040242457A1 (en) * | 2003-05-27 | 2004-12-02 | Ashby Charles R. | Use of anti-glaucoma drugs to treat visual defects associated with the use of a GABAergic agent |
| JP2006219373A (ja) | 2003-06-13 | 2006-08-24 | Daiichi Asubio Pharma Co Ltd | Pde7阻害作用を有するピリジニルピラゾロピリミジノン誘導体 |
| DK1644021T3 (da) * | 2003-06-13 | 2012-10-29 | Ironwood Pharmaceuticals Inc | Fremgangsmåder og sammensætninger til behandlingen af gastrointestinale sygdomme |
| EP2287165A3 (en) | 2003-07-14 | 2011-06-22 | Arena Pharmaceuticals, Inc. | Fused-aryl and heteroaryl derivatives as modulators of metabolism and the prophylaxis and treatment of disorders related thereto |
| US7291640B2 (en) * | 2003-09-22 | 2007-11-06 | Pfizer Inc. | Substituted triazole derivatives as oxytocin antagonists |
| US7649002B2 (en) | 2004-02-04 | 2010-01-19 | Pfizer Inc | (3,5-dimethylpiperidin-1yl)(4-phenylpyrrolidin-3-yl)methanone derivatives as MCR4 agonists |
| DE102004023069A1 (de) * | 2004-05-11 | 2005-12-08 | Bayer Healthcare Ag | Neue Darreichungsformen des PDE 5-Inhibitors Vardenafil |
| DOP2006000009A (es) | 2005-01-13 | 2006-08-15 | Arena Pharm Inc | Procedimiento para preparar eteres de pirazolo [3,4-d] pirimidina |
| DE102005001989A1 (de) * | 2005-01-15 | 2006-07-20 | Bayer Healthcare Ag | Intravenöse Formulierungen von PDE-Inhibitoren |
| GT200600042A (es) * | 2005-02-10 | 2006-09-27 | Aventis Pharma Inc | Compuestos de bis arilo y heteroarilo sustituido como antagonistas selectivos de 5ht2a |
| DE102005009240A1 (de) * | 2005-03-01 | 2006-09-07 | Bayer Healthcare Ag | Arzneiformen mit verbesserten pharmakokinetischen Eigenschaften |
| DE102005009241A1 (de) * | 2005-03-01 | 2006-09-07 | Bayer Healthcare Ag | Arzneiformen mit kontrollierter Bioverfügbarkeit |
| ATE412648T1 (de) | 2005-03-21 | 2008-11-15 | Pfizer Ltd | Substituierte triazolderivate als oxytocinantagonisten |
| US20070010525A1 (en) * | 2005-06-27 | 2007-01-11 | Meyer Jackson | Method and compositions for modulating neuropeptide hormone secretion |
| EP1917257A1 (en) * | 2005-08-10 | 2008-05-07 | Pfizer Limited | Substituted triazole derivatives as oxytocin antagonists |
| WO2007039075A2 (en) * | 2005-09-29 | 2007-04-12 | Bayer Healthcare Ag | Pde inhibitors and combinations thereof for the treatment of urological disorders |
| GB0601951D0 (en) | 2006-01-31 | 2006-03-15 | Novartis Ag | Organic compounds |
| EP2167057A1 (en) * | 2007-06-13 | 2010-03-31 | Bayer HealthCare AG | Pde inhibitors for the treatment of hearing impairment |
| AU2008326381B2 (en) * | 2007-11-21 | 2014-10-23 | Decode Genetics Ehf | Biaryl PDE4 inhibitors for treating inflammation |
| EP2674417A3 (en) * | 2007-11-21 | 2014-04-09 | Decode Genetics EHF | Biaryl PDE4 inhibitors for treating inflammation |
| KR100963644B1 (ko) * | 2007-11-23 | 2010-06-15 | 한국과학기술연구원 | 피라졸로피리미딘온 유도체 및 그의 제조방법 |
| GB0903493D0 (en) | 2009-02-27 | 2009-04-08 | Vantia Ltd | New compounds |
| AU2011305525B2 (en) | 2010-09-22 | 2016-08-18 | Arena Pharmaceuticals, Inc. | Modulators of the GPR119 receptor and the treatment of disorders related thereto |
| CN102134242B (zh) * | 2011-01-21 | 2013-08-28 | 浙江大德药业集团有限公司 | 一种用于治疗阳痿的快速长效的化合物 |
| US9402877B2 (en) | 2011-11-04 | 2016-08-02 | Xion Pharmaceuticals Corporation | Methods and compositions for oral administration of melanocortin receptor agonist compounds |
| EP3157520B1 (en) | 2014-06-23 | 2019-09-04 | Celgene Corporation | Apremilast for the treatment of a liver disease or a liver function abnormality |
| CN116850181A (zh) | 2015-01-06 | 2023-10-10 | 艾尼纳制药公司 | 治疗与s1p1受体有关的病症的方法 |
| ES2929526T3 (es) | 2015-06-22 | 2022-11-29 | Arena Pharm Inc | Sal cristalina de L-arginina del ácido (R)-2-(7-(4-ciclopentil-3-(trifluorometil)benciloxi)-1,2,3,4-tetrahidrociclo-penta[b]indol-3-il)acético para su uso en trastornos asociados al receptor S1P1 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0201188A2 (en) * | 1985-04-05 | 1986-12-17 | Warner-Lambert Company | 5-Substituted pyrazolo[4,3-d] pyrimidine-7-ones, process for preparing the compounds and pharmaceutical compositions comprising the compounds |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CH367510A (de) * | 1957-11-27 | 1963-02-28 | Ciba Geigy | Verfahren zur Herstellung neuer Sulfonamide |
| NL6914072A (zh) * | 1969-09-17 | 1971-03-19 | ||
| US4052390A (en) * | 1973-06-12 | 1977-10-04 | May & Baker Limited | Azapurinones |
| GB1561345A (en) * | 1976-10-22 | 1980-02-20 | May & Baker Ltd | 8 - azapuring - 6 - ones |
| CA1303037C (en) * | 1987-02-02 | 1992-06-09 | Smith Kline & French Laboratories Limited | Purinone derivatives as bronchodilators vasodilators and anti-allergic agents |
| GB8809481D0 (en) * | 1988-04-21 | 1988-05-25 | Smith Kline French Lab | Chemical compounds |
| DE68908786T2 (de) * | 1988-06-16 | 1994-03-17 | Smith Kline French Lab | Condensierte Pyrimidinderivate, Verfahren und Zwischenprodukte zu ihrer Herstellung und diese enthaltende pharmazeutische Zubereitungen. |
| GB8814352D0 (en) * | 1988-06-16 | 1988-07-20 | Smith Kline French Lab | Chemical compounds |
| US5075310A (en) * | 1988-07-01 | 1991-12-24 | Smith Kline & French Laboratories, Ltd. | Pyrimidone derivatives as bronchodilators |
| US4923874A (en) * | 1988-07-21 | 1990-05-08 | G. D. Searle & Co. | Use of 8-azapurin-6-one derivatives for control of hypertension |
| GB8817651D0 (en) * | 1988-07-25 | 1988-09-01 | Smith Kline French Lab | Chemical compounds |
| AU626575B2 (en) * | 1988-08-23 | 1992-08-06 | Rembrandt, Astrid Diana | The holoscopic trioptical transceiver |
| GB8827988D0 (en) * | 1988-11-30 | 1989-01-05 | Smith Kline French Lab | Chemical compounds |
| US5250534A (en) * | 1990-06-20 | 1993-10-05 | Pfizer Inc. | Pyrazolopyrimidinone antianginal agents |
| GB9013750D0 (en) * | 1990-06-20 | 1990-08-08 | Pfizer Ltd | Therapeutic agents |
-
1991
- 1991-07-09 GB GB919114760A patent/GB9114760D0/en active Pending
-
1992
- 1992-06-12 US US07/897,735 patent/US5272147A/en not_active Expired - Lifetime
- 1992-06-30 IL IL10236892A patent/IL102368A/en not_active IP Right Cessation
- 1992-07-02 DE DE69222595T patent/DE69222595T2/de not_active Expired - Lifetime
- 1992-07-02 EP EP92306137A patent/EP0526004B1/en not_active Expired - Lifetime
- 1992-07-02 DK DK92306137.8T patent/DK0526004T3/da active
- 1992-07-02 AT AT92306137T patent/ATE159019T1/de not_active IP Right Cessation
- 1992-07-02 ES ES92306137T patent/ES2107506T3/es not_active Expired - Lifetime
- 1992-07-04 TW TW081105324A patent/TW358096B/zh not_active IP Right Cessation
- 1992-07-06 CA CA002073226A patent/CA2073226C/en not_active Expired - Fee Related
- 1992-07-07 CZ CS922109A patent/CZ281316B6/cs not_active IP Right Cessation
- 1992-07-07 PL PL92295199A patent/PL170615B1/pl not_active IP Right Cessation
- 1992-07-07 PL PL92314091A patent/PL170893B1/pl not_active IP Right Cessation
- 1992-07-08 NZ NZ243472A patent/NZ243472A/en not_active IP Right Cessation
- 1992-07-08 ZA ZA925084A patent/ZA925084B/xx unknown
- 1992-07-08 KR KR1019920012115A patent/KR950011739B1/ko not_active IP Right Cessation
- 1992-07-08 MX MX9204021A patent/MX9204021A/es unknown
- 1992-07-08 BR BR929202525A patent/BR9202525A/pt not_active Application Discontinuation
- 1992-07-08 HU HU9202267A patent/HU220046B/hu not_active IP Right Cessation
- 1992-07-08 FI FI923157A patent/FI103509B/fi active
- 1992-07-08 NO NO922685A patent/NO180750C/no not_active IP Right Cessation
- 1992-07-08 RU SU5052394A patent/RU2114113C1/ru not_active IP Right Cessation
- 1992-07-08 IE IE222292A patent/IE922222A1/en not_active IP Right Cessation
- 1992-07-08 AU AU19545/92A patent/AU636816B2/en not_active Ceased
- 1992-07-09 CN CN92105591A patent/CN1034503C/zh not_active Expired - Fee Related
- 1992-07-09 EG EG37692A patent/EG19978A/xx active
- 1992-07-09 JP JP4182530A patent/JP2554824B2/ja not_active Expired - Fee Related
-
1993
- 1993-07-23 US US08/096,743 patent/US5426107A/en not_active Expired - Lifetime
-
1995
- 1995-05-03 HU HU95P/P00119P patent/HU210870A9/hu unknown
-
1996
- 1996-08-13 BR BR1100029-5A patent/BR1100029A/pt active IP Right Grant
-
1997
- 1997-11-19 GR GR970403069T patent/GR3025424T3/el unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0201188A2 (en) * | 1985-04-05 | 1986-12-17 | Warner-Lambert Company | 5-Substituted pyrazolo[4,3-d] pyrimidine-7-ones, process for preparing the compounds and pharmaceutical compositions comprising the compounds |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1034503C (zh) | 吡唑并嘧啶酮抗心绞痛剂的制备方法 | |
| RU2096411C1 (ru) | Производные бензимидазолона, смеси их изомеров или их кислотно-аддитивные соли в качестве антагониста рецептора 5-ht*00i*00a и 5-нт*002 | |
| US5719283A (en) | Intermediates useful in the synthesis of pyrazolopyrimidinone antianginal agents | |
| EP0748800B1 (en) | Pyrimidinedione, pyrimidinetrione, triazinedione derivatives as alpha-1-adrenergic receptor antagonists | |
| KR940006628B1 (ko) | 피라졸로피리미디논 안지나 치료제 | |
| RU2451015C2 (ru) | Производные 1,2,4-триазола в качестве ингибиторов сигма рецептора | |
| US5783572A (en) | Quinoxalinedione NMDA receptor antagonists | |
| CN1036569A (zh) | 制备可抑制5-脂氧合酶的4-(4-苯基-1-哌嗪基)苯基衍生物的方法 | |
| EP1268466B1 (en) | Decahydro-isoquinolines | |
| CN1333767A (zh) | 作为消炎剂的芳香族杂环化合物 | |
| EP0628032A1 (en) | Quinazolinone antianginal agents | |
| AU2003223953A1 (en) | Monocyclic aroylpyridinones as antiinflammatory agents | |
| JPS6339875A (ja) | ピリミジン誘導体 | |
| US5128343A (en) | Derivatives of pyrimidinyl-piperazinyl-alkyl azoles with anxiolytic and/or tranquilizing activity | |
| SE465928B (sv) | 1,2,4-triazolonfoereningar med antidepressiv aktivitet, foerfarande foer framstaellning av dessa och en farmaceutisk komposition | |
| PL190721B1 (pl) | Pochodna alkiloaminobenzotiazolu i alkiloaminobenzoksazolu, sposób ich wytwarzania oraz kompozycja farmaceutyczna | |
| US4950670A (en) | 6-phenyl-3-(piperazinyalalkyl)-2,4(1H,3H)-pyrimidinedione derivatives, their preparation and their application in therapy | |
| EP0650964A1 (en) | 1 2H-1-benzopyran-2-one-8-yl -piperazine derivatives | |
| JP2011515359A (ja) | 治療用トリアゾールアミド誘導体 | |
| US4472400A (en) | Triazoloquinazolones having antihistaminic and bronchospasmolytic activity | |
| CN1075480A (zh) | 新型芳基羰基氨基烷基-二氢-氧代吡啶类化合物及其生产与应用 | |
| JPH07258233A (ja) | 新規な、アミノアルキルベンゾオキサゾリノン及びベンゾチアゾリノン、それらの製造方法及びそれらを含有する製薬学的組成物 | |
| CN1079789C (zh) | 新的羟肟酸衍生物,包含它们的药物组合物及其制法 | |
| JPH02264750A (ja) | 2―アミノペンタン二酸の新規の誘導体、それらの製造方法及び中間体、それらの薬剤としての使用並びにそれらを含有する組成物 | |
| US4588725A (en) | 2-piperazinyl-quinazoline derivatives and pharmaceutical compositions containing them |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C06 | Publication | ||
| PB01 | Publication | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C15 | Extension of patent right duration from 15 to 20 years for appl. with date before 31.12.1992 and still valid on 11.12.2001 (patent law change 1993) | ||
| OR01 | Other related matters | ||
| C17 | Cessation of patent right | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 19970409 |