CN1234051A - 有含聚二有机硅氧烷脲的组分的聚合物混合物 - Google Patents
有含聚二有机硅氧烷脲的组分的聚合物混合物 Download PDFInfo
- Publication number
- CN1234051A CN1234051A CN97199049A CN97199049A CN1234051A CN 1234051 A CN1234051 A CN 1234051A CN 97199049 A CN97199049 A CN 97199049A CN 97199049 A CN97199049 A CN 97199049A CN 1234051 A CN1234051 A CN 1234051A
- Authority
- CN
- China
- Prior art keywords
- polydiorganosiloxane
- mixture
- urea
- component
- residue
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D183/00—Coating compositions based on macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing silicon, with or without sulfur, nitrogen, oxygen, or carbon only; Coating compositions based on derivatives of such polymers
- C09D183/10—Block or graft copolymers containing polysiloxane sequences
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L83/00—Compositions of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon only; Compositions of derivatives of such polymers
- C08L83/10—Block- or graft-copolymers containing polysiloxane sequences
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J183/00—Adhesives based on macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing silicon, with or without sulfur, nitrogen, oxygen, or carbon only; Adhesives based on derivatives of such polymers
- C09J183/10—Block or graft copolymers containing polysiloxane sequences
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2666/00—Composition of polymers characterized by a further compound in the blend, being organic macromolecular compounds, natural resins, waxes or and bituminous materials, non-macromolecular organic substances, inorganic substances or characterized by their function in the composition
- C08L2666/02—Organic macromolecular compounds, natural resins, waxes or and bituminous materials
- C08L2666/04—Macromolecular compounds according to groups C08L7/00 - C08L49/00, or C08L55/00 - C08L57/00; Derivatives thereof
Abstract
提供一种混合物,它包括(a)除聚二有机硅氧烷流体以外的至少一种弹性热塑性聚合物、非弹性热塑性聚合物、弹性热固性聚合物及其混合物,和(b)具有软的聚二有机硅氧烷单元,硬的多异氰酸酯残基单元和任选的软和/或硬的有机多胺残基单元和端基的含聚二有机硅氧烷脲的组分。所述硬的多异氰酸酯残基和硬的多胺残基在含聚二有机硅氧烷脲的组分中的含量小于50重量%。所述多异氰酸酯残基是失去-NCO基团的多异氰酸酯,所述多胺残基是失去-NH2基团的多胺,所述多异氰酸酯残基通过脲键与多胺残基相连。所述端基是非官能团或官能团。该含聚二有机硅氧烷脲的组分可在自由基或湿固化条件下活化。该混合物还可任选地含有增粘材料、自由基引发剂、交联剂、固化催化剂和非活性添加剂,如填料、颜料、稳定剂、抗氧剂、阻燃剂、增塑剂、相容剂等。
Description
技术领域
本发明涉及含聚二有机硅氧烷脲的材料和有机聚合物的混合物,具体涉及适合作为塑料、剥离表面、压敏粘合剂、热熔粘合剂、减震组分的混合物以及用这种混合物制得的制品。
发明的背景
聚二有机硅氧烷聚合物具有独特的性能,这种性能主要来自硅氧烷键和有机取代基的物理和化学特性。通常,聚二有机硅氧烷聚合物的杰出性能包括抗紫外光、相当低的玻璃化温度、良好的热稳定性和氧化稳定性、对许多气体具有良好的渗透性、很低的表面能、低的折射率、良好的疏水性和良好的介电性能。它们还具有很好的生物相容性,将其作为生物材料用于有血存在的体内受到人们很大的关注。聚二有机硅氧烷弹性体由于具有许多优良的性能而得到广泛的应用。但是它们有限的抗撕裂性和对低极性溶剂差的耐受性使之不适用于许多其它用途。
近年来,已公开了可自由基固化和可湿固化的液态聚二有机硅氧烷组合物在辐照并适当升温下,或在水分中能快速并完全固化,而具有优良的性能。因此。通常将使用这些组合物的后续制造步骤或修补步骤延长至发生某种程度的固化。同时,在完成固化前必须使用临时载体才能制得厚的构件,难以适当地涂覆具有不规则形状的表面。因此,仍需开发具有胶料强度(即未固化状态的强度)和受控的流动性的聚二有机硅氧烷组合物。
聚二有机硅氧烷嵌段共聚物是含聚二有机硅氧烷脲的组分,该组分可含非聚二有机硅氧烷或脲的嵌段。这些共聚物在加工成本上具有某些潜在的优点,因为其合成反应比前面提到的聚合物更快、无需催化剂并且不产生副产品。
通常聚二有机基硅氧烷压敏粘合剂在溶液中制备。常用的溶剂基的聚二有机基硅氧烷压敏粘合剂一般是带有硅烷醇官能团的高分子量聚二有机基硅氧烷(即聚二有机基硅氧烷胶)和共聚的带有硅烷醇官能团的硅酸酯树脂(即MQ树脂)的共混物,所述树脂含有R3SiO1/2单元和SiO4/2单元。当共聚的聚二有机硅氧烷树脂与聚二有机基硅氧烷相互缩合时,在粘合剂内部产生内缩合和相互缩合,使该压敏粘合剂的性能得到提高。该缩合步骤需要(1)加入催化剂,(2)在溶液中使共聚聚二有机基硅氧烷树脂与聚二有机基硅氧烷反应,以及(3)在升高的温度下使该反应进行一段时间。
业已描述了无需固化步骤的溶液聚合的聚二有机硅氧烷脲弹性体。但是,由于该组合物是在溶液中制得的,其加工的成本较高。
连续的熔融聚合方法是有利的并可用于制造例如聚氨酯弹性体和丙烯酸酯压敏粘合剂这种组合物。用于制造聚醚酰亚胺(该聚合物可含有聚二有机硅氧烷链段)的连续熔融聚合法也已有描述。近来,已描述了聚氨酯树脂并用聚二有机硅氧烷脲链段来防止用所述树脂制得的薄膜的粘连。但是,在该组合物中活性聚二有机硅氧烷的含量较少,例如小于15重量%,由于加入该聚二有机硅氧烷的目的在于使剥离更容易,因此聚二有机硅氧烷可能不完全接入主链是无害的。但是,未接入的聚二有机硅氧烷油在弹性体中起增塑剂的作用,降低压敏粘合剂的拉伸强度或降低粘性,并降低剪切性能。这种未接入的硅油还会浮散在弹性体或粘合剂的表面,污染与其接触的其它表面。
在各种用途中也使用聚合物组分的混合物。当将丙烯酸压敏粘合剂与热塑性弹性体熔融混合,随后挤出涂覆在各种基片上可观察到增强的剥离性能。也有在溶剂中将聚二有机硅氧烷脲与介电聚合物混合,形成用于静电印刷法中的显像片的介电层,使得显像片能更容易地从随后施涂的上色剂中剥离。但是,只有当聚二有机硅氧烷脲含有至少50重量%非聚二有机硅氧烷硬段时,才能获得良好的图像。
发明的概述
简单地说,本发明的一个方面是提供一种混合物,它包括(a)除聚二有机硅氧烷流体以外的至少一种热塑性聚合物、弹性热固性聚合物及其混合物,和(b)具有软的聚二有机硅氧烷单元,硬的多异氰酸酯残基单元和任选的软和/或硬的有机多胺残基单元和端基的聚合物。所述硬的多异氰酸酯残基和硬的多胺残基在含聚二有机硅氧烷脲的组分中的含量小于50重量%。所述多异氰酸酯残基是失去-NCO基团的多异氰酸酯,所述多胺残基是失去-NH2基团的多胺。所述多异氰酸酯残基通过脲键与多胺残基相连。所述端基是非官能团或官能团。所述含聚二有机硅氧烷脲的组分在自由基或湿固化条件下具有活性。该混合物还可任选地含有增粘材料、自由基引发剂、交联剂、固化催化剂和非活性添加剂,如填料、颜料、稳定剂、抗氧剂、阻燃剂、增塑剂、相容剂等。
为方便起见,下面将“除聚二有机硅氧烷流体以外的热塑性聚合物、弹性热固性聚合物及其混合物”简称为“有机聚合物”。
本发明还提供一种基本无溶剂的方法制造包括有机聚合物和含聚二有机硅氧烷脲的组分的混合物。典型的三步法包括(1)向反应容器连续地提供至少一种有机聚合物和至少一种含聚二有机硅氧烷脲的组分,(2)将这些组分混合成混合物和(3)从反应容器中送出该混合物。
或者,可用最少四步实施本发明,它包括(1)提供至少一种有机聚合物和反应组分,其中所述反应组分包括至少一种多异氰酸酯和至少一种多胺,所述多胺包括至少一种聚二有机硅氧烷胺或者至少一种聚二有机硅氧烷胺和至少一种有机胺的混合物,(2)将这些组分混合,(3)使反应组分反应,形成聚二有机硅氧烷脲嵌段共聚物,该嵌段共聚物是无官能团的或者该嵌段共聚物可在湿固化条件下或自由基条件下进一步发生反应,和(4)从反应器中送出混合物。尽管在所述的步骤中有机聚合物和反应组分在单独的步骤中提供并在单独的步骤中混合,但是应理解根据有机聚合物对异氰酸酯或胺是否具有活性,上述加料和混合次序可作各种变化。如果认为有机聚合物是非活性的,也就是说,有机聚合物与异氰酸酯或胺的反应速率明显小于异氰酸酯与胺的反应速率,从而基本上不会与异氰酸酯和胺的反应发生竞争,则对于反应组分来说有机聚合物以任何次序加入反应器。另一方面,如果有机聚合物与反应组分发生反应,也就是说有机聚合物与异氰酸酯或胺明显发生反应,实际上与异氰酸酯和胺之间的反应发生竞争,则最好在反应组分先发生反应形成含聚二有机硅氧烷脲的组分后再加入有机聚合物。这种混合方法可以是连续的方法或间歇的方法。
本发明另一方面提供一种混合物,它包括有机聚合物和含聚二有机硅氧烷脲的组分,该混合物是用基本无溶剂的混合方法制得的。
本发明还提供使用所述混合物作为涂层或层的各种制品,这种制品包括,但不限于减震制品、压敏粘合剂和热熔粘合剂制品、薄膜、涂料、具有剥离表面的制品和具有无光外观的制品。
有利的是,可将本发明用于许多组合物和制品上,因为聚二有机硅氧烷和大多数其它聚合物一般不相容,并且在高浓度聚二有机硅氧烷存在下还未观察到会形成适用的混合物。本发明出乎意料地提供含各种浓度的聚二有机硅氧烷的有用的混合物的制造方法。该组合物兼有聚二有机硅氧烷有用的低温和低表面能性能以及其它有机聚合物有用的环境温度性能。例如,与常规减震材料相比,用本发明混合物制得的减震材料表现出更加不同的减震特性和与各种表面有改进的粘性。已制得了至今未知的具有剥离性能、有光或无光泽外观的表面以及与许多基片有良好粘性的剥离涂层。另外,本发明提供的压敏粘合剂具有特定的粘合性能,兼有聚二有机硅氧烷的性能和其它聚合物材料的性能,这是本发明前本领域的普通技术人员未发现的。同时现在新的粘合剂能控制药物向皮肤的释放速率,与用已知的含聚二有机硅氧烷脲的组合物制得的现有粘合剂相比其性能有了较大的变化。
附图简述
图1是本发明透皮基质组件的剖面图;
图2是本发明透皮贮库组件的剖面图;
图3是本发明粘合剂中含透皮药物的组件的剖面图;
图4是本发明透皮多层组件的剖面图;
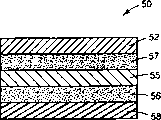
图5是本发明透皮多层组件另一个实例的剖面图;
图6是比较例3中储能模量和tgδ与温度和频率的关系图;
图7是实施例6中储能模量和tgδ与温度和频率的关系图;
图8是比较例4中储能模量和tgδ与温度和频率的关系图;
图9是实施例7中储能模量和tgδ与温度和频率的关系图;
较好实例的描述
简单地说,本发明的一个方面是提供一种混合物,它包括(a)除聚二有机硅氧烷流体以外的至少一种热塑性聚合物、热固性弹性体及其混合物,和(b)具有软的聚二有机硅氧烷单元,硬的多异氰酸酯残基单元和任选的软和/或硬的有机多胺残基单元和端基的聚合物。所述硬的多异氰酸酯残基和硬的多胺残基在含聚二有机硅氧烷脲的组分中的含量小于50重量%。所述多异氰酸酯残基是失去-NCO基团的多异氰酸酯,所述多胺残基是失去-NH2基团的多胺。所述多异氰酸酯残基通过脲键与多胺残基相连。所述端基是非官能团或官能团。该混合物还可任选地含有增粘材料、封端剂、自由基引发剂、硅烷交联剂、湿固化催化剂和非活性添加剂,如填料、颜料、稳定剂、抗氧剂、阻燃剂、增塑剂、相容剂等。
有机聚合物组分
有机聚合物组分是除聚二有机硅氧烷流体以外的热塑性聚合物、热固性弹性体及其混合物。
有机聚合物可以与聚二有机硅氧烷脲嵌段共聚物溶剂混合或熔融混合。同时,有机聚合物可以是另一种含聚二有机硅氧烷脲的组分或者是不含聚二有机硅氧烷链段的聚合物。在使用温度下,混合物一般至少具有两个区域,一个非连续区和另一个连续区,因为含聚二有机硅氧烷脲的组分与有机聚合物一般不混溶。当然,混合物可含有多于一种的有机聚合物。
热塑性材料通常是在加热至明显高于其玻璃化点以上的温度会流动并且冷却时会成为固体的材料。它们还可具有弹性性能。适用于本发明的热塑性材料一般是非弹性的,包括例如聚烯烃,如全同立构的聚丙烯、低密度聚乙烯、线型低密度聚乙烯、很低密度聚乙烯、中密度聚乙烯、高密度聚乙烯、聚丁烯,非弹性的聚烯烃共聚物或三元聚合物,如乙烯/丙烯共聚物及其掺混物;乙烯-乙酸乙烯酯共聚物,如购自杜邦化学公司的ELVAXTM260;乙烯-丙烯酸共聚物;乙烯-甲基丙烯酸共聚物,如购自杜邦化学公司的SURLYNTM1702;聚甲基丙烯酸甲酯;聚苯乙烯;乙烯-乙烯醇;聚酯;无定形聚酯;聚酰胺;氟化热塑性聚合物,如聚偏氟乙烯、聚四氟乙烯、和氟化的乙烯/丙烯共聚物;卤化的热塑性聚合物,如氯化聚乙烯。任何单独的热塑性材料可与至少一种含聚二有机硅氧烷脲的组分混合。或者,可使用热塑性材料的混合物。
具有弹性性能的热塑性材料通常被称为热塑性弹性材料。热塑性弹性材料一般定义为具有高的回弹性和低的蠕变,并且加热至高于其软化点的温度也会流动的材料,它们表现得似乎被共价交联。适用于本发明的热塑性弹性材料包括,例如线型、径向型、星型和递变苯乙烯-异戊二烯嵌段共聚物,如购自Shell ChemicalCo.,of Houston,TX的KRATONTMD1107P和购自EniChem Elastomers Americas,Inc.ofHouston,TX的EUROPRENETMSOL TE 9110;线型苯乙烯-(乙烯-丁烯)嵌段共聚物,如购自Shell Chemical Co.的KRATONTMG1657;线型苯乙烯-(乙烯-丙烯)嵌段共聚物,如购自Shell Chemical Co.的KRATONTMG1657X;线型、径向型和星型苯乙烯-丁二烯嵌段共聚物,如购自Shell Chemical Co.的KRATONTMD1118X和购自EniChem Elastomers Americas,Inc.的EUROPRENETMSOL TE6205;聚醚酯,如购自杜邦的HYTRELTMG3548,弹性乙烯-丙烯共聚物;热塑性弹性聚氨酯,如购自Morton International,Inc.,Chicago,IL的MORTHANEURETHENETM PE44-203;自粘性或增粘的聚丙烯酸酯,它具有C3-C12烷酯并可含有其它共聚单体,例如丙烯酸异辛酯和0-20重量%的丙烯酸;聚乙烯基醚;聚α-烯烃基热塑性弹性材料,如用通式(CH2CHR)x表示的材料,其中R是具有2-10个碳原子的烷基,以及基于茂金属催化剂的聚α-烯烃,如ENGAGETMEG8200,一种购自Dow Plastics Co.ofMidland,MI的乙烯/聚α-烯烃共聚物。
热固性弹性体是一种在热的影响下,通过形成共价交联的热稳定网络由易熔的并可溶的材料不可逆地转变成不易熔的并不可溶的材料的材料。适用于本发明的热固性弹性体包括,例如天然橡胶,如CV-60,一种购自Goodyear Chemical,Akron,OH的粘度受控品级的材料,和SMR-5,一种有凸纹的烟熏的片橡胶;丁基橡胶,如购自Exxon Chemical Co.的Exxon Butyl268;合成的聚异戊二烯,如购自Royal Dutch Shell of Netherlands的CARIFLEXTMIR309和购自Goodyear Tireand Rubber Co.的NATSYNTM2210;苯乙烯-丁二烯无规共聚物橡胶,如购自BFGoodrich of Akron,OH的AMERIPOLTM1011A;聚丁二烯;聚异丁烯,如购自Exxon Chemical Co.的VISTANEXTMMM L-80;聚氨酯,如美国专利2,532,011公开的聚氨基甲酸十八烷酯;无定形聚α-烯烃,如C4-C10直链或支链的聚α-烯烃;含聚二有机硅氧烷脲的组分,如美国专利5,214,119公开的聚合物。
可用增粘材料或增塑剂改性热塑性或热固性弹性体材料以改进其性能。适合于与聚合物材料一起使用的增粘材料或增塑剂最好在分子水平上与聚合物材料混溶。即溶解在任何或全部弹性材料或热塑性弹性材料的聚合物链段中。这些增粘材料或增塑剂一般与含聚二有机硅氧烷脲的组分不混溶。当含增粘材料时,按100重量份聚合物材料计,它的含量约为5-300重量份,通常高达约200重量份。适用于本发明的增粘剂包括,但不限于液态橡胶、烃树脂、松香、天然树脂如二聚或氢化的香脂和酯化的松香酸、聚萜烯、萜烯酚醛、酚醛树脂和松香酯。增塑剂的例子包括,但不限于聚丁烯、石蜡油、凡士林和某些具有长脂族侧链的邻苯二酸酯,如邻苯二甲酸双十三烷酯。
含聚二有机基硅氧烷脲的组分
总体上,可将所述组分描述为具有软的聚二有机硅氧烷单元、硬的多异氰酸酯残基单元、端基和任选的软的和/或硬的有机多胺残基单元的材料。所述多异氰酸酯残基是失去-NCO基团的多异氰酸酯,所述有机多胺残基是失去-NH基团的有机多胺,多异氰酸酯残基与聚二有机硅氧烷单元或有机多胺残基通过脲键相连。取决于聚二有机硅氧烷脲嵌段共聚物的用途,所述端基可以是非官能团或官能团。
本发明含聚二有机基硅氧烷脲的组分可用下列重复单元表示:
式中:
各个R分别是一个烷基部分,最好是具有约1-12个碳原子的烷基部分,该烷基部分可被例如三氟烷基或乙烯基类基团、乙烯基或高级烯基(可由式-R2(CH2)aCH=CH2表示,其中R2是-(CH2)b-或-(CH2)cCH=CH-,a是1、2或3;b是0、3或6;c是3、4或5)所取代;具有约6-12个碳原子的环烷基部分,该环烷基部分可被烷基、氟烷基和乙烯基所取代;或具有约6-20个碳原子的芳基部分,该芳基部分可被例如烷基、环烷基、氟烷基和乙烯基所取代;或R是美国专利第5,028,679号(在此引用参考)中所述的全氟烷基,美国专利第5,236,997号(在此引用参考)所述的含氟基团,或在第4,900,474和5,118,775号美国专利(在此引用参考)中所述的含全氟醚基团;较好的R部分至少50%为甲基,其余部分为有1-12个碳原子的单价烷基或取代的烷基、亚烯基、苯基或取代的苯基;
各个Z是一个多价基,最好是有约6-20个碳原子的亚芳基或亚芳烷基或最好是有约6-20个碳原子的亚烷基或亚环烷基,优选的Z是2,6-亚甲苯基,4,4’-亚甲基二亚苯基、3,3’-二甲氧基-4,4’-亚联苯基、四甲基-间亚二甲苯基、4,4’-亚甲基二亚环己基、3,5,5-三甲基-3-亚甲基亚环己基、1,6-亚己基、1,4-亚环己基、2,2,4-三甲基亚己基及其混合基;
各个Y是一个多价基,分别是一个最好有1-10个碳原子的亚烷基、最好有6-20个碳原子的亚芳烷基或亚芳基;
各个D分别选自氢、具有1-10个碳原子的烷基、苯基、以及包括B或Y以形成杂环的环结构基团;
B是一个多价基团,选自亚烷基、亚芳烷基、亚环烷基、亚苯基、聚环氧烷,包括例如聚环氧乙烷、聚环氧丙烷、聚四氢呋喃,及其共聚物和混合物;
m是0-约1000的数;
n是等于或大于1的数;和
p是约为5或更大的数,较好约15-2000,最好约30-1500。
使用多异氰酸酯(Z是官能度大于2的基团)和多胺(B是官能度大于2的基团),将会改变式Ⅰ的结构,使聚合物主链发生支化。使用多异氰酸酯(Z是官能度大于2的基团)和多胺(B是官能度大于2的基团),将会改变式Ⅰ的结构,使聚合物主链发生支化。
使用封端剂,将会改变式Ⅰ的结构,终止聚二有机硅氧烷脲链。
还可加入用于含聚二有机硅氧烷脲的组分的增粘材料,通常为硅酸酯树脂。在决定本发明含二有机硅氧烷脲的组分的物理性能中硅酸酯树脂起重要的作用。例如,当硅酸酯树脂的含量由低增至高浓度时,在升至较高的温度时含聚二有机硅氧烷脲的组分发生由玻璃态向橡胶态的转变。因此,在减震用途中改变硅酸酯树脂的浓度,可将该组分的最大阻尼区迁移至所需的温度范围,以适当地补充本发明混合物的有机聚合物的阻尼范围。当然M与Q的比例、D和T的含量以及树脂的分子量也会明显影响树脂的相对“硬度”,在选择树脂类型和浓度时必须予以考虑。另外,硅酸酯树脂不限于单种硅酸酯树脂,在一种组合物中组合使用多种硅酸酯树脂以获得所需的性能可能是较为有利的。
在本发明中有用的硅酸酯树脂包括由下面的结构单元M、D、T和Q及其组合组成的那些硅酸酯树脂。一般的例子包括MQ硅酸酯树脂、MQD硅酸酯树脂和MQT硅酸酯树脂,它们也可称作为共聚的硅酸酯树脂,其数均分子量较好约为100-50,000,更好的约为500-10,000,一般还具有甲基取代基。硅酸酯树脂也包括非官能和官能树脂,官能树脂有一个或多个官能团,包括例如与硅连接的氢、与硅连接的链烯基和硅烷醇。MQ硅酸酯树脂是有R’3SiO1/2单元和SiO4/2单元的共聚的硅酸酯树脂。在如Encyclopedia of Polymer Science and Enginerring,Vol.15,John Wiley&Sons,New York,(1989),pp265-270,美国专利2,676,182、3,627,851、3,772,247和5,248,739(在此引用参考)中描述了这样的树脂。具有官能团的MQ硅酸酯树脂有在美国专利4,774,310中所述的氢化甲硅烷基的硅酸酯树脂,在美国专利5,262,558中所述的有乙烯基和三氟丙基的硅酸酯树脂和美国专利4,707,531中所述有氢化甲硅烷基和乙烯基的硅酸酯树脂(在此引用参考)。一般都在溶剂中制备上述树脂。在美国专利5,319,040、5,302,685和4,935,484中描述了干的、或无溶剂的MQ硅酸酯树脂的制备(在此引用参考)。MQD硅酸酯树脂是有R’3SiO1/2单元、SiO4/2单元和R’2SiO2/2单元的三元共聚物,例如在美国专利2,736,721中所述(在此引用参考)。MQT硅酸酯树脂是有R’3SiO1/2单元、SiO4/2单元和R’SiO3/2单元的三元共聚物,例如在美国专利5,110,890(在此引用参考)和日本公开平2-36234中所述。
市售的硅酸酯树脂包括SR-545(在甲苯中的MQ树脂,购自General ElectricCo.,Silicone Resins Division,Waterford,NY);MQOH树脂(MQ树脂,购自PCR,Inc.,Gainesville,FL);MQR-32-1,MQR-32-2和MQR-32-3树脂(在甲苯中的MQD树脂,购自Shin-Etsu Silieones of America,Inc.,Torrance,CA);以及PC-403(在甲苯中的氢化官能的MQ树脂,购自Rhone-Poulenc,Latex and Specialty Polymers,Rock Hill,SC)。这些树脂一般溶解在有机溶剂中供应,购到后即可用于本发明的组合物。然而可通过本领域的各种工艺除去硅酸酯树脂中的这些有机溶剂,例如喷雾干燥、烘干等,或蒸气分离,即为本发明的组合物提供基本上100%不挥发组分含量的硅酸酯树脂。本发明含聚二有机硅氧烷脲的组分中还可以使用两种或多种硅酸酯树脂的混合物。当大部分含聚二有机硅氧烷脲的组分是非聚二有机硅氧烷时。除了或代替硅酸酯树脂,可将有机增粘剂(类似于前面所述的与有机聚合物一起使用的增粘剂)与所述组分一起使用。
当本发明混合物的含聚二有机硅氧烷脲的组分中含有增粘材料时,该组分可含有约1-80重量份增粘材料,较好含15-75重量份增粘材料。当将增粘材料加入仅含聚二有机硅氧烷脲的组分中以增加弹性时,增粘材料最好约占该组分的15-25重量%。当将增粘材料加入含聚二有机硅氧烷脲的组分中以形成混合物作为粘合剂或阻尼剂时,增粘材料最好约占混合物的30-70重量%。在混合物中含聚二有机硅氧烷脲的组分和硅酸酯树脂的总重量份数等于100。增粘材料的最佳含量取决于诸如所使用的反应剂的种类和用量、聚二有机硅氧烷脲嵌段共聚物的硬段和软段的分子量、有机聚合物的种类和用量以及本发明组合物所需的用途这些因素。
其它添加剂
可在有机聚合物、聚二有机硅氧烷脲嵌段的有机聚合物或者在本发明组合物的两种组分中均混入填料、增塑剂和其它合适的改性剂。所述填料包括,如热解法二氧化硅(fumed silica)、碳纤维、炭黑、玻璃珠、玻璃泡、玻璃纤维、矿物纤维、粘土颗粒、有机纤维(如尼龙、KEVLARTM)、金属颗粒等,每100份有机聚合物和聚二有机基硅氧烷脲嵌段共聚物组分可加入约100份的填料,只要加入添加剂时这些添加剂不会损害最终聚合物产品的功能和官能度即可。其它添加剂如染料、颜料、阻燃剂、稳定剂、抗氧剂、相容剂,抗微生物剂(如氧化锌)、导电体、导热体(如氧化铝、氮化硼、氮化铝、镍颗粒)等可以本发明组合物的约1-50体积%的量掺混到这些体系中。
含聚二有机硅氧烷脲的组分的活性组分
在反应中不同的多异氰酸酯会以不同的方式改变含聚二有机硅氧烷脲的组分的性能。例如,如果使用聚碳化二亚胺改性的二苯基甲烷二异氰酸酯,如ISONATETM143L(购自Dow Chemical Co.),所得的含聚二有机硅氧烷脲的组分与用其它二异氰酸酯制得的共聚物相比,具有更强的耐溶剂性能。如果使用四甲基-间亚二甲苯基二异氰酸酯,所得的嵌段共聚物具有相当低的熔体粘度,使其特别适用于注模。
本发明方法中所用的二异氰酸酯可用下式表示:
OCN-Z-NCO
(Ⅱ)
可以与多胺,尤其是式Ⅱ的聚二有机硅氧烷二胺反应的任何二异氰酸酯均可用于本发明。这些二异氰酸酯的例子包括但不限于芳族的二异氰酸酯,如2,6-甲苯二异氰酸酯,2,5-甲苯二异氰酸酯、2,4-甲苯二异氰酸酯、间亚苯基二异氰酸酯、对亚苯基二异氰酸酯、亚甲基二(邻氯苯基二异氰酸酯)、亚甲基二亚苯基-4,4’-二异氰酸酯、聚碳化二亚胺改性的亚甲基二亚苯基二异氰酸酯、(4,4’-二异氰酸基-3,3’,5,5’-四乙基)二苯基甲烷、4,4’-二异氰酸基-3,3’-二甲氧基联苯基(邻-二茴香胺二异氰酸酯)、5-氯-2,4-甲苯二异氰酸酯、1-氯甲基-2,4-二异氰酸基苯;芳族-脂族二异氰酸酯如间亚二甲苯基二异氰酸酯、四甲基-间-亚二甲苯基二异氰酸酯;脂族二异氰酸酯,如1,4-二异氰酸基丁烷、1,6-二异氰酸基己烷、1,12-二异氰酸基十二烷、2-甲基-1,5-二异氰酸基戊烷;以及环脂族二异氰酸酯如亚甲基二亚环己基-4,4’-二异氰酸酯、3-异氰酸甲酯基-3,5,5-三甲基环己基异氰酸酯(异佛尔酮二异氰酸酯)、2,2,4-三甲基己基二异氰酸酯和亚环己基-1,4-二异氰酸酯及其混合物。
较好的二异氰酸酯包括2,6-甲苯二异氰酸酯、亚甲基二亚苯基-4,4’-二异氰酸酯、聚碳化二亚胺改性的亚甲基二苯基二异氰酸酯、4,4’-二异氰酸基-3,3’-二甲氧基联苯基(邻-二茴香胺二异氰酸酯)、四甲基-间亚二甲苯基二异氰酸酯、亚甲基二亚环己基-4,4’-二异氰酸酯、3-异氰酸甲酯基-3,5,5-三甲基环己基异氰酸酯(异佛尔酮二异氰酸酯)、1,6-二异氰酸基己烷、2,2,4-三甲基己基二异氰酸酯以及亚环己基-1,4-二异氰酸酯。特别好的是四甲基-间亚二甲苯基二异氰酸酯。使用四甲基-间亚二甲苯基二异氰酸酯制备的含聚二有机硅氧烷脲的组分一般具有比使用其它二异氰酸酯制备的类似共聚物更低的熔体粘度以及更高的模量。
可以与多胺,特别是式Ⅱ的聚二有机硅氧烷二胺反应的任何三异氰酸酯均可用于本发明。这些三异氰酸酯的例子包括但不限于多官能团的异氰酸酯,如那些由缩二脲、异氰脲酸酯、加合物等制备的多官能团异氰酸酯。一些可购得的多异氰酸酯包括Bayer的DESMODURTM和MONDURTM系列以及Dow Plastics的PAPITM系列。
较好的三异氰酸酯包括DESMODURTMN-3300和MONDURTM489。
在本发明的方法中有用的聚二有机硅氧烷二胺可用下式表示:
其中各R、Y、D和p的定义如上所述。本发明中所用的聚二有机硅氧烷二胺的数均分子量一般约大于700。
本发明中所用的聚二有机硅氧烷二胺(也称作硅氧烷二胺)是上式Ⅲ范围内任何一种,包括那些分子量在约700至150,000范围内的聚合物。聚硅氧烷二胺在下述文献中有所揭示,如美国专利No.3,890,269、美国专利No.4,661,577、美国专利No.5,026,890、美国专利5,214,119、美国专利No.5,276,122、美国专利5,461,134和美国专利5,512,650(上述文献均在此引作参考)。
聚二有机硅氧烷二胺可购自Shin Etsu Silicones ofAmerica,Inc.,Torrance,CA,以及Huels America,Inc.。较好的是如美国专利No.5,214,119(于此引用参考)中所述制备的基本纯的聚二有机硅氧烷二胺。具有如此高纯度的聚二有机硅氧烷二胺是如下制备的:将环状有机硅烷与二(氨基烷基)二硅氧烷反应,使用无水的氨基烷基官能团的甲硅烷醇盐催化剂如四甲铵-3-氨基丙基二甲基甲硅烷醇盐,用量最好少于环状有机硅氧烷的总重量的0.15%(重量),反应分两步进行。最好的聚二有机硅氧烷二胺是使用铯和铷催化剂根据美国专利5,512,650公开的方法制得。
本发明所用的聚二有机硅氧烷二胺的例子包括但不限于聚二甲基硅氧烷二胺、聚二苯基硅氧烷二胺、聚三氟丙基甲基硅氧烷二胺、聚苯基甲基硅氧烷二胺、聚二乙基硅氧烷二胺、聚二乙烯基硅氧烷二胺、聚乙烯基甲基硅氧烷二胺、聚(5-己烯基)甲基硅氧烷二胺,以及它们的共聚物和混合物。
本发明所用的有机多胺的例子包括但不限于聚氧化烯二胺,如来自Huntsman的D-230、D-400、D-2000、D-4000、DU-700、ED-2001和EDR-148,聚氧化烯三胺,如来自Hunstman的T-3000和T-5000,聚亚烷基如来自DuPont的DYTEKTM A和DYTEKTM EP,及其混合物。
多异氰酸酯与多胺的反应一旦发生,脲键中的活性氢仍可以与过量的异氰酸酯反应。增加异氰酸酯与胺的比率,会促进缩二脲部分的生成,尤其是在高温下,这将会导致聚合物的支化或交联。生成低量至中量的缩二脲,对剪切性能和耐溶剂性能是有利的。
在含聚二有机硅氧烷脲的组分中的异氰酸酯残基的性能影响硬挺性和流动性,还会影响混合物的性能。如果使用足量的含二有机硅氧烷脲的组分,与由亚甲基二亚环己基-4,4’-二异氰酸酯、3-异氰酸甲酯基-3,5,5-三甲基环己基异氰酸酯和间亚二甲苯基二异氰酸酯形成的残基相比,由形成可结晶脲的二异氰酸酯(如四甲基-间亚二甲苯基二异氰酸酯、1,12-十二烷二异氰酸酯、邻-二茴香胺二异氰酸酯)形成的异氰酸酯残基能使混合物更具硬挺性。
可根据需要加入任选的封端剂,使含聚二有机硅氧烷脲的组分具有非官能的湿固化部分或可自由基固化的部分。该试剂能与胺或异氰酸酯反应。
如有必要,可使用交联剂(如硅烷试剂)交联可湿固化的含聚二有机硅氧烷脲的组分,或者将光引发剂用于可自由基固化的含聚二有机硅氧烷脲的组分。使用时,这些组分的用量应适合于所需的目的,一般的用量约为可聚合组合物的总重量的0.1-5%。
制造方法
本发明的组合物和结构物可用现有技术中已知的溶剂方法、无溶剂方法或两者的结合而制备。本领域的普通技术人员可以期望最适宜的材料用于具体用途,该材料与结构和含聚二有机硅氧烷脲的组分比例、有机聚合物的结构和比例、任选的引发剂的结构和是否加入填料、添加剂或性能改性剂有关。另外,根据所使用的方法可制得性能各异的组合物。同时,可在含聚二有机硅氧烷脲的组分与有机聚合物混合前制得该组分,或者在有机聚合物的存在下制备该组分。在后面一种情况下,有机聚合物必须不与胺或异氰酸酯发生反应,也就是说,有机聚合物与异氰酸酯或胺的反应速度明显小于异氰酸酯与胺的反应速度,从而基本不与异氰酸酯和胺之间的反应竞争。
基于溶剂的方法是本领域熟知的。用于制备适用于本发明的含聚二有机硅氧烷脲的组分的基于溶剂的方法的例子包括:Tyagi等的“SegmentedOrganosiloxane Copolymers:2.Thermal and Mechanical Properties of Siloxane-UreaCopolymers,”Polymer,vol.25,1984年12月和美国专利No.5,214,119(Leir等),这些文献在此引为参考。此时可将有机聚合物加入至聚二有机硅氧烷脲嵌段聚合物的溶剂溶液中,形成适合于作为,例如剥离表面、密封垫圈、密封胶和封装化合物的组合物。如果加入增粘材料,形成的混合物可用于结构物中和用于使本发明制品粘结、防腐或减震的方法中。
用于制造本发明混合物的另一种特别适用的方法是无溶剂法。可在加入聚合物组分形成混合物前制得含聚二有机硅氧烷脲的组分,或者在有机聚合物的存在下制得该组分。在这两种情况下,当在基本无溶剂的条件下制备含聚二有机硅氧烷脲的组分时,可使用任何反应器,只要该反应器能使反应的异氰酸酯反应组分和胺反应组分充分混合即可。可采用间歇的方法使用例如装有机械搅拌器的烧瓶进行反应,条件是反应产物在反应温度下具有足够低的粘度以便混合。另外,可采用连续的方法使用例如单螺杆或双螺杆挤出机进行反应。反应器较好是擦过表面的反向旋转或同向旋转双螺杆挤出机。反应器最好是擦过表面的反应器,其螺杆的螺纹棱面和机筒之间具有相对紧密的间隙,该间隙值通常约0.1-2mm。使用的螺杆在大部分反应发生的区域较好相互间完全或部分啮合,或者完全或部分擦过。在反应器中制造含聚二有机硅氧烷脲的组分的总停留时间通常约为5秒-20分钟,更通常约为15秒-8分钟。异氰酸酯和胺反应剂之间的反应较快,可在室温下进行。因此,制造含聚二有机硅氧烷脲的组分可容易地在例如一个长径比仅为5∶1的双螺杆挤出机中进行。140-250℃的温度一般足以将含聚二有机硅氧烷脲的组分从反应器中送出。
在多胺和多异氰酸酯的反应过程中不存在溶剂能产生非常有效的反应。使用本发明方法的平均停留时间比溶剂聚合所需的时间通常短10-1000倍。如果需要,在本方法中可加入少量(如组合物总量的约0.5-5%)非活性溶剂,作为载体用于将无溶剂时是固体的材料注入反应室,或者用于增加在反应室中无溶剂时低流动速率物流的稳定性。
加料的速率也是重要的,尤其是在使用含聚二有机硅氧烷脲的反应试剂时。由于在多胺和多异氰酸酯之间快速发生反应,因此两种反应试剂最好以相同的速率(尤其是使用高分子量多胺,即分子量约为50,000和更高时)加入挤出机。这种加料方法通常能减少最终产品不合需求的不一致。确保将很低流量的多异氰酸酯物流连续加入挤出机的一种方法是使多异氰酸酯的进料线与螺杆经过的螺纹接触或非常接近于接触。另一种方法是使用连续的喷雾注射组件,该组件将多异氰酸酯细滴的连续物流喷入反应器。
制得的含聚二有机硅氧烷脲的组分的分子量可比用溶剂法制得的该组分可能的分子量更高。在溶剂法中用分子量高于20,000的聚二有机硅氧烷二胺制备含聚二有机硅氧烷脲的组分的转化率难以达到无溶剂法中获得的转化率。
有机聚合物一般以熔融物流的方式加入含聚二有机硅氧烷脲的组分中,或加入含聚二有机硅氧烷脲的组分的一种反应试剂中。有时在含聚二有机硅氧烷脲的组分以(1)粒料、(2)反应试剂或(3)来自辅助容器的单独的熔融物流的形式加入之前,该聚合物材料需要在一个单独的容器中进行熔融。较好使用单独的容器的例子包括,例如(1)最好使添加剂浓缩在有机聚合物中。(2)有机聚合物需要高的加工温度和(3)有机聚合物包括弹性热固性材料。
在形成混合物时各种组分的加料次序是重要的。当有机聚合物如前面所述基本不与多异氰酸酯和多胺发生反应时,可使用任何加料次序。可将含聚二有机硅氧烷脲的组分加入有机聚合物中,反过来也一样,或者可在有机聚合物的存在下制得含聚二有机硅氧烷脲的组分。但是,如果有机聚合物会与异氰酸酯或胺发生反应,则必须在形成含聚二有机硅氧烷脲的组分以后加入有机聚合物。并且,如果处理有机聚合物的温度会破坏含聚二有机硅氧烷脲的组分,则有机聚合物最好在单独的容器中充分加热至可加工的状态,随后加至含聚二有机硅氧烷脲的组分的熔融物流中。通常可在任何加工步骤中加入其它添加剂,如增塑材料、增粘材料、颜料、填料、引发剂等,因为它们一般不与反应试剂发生反应,但是通常在形成大量含聚二有机硅氧烷脲的组分后才加入添加剂。
当将非热塑性弹性材料的有机聚合物与含聚二有机硅氧烷脲的组分混合时,有机聚合物一般需要特殊的条件进行熔融处理。使非热塑性弹性材料可熔融处理的两种方法为(1)用增粘或增塑材料溶胀该非热塑性弹性材料,降低其表观熔体粘度或者(2)如美国专利5,539,033(在此引为参考)所述塑炼该材料。
四种方法因素会影响无溶剂法制得的混合物的最终性能。第一,含聚二有机硅氧烷脲的组分的性能受到该含聚二有机硅氧烷脲的组分是用溶剂法制得还是用基本无溶剂的方法制得的影响;第二,如果经受太多的热量和剪力,含聚二有机硅氧烷脲的组分会降解;第三,混合物的稳定性受到含聚二有机硅氧烷脲的组分与有机聚合物的混合方法的影响;第四,用混合物制得的制品的形态由加工参数和混合物中组分特性的相互作用所决定。
在第一个因素中,可使用溶剂或无溶剂的方法先制得含聚二有机硅氧烷脲的组分,或者在有机聚合物的存在下制得该组分。在溶剂中制备含聚二有机硅氧烷脲的组分在上面已经公开。在基本无溶剂的条件下制备含聚二有机硅氧烷脲的组分的方法可使制得的含聚二有机硅氧烷脲的组分具有高的分子量,或者制得共价或非共价交联的产物,使之不溶于普通的有机溶剂。该方法还可制得高分子量的含聚二有机硅氧烷脲的组分,尤其当使用分子量约超过20,000的聚二有机硅氧烷二胺时。
第二,如果在剪切条件下,尤其在氧存在下将含聚二有机硅氧烷脲的组分过度加热,它会降解。当在有机聚合物的存在下,尤其在惰性气氛中制造混合物时,含聚二有机硅氧烷脲的组分受到的热量和剪切影响最少。
第三,混合物的稳定性受到含聚二有机硅氧烷脲的组分与有机聚合物的混合方法的影响。聚二有机硅氧烷与大多数其它聚合材料通常不混溶。但是,本发明人发现当均处于熔融状态时,多种聚合物可与含聚二有机硅氧烷脲的组分混合。应注意使一种组分软化的所需的条件不能使另一种组分降解。混合温度最好高于混合物的混合和传输温度,比含聚二有机硅氧烷脲的组分的降解温度低,约为140-250℃,最好约为160-220℃。组分可在其中充分地进行加热并在熔融状态混合的任何容器均适用于制造本发明混合物。
第四,加工步骤影响用本发明混合物制得的制品的形态。混合物一般至少具有两个区域,一个非连续区和另一个连续区,因为含聚二有机硅氧烷脲的组分一般与有机聚合物不混溶。包含次要相的组分通常形成非连续区,其形状从球状向椭球状至带状至纤维状变化。包含主要相的组分通常形成包围非连续区的连续区。当混合物形成制品(如薄膜或涂层)时,如果该混合物受到足够的剪切或拉力,混合物的非连续区通常会拉长。当混合物不再受到拉力或剪切力时,如果在使用温度至少一种组分具有足够的粘度,则非连续区一般会保持拉长状态,阻止拉长区松弛至球状。拉长的形态通常是稳定的,除非将混合物再加热至高于组分的软化点的温度。
尽管可使用基于溶剂的方法和无溶剂的方法制造本发明混合物,但在某些情况下组合两种方法更好些。对于后一种情况,含聚二有机硅氧烷脲的组分可以用基于溶剂的方法制得,然后干燥并与有机聚合物熔融混合。
制品的类型
根据具体的配方,本发明的组合物可用于制备各种制品。本发明组合物可用作例如剥离膜、压敏粘合带、压敏粘合转移带、压敏粘合医用带(包括透皮给药组件)或直接涂覆在所需制品上的压敏粘合涂层。
压敏粘合剂制品是通过用已知的热熔或溶剂涂覆方法施用压敏粘合剂而制得的。可以使用任何适宜的基材,包括(但不限于)例如布料和玻璃纤维布、镀金属的膜和箔、聚合物薄膜、非织造物、纸张和涂覆聚合物的纸张以及泡沫背衬。聚合物薄膜包括(但不限于)聚烯烃,如聚丙烯、聚乙烯、低密度聚乙烯、线型低密度聚乙烯和高密度聚乙烯;聚酯,如聚对苯二甲酸乙二醇酯;聚碳酸酯;乙酸纤维素;聚酰亚胺,如KAPTONTM。通常由无规取向的纤维制得的非织造物包括(但不限于)尼龙、聚丙烯、乙烯-乙酸乙烯酯共聚物、聚氨酯、人造纤维等等。泡沫背衬包括(但不限于)丙烯酸类、硅氧烷、聚氨酯、聚乙烯、氯丁橡胶和聚丙烯,可以带有填料或不带填料。多层背衬(如聚乙烯-铝膜复合物)也是适宜的。
对于压敏粘合带,这些材料通常先制成带有一层涂覆在背衬上的压敏粘合材料的带状结构。随后可将露出的压敏粘合剂涂覆表面施加于一个以后将剥离的表面上,或者直接施用于所需的基材。
某些压敏粘合剂制品使用剥离衬里(即转移带),它是将所述组合物涂覆在两片带有剥离涂层的衬里之间制得的。所述剥离衬里常包括聚合物材料(如聚酯、聚乙烯、聚烯烃等),或者有剥离涂层的纸或涂覆聚乙烯的纸。较好的是先在每片剥离衬里上涂覆或底涂本发明粘合剂材料的剥离材料。当混合物含有大量增粘的含聚二有机硅氧烷脲的组分,适用的剥离衬里包括那些适用于硅氧烷粘合剂的衬里材料。多氟多醚涂覆的衬里的例子描述在欧洲专利公报No.433070中。其它适用的剥离衬里的剥离涂料组合物描述在欧洲专利公报No.378420、美国专利No.4,889,753和欧洲专利公报No.311262中。市售的衬里和组合物包括购自DowCorning Corp.,Midland,MI的Dow Corning SYL-OFFTMQ2-7785含氟硅氧烷剥离涂料,购自Shin-Etsu Silicones of America,Inc.,Torrance,CA的X-70-029NS含氟硅氧烷剥离涂料,购自Release International,Bedford Park,IL的S TAKE-OFFTM2402含氟硅氧烷剥离衬里等。
本发明组合物还适用于医疗用途,包括透皮给药组件。透皮给药组件设计成使治疗有效量的药物透过或送至病人的皮肤中。透皮给药具有明显的优点;与注射不同,它是非创伤性的;与口服给药不同,它避免了肝的首过代谢,它使对肠胃道的影响减到最小,并且它提供稳定的血中浓度。
已知有许多透皮给药组件。现有技术中已知的组件包括基质组件,其中药物被置于非粘性聚合物材料中;贮库组件,其中药物被置于液体中,并通过控速膜送至皮肤;粘合剂中含药物组件,其中药物被置于粘合剂聚合物中;以及更复杂的多层组件,包括若干不同的层,如含药物层,含赋形剂层,用于控制药物和赋形剂释放速率的层,以及使组件粘附于皮肤的层。
所有这些组件都包括药物制剂,保持与病人皮肤接触的粘合剂,在储存期间保护所述组件并在将该组件用于皮肤之前除去的剥离衬里,以及在使用时保护组件不受外部影响的背衬。
基质组件如图1所示。组件10包括背衬12,含有药物和任选的赋形剂的基质14,围绕基质14的环状粘合剂层16,以及剥离衬里18。
贮库组件如图2所示。组件20包括背衬22,含有药物和任选的赋形剂的液体制剂24,用于控制药物和赋形剂释放至皮肤的速率的膜25,粘合剂层26,以及剥离衬里28。粘合剂层也可以以环状存在,如基质组件中所述(参见图2)。
在粘合剂中含药物的组件如图3所示。组件30包括背衬32,含有药物和任选的赋形剂的粘合剂层37,以及剥离衬里38。
多层组件如图4所示。组件40包括背衬42,含有药物和任选的赋形剂的粘合剂层47,用于控制药物和赋形剂释放至皮肤的速率的第二粘合剂层43,以及剥离衬里48。
多层组件的第二个实例如图5所示。组件50包括背衬52,含有药物和任选的赋形剂的粘合剂层57,膜55,第二粘合剂层56,以及剥离衬里58。可选择所述的膜来控制药物和赋形剂释放至皮肤的速率,或使组件具有物理稳定性。
对皮肤的粘合性是任何透皮给药系统的关键要求。因为药物的释放直接与皮肤接触的面积成比例,因此在除去前该组件必须具有并保持足够的与皮肤的粘性。用于皮肤接触层的粘合剂最好具有下述性能:良好的初始皮肤粘性,即粘性;在使用期间具有足够的粘性;能从皮肤上干净剥离;以及与皮肤的相容性(无刺激和不致敏)。当粘合剂与给定组件中所用的具体药物和赋形剂接触时,能保持这些性能是重要的。
在含有药物和赋形剂或者将药物和赋形剂透过的层中使用的粘合剂必须与该药物和赋形剂相容。较好的是这些粘合剂不与所述药物和赋形剂发生化学反应。在许多情况下,最好是药物溶解在粘合剂中而不是分散于其中。通常需要甚至必须定制粘合剂以用于具体的药物/赋形剂混合物。
透皮给药组件可制成制品的形式,如带、贴片、片、敷料或本领域普通技术人员已知的任何其它形式。一般该组件以贴片的形式,其尺寸适合于释放预定量的药物。适宜的剥离衬里包括上面在制备压敏粘合剂制品中所述的那些衬里。
本发明的组合物还适用于将路面标记片材和路面标记物粘附在路面(如混凝土和沥青)上。路面标记片材通常包括面层、复合层压层和用于与路面粘结的一层或多层粘合层。应选择用于面层的材料和粘合剂使它们能足够牢固地粘合在一起,从而在路面标记所处的条件下不会脱层。面层通常是柔性的聚合物层,该层较好的是耐久的和耐磨的。可制备面层的材料的说明性实例包括(但不限于)聚乙烯基类、聚氨酯类、环氧树脂类、聚胺类、聚脲类和聚酯类。也可以使用这些材料的混合物。适宜的聚合物材料可以是热塑性或热固性弹性聚合物。
一般来说,面层还包括许多嵌入面层并且一些颗粒从上表面上突出的逆向反射颗粒和/或防滑颗粒。可以任选地在面层的下表面施加基片,使之具有所需的适合性和强度。该基片可以含有颗粒状填料,以降低成本和改进性能(如表面硬度或挠性)。可以任选地向面层或基片中加入颜料,使之具有所需的色彩。
路面标记片材通常具有一层施涂于下表面上的橡胶/树脂粘合剂,用于与路面相粘结。如果不存在基层,则可将本发明压敏粘合剂混合物直接施涂于面层的下表面上,或者如果存在基层,则可将该压敏粘合剂混合物施涂于基层的下表面。另外,该粘合剂可有利地施涂于橡胶树脂压敏粘合剂的下表面。
含聚二有机硅氧烷脲的组分的聚合物混合物较好以100%固体可热熔涂覆的组合物形式施涂,并且可以用各种方法施涂,包括刮刀涂覆或挤压涂覆。或者,还可以将本发明压敏粘合剂制成在有剥离涂覆层的衬里(即转移带)之间的粘合层。可根据需要除去一层有剥离涂覆层的衬里,将粘合剂粘附在路面标记材料的面层、基层或橡胶树脂压敏粘合剂上。随后可在施用于路面之前,除去在路面标记材料的下表面上的另一层剥离衬里。
本发明的路面标记片材对于各种路面具有优良的粘性,长期稳定的剥离力值,在宽的温度范围内及高湿度条件下具有优良的性能。
本发明的组合物还可用于压敏粘合剂中,所述粘合剂能容易地粘附在经处理的或未经处理的表面(尤其是金属)上,形成高度适合的连续界面硅氧烷涂层,防止会腐蚀未经保护表面的环境污染物的进入。本发明满足了市场对防护性涂料的需求,该涂料能施涂在控制实验室的外面或在工厂条件下施用。该涂料能粘附于冷的、潮湿的或生锈的金属以及已有的防护性涂层(如在油管和气管上的环氧树脂、聚乙烯和聚丙烯)上。不完全的应用实例包括:永久修复涂层缺陷或露胶;涂覆已拆开的管端以便连接;保护现场装配前必须裸露的部件;用一种可除去的方法在进一步加工前防止裸露的金属快速锈蚀;以及在相邻的涂覆或未涂覆的钢部件(如在钢筋网中)之间作为保护粘合剂。
压敏粘合剂贴片通常包括保护性含聚二有机硅氧烷脲的压敏粘合剂混合物和任选的隔离或边缘(edge)粘合剂,适合性隔离层或背衬材料层,或这些材料的组合。对于某些用途,较好的是该背衬不屏蔽电力线,使得敞开结构的背衬好于例如聚乙烯或PVC的实心薄膜。当贴合某些表面时,能更好地与表面外形匹配的锥形或异形粘合剂层是更理想的。
本发明的组合物也可用作热收缩管的压敏粘合剂或热熔粘合剂。这些结构物提供能承受热收缩加工时所经历的高温并在冷却后形成环境密封的单一制品。这些材料的流变性、热稳定性、粘性和透明度使它们能适用于该用途。
本发明的组合物也可涂覆在不同的剥离衬里上,即该衬里的一面涂有第一剥离涂层,在反面涂有第二剥离涂层。这两层剥离涂层最好具有不同的剥离值。例如,一层剥离涂层的剥离值可为5克/cm(即从涂层上除去1cm宽的条状材料需要5克力),而第二剥离涂层的剥离值可为15克/cm。该材料可涂覆在有更高剥离值的剥离衬里涂层上。得到的带子可绕成卷。当该带子开卷时,压敏粘合剂粘附于有更高剥离值的剥离涂层上。该带子施用于基材之后,可除去该剥离衬里以露出粘合剂表面备用。
热熔粘合剂是能用于将非粘性表面粘合在一起形成复合物的组合物,当施用于基材上时,热熔粘合剂应具有足够的流动性,使表面完全湿润而不留空白处,即使表面是粗糙的也如此。因此,粘合剂在施用时必须是低粘度的。然而,该粘合剂通常凝结成固体而发挥足够的粘合强度,以在受力情况下仍然能粘附在基材上。
对于热熔粘合剂,从液态向固态的转变可以通过多种方式完成。第一种方式,热熔粘合剂可以是热塑性的,当受热时软化并熔化,当冷却时又会硬化。这样加热产生足够高的流动性以达到充分湿润。或者,该热熔粘合剂可以溶解在溶剂或载体中,将粘合剂的粘度降低至足以达到满意的湿润,当除去溶剂或载体时提高粘合剂的粘度。如果需要,该粘合剂可以是热活化的。
本发明的组合物也可单独用作减震材料,即自由层处理,或与刚性层结合,即作为受约束层处理的一部分。如果将减震材料夹在欲减震的结构物/组件和相对刚性层(如金属薄板)之间使用最为有效。这迫使粘弹性材料在平板振动时发生剪切变形,比材料拉伸和压缩变形时消散更多的能量,与自由层处理时的情况相同。较好的是受约束层结构由一层或多层刚性层和一层或多层减震材料的层压物组成。
受约束层结构可用多种方法制备。在一种方法中,用常规的溶液涂覆法或现有技术中已知的热熔涂覆技术将减震材料层涂覆在剥离衬里上。将得到的粘弹性材料层转移至硬背衬上并与其粘附,由此得到受约束层结构。在另一种方法中,用常规的溶液涂覆法或现有技术中已知的热熔涂覆技术将减震材料层直接涂覆在硬背衬上。在每一种情况中,受约束层结构均随后粘合于需要减震的结构上。该结构可用任何方法联结,只要受约束层通过粘弹性材料界面固定于振动结构上(即不发生机械联结)即可。当该结构随后在内力或外力的影响下震动时,震动即被减弱。
本发明的减震材料的另一用途是在用于双向减震组件,参见Neilsen,E.J。等的“Viscoelastic Damper Overview For Seismic and Wind Applications,”StructuralEngineering Association of California,Tahoe Olympiad,1994年10月。为了结构物减震,双向或大位移减震是将结构(如建筑物)的次音速振动转变成粘弹性材料的剪切变形。在该用途中,在使用温度下具有最大减震能力的材料的剪切储能模量G’较好约为6.9×103-3.45×107Pa,更好为3.45×104-1.4×107Pa,最好为3.45×105-6.9×106Pa,且在使用的温度和频率范围内tanδ尽可能高。在使用的温度和频率范围内,该材料拉伸伸长至少为100%或剪切应变能力至少为100%。
当减震材料具有压敏粘合性能时,该材料通常可粘附于刚性层而不使用另外的粘合剂。然而,有时需要使用高强度粘合剂(如丙烯酸粘合剂、环氧粘合剂或硅氧烷粘合剂,所有这些均是本领域的普通技术人员熟知的)薄层(如20-50μm厚),以将本发明的减震组合物粘结于结构物上。
对于大多数应用,减震材料层的厚度至少为0.01mm至约100mm,更好为0.05至100mm。可用现有技术中已知的任何技术(如喷涂、浸涂、刮刀涂覆或帘式淋涂、或模塑、层压、浇铸或挤塑)施涂该减震材料。
如上所述,刚性层是本发明的受约束层减震结构的主要部分。适用于刚性层的材料的劲度较好至少约为减震材料的劲度(即储能模量)的100倍,该刚性层劲度在伸长时测定。根据刚性层的模量,可通过调节该层的厚度(如从约25微米至5厘米)来改变所需的刚性层的劲度。用于受约束层结构的适宜的刚性材料的实例包括金属(如铁、钢、镍、铝、铬、钴和铜及其合金)以及刚性聚合物材料(如聚苯乙烯、聚酯、聚氯乙烯、聚氨酯、聚碳酸酯、聚酰亚胺和聚环氧化物),纤维增强塑料(如玻璃纤维增强聚酯、陶瓷纤维增强聚酯和金属纤维增强的聚酯),玻璃和陶瓷。
本发明的减震组合物对在很宽的温度和频率范围内要求有效减震的各种用途都是有用的,它还应满足在规定的温度范围内,对最小和/或最大模量的要求。经常要求最大减震范围(即损耗因子接近最大值的点)出现在所需的减震温度和频率范围的中央。设计用于特定用途的最佳减震材料,需要了解聚二有机硅氧烷脲嵌段共聚物、有机聚合物、硅酸酯树脂、任选的聚二有机硅氧烷低聚脲嵌段共聚物和填料以及它们的浓度对减震性能的影响。
根据所用的具体配方,本发明的组合物可以是压敏粘合剂材料、热活化粘合剂、减震材料和非粘性材料。使用非粘性减震材料需要使用粘结剂(即根据具体用途的几何形状,将减震材料粘结于受约束层和/或共振结构上的材料)。
提供压敏粘合性能的减震材料通常如下施用:首先制备一个带结构,它含有一层涂覆在两层衬里之间的减震材料,在至少一层衬里上涂有剥离材料。具有压敏粘合剂特性的本发明减震材料能很好地与聚酯、聚碳酸酯、聚烯烃(如聚乙烯和聚丙烯)以及TEFLONTM粘结,后两类材料是已知通常难以与粘合剂粘附的材料。
通过以下非限定本发明范围的实施例将进一步说明本发明。在实施例中,除非另有说明,否则所有的份和百分数均以重量计。所有的分子量均为数均分子量,单位为克/摩尔。
测试方法
以下测试方法用来表征以下实施例所制备的含聚二有机硅氧烷脲的组分:
机械性能
含聚二有机硅氧烷脲的组分的机械性能测试如下:制得共聚物在四氢呋喃或者50/50的甲苯/异丙醇中的10%的溶液,将溶液倒入陪替氏培养皿中,蒸发溶剂来制备厚度约为0.4至1.5mm的薄膜。
在INSTRONTM1122型拉伸试验机上测试机械性能。该试验是根据ASTMD412-83修改方法进行的。根据Method B(切割环状样品)来制备样品。1型环(圆周长为5.1cm)是通过特殊设计的精密切环机制成的。以比0.5%更高的精度将INSTRONTM模拟输出信号发送给数字式伏特计,数字读数由计算机记录下来。ASTM测试的修改如下:十字头速率为12.7cm/min;测试夹具轴(上下夹片)以每分钟30转的转速同向转动,以保持整个环的应变一致。然后计算得到模量、最大应力和断裂时的伸长率。
180°剥离
在聚酯薄膜上的含聚二有机硅氧烷脲的组分基的压敏粘合剂涂层上覆盖一剥离衬里,并切割成12.7毫米(0.5英寸)×15厘米(6英寸)的长条。去除剥离衬里,将该长条粘附于10厘米(4英寸)×20厘米(8英寸)的清洁的经溶剂洗涤的玻璃试验板,使用2公斤(4-1/2磅)辊在该长条上碾压两次。在室温下对该粘合组件暂停加压约20分钟,使用I-Mass剥离测试仪以30.5厘米/分钟(12英寸/分钟)的分离速率测定180°剥离,数据收集时间为10秒钟。测定两个样品;所给出的粘合力数据是两个样品的平均值。压敏粘合带的180°剥离较好至少约为5.5N/dm(5盎司/英寸),更好至少约为21.8N/dm(20盎司/英寸)。
剪切强度
在聚酯薄膜上的含聚二有机硅氧烷脲的组分基的压敏粘合剂的涂层上覆盖剥离衬里,并切割成12.7毫米(0.5英寸)×15厘米(6英寸)的长条。去除剥离衬里,将该长条粘附于不锈钢板,使每一长条的12.7毫米×12.7毫米的部分与该板牢固接触,带的一端则是自由的。将粘有涂覆长条的板放在支架上,使板与伸出的带的自由端形成178°的角度,在涂覆条的自由端悬挂1kg的负荷以向该自由端施加拉伸力。使用比180°小2°的角度以抵消任何剥离力,从而确保测得的只是剪切力,以便更精确地测定受试带的把握力。将每一条带样品从试验板分离所经过的时间记作剪切强度。除非另有说明,否则所有在此记录为剪切断裂的均指粘合剂的粘结断裂。
减震性能(储能模量和损耗因子)
使用下列方法中的一种制备厚度约750微米的试样:
1)使用孔板设置在250-380微米之间的刮刀涂覆机在50微米的聚对苯二甲酸乙二醇酯剥离衬里上涂覆减震材料的溶液,在70℃将其干燥1分钟,随后在175℃干燥10分钟。通过一个夹辊将获得的数层减震材料层在压力下层压在一起,制得适当厚度的试样,
2)在浅容器的底部将减震材料的溶液直接流延在剥离衬里上,在环境条件下将其至少干燥2天以获得适当厚度的试样,或者
3)在160℃,在两片平行的衬有涂覆含氟硅氧烷的50微米厚聚对苯二甲酸乙二醇酯剥离衬里的铝板之间熔融模压纯(neat)减震材料试样,获得适当厚度的试样。
使用Polymer Laboratories Dynamic Mechanical Thermal Analyzer(DMTA)MarkⅡ及在热扫描期间的多路调制频率技术,测定储能模量(G’)和损耗因子(tanδ),即在频率和温度同时变化时测定性能。在应变设定为1使温度以2℃/分钟的速率从-100℃连续变化至200℃。记录在1.0Hz频率的测量值,每隔约3℃至5℃进行一次测量并内插,得到为满足报告所需的每隔10℃的测定结果。
在这些实施例中,储能模量G’实用范围(utility window)指储能模量为3.45×105Pa至6.9×106Pa的温度范围。损耗因子tanδ实用范围是指损耗因子大于或等于0.4的温度范围。有用的温度范围是指储能模量为3.45×105-6.9×106Pa,且损耗因子tanδ大于0.4的温度范围。当这样表明时,熔体流动是指样品在高温下显示熔体流动。熔体流动对于减震用途通常是不合乎要求的。因此,显示熔体流动的材料必须在低于熔体流动的温度下使用。
在下面的实施例中,所有的多异氰酸酯按购到时的形式使用,用多异氰酸酯供应商给出的多异氰酸酯分子量和酸滴定测得的多胺分子量计算多异氰酸酯、多胺的异氰酸酯对胺的比例。
实施例
在下面的实施例中,所有的二异氰酸酯按购到时的形式使用,用二异氰酸酯供应商给出的二异氰酸酯分子量和酸滴定测得的二胺分子量计算二异氰酸酯对二胺的比例。除非另有说明,否则实施例中所有的份和百分数均以重量计。所有具有不同分子量的聚二有机硅氧烷二胺是使用与美国专利5,512,650所公开的相同方法制得的。所有给出的分子量均是以g/摩尔为单位的数均分子量。
实施例1-4和比较例C1-C2
实施例1-4的薄膜包括不同比例的含聚二有机硅氧烷脲的组分和各种热塑性组分。
在实施例1中,将聚二甲基硅氧烷二胺(分子量50,200)以83.2g/min的速率注入Rerstorff十段、40mm直径、1600mm长的同向旋转双螺杆挤出机的第2段中,将亚甲基二亚环己基-4,4’-二异氰酸酯以0.709g/min的速率注入第4段中。在19.05mm直径的单螺杆挤出机中熔融粒状的有机聚合物MORTHANETM PE44-203(购自Morton International Inc.,Chicago,IL),随后将其以35.0g/min的速率注入Berstorff挤出机的第6段中。Berstorff挤出机的螺杆以100转/分钟的速率旋转。随后将混合物通入计量泵、颈管和152mm宽的平膜模头。Berstorff挤出机和模头的温度分布为:第1段=20℃、第2-5段为150℃、第6-9段为180℃、第10段=200℃;端盖、熔融泵、颈管和模头=180℃;形成的混合物从模头中挤出成250微米(10mil)厚的薄膜。
实施例2的薄膜用与实施例1相同的方法制得,但是以10.5g/min的速率加入聚二有机硅氧烷二胺,以0.0896g/min的速率加入亚甲基二亚环己基-4,4’-二异氰酸酯,以42.0g/min的速率加入有机聚合物。
在实施例3中,以18.9g/min的速率将聚二有机硅氧烷二胺注入Berstorff六段、25mm直径、737.5mm长同向旋转双螺杆挤出机的第2段中,亚甲基二亚环己基-4,4’-二异氰酸酯以0.0985g/min的速率注入第2段中。在25.4mm直径的单螺杆挤出机中熔融粒状的有机聚合物KRATONTM1107(购自Shell Chemical Co.,Houston,TX)并随后以18.7g/min的速率注入Berstorff挤出机的第4段中。Berstorff挤出机的螺杆以75转/分钟的速率旋转。随后将混合物通入计量泵、颈管和152mm宽的平膜模头。Berstorff挤出机的温度分布为:第1段=29℃、第2段=80℃、第3段=150℃、第4段=180℃、第5、6段、端盖、熔融泵、颈管和模头=200℃;形成的混合物从模头中挤出成60微米(2.4mil)厚的薄膜。
实施例4的薄膜用与实施例3相同的方法制得,但是以9.5g/min的速率加入聚二有机硅氧烷二胺,以0.0450g/min的速率加入亚甲基二亚环己基-4,4’-二异氰酸酯,以43.2g/min的速率加入有机聚合物。
比较例C1的薄膜用与实施例1相同的方法制得,但是以42g/min的速率仅加入有机聚合物,薄膜的厚度为200微米(8mil)。
比较例C2的薄膜用与比较例C1相似的方法制得,但是用有机聚合物KRATONTM1107代替MORTHANETMPE44-203,加料速率为50g/min,薄膜厚240微米(9.5mil)。
测试所有薄膜的拉伸模量、断裂应力和断裂伸长率。结果列于表1a和表1b。
表1a
| 实施例 | 模量(MN/m2) | 断裂应力(MN/m2) | 断裂伸长率(%) |
| 1 | N/A | N/A | N/A |
| 2 | 7.82 | 2.53 | 160 |
| C1 | 16.1 | 5.48 | 360 |
| 3 | 0.39 | 2.7 | 1250 |
| 4 | 0.62 | 8.4 | 1290 |
| C2 | 0.8 | 6.04 | * |
(N/A-未测试)(*-未观察到断裂)
将两种压敏粘合带(#375盒密封带和#845书籍粘合带,均购自美国3M公司)置于聚二甲基硅氧烷聚脲嵌段共聚物混合物剥离表面上并用1.13kg(2.5磅)的压辊碾压。在50%相对湿度和22.2℃下将试样至少保持4小时后以229cm/min(90英寸/min)的速度和180°的剥离角测定剥离值。随后将剥离的粘合带置于用乙酸乙酯彻底清洗的玻璃上。用相同的方法测定与玻璃的重复粘性值。随后用相同的方法测定与玻璃的重复粘性值。将所述与玻璃的重复粘性值与同样的粘合带在与比较例C1的试样接触后与玻璃的粘性进行比较。与比较例C1试样接触后粘合带的粘性相比,计算粘合带与剥离层接触后对玻璃的重复粘性的保留百分数。初始剥离值和重复粘性的保留百分数列于表1b。
表1b
| 实施例 | 375盒密封带 | 345书籍粘合带 | ||
| 剥离(N/dm) | 重复粘性% | 剥离(N/dm) | 重复粘性% | |
| 1 | 0.4 | 93 | 0.8 | 88 |
| 2 | 5.8 | 94 | 14 | 98 |
| C1 | 11 | 100 | 26 | 100 |
这些新的含聚二甲基硅氧烷脲的混合物具有适用的拉伸和剥离性能。实施例1和2表明与比较例1相比具有更低的与压敏粘合剂的粘性,但仍保持优良的重复粘性值。实施例4表明与比较例2的纯聚合物相比它具有更高的断裂应力。
实施例5-7和比较例C3-C4
实施例5-7的增粘薄膜含有含聚二有机硅氧烷脲的组分和热塑性组分,这两种组分或其中之一是增粘的。
实施例5的增粘薄膜用与实施例1相同的方法制得,但是使用不同的有机聚合物和不同的加工条件。以17.5g/min的速率将GE Silicones SR 1000增粘树脂加入第1段,以17.5g/min的速率将聚二甲基硅氧烷二胺注入第2段,以0.100g/min的速率将亚甲基二亚环己基-4,4’-二异氰酸酯注入第4段,以35.0g/min的速率通过单螺杆挤出机将KratonTM1107加入双螺杆挤出机的第6段。将双螺杆挤出机、齿轮泵、颈管和模头的温度设定在180℃,双螺杆挤出机的螺杆转速设定为40转/分钟。将形成的混合物挤出在50微米厚的聚酯基片上形成50微米厚的薄膜。
在实施例6中,用与实施例5相同的方法制得增粘薄膜,但是以35.0g/min的速率使用热熔粘合剂HL 2542X(KratonTM基压敏粘合剂,购自H.B.Fuller,St.Paul,MN)代替KratonTM,双螺杆挤出机的螺杆转速为120转/分,双螺杆挤出机的第2段-第6段的温度为160℃。在剥离衬里之间挤出50微米厚的混合物薄膜,随后层压数次形成总厚1mm的层压物用于动态机械试验。
在实施例7中,用与实施例5相同的方法制得增粘薄膜,但是不使用SR1000,以35.0g/min的速率加入聚二甲基硅氧烷二胺,以0.201g/min的速率加入亚甲基二亚环己基-4,4’-二异氰酸酯,双螺杆挤出机的螺杆转速为40转/分钟。如实施例6制得1mm厚的薄膜用于动态机械试验。
用与实施例5相同的方法制得比较例C3的增粘薄膜,但是不加入有机聚合物,并将其余三种物流的流动速率加倍,挤出机螺杆的旋转速率为120转/分钟。如实施例6制得1mm厚的薄膜用于动态机械试验。
用与实施例6相同的方法制得比较例C4的增粘薄膜,但是仅加入HL2542X,速率为50.0g/min。如实施例6制得1mm厚的薄膜用于动态机械试验。
试验所有涂覆在聚酯上的增粘薄膜的180°剥离和剪切强度。结果列于表2。
表2
| 实施例 | 180°剥离(N/m2) | 剪切强度(min) |
| 5 | 36 | 3 |
| C3 | CF | 8 |
| 6 | 77 | 10 |
| C4 | 142 | >100 |
| 7 | 15 | 1 |
这些含聚二甲基硅氧烷脲的混合物表现出适用的剥离和剪切性能。具体地说,实施例6表明向聚二甲基硅氧烷脲压敏粘合剂加入另一种压敏粘合剂,增强了聚二甲基硅氧烷脲压敏粘合剂的剪切性能。
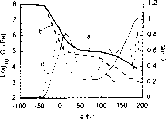
试验挤出于剥离衬里之间随后再层压的试样的减震特性。测得储能模量G’和损耗因子tanδ,结果分别列于图6-图9。
这些新的含聚二甲基硅氧烷脲的混合物具有适用的能量缓冲性能。图7和图9表明通过将增粘的聚二甲基硅氧烷脲或聚二甲基硅氧烷脲弹性体与增粘的有机体系相混合,可形成大范围的阻尼性能。例如,可将增粘的有机体系的适用的温度范围(tanδ>0.4)扩大约20℃而不会对储能模量有不良影响。
现在参照附图,图6表示比较例3储能模量(粗线)和tanδ(细线)与温度和频率的关系。实线表示10Hz,虚线表示0.1Hz。
图7表示实施例6储能模量(粗线)和tanδ(细线)与温度和频率的关系。实线表示10Hz,虚线表示0.1Hz。
图8表示比较例4储能模量(粗线)和tanδ(细线)与温度和频率的关系。实线表示10Hz,虚线表示0.1Hz。
图9表示实施例7储能模量(粗线)和tanδ(细线)与温度和频率的关系。实线表示10Hz,虚线表示0.1Hz。
实施例8-12
实施例8-12的增粘薄膜含有具有不同分子量的增粘的含聚二有机硅氧烷脲的组分和热塑性组分。
实施例8的增粘薄膜用与实施例6相同的方法制得,但是使用不同的有机聚合物、不同的流动速率和加工条件。以15.0g/min的加料速率用有机聚合物ENGAGETM8200(购自Dow Chemical Co,Midland,MI)代替KRATONTM1107.分别以30.0g/min、30.0g/min和0.172g/min的加料速率加入SR1000、聚二甲基硅氧烷二胺和亚甲基二亚环己基-4,4’-二异氰酸酯。挤出机的螺杆转速为120转/分钟。在聚酯基片上制得仅50微米厚的增粘薄膜层。
在实施例9中,用与实施例8相同的方法制得增粘薄膜,但是分别以17.5g/min的速率加入SR1000和聚二甲基硅氧烷二胺,以0.100g/min的速率加入亚甲基二亚环己基-4,4’-二异氰酸酯,以35.0g/min的速率加入Engage8200。
在实施例10中,以30.1g/min的速率将购自GE Silicones的MQ增粘树脂(1170-002,批号EF002)加入Berstorff十段、40mm直径、1600mm长同向旋转双螺杆挤出机的第1段中。以29.9g/min的速率将聚二甲基硅氧烷二胺(分子量34,800)注入第2段中,以0.236g/min的速率将亚甲基二亚环己基-4,4’-二异氰酸酯加入第5段中。Berstoff挤出机的温度分布为:第1段=85℃、第2-5段=60℃、第6段=120℃,第7-10段、端盖和熔融泵=180℃。Berstorff挤出机的螺杆转速为100转/分。将增粘的含聚二有机硅氧烷脲的组分收集在剥离衬里上并冷却。将3重量份该增粘的含聚二有机硅氧烷脲的组分与1重量份KRAT0NTM1107一起加入19.05mm直径的单螺杆挤出机中。第1段用水冷却,第2段和第3段及127mm宽的平膜模头的温度为200℃。螺杆转速为40转/分。形成的薄膜厚50微米。
用与实施例10相同的方法制得实施例11的增粘薄膜,但是使用不同的聚二有机硅氧烷二胺和流动速率。以44.0g/min的速率使用聚二甲基硅氧烷二胺(分子量105,000)。以53.8g/min的速率加入MQ树脂,以0.121g/min的速率加入亚甲基二亚环己基-4,4’-二异氰酸酯。
在实施例12中,用与实施例10相同的方法制得增粘薄膜,但是使用有机聚合物PetrotheneTM3150(购自Quantum Chemical Corp.,Cincinnati,OH的高密度聚乙烯)。
试验所有增粘薄膜的180°剥离和剪切强度。结果列于表3。
表3
| 实施例 | 180°剥离(N/m2) | 剪切强度(min) |
| 8 | 58 | 3 |
| 9 | 53 | 2 |
| 10 | 36 | 97 |
| 11 | 61 | 260 |
| 12 | 7 | 220 |
这些含聚二甲基硅氧烷脲的混合物表明了适用的剥离和剪切性能。实施例8和9表明在混合物中高达50%的聚乙烯基的热塑性弹性体仍能形成优良的剥离性能。实施例10和11表明在制造具有优良剥离性能和剪切性能的含聚二甲基硅氧烷脲的混合物时可使用宽分子量范围的硅氧烷二胺。含聚乙烯热塑性聚合物的实施例12具有适用的剥离和剪切性能。
实施例13-14和比较例C5-C6
用下列方法制得增粘的聚二甲基硅氧烷脲。以58.1g/min的速率将MQ增粘树脂加入与比较例1相同的40mmBerstorff双螺杆挤出机的第1段中。以58.1g/min的速率将分子量为52,000的聚二甲基硅氧烷二胺注入第2段中。以0.313g/min的速率将亚甲基二亚环己基-4,4’-二异氰酸酯加入第8段中。使二异氰酸酯的加料线轻微地掠过挤出机螺杆,使螺杆每一转动均有少量二异氰酸酯带入挤出机。螺杆的转速为300转/分钟,挤出机的温度分布为:第1段=28℃、第2段=100℃、第3段=71℃、第4段=77℃、第5段=87℃、第6段=78℃、第7段=85℃、第8段=77℃、第9段=117℃、第10段=144℃、端盖=150℃、熔融泵=180℃。随后将该增粘的聚二甲基硅氧烷脲加入实施例5的双螺杆挤出机的第8段中,该挤出机的第8段-第10段、端盖、熔融泵、颈管和模头的温度为180℃。螺杆旋转速度为40转/分。如实施例5那样将该增粘的聚二甲基硅氧烷脲以120微米的厚度涂覆在聚酯上。压敏粘合剂的试验结果列于表4。
实施例13
如下制得天然橡胶基压敏粘合剂体系:将35.2重量份CV60橡胶(天然橡胶,购自Goodyear Chemical,Akron,OH)、35.2份EscorezTM1304(Exxon ChemicalCorp.,Houston,TX)、28.8份SYNPOLTM1011A(Ameripol Synpol)与0.7份IRGANOXTM1010(Ciba-Geigy,Hawthorne,NY)相混合。以90重量份增粘的聚二甲基硅氧烷脲对10重量份天然橡胶粘合剂的比例将比较例5的增粘的聚二甲基硅氧烷脲与天然橡胶基粘合剂干混。随后用与比较例6相同的方法将该增粘的共混物加入双螺杆挤出机的第8段中并涂覆在聚酯背衬上。压敏粘合剂的试验结果列于表4。
实施例14
如实施例13制得增粘的聚合物混合物,但是使用10重量份增粘的聚二甲基硅氧烷脲和90重量份天然橡胶基粘合剂。双螺杆挤出机和模头设定在下列温度:第8段-第10段、端盖、熔融泵和径管=160℃,模头=180℃。螺杆转速为50转/分钟。压敏粘合剂试验结果列于表4。
比较例6
如实施例13制得增粘的聚合物混合物,但是使用100重量份天然橡胶基粘合剂。双螺杆挤出机和模头设定在下列温度:第8段-第10段、端盖、熔融泵和径管=140℃,模头=180℃。螺杆转速为40转/分钟。压敏粘合剂试验结果列于表4。
对所有的增粘薄膜均试验其180°剥离和剪切强度。结果列于表4。
表4
| 实施例 | 180°剥离(N/m2) | 剪切强度(min) |
| C5 | 91 | 31 |
| 13 | 105 | 27 |
| 14 | 40 | 20 |
| C6 | 73 | 44 |
由表4的数据可见,聚二甲基硅氧烷脲、热固性聚合物和增粘剂的混合物具有很适用的剥离和剪切性能。与比较例相比实施例13具有异常高的剥离值,表明在某些体系中获得了协同的性能。
实施例15-17和比较例C8-C9
下列方法表明,本发明粘合剂能控制多层透皮给药组件的给药速率。
实施例15-17和比较例C6-C9的透皮给药组件分别使用实施例5-7和比较例C4-C5的增粘薄膜作为速率控制粘合剂。在各种情况下,在两层剥离衬里之间制得试样,并在将该试样用于下列方法之前除去其中的一层剥离衬里。
各个试验片由4层组成:背衬、含药物的第一粘合剂层、控制速率的第二粘合剂层和剥离衬里。将丙烯酸酯粘合剂共聚物((57.5/39/3.5)丙烯酸异辛酯/丙烯酸2-羟基乙酯/ELVACITETM(ICI丙烯酸类)1020聚甲基丙烯酸基酯大单体,在乙酸乙酯中的50%固体)和苯巴比妥组合在一起并混合成均匀的涂覆制剂。将该制剂涂覆在背衬(1109 SCOTCHPAKTMtan,聚酯薄膜层压物,购自美国3M公司)上,随后在43℃干燥15分钟。形成的涂层含5重量%苯巴比妥且厚度为5mil(127微米)。将露出的表面层压在载带在剥离衬里上的速率控制粘合剂层上。从得到的层压物上冲切试验片(圆形,5cm2)。
为防止试验片的周边释放药物,在各个试验片上同心地排列一层粘合剂覆盖层。将粘合剂覆盖层(圆形、25cm2、涂覆在背衬上的1mil(25微米)聚异丁烯层)层压在试验片的背衬上,使得该试验片和覆盖层同心地排列在一起。从试验片上除去剥离衬里。将环状覆盖层(25cm2、内径22mm,涂覆在背衬上的1mil(25微米)聚异丁烯层)置于试验片/覆盖层的层压物的中央,随后将粘合剂表面层压在一起,形成环绕试验片周围的密封。将剥离衬里放回试验片,随后以试验片据中的方式冲切整个组件(圆形12.5cm2)。将该组件加热密封在金属薄片盒中并使之平衡8天。
随后将该组件从金属薄片盒中取出并用双面涂覆的粘合带将其一端固定在玻璃板上,使得该组件的背衬与双面涂覆的粘合带直接接触。从试验片上除去剥离衬里。将该玻璃载片置于带磁力搅拌器的120ml长形大口玻璃瓶中。将6升HPLC级水、2.2835g磷酸氢二钠一水合物、9.7538g磷酸二氢钠七水合物、和46.4502g氯化钠混合在一起制得释放液。向该释放液中加入叠氮化钠(0.04%)以便在试验过程中抑制细菌的生长。将100ml32℃的释放液加入所述玻璃瓶中,使试验片完全浸没在该剥离液中。盖上玻璃瓶,随后将其置于32℃的控温室中。在整个实验过程中均搅拌该释放液。
在规定的时刻(1小时、8小时、24小时、72小时和168小时),打开盖子,取出1.0ml释放液试样置于HPLC试样瓶中。使用高性能逆相液相色谱法(Waters LC1 Module Plus,柱:15cm×4.6mm内径的Supelcosil LC-ABZ,5微米粒径;流动相:75%25mM磷酸氢二钾缓冲液/25%丙烯腈(v/v);流动速率:2.0ml/min;检测器:UV,254nm 0.005AUFS;运行时间:10分钟;注射体积20微升)定量测定试样中苯巴比妥的含量。
使用下面计算式得到释放百分数:
式中,
Ri=在时刻“i”试样中释放的苯巴比妥百分数
i=时刻的序数(数值:1,2,3…n)
Ci=在时刻i,HPLC分析的试样浓度(微克/毫升)
C0=0
T.C.=以微克/cm2为单位的理论苯巴比妥含量
S.A.=试验片以cm2为单位的表面积。
下表列出了用于速率控制层的粘合剂的实施例号、层厚和在各时刻释放的累积释放的百分数。各个数据均是三个单独的试验片测量的平均值。空缺表示在该时间点未取样。
表5
苯巴比妥的释放速率百分数
| 实施例 | 厚度(微米(mils)) | 1小时 | 8小时 | 24小时 | 72小时 | 168小时 |
| C8 | 51.0(2.0) | 0 | 0 | 0 | 0 | 0.6 |
| 21 | 51.0(2.0) | 0 | 0 | 1.3 | 7 | 18 |
| 22 | 48.3(1.9) | 0 | 0 | 9 | 9 | 20 |
| 23 | 55.9(2.2) | 0 | 0 | 0 | 0.6 | 2.5 |
| C9 | 51.0(2.0) | - | - | - | 14 | 33 |
上表的结果表明,本发明粘合剂能影响苯巴比妥从多层透皮给药组件中释放的速率。
实施例18-19和比较例C10
粘合剂试样的制备:
在实施例18中,将聚二甲基硅氧烷二胺(分子量52,900)以33.5g/min的速率注入Rerstorff十段、40mm直径、1600mm长的同向旋转双螺杆挤出机的第1段的后部中。将MQ增粘树脂粉以40.3g/min的速率加入第1段的前部。将亚甲基二亚环己基-4,4’-二异氰酸酯以0.155g/min的速率注入第4段中。挤出机的温度分布为:第1段=20℃、第2段和第3段为50℃、第4段为60℃、第5段为100℃、第6段为130℃、第7段为160℃、第8段为180℃、第9段、第10段和端盖为160℃。螺杆旋转速度为80转/分钟。对第8段抽真空。以线料收集该材料并在空气中冷却之。
在这些实验中使用两种全扩链的RExSi粘合剂。实施例18对每份含聚二甲基硅氧烷脲的弹性体用1.2份MQ树脂增粘,实施例19对每份含聚二甲基硅氧烷脲的弹性体仅用1份MQ树脂增粘。未加入阻燃添加剂。
将250g含聚二甲基硅氧烷脲的弹性体粘合剂与250g阻燃的嵌段共聚物粘合剂试样(HL2886,购自H.B.Fuller,St.Paul,MN)熔融混合(用布拉本德仪的加热(150℃)混合筒附件)2-3分钟。然后使部分混合的试样通过25mm加热至170℃并以50rpm运转的锥形挤出机,使之混合均匀。
比较例10是一种阻燃嵌段共聚物粘合剂(HL2886,购自H.B.Fuller,St.Paul,MN)。
使用单螺杆Haake挤出机和冷拉模将该热熔粘合剂以250微米(10mil)的厚度涂覆在SJ-3419(平背衬的阻燃钩形带,购自3M)上。挤出机料筒具有三个控制温度分别设定在室温、121℃(250°F)和135℃(275°F)。模头温度设定在149℃(300°F)。将挤出机设定在50rpm,将料筒保持满料,调节料片速度以形成250微米(10mil)厚的均匀涂层。冷却的粘合剂无需任何附加的固化步骤,因为两种组分已物理地交联。
试验该涂覆粘合剂的试样在30cm/min剥离速率下的90°剥离和在涂覆环氧的不锈钢板(BMS10-11,购自Boeing,Seattle,WA)上的90°蠕变(如下所述)。还试验试样的阻燃性。
直立燃烧试验
在5cm(2”)宽×17.5cm(7”)长的试样的边缘将其垂直支承。将该试样置于Bunsen燃烧器上,调节标称内径1.2cm管使火焰高3.75cm(1.5”)。试样的下边缘必须距燃烧器的顶端1.8cm(3/4”)。施加火焰60秒并熄火。记录燃烧时间(试样持续燃烧的时间)、燃烧长度和液滴燃烧时间(滴下物持续燃烧时间)。最少试验三个试样并将结果取平均。
蠕变试验
用2.5kg的压辊碾压8次将2.54宽×15.2cm长的试样层压在涂覆环氧的不锈钢板(BMS10-11,购自Boeing,Seattle,WA)上。在室温将试样放置(dwell)24小时。将试验板固定在水平位置,拉去一部分粘合带并施加908g负荷。在试验板上以90°拉去粘合带1小时后,测定剥离前缘的水平位移。
表6
| 实施例 | 90°剥离(N/dm) | 蠕变(cm) | 燃烧试验 |
| C10 | 70.5 | 1.04 | 12.79cm燃烧长度,无燃烧下滴物 |
| 18 | 120.4 | 1.9 | 13.3燃烧长度,约8秒燃烧下滴物 |
| 19 | 107.5 | 1.58 | 15.8cm燃烧长度,略超过5秒的燃烧下滴物 |
用非阻燃的RExSi粘合剂代替50%阻燃的嵌段共聚物粘合剂增加了共混料的剥离性能,未对燃烧试验性能产生重大影响。尽管燃烧试验性能稍有下降,但是通过减少50%阻燃剂可明显减少产生的毒性烟雾的量。
实施例20-22
实施例20-22的增粘薄膜含有不同比例的增粘的含聚二有机硅氧烷脲的组分和不同的有机聚合物。
先如下制得增粘的含聚二有机硅氧烷脲的组分:将聚二甲基硅氧烷二胺(分子量52,900)以15.5g/min的速率加入Leistritz十段反向旋转的完全啮合的双螺杆挤出机的第1段,将MQ增粘树脂粉以18.4g/min的速率加入第2段。将二环己基甲烷-4,4’-二异氰酸酯以0.072g/min的速率加入开口第6段中,使加料线擦过螺杆。各个160mm长的段的温度分布为:第1段=25℃、第2段的温度不控制、第3段为35℃、第4段和第5段为50℃、第6段为100℃、第7段为170℃、第8段至第10段为180℃、端盖为170℃。螺杆旋转速度为50转/分钟。以线料收集该材料并在空气中冷却之。
在实施例20中,将9g增粘的含聚二有机硅氧烷脲的组分和34g丙烯酸类压敏粘合剂组分(95重量%丙烯酸异辛酯/5重量%丙烯酸,根据美国专利RE24,906(Ulrich)进行水乳液聚合并干燥,该专利在此引为参考)加入在150℃运行的50克BRABENDERTMσ形浆式混合机中。将组分在约50rpm混合3分钟。将约1-5g混合的组合物置于一张有剥离涂层的纸张和75微米厚的双取向聚对苯二甲酸乙二醇酯薄膜之间制得压敏膜。随后将该组件置于具有两片尺寸为152mm(6英寸)×152mm(6英寸)钢板的液压平板式压机中的铝板之间。在138℃(280°F)施加约28MPa(4000psi)并使用12秒停留时间使得压敏粘合剂涂层具有约100微米(4mil)的厚度。
用与实施例20相同的方法制得实施例21和22的增粘薄膜,但是增粘的含聚二有机硅氧烷脲的组分和聚合物组分的量分别为13.5g/31.5g和18g/27g。
试验增粘薄膜与玻璃的180°剥离。结果列于表7。
表7
实施例 180°剥离
20 10.4
21 9.7
22 17.2
在不偏离本发明精神和范围的前提下的各种改进和变化对本领域的普通技术人员是显而易见的,本发明不限于上述仅用于说明目的的内容。
Claims (22)
1.一种混合物,它包括(a)除聚二有机硅氧烷流体以外的至少一种热塑性聚合物、弹性热固性聚合物及其混合物,和(b)具有软的聚二有机硅氧烷单元,硬的多异氰酸酯残基单元和任选的软和/或硬的有机多胺残基单元和端基的聚合物。
2.如权利要求1所述的混合物,其特征在于所述硬的多异氰酸酯残基和硬的多胺残基在含聚二有机硅氧烷脲的组分中的含量小于50重量%。
3.如权利要求1所述的混合物,其特征在于所述含聚二有机硅氧烷脲的组分是(a)至少一种多胺,该多胺包括至少一种聚二有机硅氧烷二胺,或至少一种聚二有机硅氧烷二胺和至少一种有机多胺的混合物与(b)至少一种多异氰酸酯的反应产物。
4.如权利要求3所述的混合物,其特征在于所述含聚二有机基硅氧烷脲的组分可用下列重复单元表示:
其中:
各个R分别是一个具有1-12个碳原子的烷基部分,该烷基部分可被例如三氟烷基或乙烯基类基团、乙烯基或由式-R2(CH2)aCH=CH2表示的高级烯基所取代,其中R2是-(CH2)b-或-(CH2)cCH=CH-,a是1、2或3;b是0、3或6;c是3、4或5;具有6-12个碳原子的环烷基部分,该环烷基部分可被烷基、氟烷基和乙烯基所取代;或芳基部分,且可被烷基、环烷基、氟烷基和乙烯基所取代;或R是全氟烷基、含氟基团或含全氟醚基团;
各个Z是一个具有6-20个碳原子的亚芳基或亚芳烷基、具有6-20个碳原子的亚烷基或亚环烷基的多价基团;
各个Y是一个分别为具有1-10个碳原子的亚烷基、具有6-20个碳原子的亚芳烷基或亚芳基的多价基团;
各个D分别选自氢、具有1-10个碳原子的烷基、苯基、以及包括B或Y以形成杂环的环结构基团;
B是一个多价基团,选自亚烷基、亚芳烷基、亚环烷基、亚苯基、聚环氧烷,及其共聚物和混合物;
m是0-1000的数;
n是等于或大于1的数;和
p是为5或更大的数。
5.如权利要求4所述的混合物,其特征在于含聚二有机硅氧烷脲的组分的R部分中至少50%为甲基,其余部分为单价烷基或取代的烷基、亚烯基、苯基或取代的苯基;
6.如权利要求4所述的混合物,其特征在于Z是2,6-亚甲苯基,4,4’-亚甲基二亚苯基、3,3’-二甲氧基-4,4’-亚联苯基、四甲基-间亚二甲苯基、4,4’-亚甲基二亚环己基、3,5,5-三甲基-3-亚甲基亚环己基、1,6-亚己基或1,4-亚环己基。
7.如权利要求1所述的混合物,其特征在于所述含聚二有机硅氧烷脲的组分包括软的聚二有机硅氧烷单元、硬的多异氰酸酯残基单元,任选的软的和/或硬的有机多胺残基单元,和端基,其中所述多异氰酸酯残基是失去-NCO基团的多异氰酸酯,所述多胺残基是失去-NH2基团的多胺,所述多异氰酸酯残基通过脲键与多胺残基或聚二有机硅氧烷残基相连,所述端基是非官能的封端基团或官能的封端基团。
8.如权利要求1所述的混合物,其特征在于它还包括至少一种增粘材料。
9.如权利要求8所述的混合物,其特征在于所述增粘材料是硅酸酯树脂或有机增粘剂。
10.一种减震受束层结构,包括至少一种刚性基片和至少一层含权利要求8所述的增粘组合物的层,其中所述增粘组合物是固定在基片上的。
11.一种减震复合物,包括挠性基片和涂覆该基片上的权利要求8所述的组合物。
12.一种双向减震受束层结构,包括至少两层刚性部件和权利要求8所述的增粘组合物,各个刚性部件的一个主表面贴近另一个刚性部件的主表面,并且部件间的间隔小,其特征在于所述增粘组合物位于小间隔的刚性部件之间并粘合在刚性部件上。
13.一种压敏粘合剂涂覆制品,包括挠性基片和涂覆在该基片上的权利要求8所述的增粘组合物。
14.一种压敏粘合剂制品,包括一层用权利要求1所述组合物制得的层,该层的一个表面无粘性,另一个表面具有粘性。
15.一种有剥离涂层制品,包括挠性基片和涂覆于该基片上的权利要求1所述的组合物。
16.一种包括有机聚合物和含聚二有机硅氧烷脲嵌段共聚物的混合物的制备方法,包括下列步骤:
(a)向反应容器连续地提供至少一种含聚二有机硅氧烷脲的组分和至少一种有机聚合物,
(b)将这些组分混合成混合物,和
(c)从反应容器中送出该混合物。
17.如权利要求16所述的方法,其特征在于所述混合在基本无溶剂的条件下进行。
18.一种包括有机聚合物和含聚二有机硅氧烷脲共聚物的混合物的制备方法,包括下列步骤:
(a)连续地向反应器中提供反应组分和至少一种不与胺官能团或异氰酸酯官能团反应的有机聚合物,其中所述反应组分包括至少一种多异氰酸酯和至少一种多胺,所述多胺包括至少一种聚二有机硅氧烷胺或者至少一种聚二有机硅氧烷胺和至少一种有机胺的混合物;
(b)将这些组分混合;
(c)使反应组分反应,形成聚二有机硅氧烷脲嵌段共聚物;
(d)从反应器中送出混合物。
19.一种压敏粘合带的制备方法,包括下列步骤:
(a)向反应容器连续地提供至少一种有机聚合物、至少一种聚二有机硅氧烷脲组分和至少一种增粘材料;
(b)将这些组分混合成压敏粘合剂,和
(c)从反应容器中送出该粘合剂,并将该压敏粘合剂与成膜聚合物树脂共挤出。
20.一种压敏粘合带的制备方法,包括下列步骤:
(a)连续地向反应器中提供反应组分、至少一种不与胺官能团或异氰酸酯官能团反应的有机聚合物和至少一种增粘材料,其中所述反应组分包括至少一种多异氰酸酯和至少一种多胺,所述多胺包括至少一种聚二有机硅氧烷胺或者至少一种聚二有机硅氧烷胺和至少一种有机胺的混合物;
(b)将这些组分混合;
(c)使反应组分反应,形成聚二有机硅氧烷脲嵌段共聚物;
(d)从反应器中送出压敏粘合剂,并将该压敏粘合剂与成膜聚合物树脂共挤出。
21.用权利要求16的方法制得的权利要求1的混合物。
22.用权利要求18的方法制得的权利要求1的混合物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/735,836 US6846893B1 (en) | 1996-10-23 | 1996-10-23 | Polymer mixtures containing polydiorganosiloxane urea-containing components |
| US08/735,836 | 1996-10-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN1234051A true CN1234051A (zh) | 1999-11-03 |
Family
ID=24957391
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN97199049A Pending CN1234051A (zh) | 1996-10-23 | 1997-09-25 | 有含聚二有机硅氧烷脲的组分的聚合物混合物 |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US6846893B1 (zh) |
| EP (1) | EP0934360B1 (zh) |
| JP (1) | JP2001508818A (zh) |
| KR (1) | KR20000052713A (zh) |
| CN (1) | CN1234051A (zh) |
| AU (1) | AU4501097A (zh) |
| BR (1) | BR9712551A (zh) |
| CA (1) | CA2268113A1 (zh) |
| DE (1) | DE69734721T2 (zh) |
| ES (1) | ES2251033T3 (zh) |
| WO (1) | WO1998017726A1 (zh) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102449087A (zh) * | 2009-05-28 | 2012-05-09 | 宝洁公司 | 防粘片材料 |
| CN101346448B (zh) * | 2005-12-23 | 2012-10-03 | 3M创新有限公司 | 粘合剂组合物 |
| CN101701131B (zh) * | 2009-09-08 | 2013-02-13 | 南京南大波平电子信息有限公司 | 一种智能主动型抗结冰涂层材料及其制备方法和应用 |
| CN104781354A (zh) * | 2012-07-03 | 2015-07-15 | 3M创新有限公司 | 硅氧烷基管涂层 |
| CN108384453A (zh) * | 2018-03-21 | 2018-08-10 | 广州市维思涂料科技有限公司 | 一种用于金属彩钢板表面的水性涂料及其制备方法 |
| CN109476845A (zh) * | 2016-07-13 | 2019-03-15 | 瓦克化学股份公司 | 含有硅氧烷-有机共聚物的聚合物组合物 |
| CN109790386A (zh) * | 2016-10-07 | 2019-05-21 | 瓦克化学股份公司 | 含有硅氧烷-有机共聚物的聚合物组合物 |
| CN111344373A (zh) * | 2017-10-09 | 2020-06-26 | 3M创新有限公司 | 用于微型扬声器膜片的阻尼粘合剂层 |
Families Citing this family (63)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7749111B1 (en) | 1999-01-11 | 2010-07-06 | Lifetime Products, Inc. | System and method for bonding an acrylic surface to a frame |
| US6569521B1 (en) | 2000-07-06 | 2003-05-27 | 3M Innovative Properties Company | Stretch releasing pressure sensitive adhesive tape and articles |
| US7012110B2 (en) | 2001-12-18 | 2006-03-14 | 3M Innovative Properties Company | Silicone pressure sensitive adhesives prepared using processing aids, articles, and methods |
| US7090922B2 (en) | 2001-12-18 | 2006-08-15 | 3M Innovative Properties Company | Silicone priming compositions, articles, and methods |
| US6730397B2 (en) | 2001-12-18 | 2004-05-04 | 3M Innovative Properties Company | Silicone pressure sensitive adhesives, articles and methods |
| NZ569215A (en) * | 2003-03-11 | 2009-05-31 | Fuller H B Licensing Financ | A moisture curable sealant composition |
| US8080308B2 (en) * | 2003-03-11 | 2011-12-20 | H.B. Fuller Company | One-part moisture curable hot melt silane functional poly-alpha-olefin sealant composition |
| US6930148B2 (en) * | 2003-04-08 | 2005-08-16 | Texas Petrochemicals Lp | Enhanced polyisobutylene modified hot melt adhesive formulation |
| US7666338B2 (en) * | 2004-03-25 | 2010-02-23 | Ppg Industries Ohio, Inc. | Focused heat extrusion process for manufacturing powder coating compositions |
| US7605194B2 (en) | 2003-06-24 | 2009-10-20 | Ppg Industries Ohio, Inc. | Aqueous dispersions of polymer-enclosed particles, related coating compositions and coated substrates |
| US7172800B2 (en) * | 2003-11-03 | 2007-02-06 | Material Sciences Corporation | Sheet molding compound damper component, and methods for making and using the same |
| US7407709B2 (en) * | 2003-12-22 | 2008-08-05 | 3M Innovative Properties Company | Silicone pressure sensitive adhesive and articles |
| WO2006014273A1 (en) * | 2004-07-02 | 2006-02-09 | Polyone Corporation | Soil-resistant thermoplastic elastomer compositions and related methods |
| JP5086235B2 (ja) * | 2005-03-09 | 2012-11-28 | クティセンセ アクティーゼルスカブ | マイクロ電子システムを内部に埋め込んだ三次元接着デバイス |
| US20070149745A1 (en) * | 2005-12-23 | 2007-06-28 | 3M Innovative Properties Company | Polydiorganosiloxane-containing materials with oxalylamino groups |
| US7501184B2 (en) | 2005-12-23 | 2009-03-10 | 3M Innovative Properties Company | Polydiorganosiloxane polyoxamide copolymers |
| EP1963899B1 (en) | 2005-12-23 | 2013-08-28 | 3M Innovative Properties Company | Films including thermoplastic silicone block copolymers |
| JP5015953B2 (ja) * | 2005-12-23 | 2012-09-05 | スリーエム イノベイティブ プロパティズ カンパニー | 熱可塑性シリコーンブロックコポリマー類を含む多層膜 |
| US7560166B2 (en) * | 2005-12-28 | 2009-07-14 | 3M Innovative Properties Company | Adhesive article, composite article, and methods of making the same |
| US20080058460A1 (en) * | 2006-09-05 | 2008-03-06 | Dow Corning Corporation | Silicone hot melt additive for thermoplastics |
| US20080058449A1 (en) * | 2006-09-05 | 2008-03-06 | Dow Corning Corporation | Silicon hot melt additive for fluoroplastics |
| US8101702B2 (en) * | 2007-01-12 | 2012-01-24 | Dow Corning Corporation | Silicone-containing composition |
| US8334037B2 (en) | 2007-05-11 | 2012-12-18 | 3M Innovative Properties Company | Multi-layer assembly, multi-layer stretch releasing pressure-sensitive adhesive assembly, and methods of making and using the same |
| US7833577B2 (en) * | 2007-05-11 | 2010-11-16 | 3M Innovative Properties Company | Methods of making a pressure-sensitive adhesive assembly |
| US7705101B2 (en) | 2007-06-22 | 2010-04-27 | 3M Innovative Properties Company | Branched polydiorganosiloxane polyamide copolymers |
| US20080318065A1 (en) | 2007-06-22 | 2008-12-25 | Sherman Audrey A | Mixtures of polydiorganosiloxane polyamide-containing components and organic polymers |
| US8063166B2 (en) | 2007-06-22 | 2011-11-22 | 3M Innovative Properties Company | Polydiorganosiloxane polyamide copolymers having organic soft segments |
| US7705103B2 (en) * | 2007-06-22 | 2010-04-27 | 3M Innovative Properties Company | Polydiorganosiloxane polyoxamide copolymers |
| US7507849B2 (en) * | 2007-06-22 | 2009-03-24 | 3M Innovative Properties Company | Cyclic silazanes containing an oxamido ester group and methods of making these compounds |
| KR20160008653A (ko) | 2008-01-11 | 2016-01-22 | 쓰리엠 이노베이티브 프로퍼티즈 컴파니 | 광학적으로 투명한 연신 해제 감압 접착제 |
| DE102008000465A1 (de) * | 2008-02-29 | 2009-09-03 | Wacker Chemie Ag | Polymerblends enthaltend Polyorganosiloxan-Polyharnstoffcopolymere |
| US8673419B2 (en) * | 2008-03-14 | 2014-03-18 | 3M Innovative Properties Company | Stretch releasable adhesive tape |
| US8431671B2 (en) * | 2008-03-26 | 2013-04-30 | 3M Innovative Properties Company | Structured polydiorganosiloxane polyamide containing devices and methods |
| DE102009046850A1 (de) * | 2009-11-18 | 2011-05-19 | Wacker Chemie Ag | Siloxan-Copolymere enthaltende Zusammensetzungen |
| JP6275123B2 (ja) | 2012-05-18 | 2018-02-07 | スリーエム イノベイティブ プロパティズ カンパニー | 医療用接着物品 |
| CA2876351C (en) | 2012-06-11 | 2021-03-23 | 3M Innovative Properties Company | Melt-processable compositions having silicone-containing polymeric process additive and synergist |
| US10239301B2 (en) | 2012-07-03 | 2019-03-26 | 3M Innovative Properties Company | Heat-activatable siloxane-based adhesives |
| KR101676985B1 (ko) * | 2013-01-15 | 2016-11-16 | 도요 고무 고교 가부시키가이샤 | 고분자 액츄에이터 |
| EP2996873B1 (en) | 2013-05-17 | 2017-02-22 | 3M Innovative Properties Company | Reaction mixture, porous particles and methods of making |
| EP2996682B1 (en) | 2013-05-17 | 2018-07-04 | 3M Innovative Properties Company | Release of biologically active agents from polymeric composite particles |
| JP6452682B2 (ja) | 2013-06-24 | 2019-01-16 | スリーエム イノベイティブ プロパティズ カンパニー | 感圧接着剤のストライプを備える物品 |
| KR102220785B1 (ko) | 2013-06-24 | 2021-02-26 | 쓰리엠 이노베이티브 프로퍼티즈 캄파니 | 표면-부화된 스트라이프를 갖는 감압성 접착제 층 및 제조 방법 |
| EP3237502A1 (en) | 2014-12-23 | 2017-11-01 | 3M Innovative Properties Company | Curable and cured epoxy resin compositions |
| US10723894B2 (en) | 2014-12-23 | 2020-07-28 | 3M Innovative Properties Company | Tie layers prepared from particle-containing waterborne suspensions |
| KR102444532B1 (ko) | 2014-12-23 | 2022-09-16 | 쓰리엠 이노베이티브 프로퍼티즈 컴파니 | 양면 다층 접착제 |
| BR112017021396A2 (pt) | 2015-04-06 | 2018-07-03 | 3M Innovative Properties Company | composição em gel, filme e método para aplicação de uma composição em gel |
| KR102024481B1 (ko) | 2015-06-03 | 2019-09-23 | 쓰리엠 이노베이티브 프로퍼티즈 컴파니 | 가요성 디스플레이용 조립체 층 |
| KR20180015224A (ko) | 2015-06-03 | 2018-02-12 | 쓰리엠 이노베이티브 프로퍼티즈 컴파니 | 가요성 디스플레이 응용을 위한 실리콘-기반 조립체 층 |
| WO2016196541A1 (en) | 2015-06-03 | 2016-12-08 | 3M Innovative Properties Company | Acrylic-based flexible assembly layer |
| JP6579705B2 (ja) * | 2015-09-29 | 2019-09-25 | クミ化成株式会社 | 構造体及びその製造方法 |
| WO2018017554A1 (en) | 2016-07-22 | 2018-01-25 | 3M Innovative Properties Company | Siloxane-based adhesive layers as ceramic precursors |
| US20190381208A1 (en) | 2016-10-13 | 2019-12-19 | 3M Innovative Properties Company | Removable Film Forming Gel Compositions Featuring Adhesion Promoters |
| EP3700994A4 (en) | 2017-10-26 | 2021-08-04 | 3M Innovative Properties Company | COMPOSITION OF A SILICONE BASED ADHESIVE AND CELLULOSE NANOCRYSTALS, PROCESS AND ARTICLE |
| WO2020243506A1 (en) | 2019-05-31 | 2020-12-03 | Kindeva Drug Delivery | Removable film-forming gel compositions featuring adhesion promoters |
| WO2021014333A1 (en) | 2019-07-25 | 2021-01-28 | 3M Innovative Properties Company | Fluid-managing medical adhesive articles with microstructured surfaces |
| WO2021028821A1 (en) | 2019-08-15 | 2021-02-18 | 3M Innovative Properties Company | Core-sheath filament with a silicone-containing block copolymer core |
| JP2023502413A (ja) | 2019-11-20 | 2023-01-24 | スリーエム イノベイティブ プロパティズ カンパニー | 重ね貼り時に高い光学的透明度を有する医療用テープ |
| CN115426992A (zh) | 2020-04-13 | 2022-12-02 | 3M创新有限公司 | 具有低有效弹性模量的医用粘合剂制品 |
| US20230372158A1 (en) | 2020-10-21 | 2023-11-23 | 3M Innovative Properties Company | Packaged medical articles with reduced packaging |
| WO2022123489A1 (en) | 2020-12-11 | 2022-06-16 | 3M Innovative Properties Company | Perforated tapes for medical applications |
| WO2022137062A1 (en) | 2020-12-21 | 2022-06-30 | 3M Innovative Properties Company | Dual-sided adhesive tapes with on-demand adhesion |
| DE102021205464B4 (de) | 2021-05-28 | 2023-05-11 | Tesa Se | Klebeband mit unvernetzter Silikon-Haftklebmasse und Verwendung |
| CN117750951A (zh) | 2021-07-29 | 2024-03-22 | 3M创新有限公司 | 包含水杨酸的成膜组合物和使用方法 |
Family Cites Families (76)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2532011A (en) | 1946-09-07 | 1950-11-28 | Minnesota Mining & Mfg | Liners and adhesive tapes having low adhesion polyvinyl carbamate coatings |
| US2736721A (en) | 1952-10-08 | 1956-02-28 | Optionally | |
| US2672182A (en) | 1952-11-15 | 1954-03-16 | George W Gwin | Child's auxiliary serving tray |
| US2814601A (en) | 1954-04-29 | 1957-11-26 | Dow Corning | Organopolysiloxane adhesive and pressure-sensitive adhesive tape containing same |
| DE1017883B (de) | 1954-07-08 | 1957-10-17 | Fellows Gear Shaper Co | Schalt- und Vorschubeinrichtung fuer Zahnradherstellungsmaschinen |
| US3528940A (en) | 1966-12-15 | 1970-09-15 | Gen Electric | Silicone pressure-sensitive adhesive of improved strength |
| US3562352A (en) | 1968-09-06 | 1971-02-09 | Avco Corp | Polysiloxane-polyurethane block copolymers |
| US3627851A (en) | 1970-10-23 | 1971-12-14 | Dow Corning | Flexible coating composition |
| BE786656A (fr) | 1971-07-30 | 1973-01-24 | Ici Ltd | Siloxanes |
| US3890269A (en) | 1972-08-11 | 1975-06-17 | Stauffer Chemical Co | Process for preparing aminofunctional polysiloxane polymers |
| US4002794A (en) * | 1975-07-18 | 1977-01-11 | Nashua Corporation | Adhesive material and articles incorporating same |
| US4117192A (en) | 1976-02-17 | 1978-09-26 | Minnesota Mining And Manufacturing Company | Deformable retroreflective pavement-marking sheet material |
| US4675361A (en) * | 1980-02-29 | 1987-06-23 | Thoratec Laboratories Corp. | Polymer systems suitable for blood-contacting surfaces of a biomedical device, and methods for forming |
| US4299874A (en) | 1980-03-31 | 1981-11-10 | Minnesota Mining And Manufacturing Company | Removable pavement-marking sheet material |
| CA1202508A (en) | 1981-05-07 | 1986-04-01 | Norio Murata | Protective packaging assembly and method for optical fibers |
| EP0068385B1 (en) * | 1981-06-22 | 1986-09-24 | Kanegafuchi Kagaku Kogyo Kabushiki Kaisha | Thermoplastic elastomers for medical use as moulded articles brought into direct contact with blood |
| FR2513644B1 (fr) | 1981-09-30 | 1985-06-21 | Rhone Poulenc Spec Chim | Copolymeres sequences polysiloxaniques et polyurethannes utilisables notamment comme elastomeres thermoplastiques |
| US4777276A (en) | 1981-10-29 | 1988-10-11 | Minnesota Mining And Manufacturing Company | Acrylamidoacylated oligomers |
| DE3143994A1 (de) | 1981-11-05 | 1983-05-11 | Bayer Ag, 5090 Leverkusen | Verfahren zur herstellung von duennwandigen gegenstaenden aus thermoplastischen polyurethanen oder polyurethanharnstoffen durch extrusion |
| US4490432A (en) | 1982-04-23 | 1984-12-25 | Minnesota Mining And Manufacturing Company | Reinforced pavement-marking sheet material |
| US4447493A (en) | 1982-07-26 | 1984-05-08 | Minnesota Mining And Manufacturing Company | Vibration-damping constrained-layer constructions |
| US4605712A (en) | 1984-09-24 | 1986-08-12 | Ciba-Geigy Corporation | Unsaturated polysiloxanes and polymers thereof |
| US4563539A (en) | 1984-12-18 | 1986-01-07 | Dow Corning Corporation | Acrylofunctional silicones |
| US4539345A (en) | 1985-02-04 | 1985-09-03 | Minnesota Mining And Manufacturing Company | Moisture-curable polyurethane composition |
| JPS61195129A (ja) | 1985-02-22 | 1986-08-29 | Toray Silicone Co Ltd | 有機けい素重合体の製造方法 |
| US4661577A (en) | 1985-10-01 | 1987-04-28 | General Electric Company | Aminofunctional polysiloxanes |
| US4736048A (en) | 1986-06-04 | 1988-04-05 | Dow Corning Corporation | Pressure sensitive adhesive release liner and fluorosilicone compounds, compositions and method therefor |
| US4889753A (en) | 1986-06-04 | 1989-12-26 | Dow Corning Corporation | Pressure sensitive adhesive release liner and fluorosilicone compounds, compositions and method therefor |
| US5214119A (en) * | 1986-06-20 | 1993-05-25 | Minnesota Mining And Manufacturing Company | Block copolymer, method of making the same, dimaine precursors of the same, method of making such diamines and end products comprising the block copolymer |
| US5512650A (en) | 1986-06-20 | 1996-04-30 | Minnesota Mining And Manufacturing Company | Block copolymer, method of making the same, diamine precursors of the same, method of making such diamines and end products comprising the block copolymer |
| DE3752135T2 (de) | 1986-06-20 | 1998-04-16 | Minnesota Mining & Mfg | Blockcopolymer, Verfahren zu seiner Herstellung, Diaminvorprodukte für dieses Verfahren bzw. Verfahren zu deren Herstellung sowie das Blockcopolymer enthaltende Endprodukte |
| GB8615862D0 (en) | 1986-06-28 | 1986-08-06 | Dow Corning Ltd | Making siloxane resins |
| DE3717073A1 (de) | 1987-05-21 | 1988-12-08 | Wacker Chemie Gmbh | Siliconharzpulver und verfahren zu deren herstellung |
| US4933396A (en) | 1987-06-26 | 1990-06-12 | Minnesota Mining And Manufacturing Company | Method of making purely primary diamines |
| JPS6422310A (en) | 1987-07-17 | 1989-01-25 | Shinetsu Chemical Co | Antifoaming agent |
| US5221724A (en) * | 1987-08-12 | 1993-06-22 | Wisconsin Alumni Research Foundation | Polysiloxane polyurea urethanes |
| JPS6474268A (en) | 1987-09-14 | 1989-03-20 | Shinetsu Chemical Co | Curable silicone composition |
| US4908208A (en) | 1988-04-22 | 1990-03-13 | Dow Corning Corporation | Matrix for release of active ingredients |
| US5026890A (en) | 1988-05-20 | 1991-06-25 | General Electric Company | Method and intermediates for preparation of bis(aminoalkyl)polydiorganosiloxanes |
| JPH0813888B2 (ja) | 1988-07-27 | 1996-02-14 | 信越化学工業株式会社 | シリコーン樹脂およびその製造方法 |
| JPH0623255B2 (ja) | 1988-10-05 | 1994-03-30 | 信越化学工業株式会社 | パーフルオロアルキル基含有オルガノポリシロキサンの製造方法 |
| US4948859A (en) | 1988-10-28 | 1990-08-14 | Minnesota Mining And Manufacturing Company | Extruder polymerization of polyurethanes |
| US4968766A (en) | 1989-01-12 | 1990-11-06 | Dow Corning Corporation | Fluorosilicone compounds and compositions for adhesive release liners |
| JPH0618880B2 (ja) | 1989-02-21 | 1994-03-16 | 信越化学工業株式会社 | フルオロオルガノポリシロキサン及びその製造方法 |
| GB8906626D0 (en) | 1989-03-22 | 1989-05-04 | Dow Corning | Method of making organosiloxane resins |
| US5237082A (en) | 1989-09-22 | 1993-08-17 | Minnesota Mining And Manufacturing Company | Radiation-curable silicone elastomers and pressure sensitive adhesives |
| US5091483A (en) | 1989-09-22 | 1992-02-25 | Minnesota Mining And Manufacturing Company | Radiation-curable silicone elastomers and pressure sensitive adhesives |
| AU632869B2 (en) | 1989-12-14 | 1993-01-14 | Minnesota Mining And Manufacturing Company | Fluorocarbon-based coating compositions and articles derived therefrom |
| DK0455585T3 (da) | 1990-04-26 | 1994-05-02 | Ciba Geigy Ag | Umættede urinstofpolysiloxaner |
| US5194113A (en) | 1990-12-24 | 1993-03-16 | Minnesota Mining And Manufacturing Company | Process for making conformable thermoplastic marking sheet |
| JP2669948B2 (ja) | 1991-02-18 | 1997-10-29 | 信越化学工業株式会社 | 硬化性フルオロシリコ−ンポリマ−組成物 |
| CA2103799A1 (en) | 1991-02-28 | 1992-09-17 | James M. Kaczmarczik | Pavement markers with silicone adhesive |
| JPH0739160B2 (ja) | 1991-04-24 | 1995-05-01 | ニチアス株式会社 | 制振材 |
| JP2684130B2 (ja) | 1991-08-15 | 1997-12-03 | 信越化学工業株式会社 | アミノ基含有ポリシロキサンの製造方法 |
| JPH0551459A (ja) | 1991-08-22 | 1993-03-02 | Toray Dow Corning Silicone Co Ltd | 有機ケイ素重合体の製造方法 |
| US5248739A (en) | 1991-10-18 | 1993-09-28 | Dow Corning Corporation | Silicone pressure sensitive adhesives having enhanced adhesion to low energy substrates |
| JP2646046B2 (ja) | 1991-10-21 | 1997-08-25 | 信越化学工業株式会社 | 高減衰性シリコーン組成物及びその硬化物 |
| JPH07119081B2 (ja) | 1991-10-30 | 1995-12-20 | ニチアス株式会社 | 制振材 |
| JP3186143B2 (ja) | 1991-11-11 | 2001-07-11 | 日東電工株式会社 | 耐熱性の良好な制振性感圧接着剤組成物とその製法 |
| US5286815A (en) | 1992-02-07 | 1994-02-15 | Minnesota Mining And Manufacturing Company | Moisture curable polysiloxane release coating compositions |
| US5589563A (en) * | 1992-04-24 | 1996-12-31 | The Polymer Technology Group | Surface-modifying endgroups for biomedical polymers |
| JP2666661B2 (ja) | 1992-06-18 | 1997-10-22 | 信越化学工業株式会社 | オルガノポリシロキサンパウダーの製造方法 |
| US5539033A (en) | 1992-11-06 | 1996-07-23 | Minnesota Mining And Manufacturing Company | Solventless compounding and coating of non-thermoplastic hydrocarbon elastomers |
| DE4243799A1 (de) * | 1992-12-23 | 1994-06-30 | Bayer Ag | Siloxanblockcopolymer-modifizierte thermoplastische Polyurethane |
| US5319040A (en) | 1993-03-12 | 1994-06-07 | General Electric Company | Method for making substantially silanol-free silicone resin powder, product and use |
| US5468815A (en) | 1994-01-12 | 1995-11-21 | Minnesota Mining And Manufacturing | Low coefficient of friction silicone release formulations incorporating higher alkenyl-functional silicone gums |
| DE69524880T2 (de) | 1994-10-04 | 2002-08-22 | Minnesota Mining & Mfg | Reaktive, zweiteilige polyurethanzusammensetzungen, sowie daraus hergestellte gegebenenfalls selbstheilende und kratzfeste beschichtungen |
| US5670598A (en) * | 1995-03-24 | 1997-09-23 | Minnesota Mining And Manufacturing Company | Diblock and triblock polydiorganosiloxane-polyurea block copolymers |
| BR9608034A (pt) | 1995-04-25 | 1999-01-12 | Minnesota Mining & Mfg | Copolímeros segmentados de polidiorganossiloxano oligouréia e processo para preparação dos mesmos |
| CA2219787A1 (en) | 1995-04-25 | 1996-10-31 | Mieczyslaw H. Mazurek | Polydiorganosiloxane polyurea segmented copolymers and a process for making same |
| WO1996034028A1 (en) | 1995-04-25 | 1996-10-31 | Minnesota Mining And Manufacturing Company | Tackified polydiorganosiloxane oligourea segmented copolymers and a process for making same |
| WO1996035458A2 (en) | 1995-04-25 | 1996-11-14 | Minnesota Mining And Manufacturing Company | Tackified polydiorganosiloxane polyurea segmented copolymers and a process for making same |
| US5728469A (en) * | 1995-06-06 | 1998-03-17 | Avery Dennison Corporation | Block copolymer release surface for pressure sensitive adhesives |
| US5663227A (en) * | 1996-03-14 | 1997-09-02 | United States Postal Service | Release agent for linerless pressure sensitive postage stamps |
| US6664359B1 (en) | 1996-04-25 | 2003-12-16 | 3M Innovative Properties Company | Tackified polydiorganosiloxane polyurea segmented copolymers and a process for making same |
| US6407195B2 (en) | 1996-04-25 | 2002-06-18 | 3M Innovative Properties Company | Tackified polydiorganosiloxane oligourea segmented copolymers and a process for making same |
-
1996
- 1996-10-23 US US08/735,836 patent/US6846893B1/en not_active Expired - Fee Related
-
1997
- 1997-09-25 JP JP51936698A patent/JP2001508818A/ja not_active Ceased
- 1997-09-25 ES ES97943568T patent/ES2251033T3/es not_active Expired - Lifetime
- 1997-09-25 BR BR9712551-2A patent/BR9712551A/pt not_active IP Right Cessation
- 1997-09-25 CN CN97199049A patent/CN1234051A/zh active Pending
- 1997-09-25 KR KR1019990703511A patent/KR20000052713A/ko not_active Application Discontinuation
- 1997-09-25 AU AU45010/97A patent/AU4501097A/en not_active Abandoned
- 1997-09-25 DE DE69734721T patent/DE69734721T2/de not_active Expired - Lifetime
- 1997-09-25 EP EP97943568A patent/EP0934360B1/en not_active Expired - Lifetime
- 1997-09-25 CA CA002268113A patent/CA2268113A1/en not_active Abandoned
- 1997-09-25 WO PCT/US1997/017200 patent/WO1998017726A1/en active IP Right Grant
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101346448B (zh) * | 2005-12-23 | 2012-10-03 | 3M创新有限公司 | 粘合剂组合物 |
| CN102449087A (zh) * | 2009-05-28 | 2012-05-09 | 宝洁公司 | 防粘片材料 |
| CN101701131B (zh) * | 2009-09-08 | 2013-02-13 | 南京南大波平电子信息有限公司 | 一种智能主动型抗结冰涂层材料及其制备方法和应用 |
| CN104781354A (zh) * | 2012-07-03 | 2015-07-15 | 3M创新有限公司 | 硅氧烷基管涂层 |
| CN104781354B (zh) * | 2012-07-03 | 2017-07-11 | 3M创新有限公司 | 硅氧烷基管涂层 |
| CN109476845A (zh) * | 2016-07-13 | 2019-03-15 | 瓦克化学股份公司 | 含有硅氧烷-有机共聚物的聚合物组合物 |
| CN109476845B (zh) * | 2016-07-13 | 2021-12-31 | 瓦克化学股份公司 | 含有硅氧烷-有机共聚物的聚合物组合物 |
| CN109790386A (zh) * | 2016-10-07 | 2019-05-21 | 瓦克化学股份公司 | 含有硅氧烷-有机共聚物的聚合物组合物 |
| CN111344373A (zh) * | 2017-10-09 | 2020-06-26 | 3M创新有限公司 | 用于微型扬声器膜片的阻尼粘合剂层 |
| CN108384453A (zh) * | 2018-03-21 | 2018-08-10 | 广州市维思涂料科技有限公司 | 一种用于金属彩钢板表面的水性涂料及其制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| DE69734721D1 (de) | 2005-12-29 |
| AU4501097A (en) | 1998-05-15 |
| EP0934360B1 (en) | 2005-11-23 |
| US6846893B1 (en) | 2005-01-25 |
| KR20000052713A (ko) | 2000-08-25 |
| ES2251033T3 (es) | 2006-04-16 |
| JP2001508818A (ja) | 2001-07-03 |
| WO1998017726A1 (en) | 1998-04-30 |
| DE69734721T2 (de) | 2006-07-20 |
| EP0934360A1 (en) | 1999-08-11 |
| CA2268113A1 (en) | 1998-04-30 |
| BR9712551A (pt) | 1999-10-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1234051A (zh) | 有含聚二有机硅氧烷脲的组分的聚合物混合物 | |
| CN101688045B (zh) | 含有聚二有机硅氧烷-聚酰胺的组分与有机聚合物的混合物 | |
| JP4709176B2 (ja) | 硬化性かつ粘着性のポリジオルガノシロキサンオリゴウレアセグメント化共重合体 | |
| US6664359B1 (en) | Tackified polydiorganosiloxane polyurea segmented copolymers and a process for making same | |
| CN1264867C (zh) | 有机硼烷·胺加成物引发剂体系和由其制得的可聚合组合物 | |
| CN101687997B (zh) | 带支链聚二有机硅氧烷-聚酰胺共聚物 | |
| US5292586A (en) | Solventless or high solids-containing silicone pressure sensitive adhesive compositions | |
| CN101687998B (zh) | 具有有机软链段的聚二有机硅氧烷-聚酰胺共聚物 | |
| CN100351294C (zh) | 光学透明和抗静电的压敏粘合剂 | |
| CN1604949A (zh) | 有机硅压敏粘合剂、制品及其制备和使用方法 | |
| CN1181764A (zh) | 聚二有机硅氧烷聚脲嵌段共聚物及其制备方法 | |
| US6387487B1 (en) | Dual cure, low-solvent silicone pressure sensitive adhesives | |
| CN1454247A (zh) | 可拉伸剥离压敏粘合带及制品 | |
| WO1996035458A2 (en) | Tackified polydiorganosiloxane polyurea segmented copolymers and a process for making same | |
| CN1732240A (zh) | 剥离组合物和由其制得的制品 | |
| CN108603090A (zh) | 粘合剂组合物 | |
| CN1897987A (zh) | 有机硅压敏粘合剂和制品 | |
| JP4558997B2 (ja) | ポリウレア系接着剤、それから製造される物品、ならびにそれを製造および使用する方法 | |
| CN1402743A (zh) | 改进的反应性热熔粘合剂 | |
| CN1272866A (zh) | 共混的压敏粘合剂 | |
| CN1942548A (zh) | 湿气固化型聚氨酯热熔粘合剂 | |
| JP4267690B2 (ja) | 粘着性ポリジオルガノシロキサンポリ尿素セグメントコポリマーおよびその製造方法 | |
| EP2284239A1 (en) | Bonder article | |
| WO2023087854A1 (zh) | 一种防污涂料及其使用方法和应用 | |
| CN1495241A (zh) | 压敏粘合剂制品 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |