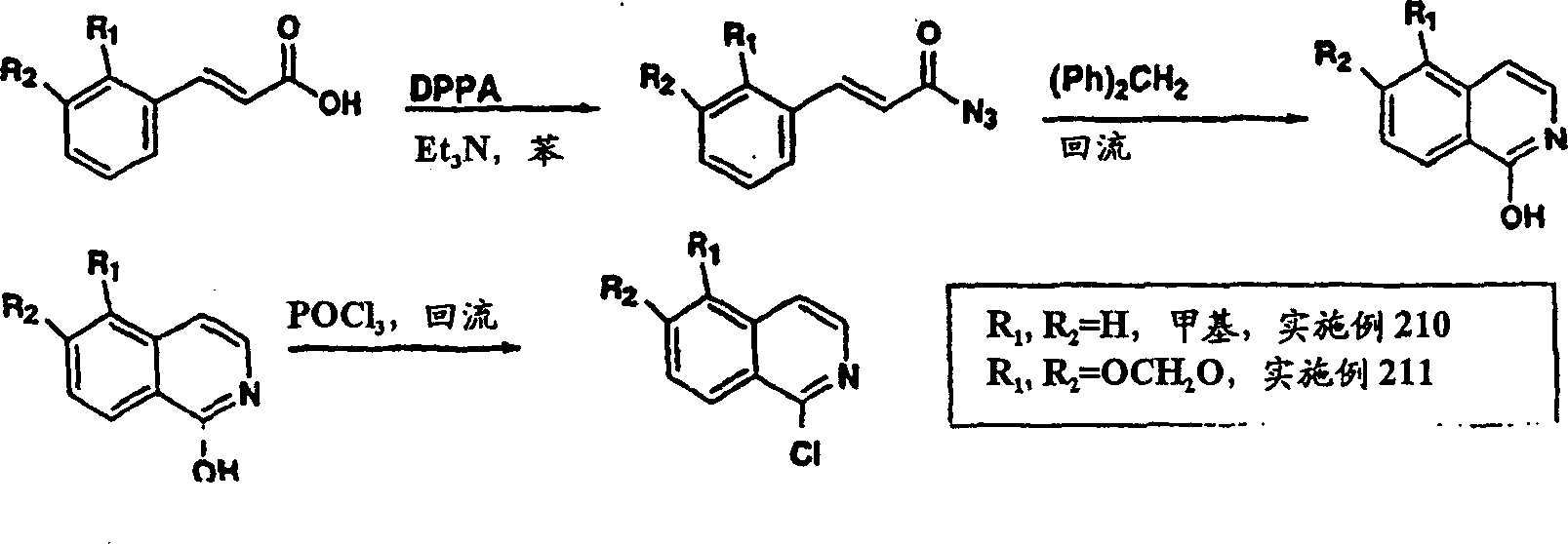
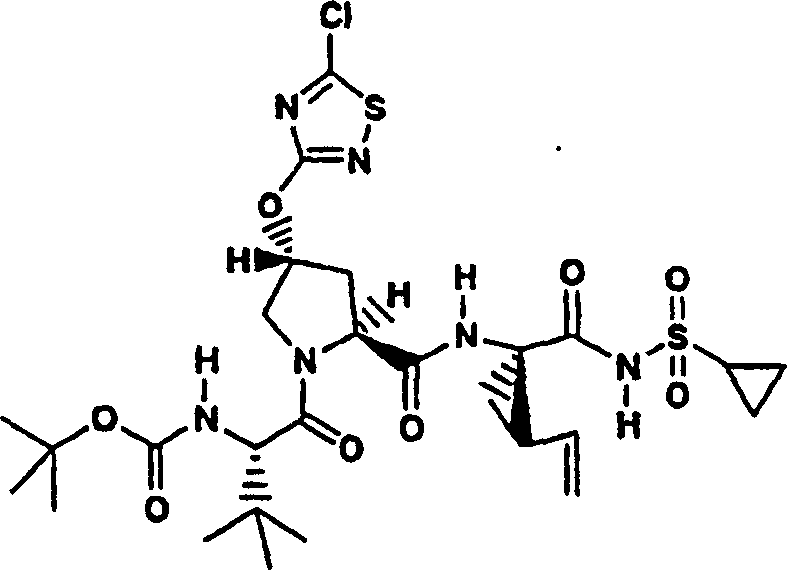
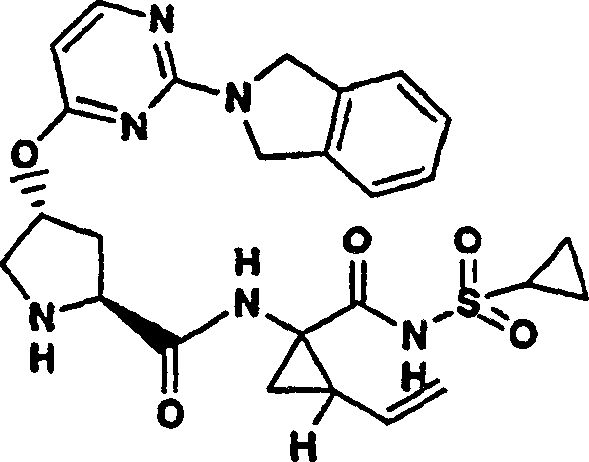
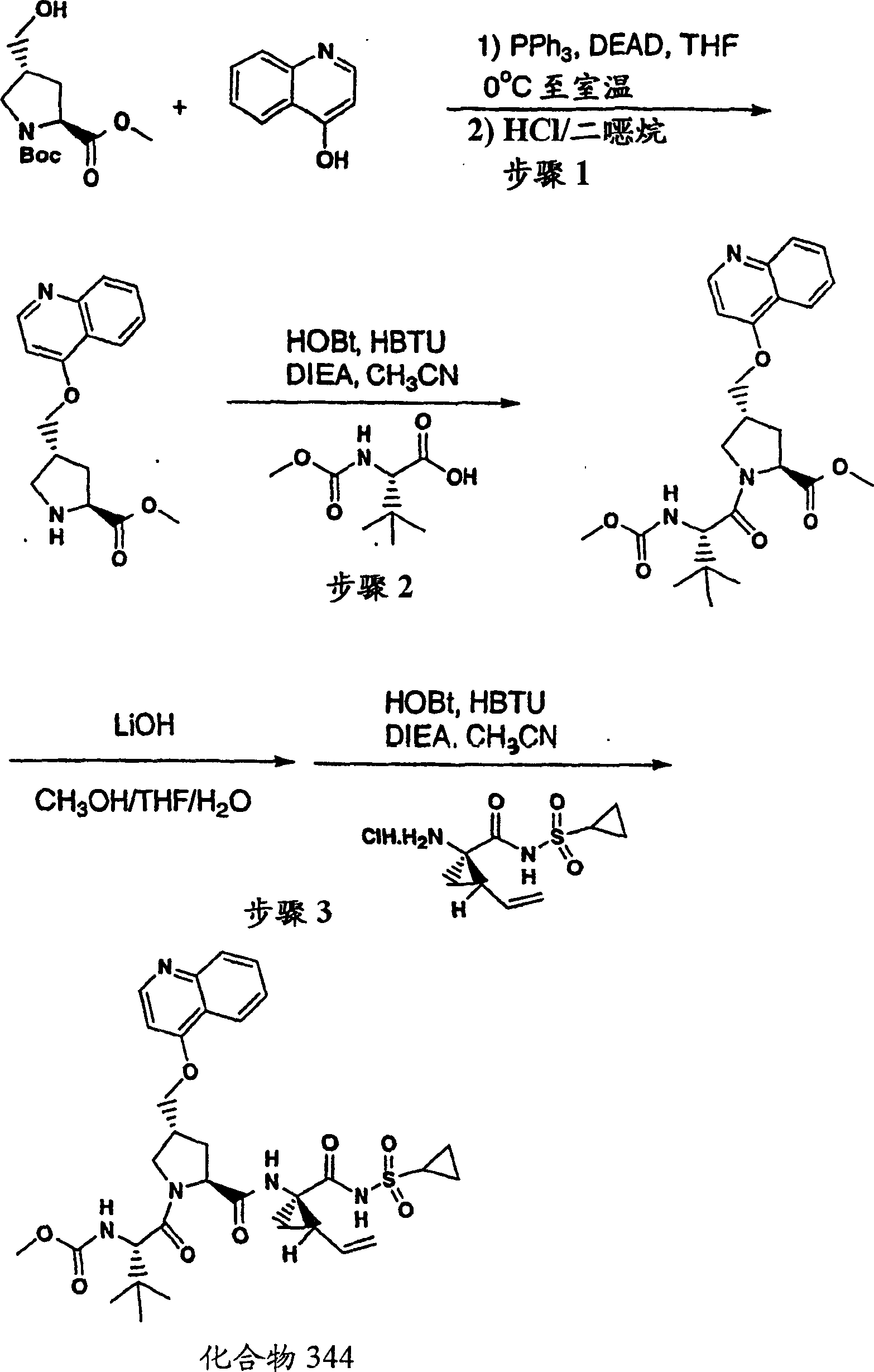
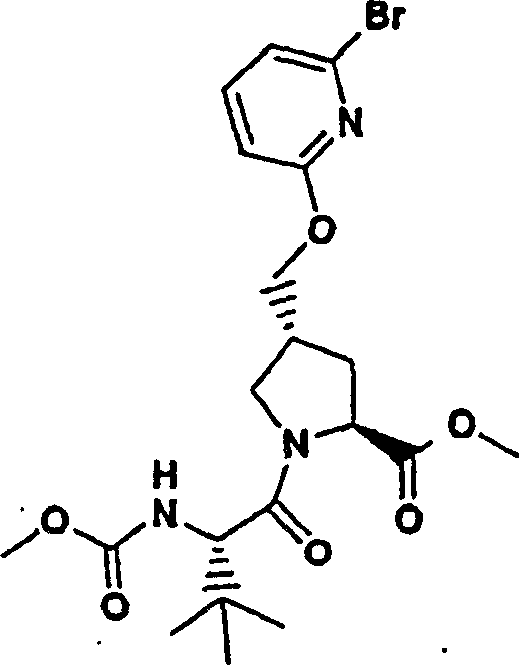
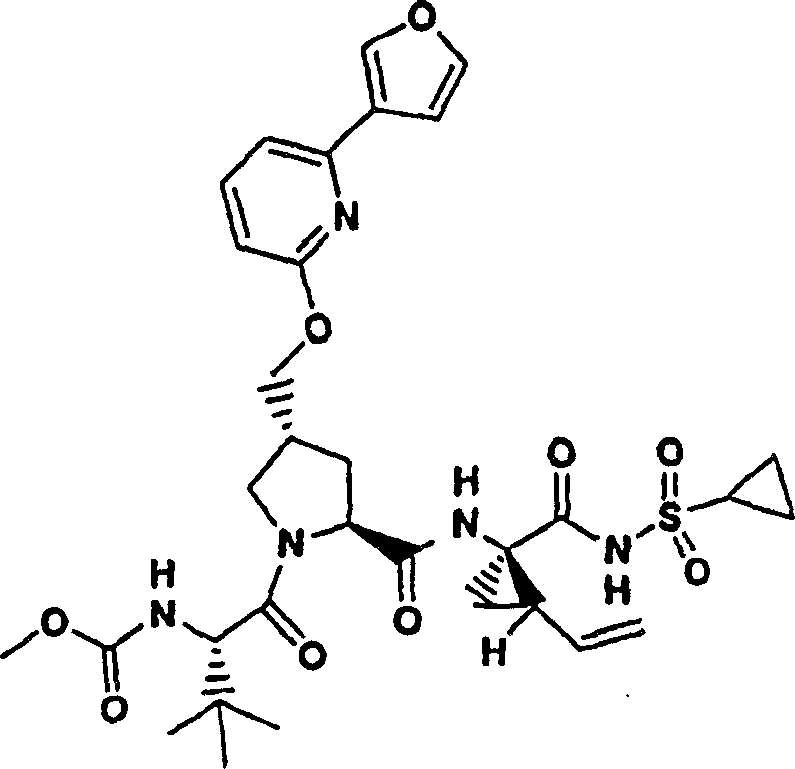
实施例
以下的具体实例阐明本发明化合物的合成法,但不对本发明的宗旨或者范围构成限制。为制备本发明所包含的化合物,可将这些方法进行改变,但这些改变没有具体公开。另外,以某些不同方式生成相同化合物的不同方法对本领域技术人员来讲也是显而易见的。
除非另外指明,否则溶液百分比表示重量对体积的关系,溶液比率表示体积对体积的关系。在Bruker 300、400或者500MHz波谱仪上记录核磁共振(NMR)光谱;化学位移(δ)以每百万分之一报道。按照Still的快速层析技术(W.C.Still等,J.Org.Chem.,(1978),43,2923),于硅胶(SiO2)上进行快速层析法。
采用SPD-10AV UV-Vis检测器,在Shimadzu LC-10AS液相色谱仪上记录所有的液相层析(LC)数据,在电喷雾模式(ES+)下,用Micromass Platform测定质谱(MS)数据。
除非另外指明,否则采用具有以下条件的7个方法学中的一种,通过LC/MS,分析每个化合物。
柱:(方法A)-YMC ODS S7 C18 3.0×50mm
(方法B)-YMC ODS-A S7 C18 3.0×50mm
(方法C)-YMC S7 C18 3.0×50mm
(方法D)-YMC Xterra ODS S7 3.0×50mm
(方法E)-YMC Xterra ODS S7 3.0×50mm
(方法F)-YMC ODS-A S7 C18 3.0×50mm
(方法G)-YMC C18 S5 4.6×50mm]
梯度:100%溶剂A/0%溶剂B至
0%溶剂A/100%溶剂B
梯度时间:2分钟(A、B、D、F、G);8分钟(C、E)。
保留时间:1分钟(A、B、D、F、G);2分钟(C、E)。
流速:5mL/min
检测器波长:220nm
溶剂A:10%MeOH/90%H2O/0.1%TFA
溶剂B:10%H2O/90%MeOH/0.1%TFA。
在本申请中使用的缩写,具体包括在以下示例性实施例中的缩写为本领域技术人员所熟知。其中一些所用的缩写如下所示:
rt 室温
Boc 叔丁氧基羰基
DMSO 二甲基亚砜
EtOAc 乙酸乙酯
t-BuOK 叔丁醇钾
Et2O 乙醚
TBME 叔丁基甲基醚
THF 四氢呋喃
CDI 羰基二咪唑
DBU 1,8-二氮杂双环[5.4.0]十一碳-7-烯
TFA 三氟乙酸
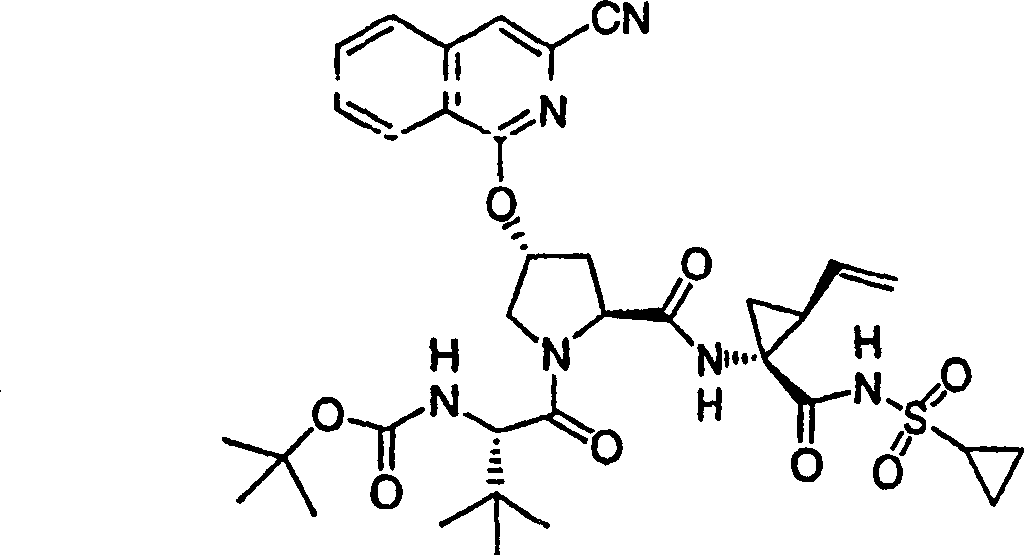
NMM N-甲基吗啉
HATU O-7-氮杂苯并三唑-1-基
HBTU 六氟磷酸O-{1H-苯并三唑-1-基)-
N,N,N′,N′-四甲基脲_
HOBT N-羟基苯并三唑
PyBrop 六氟磷酸溴-双吡咯烷-_
DMF 二甲基甲酰胺
MeOH 甲醇
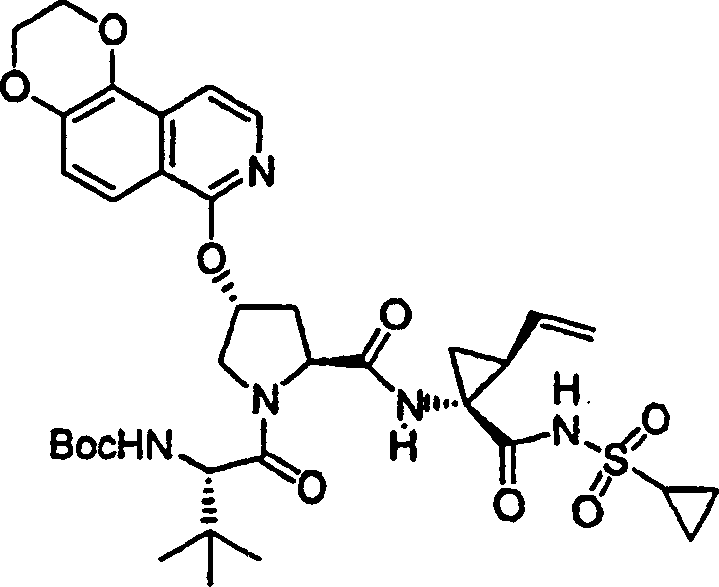
EDTA 乙二胺四乙酸
HRMS 高分辨率质谱
DMAP 4-二甲基氨基吡啶
DIPEA 二异丙基乙胺
DCM 二氯甲烷
DCE 二氯乙烷
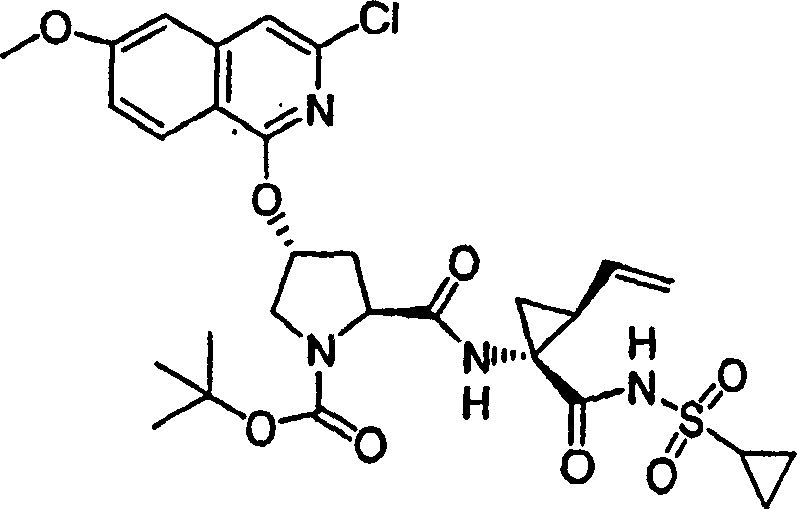
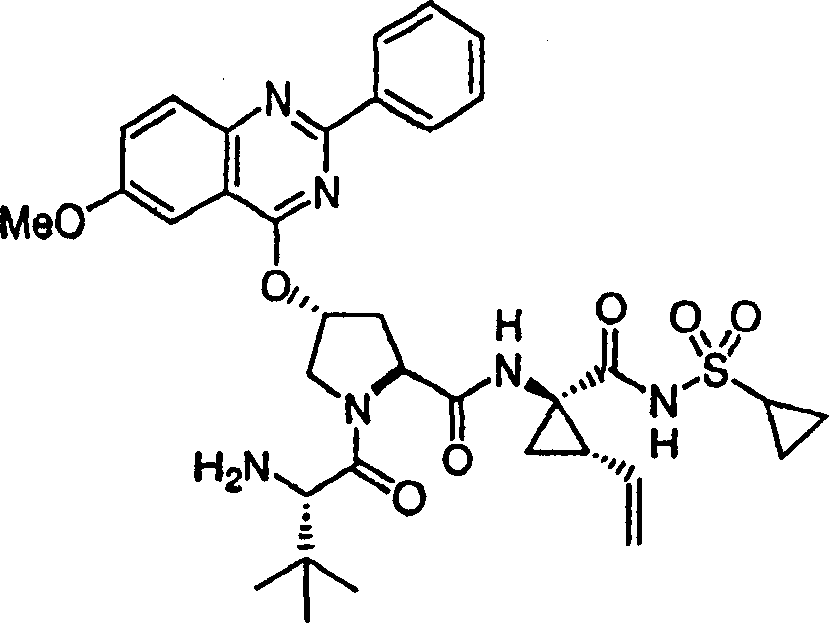
以下实施例描述的本发明化合物和化学中间体按照以下方法制备。应注意以下示例性部分分成各部分存在。各部分标记为部分A至K。在本申请的整个实施例部分,实施例编号和化合物编号并不连续,因此,各部分在编号上是“中断”的。在各部分中的编号通常是连续的。部分L描述化合物的生物活性。部分M描述可使用本发明方法制备的其它化合物的子集。
部分A:
各中间体的制备:
各P1中间体的制备:
在该部分描述的P1中间体可在本文描述的方法中用于制备式I化合物。
I P1组件:
1.外消旋(1R,2S)/(1S,2R)-1-氨基-2-乙烯基环丙烷甲酸乙酯的制备
方法A
步骤1
将甘氨酸乙酯盐酸盐(303.8g,2.16mole)悬浮在叔丁基甲基醚(1.6L)中。加入苯甲醛(231g,2.16mole)和无水硫酸钠(154.6g,1.09mole)并使用冰水浴将混合物冷却至0℃。在30分钟内滴加三乙胺(455mL,3.26mole)并将混合物在室温下搅拌48。随后加入冰冷却的水(1L)猝灭反应物并分离出有机层。水相用叔丁基甲基醚(0.5L)萃取并将合并的有机相用饱和碳酸氢钠水溶液(1L)和盐水(1L)的混合物洗涤。溶液经硫酸镁干燥,真空浓缩,得到392.4g N-苄基亚胺产物,为粘稠黄色油状物,该产物直接用于下一步骤中。
1HNMR(CDCl3,300MHz)δ1.32(t,J=7.1Hz,3H),4.24(q,J=7.1Hz,2H),4.41(d,J=1.1Hz,2H),7.39-7.47(m,3H),7.78-7.81(m,2H),8.31(s,1H).
步骤2
在60分钟内,向叔丁醇锂(84.06g,1.05mol)的无水甲苯(1.2L)悬浮液中滴加(N-苄基亚胺)甘氨酸乙酯(100.4g,0.526mol)和反式-1,4-二溴-2-丁烯(107.0g,0.500mol)在无水甲苯(0.6L)中的混合物。完成加入后,加入水(1L)和叔丁基甲基醚(TBME,1L)猝灭深红色混合物。分离出水相并用TBME(1L)进行第二次萃取。合并有机相,加入1NHCl(1L)并将混合物在室温下搅拌2h。分离出有机相并用水(0.8L)萃取。随后合并水相,用盐(700g)饱和,加入TBME(1L)并将混合物冷却至0℃。滴加10N NaOH将搅拌着的混合物碱化至pH为14,分离有机层,水相用TBME(2×500mL)萃取。将合并的有机萃取液干燥(硫酸镁)并浓缩至1L的体积。向该游离胺溶液中加入BOC2O或二碳酸二叔丁酯(131.0g,0.6mol),将混合物在室温下搅拌4天。向反应物中加入另外的二碳酸二叔丁酯(50g,0.23mol),将混合物回流3h,随后冷却至室温过夜。反应混合物经硫酸镁干燥并真空浓缩,得到80g粗物质。该剩余物经快速层析纯化(2.5Kg SiO2,洗脱液:1-2%甲醇/二氯甲烷),得到57g(53%)外消旋的N-Boc-(1R,2S)/(1S,2R)-1-氨基-2-乙烯基环丙烷甲酸乙酯,为黄色油状物,该油状物在冰箱中静置时固化:
1H NMR(CDCl3,300MHz)δ1.26(t,J=7.1Hz,3H),1.46(s,9H),1.43-1.49(m,1H),1.76-1.82(br m,1H),2.14(q,J=8.6Hz,1H),4.18(q,J=7.2Hz,2H),5.12(ddJ=10.3,1.7Hz,1H),5.25(br s,1H),5.29(dd,J=17.6,1.7Hz,1H),5.77(ddd,J=17.6,10.3,8.9Hz,1H);MS m/z 254.16(M-1)
步骤3外消旋(1R,2S)/(1S,2R)1-氨基-2-乙烯基环丙烷甲酸乙酯盐酸盐的制备
将N-Boc-(1R,2S)/(1S,2R)-1-氨基-2-乙烯基环丙烷甲酸乙酯(9.39g,36.8mmol)溶于4N HCl/二_烷(90ml,360mmol)中并在室温下搅拌2h。将反应混合物浓缩,得到(1R,2S)/(1S,2R)-1-氨基-2-乙烯基环丙烷甲酸乙酯盐酸盐,定量收率(7g,100%)。
1H NMR(甲醇-d4)δ1.32(t,J=7.1,3H),1.72(dd,J=10.2,6.6Hz,1H),1.81(dd,J=8.3,6.6Hz,1H),2.38(q,J=8.3Hz,1H),4.26-4.34(m,2H),5.24(dd,10.3,1.3Hz,1H)5.40(d,J=17.2,1H),5.69-5.81(m,1H).
外消旋N-Boc-1-氨基-2-乙烯基环丙烷甲酸乙酯盐酸盐的另一种制备方法
在-78℃下,向叔丁醇钾(11.55g,102.9mmol)的THF(450mL)溶液中加入商品N,N-二苄基亚胺的甘氨酸乙酯(25.0g,93.53mmol)的THF(112mL)溶液。使反应混合物升温至0℃,搅拌40min,随后再次冷却至-78℃。向该溶液中加入反式-1,4-二溴-2-丁烯(20.0g,93.50mmol),将混合物在0℃下搅拌1h,并再次冷却至-78℃。加入叔丁醇钾(11.55g,102.9mmol),立即将混合物升温至0℃,并再搅拌1小后,接着真空浓缩。将粗产物溶于乙醚(530mL),加入1N盐酸(106mL,106mmol)并在室温下将所得的两相混合物搅拌3.5h。分离各层,水层用乙醚(2×)洗涤并用饱和碳酸氢钠水溶液碱化。所需的胺用乙醚萃取(3×)并将合并的有机萃取液用盐水洗涤,干燥(硫酸镁)并真空浓缩,得到游离胺。该物质用4N HCl的二_烷(100mL,400mmol)溶液处理并浓缩,得到(1R,2S)/(1S,2R)-1-氨基-2-乙烯基环丙烷甲酸乙酯盐酸盐,为棕色半固体物(5.3g,收率34%),与按照方法A得到的物质相同,不同之处在于存在少量未确定的芳族杂质(8%)。
N-Boc-(1R,2S)/(1S,2R)-1-氨基-2-乙烯基环丙烷甲酸乙酯的拆分
拆分A
向磷酸钠缓冲水溶液(0.1M,4.25L,pH 8,装于12L夹套式反应器中,保温39℃,以300rpm的速率搅拌)中加入511g Acalase 2.4L(约425mL)(Novozymes NoRth America Inc.)。当混合物的温度达到39℃时,加入50%NaOH水溶液将pH调节至8.0。随后在40分钟内加入所述外消旋N-Boc-(1R,2S)/(1S,2R)-1-氨基-2-乙烯基环丙烷甲酸乙酯(85g)在850mL DMSO中的溶液。接着保持反应温度在40℃下24.5h,在此期间,在第1.5h和19.5h的时间点,使用50%NaOH水溶液将混合物的pH调节至8.0。24.5h后,测得所述酯的对映体过量为97.2%,将反应物冷却至室温(26℃),搅拌过夜(16h),此后,测定所述酯的对映体过量为100%。接着用50%NaOH将反应混合物的pH调节至8.5,并将所得的混合物用MTBE萃取(2×2L)。随后将合并的MTBE萃取液用5%碳酸氢钠(3×100mL)、水(3×100mL)洗涤,并真空蒸发,得到对映异构体纯的N-Boc-(1R,2S)/-1-氨基-2-乙烯基环丙烷甲酸乙酯,为浅黄色固体物(42.55g;纯度:97%,210nm下测定,不含酸;100%对映异构体过量(″ee″))。
随后用50%硫酸将来自萃取方法的水层酸化至pH为2,并用MTBE萃取(2×2L)。MTBE萃取液用水洗涤(3×100mL)并蒸发,得到所述酸,为浅黄色固体物(42.74g;纯度:99%,210nm下测定,不含酯)。
1R,2S-酯 1S,2R-酸
| |
酯 |
酸 |
|
高分辨率质谱 |
(+)ESI,C13H22NO4,[M+H]+,计算值256.1549,实测值256.1542 |
(-)ESI,C11H16NO4,[M-H]-,计算值226.1079,实测值226.1089 |
|
NMR观察化学位移溶剂:CDCl3(质子δ7.24ppm,C-13δ77.0PPM)Bruker DRX-500C:峰500.032MHz,碳125.746MHz |
|
位置 |
位置(峰)ppm |
C-13ppm |
碳(峰)ppm |
C-13ppm |
|
1 |
---- |
40.9 |
---- |
40.7 |
|
2 |
2.10(q,J=9.0Hz) |
34.1 |
2.17(q,J=9.0Hz) |
35.0 |
|
3a |
1.76(br) |
23.2 |
1.79(br) |
23.4 |
|
3b |
1.46(br) |
1.51,(br) |
|
4 |
---- |
170.8 |
---- |
175.8 |
|
5 |
5.74(ddd,J=9.0,10.0,17.0Hz) |
133.7 |
5.75(m) |
133.4 |
|
6a |
5.25(d,J=17.0Hz) |
117.6 |
5.28(d,J=17.0Hz) |
118.1 |
| 6b |
5.08(dd,J=10.0,1.5Hz) |
5.12(d,J=10.5Hz) |
|
7 |
---- |
155.8 |
---- |
156.2 |
|
8 |
---- |
80.0 |
---- |
80.6 |
|
9 |
1.43(s) |
28.3 |
1.43(s) |
28.3 |
|
10 |
4.16(m) |
61.3 |
---- |
---- |
|
11 |
1.23(t,J=7.5Hz) |
14.2 |
---- |
---- |
拆分B
向在24孔板(容量:10毫升/孔)的一个孔中的0.5mL 100mMHeps·Na缓冲液(pH 8.5)内加入0.1mL Savinase 16.0L(蛋白酶,来自Bacillus clausii)(Novozymes NoRth America Inc.)和所述外消旋N-Boc-(1R,2S)/(1S,2R)-1-氨基-2-乙烯基环丙烷甲酸乙酯(10mg)在0.1mLDMSO中的溶液。将所述板密封并在250rpm、40℃下保温。18h后,测定所述酯的对映体过量为44.3%,接着:取出0.1mL反应混合物并与1mL乙醇充分混合;离心后,用手性HPLC分析10μL上层清液。向剩余的反应混合物中加入0.1mL DMSO,并将所述板在250rpm、40℃下再保温3天,随后向孔中加入4mL乙醇。离心后,用手性HPLC分析10μL上层清液,测定所述酯的对映体过量为100%。
拆分C
向在24孔板(容量:10毫升/孔)的一个孔中的0.5mL 100mMHeps·Na缓冲液(pH 8.5)内加入0.1mL Esperase 8.0L(蛋白酶,来自Bacillus halodurans)(Novozymes NoRth America Inc.)和所述外消旋N-Boc-(1R,2S)/(1S,2R)-1-氨基-2-乙烯基环丙烷甲酸乙酯(10mg)在0.1mL DMSO中的溶液。将所述板密封并在250rpm、40℃下保温。18h后,测定所述酯的对映体过量为39.6%,接着:取出0.1mL反应混合物并与1mL乙醇充分混合;离心后,用手性HPLC分析10μL上层清液。向剩余的反应混合物中加入0.1mL DMSO,并将所述板在250rpm、40℃下再保温3天,随后向孔中加入4mL乙醇。离心后,用手性HPLC分析10μL上层清液,测定所述酯的对映体过量为100%。
按照以下方式分析样品:
1)样品制备:将约0.5ml反应混合物与10体积EtOH充分混合。离心后,将10μL上层清液注射入HPLC柱中。
2)转化率测定:
柱:YMC ODS A,4.6×50mm,S-5μm
溶剂:A,1mM盐酸;B,MeCN
梯度:30%B,1min;30-45%B,0.5min内;45%B,1.5min;45-30%B,0.5min内。
流速:2ml/min
UV检测:210nm
保留时间:酸,1.2min;酯,2.8min。
3)测定所述酯的对映体过量:
柱:CHIRACEL OD-RH,4.6×150mm,S-5μm
流动相:MeCN/50mM高氯酸水溶液(67/33)。
流速:0.75ml/min。
UV检测:210nm。
保留时间:
(1S,2R)异构体,酸形式:5.2min;
外消旋混合物:18.5min和20.0min;
(1R,2S)异构体,酯形式:18.5min。
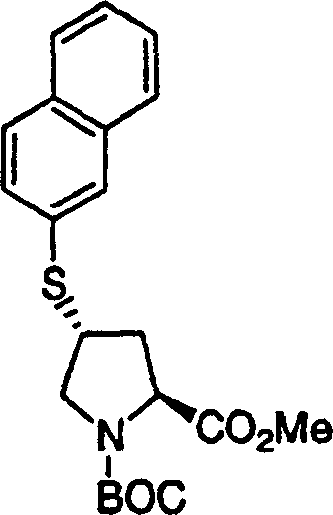
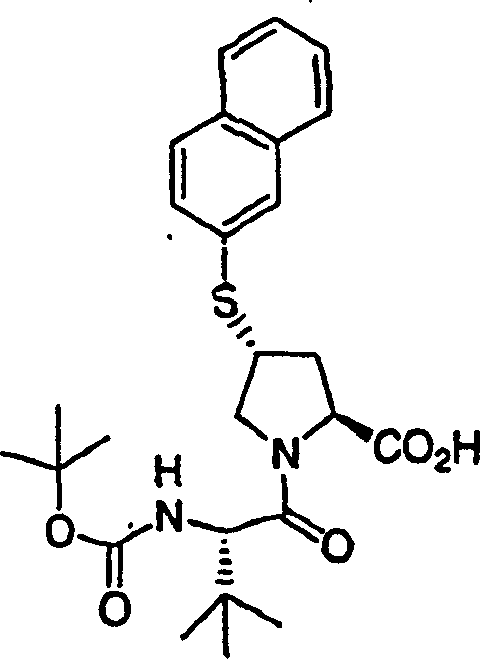
2.N-Boc-(1R,2S)-1-氨基-2-环丙基环丙烷甲酸乙酯的制备
用乙酸钯(5mg,0.022mmol)处理N-Boc-(1R,2S)-1-氨基-2-乙烯基环丙烷甲酸(255mg,1.0mmol)的乙醚溶液(10mL)。将所得橙/红色溶液置于氮气气氛中。在1小时内,滴加重氮甲烷的乙醚溶液。将所得的溶液在室温下搅拌18h。使用氮气流将过量的重氮甲烷除去。将所得的溶液通过旋转蒸发浓缩,得到粗产物。快速层析(10%EtOAc/己烷)纯化,得到210mg(78%)N-Boc-(1R,2S)-1-氨基-2-环丙基环丙烷甲酸乙酯,为无色油状物。LC-MS(保留时间:2.13,类似于方法A,不同之处在于梯度时间为3min,Xterra MS C18 S73.0×50mm柱),MS m/e 270(M++1)。
3.1-叔丁氧基羰基氨基-环丙烷-甲酸为通过商业渠道获得的商品
4.1-氨基环丁烷甲酸甲酯-盐酸盐的制备
将1-氨基环丁烷甲酸(100mg,0.869mmol)(Tocris)溶于10mL甲醇中,鼓泡通入HCl气体2h。将反应混合物搅拌18h,随后真空浓缩,得到144mg黄色油状物。在10mL乙醚中研磨,得到100mg题述产物,为白色固体物。
1H NMR(CDCl3)δ2.10-2.25(m,1H),2.28-2.42(m,1H),2.64-2.82(m,4H),3.87(s,3H),9.21(br s,3H).
5.下述外消旋(1R,2R)/(1S,2S)1-氨基-2-乙基环丙烷甲酸叔丁酯的制备
乙基与羧基呈顺式构型
步骤1:下述2-乙基环丙烷-1,1-二甲酸二-叔丁酯的制备
向苄基三乙基氯化铵(21.0g,92.2mmol)在50%氢氧化钠水溶液(92.4g,185mL H2O)中的悬浮液内加入1,2-二溴丁烷(30.0g,138.9mmol)和丙二酸二叔丁酯(20.0g,92.5mmol)。将反应混合物在室温下剧烈搅拌18h,随后加入冰水混合物。粗产物用二氯甲烷萃取(3×),依次用水(3×)和盐水洗涤,合并各有机萃取液。干燥有机层(硫酸镁)、过滤并真空浓缩。所得的剩余物经快速层析纯化(100g SiO2,3%乙醚/己烷),得到题述产物(18.3g,67.8mmol,收率73%),该产物直接用于下一步反应中。
步骤2:下述外消旋2-乙基环丙烷-1,1-二甲酸叔丁酯的制备
0℃下,将步骤1的产物(18.3g,67.8mmol)加入叔丁醇钾(33.55g,299.0mmol)的无水乙醚(500mL)悬浮液中,接着加入H2O(1.35mL,75.0mmol),并在室温下剧烈搅拌过夜。将反应混合物倒入冰水混合物中并用乙醚洗涤(3×)。在0℃下用10%柠檬酸水溶液酸化水层,并用EtOAc萃取(3×)。合并的有机层用水(2×)和盐水洗涤,干燥(硫酸镁)并真空浓缩,得到题述产物,为浅黄色油状物(10g,46.8mmol,69%收率)。
步骤3:下述(1R,2R)/(1S,2S)2-乙基-1-(2-三甲基甲硅烷基乙氧基羰基氨基)环丙烷-甲酸叔丁酯的制备
向步骤2的产物(10g,46.8mmol)和3g新鲜制备的活化4_分子筛的无水苯(160mL)悬浮液中加入三乙胺(7.50mL,53.8mmol)和DPPA(11mL,10.21mmol)。反应混合物回流3.5h,随后加入2-三甲基甲硅烷基-乙醇(13.5mL,94.2mmol),将反应混合物回流过夜。将反应混合物过滤,用乙醚稀释,依次用10%柠檬酸水溶液、水、饱和碳酸氢钠水溶液、水(2×)、盐水(2×)洗涤,干燥(硫酸镁)并真空浓缩。剩余物悬浮于10g Aldrich聚异氰酸酯净化树脂在120mL二氯甲烷中的溶液中,室温下搅拌过夜,过滤,得到题述产物(8g,24.3mmol;52%),为浅黄色油状物:
1H NMR(CDCl3)δ0.03(s,9H),0.97(m,5H),1.20(bm,1H),1.45(s,9H),1.40-1.70(m,4H),4.16(m,2H),5.30(bs,1H).
步骤4:下述外消旋(1R,2R)/(1S,2S)1-氨基-2-乙基环丙烷甲酸叔丁酯的制备。
乙基与羧基呈顺式构型
向步骤3的产物(3g,9mmol)中加入1.0M TBAF的THF溶液(9.3mL,9.3mmol),将混合物加热回流1.5h,冷却至室温,随后用500mlEtOAc萃取。溶液依次用水(2×100mL)和盐水(2×100mL)洗涤,干燥(硫酸镁),真空浓缩得到题述中间体。
II P1′组件:
使用本文中描述的方法,以下制备的P1′组件可用于制备式I化合物。
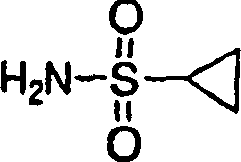
1.环丙基磺酰胺的制备:
步骤1:N-叔丁基-(3-氯)丙基磺酰胺的制备
将叔丁基胺(3.0mol,315.3mL)溶于THF(2.5L)中。将溶液冷却至-20℃。缓慢加入3-氯丙烷磺酰氯(1.5mol,182.4mL)。将反应混合物升温至室温并搅拌24h。将混合物过滤,滤液真空浓缩。将剩余物溶于二氯甲烷(2.0L)中,所得的溶液用1N HCl(1.0L)、水(1.0L)、盐水(1.0L)洗涤并经硫酸钠干燥。过滤并真空浓缩,得到浅黄色固体物,在己烷中结晶,得到产物,为白色固体物(316.0g,99%)。
1H NMR(CDCl3)δ1.38(s,9H),2.30-2.27(m,2H),3.22(t,J=7.35Hz,2H),3.68(t,J=6.2Hz,2H),4.35(b,1H).
步骤2:环丙烷磺酸叔丁基酰胺的制备
在-78℃下,向N-叔丁基-(3-氯)丙基磺酰胺(2.14g,10.0mmol)的THF(100mL)溶液中加入n-BuLi(2.5M己烷溶液,8.0mL,20.0mmol)。在1h内,将反应混合物升温至室温。真空除去挥发份。使剩余物在乙酸乙酯和水(200mL,200mL)间分配。分离出的有机相用盐水洗涤、经硫酸钠干燥、过滤并真空浓缩。剩余物在己烷中重结晶,得到所需的产物,为白色固体物(1.0g,56%)。
1H NMR(CDCl3)δ0.98-1.00(m,2H),1.18-1.19(m,2H),1.39(s,9H),2.48-2.51(m,1H),4.19(b,1H)。
步骤3:环丙基磺酰胺的制备
将环丙烷磺酸叔丁基酰胺(110.0g,0.62mol)的TFA(500mL)溶液在室温下搅拌16h。真空除去挥发份。剩余物在EtOAC/己烷(60mL/240mL)中重结晶,得到所需的产物,为白色固体物(68.5g,91%)。
1H NMR(DMSO-d6)δ0.84-0.88(m,2H),0.95-0.98(m,2H),2.41-2.58(m,1H),6.56(b,2H).
2.环丙基磺酰胺作为选择的制备方法
向100ml冷却至0℃的THF溶液中鼓入氨气,直至饱和。向该溶液中加入5g(28.45mmol)环丙基磺酰氯(购自Array Biopharma)在50mL THF中的溶液中,将溶液在室温下加热过夜并再搅拌一天。将混合物浓缩,直至剩余1-2mL溶剂,装入30g硅胶柱层析(洗脱液:30%-60%EtOAc/己烷)中,得到3.45g(100%)环丙基磺酰胺,为白色固体物。
1H NMR(甲醇-d4)δ0.94-1.07(m,4H),2.52-2.60(m,1H);13C NMR(甲醇-d4)δ5.92,33.01。
3.环丁基磺酰胺的制备
向冷却至-78℃的5.0g(37.0mmol)环丁基溴在30ml无水乙醚中的溶液内加入44mL(74.8mmol)1.7M叔丁基锂的戊烷溶液,在1.5h内,将溶液缓慢升温至-35℃。将该混合物经插套管缓慢加入5.0g(37.0mmol)冷却至-40℃的新鲜蒸馏的磺酰氯在100ml己烷中的溶液内,在1h内升温至0℃并小心真空浓缩。将该混合物再次溶于乙醚中,用一些冰冷却水洗涤一次,干燥(硫酸镁)并小心浓缩。将该混合物再次溶于20ml THF中,滴加入500ml饱和氨的THF溶液中,搅拌过夜。将混合物真空浓缩至得到粗品黄色固体物,在尽可能少量的二氯甲烷的己烷溶液(含1-2滴甲醇)中重结晶,得到1.90g(38%)环丁基磺酰胺,为白色固体物。
1H NMR(CDCl3)δ1.95-2.06(m,2H),2.30-2.54(m,4H),3.86(p,J=8Hz,1H),4.75(brs,2H);13C NMR(CDCl3)δ16.43,23.93,56.29.HRMS m/z(M-H)-C4H8NSO2的计算值:134.0276,实测值134.0282。
4环戊基磺酰胺的制备
将18.5mL(37.0mmol)2M氯化环戊基-镁的乙醚溶液滴加入3.0mL(37.0mmol)新鲜蒸馏的磺酰氯(得自Aldrich)在100ml己烷中的溶液(冷却至-78℃)内。在1小时内将混合物升温至0℃并小心真空浓缩。将该混合物再次溶于乙醚(200mL)中,用一些冰冷却的水(200mL)洗涤一次,干燥(硫酸镁)并小心浓缩,将该混合物再次溶于35ml THF中,滴加入500ml饱和氨的THF溶液并搅拌过夜。将混合物真空浓缩至得到粗品黄色固体物,剩余物经50g硅胶过滤,使用70%EtOAc-己烷为洗脱液,随后将溶液浓缩。剩余物在尽可能少量的二氯甲烷的己烷溶液(含1-2滴甲醇)中重结晶,得到2.49g(41%)环戊基磺酰胺,为白色固体物。
1H NMR(CDCl3)δ1.58-1.72(m,2H),1.74-1.88(m,2H),1.94-2.14(m,4H),3.48-3.59(m,1H),4.80(bs,2H);13C NMR(CDCl3)δ25.90,28.33,63.54;MS m/e 148(M-H)-.
5.环己基磺酰胺的制备
将18.5mL(37.0mmol)2M氯化环己基镁(TCI Americas)的乙酸溶液滴加入3.0mL(37.0mmol)新鲜蒸馏的磺酰氯在100ml己烷中的溶液(冷却至-78℃)内。在1小时内将混和物升温至0℃并小心真空浓缩。该混合物再次溶于乙醚(200mL),用一些冰冷却的水(200mL)洗涤一次,干燥(硫酸镁)并小心浓缩。将该混合物再次溶于35ml THF中,滴加入500ml氨饱和的THF溶液并搅拌过夜。将混合物真空浓缩至得到粗品黄色固体物,剩余物经50g硅胶过滤,使用70%EtOAc-己烷为洗脱液,浓缩。剩余物在尽可能少量的二氯甲烷的己烷溶液(含1-2滴甲醇)中重结晶,得到1.66g(30%)环己基-磺酰胺,为白色固体物。
1H NMR(CDCl3)δ1.11-1.37(m,3H),1.43-1.56(m,2H),1.67-1.76(m,1H),1.86-1.96(m,2H),2.18-2.28(m,2H),2.91(tt,J=12,3.5Hz,1H),4.70(bs,2H);13CH NMR(CDCl3)δ25.04,25.04,26.56,62.74;MS m/e 162(M-1)-.
6.新戊基磺酰胺的制备
按照制备环己基磺酰胺的方法,将49mL(37mmol)0.75M氯化新戊基镁(Alfa)的乙醚溶液转化为1.52g(27%)新戊基磺酰胺,为白色固体物。
1H NMR(CDCl3)δ1.17(s,9H),3.12(s,2H),4.74(brs,2H);13C NMR(CDCl3)δ29.46,31.51,67.38;MS m/e 150(M-1)-.
7.环丁基甲基(carbinyl)-磺酰胺的制备
将12.3g(83mmol)环丁基甲基溴(Aldrich)和13.7g(91mmol)碘化钠在150ml丙酮中的溶液回流过夜,随后冷却至室温。过滤出无机固体物,分别在常压及150托、80℃下蒸馏出丙酮和环丙基甲基碘(8.41g,46%)。
将冷却至-78℃的4.0g(21.98mmol)环丁基甲基碘在30ml无水乙醚(乙醚)中的溶液经插管装入17mL(21.98mmol)1.3M仲丁基锂的环己烷溶液中,将所得溶液搅拌5min。向该混合物中经插管加入3.0g(21.98mmol)新鲜蒸馏的磺酰氯在110ml己烷中的溶液(冷却至-78℃),在1小时内将混合物升温至室温,随后小心真空浓缩。该混合物再次溶于乙醚,用一些冰冷却的水洗涤一次,干燥(硫酸镁)并小心浓缩,将该混合物再次溶于30ml THF中,滴加入500ml氨饱和的THF溶液并搅拌过夜。将混合物真空浓缩至得到粗品黄色固体物,在尽可能少量的二氯甲烷的己烷溶液(含1-2滴甲醇)中重结晶,得到1.39g(42%)环丁基甲磺酰胺,为白色固体物。
1H NMR(CDCl3)δ1.81-2.03(m,4H),2.14-2.28(m,2H),2.81-2.92(m,1H),3.22(d,J=7Hz,2H),4.74(brs,2H);13C NMR(CDCl3)δ19.10,28.21,30.64,60.93;MS m/e 148(M-1)-.时间:1.73,甲醇B),818(M++H)
8:环丙基甲磺酰胺的制备
使用用于制备环丁基甲磺酰胺的方法,由环丙基甲基溴(Aldrich)制备环丙基甲磺酰胺(还可参见JACS 1981,第442-445页)。
1H NMR(CDCl3)δ0.39-0.44(m,2H),0.67-0.76(m,2H),1.13-1.27(m,1H),3.03(d,J=7.3Hz,2H),4.74(brs,2H);13CNMR(CDCl3)δ4.33,5.61,59.93;MS m/e 134(M-1).
III用作构造构成式I化合物的P2组件的原料的杂环
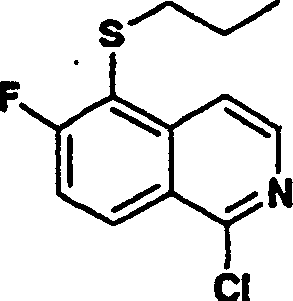
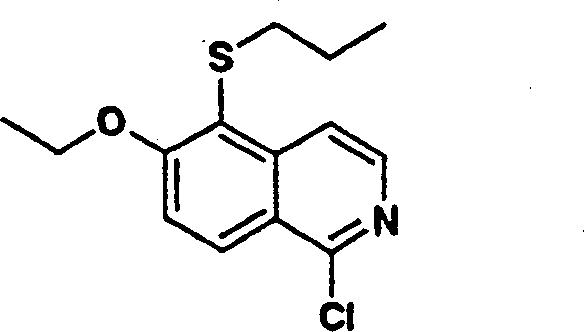
1.异喹啉
使用上述的两种方法,可将异喹啉(1)及其取代的类似物引入P2组件中,这些方法将在本文详细描述。随后使用用于异喹啉类似物的类似方法,可将所述P2组件(3)转化为式I化合物。
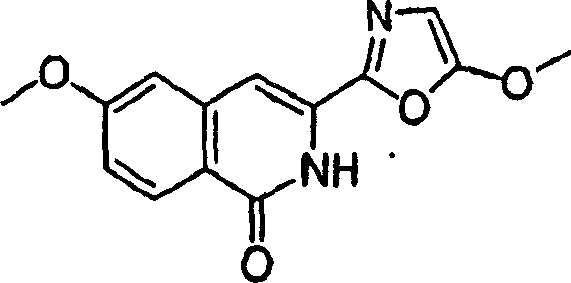
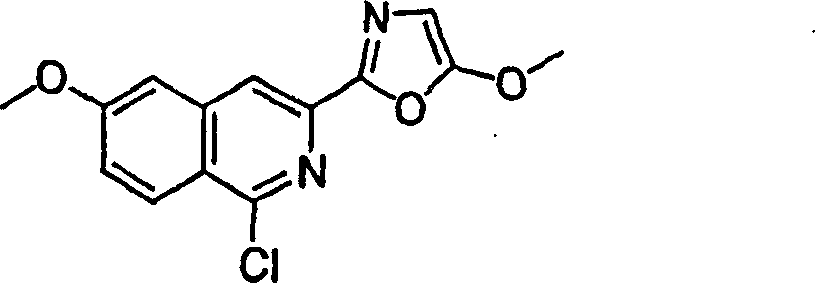
2.异_唑吡啶和_唑吡啶(1)。
可使用已知化学方法制备异_唑和_唑杂环(1)及其类似物,并随后使用与部分B所示的,用于类似的异_唑吡啶中间体的化学方法引入到式I化合物中。
部分B:
在部分B中,使用以下条件进行LC/MS分析。
柱:
方法A:YMC ODS-A C18 S7(4.6×33mm)。
方法B:YMC Xterra ODS S7(3.0×50mm)。
方法C:Xterra ms C18(4.6×33mm)。
方法D:YMC ODS-A C18 S3(4.6×33mm)。
梯度:100%溶剂A/0%溶剂B至0%溶剂A/100%溶剂B
梯度时间:3min。
保留时间:1min。
流速:5mL/min。
检测器波长:220nm。
溶剂:溶剂A:10%甲醇/90%水/0.1%TFA。溶剂B:90%甲醇/10%水/0.1%TFA。
以下条件用于制备型HPLC分离。
柱:Phenomenex-Luna 30×100mm,S5
梯度:60%溶剂A/40%溶剂B至0%溶剂A/100%溶剂B
梯度时间:15min。
保留时间:20min。
流速:30mL/min。
检测器波长:220nm。
溶剂:溶剂A:10%甲醇/90%水/0.1%TFA。溶剂B:90%甲醇/10%水/0.1%TFA。
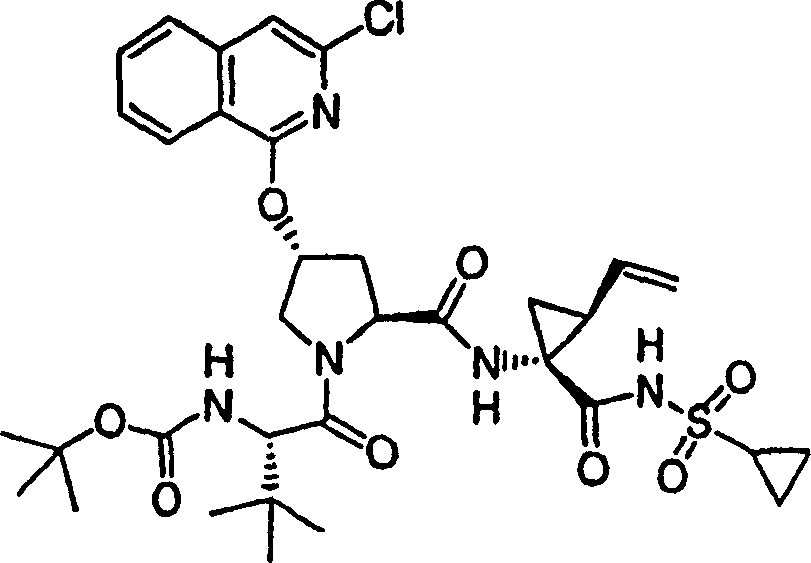
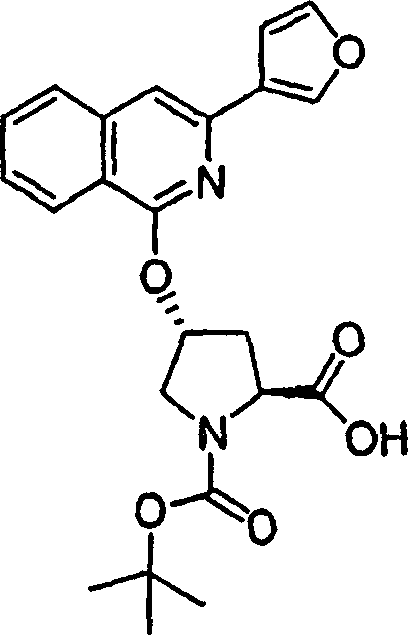
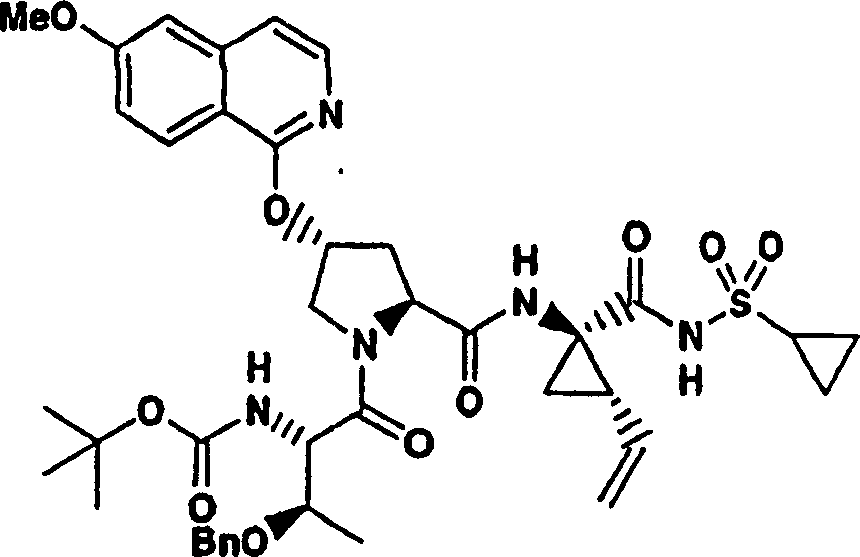
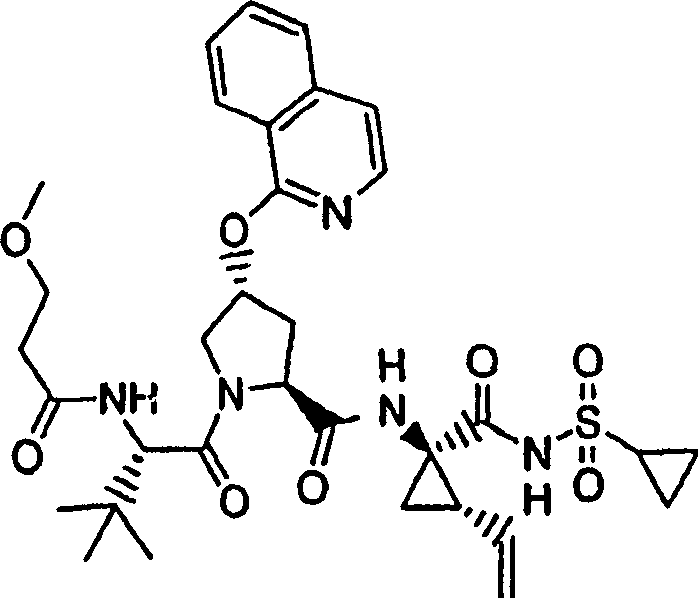
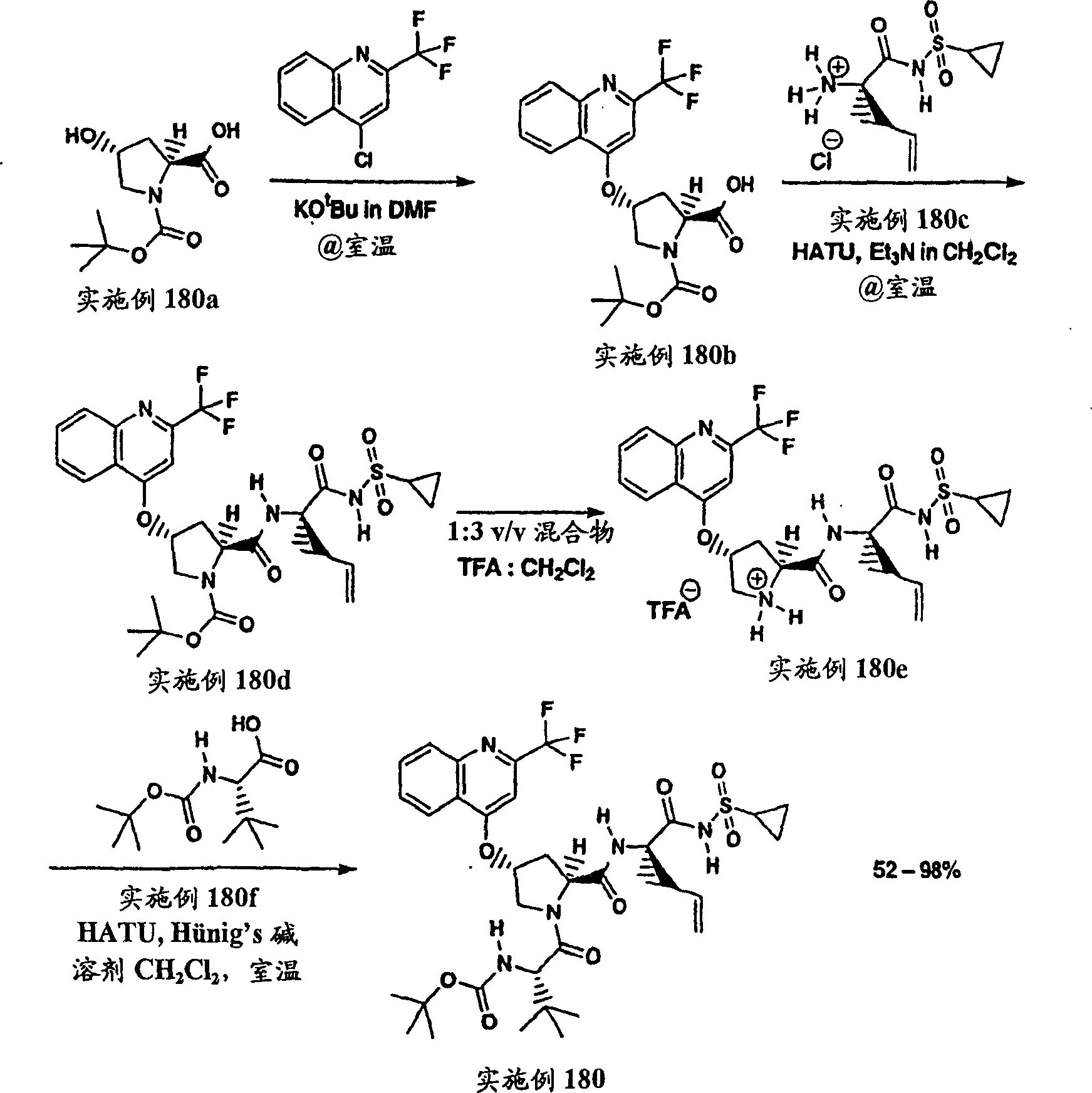
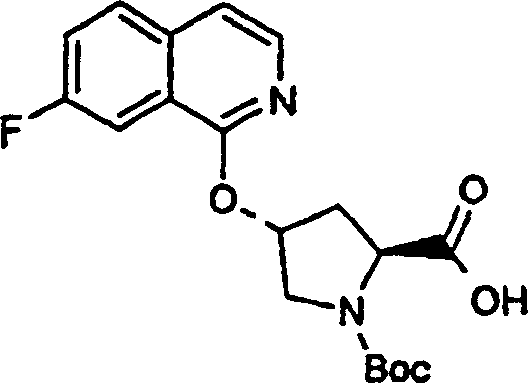
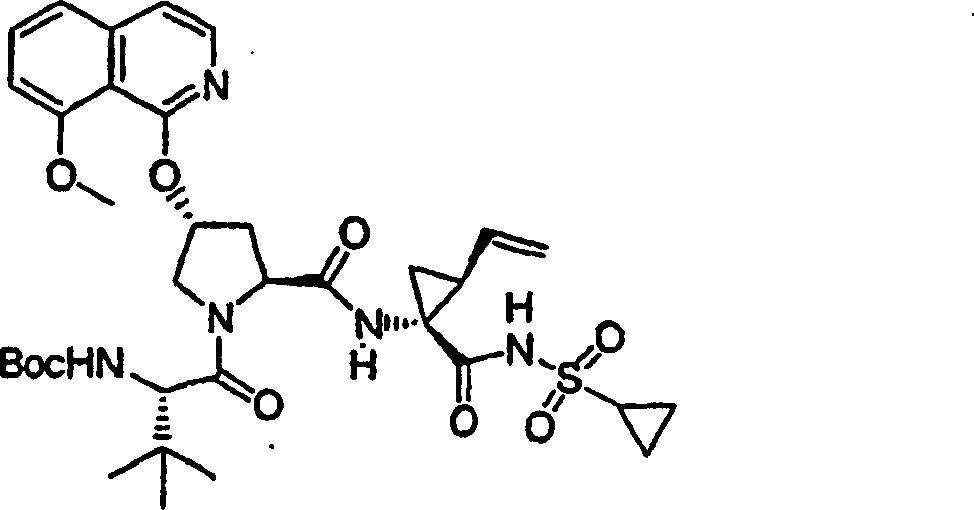
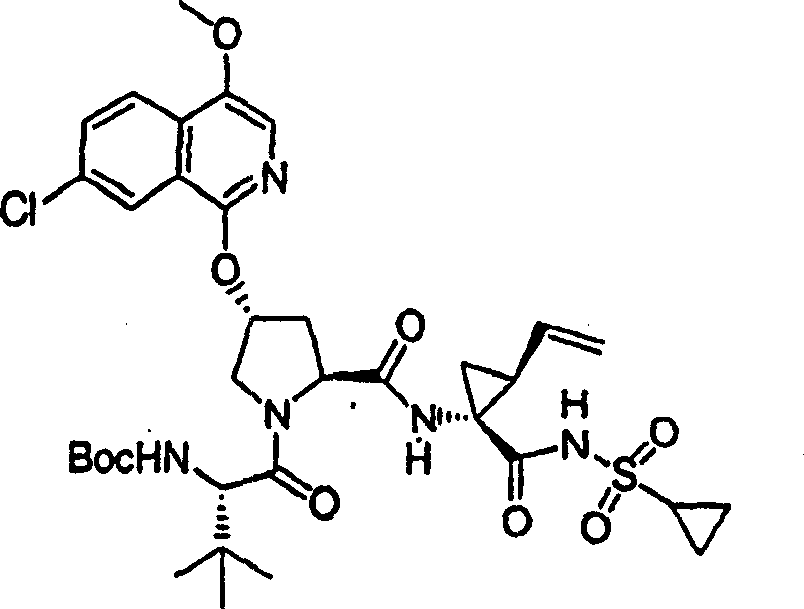
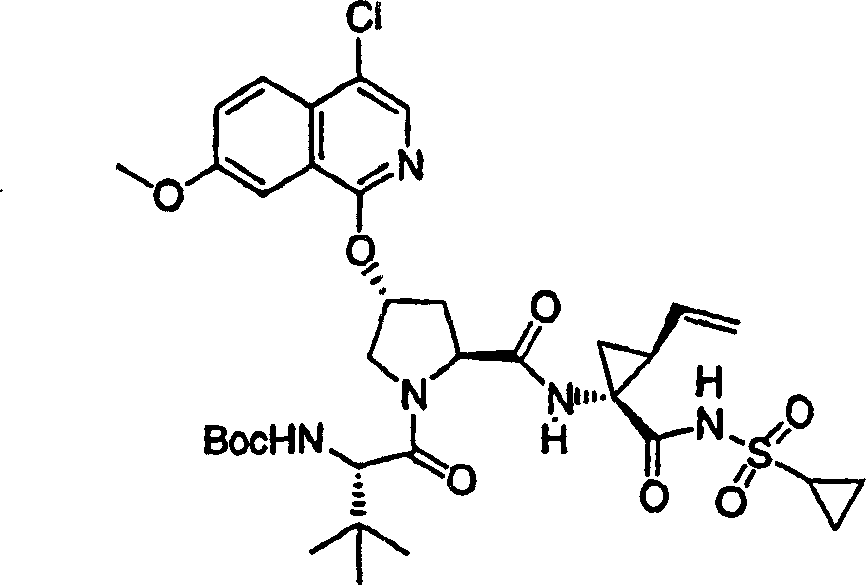
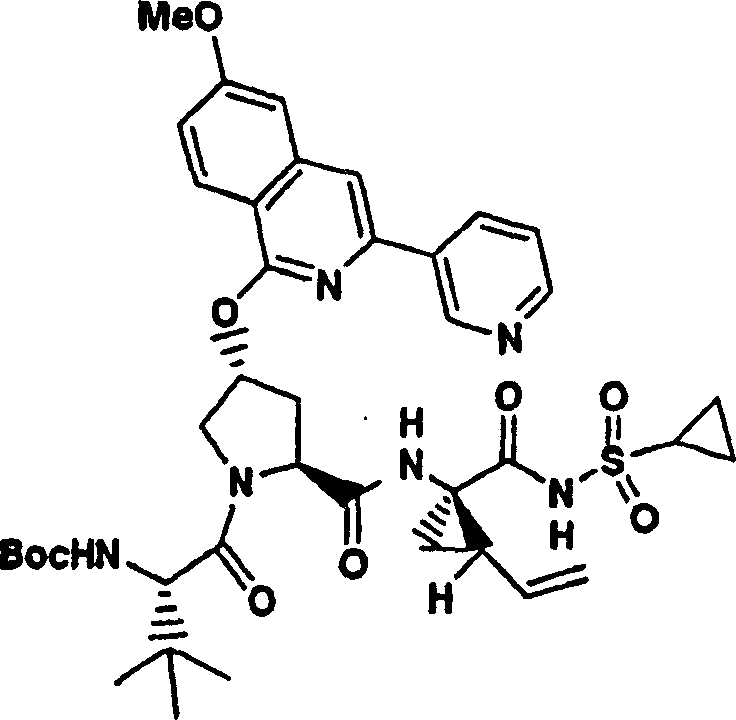
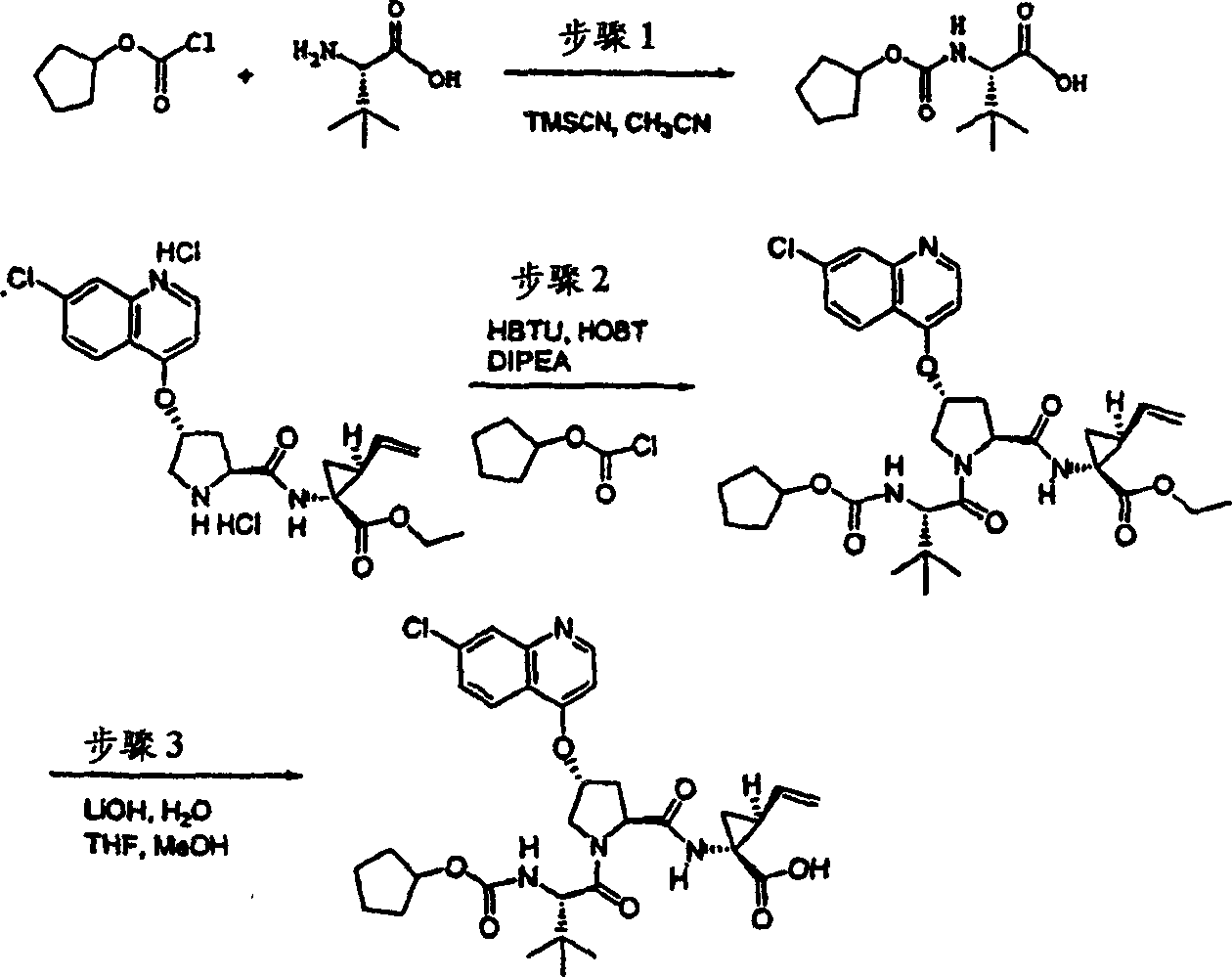
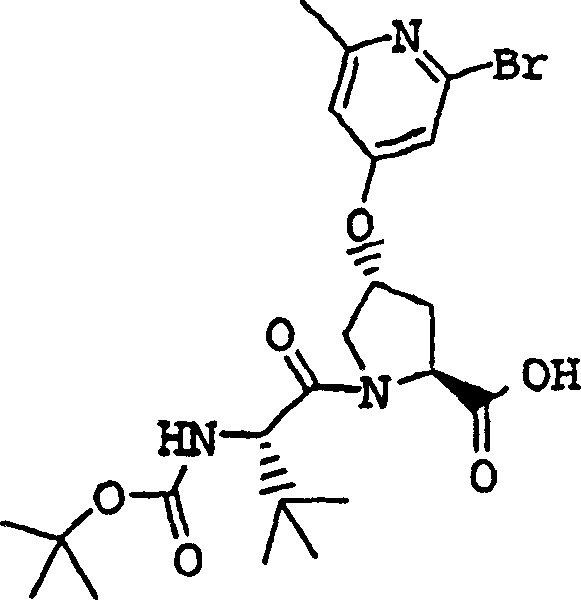
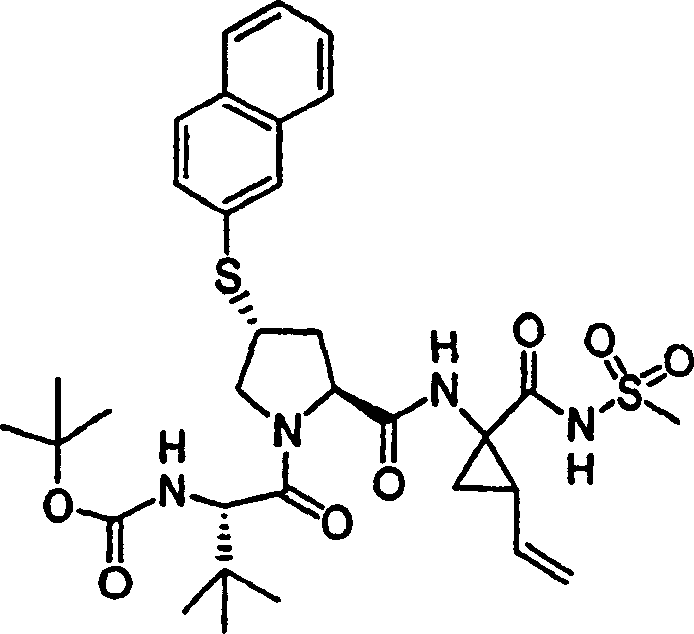
实施例1:化合物1的制备
化合物1
方案1
步骤1:
将3,5-二甲基-4-硝基-异_唑(1.42g,10.0mmol)、苯乙醛(1.32g,11.0mmol)在哌啶(1mL)和乙醇(10mL)中的混合物加热回流16h。冷却至环境温度后,过滤收集沉淀出的产物。滤饼用冷乙醇充分洗涤,得到1.20g(53%)所需的产物,为白色固体物。
1H NMR(CDCl3)δ2.87(s,3H),7.46-7.50(m,3H),7.56(d,J=8.5Hz,1H),7.7-7.80(m,2H);
LC-MS(保留时间:1.19min,方法B),MS m/z 227(M++H)。
步骤2:
将3-甲基-5-苯基-异_唑并[4,5-b]吡啶-4-氧化物(1.00g,4.40mmol)和磷酰氯(2.71g,17.7mmol)在氯仿(10mL)中的溶液加热回流1h。冷却至环境温度后,最终的溶液用氯仿(50mL)稀释,用碳酸氢钠水溶液(两次,每次50mL)和盐水洗涤,经硫酸镁干燥,过滤,蒸发。剩余物经快速层析纯化(4∶1己烷-EtOAc),得到790mg(73%)所需的产物,为白色固体物。
1H NMR(CDCl3)δ2.72(s,3H),7.46-7.54(m,3H),7.91(s,1H),8.00-8.03(m,2H);LC-MS(保留时间:1.76min,方法B),MS m/z 245,247(M++H)。
方案2
步骤3:
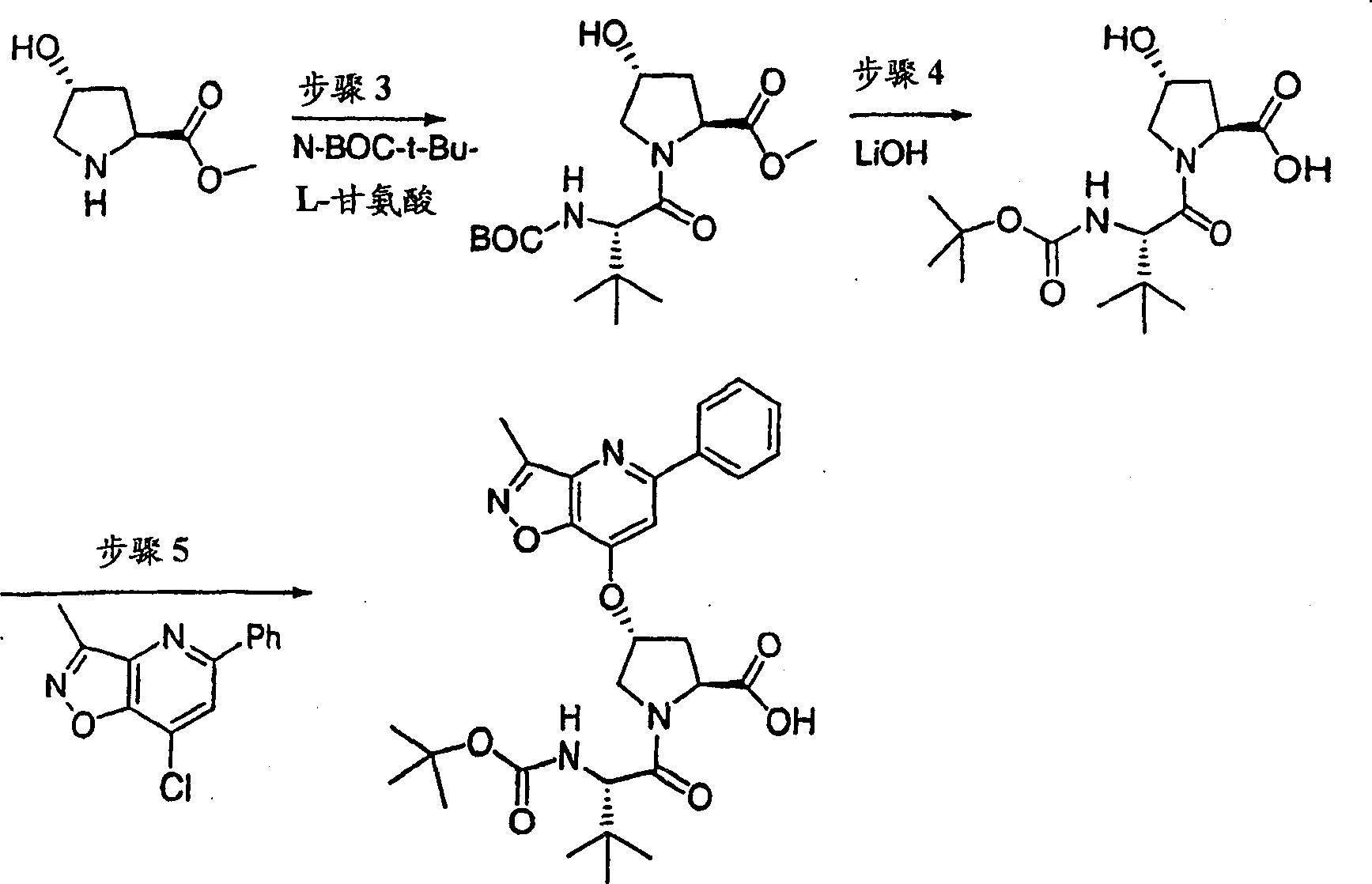
在0℃下,向4-羟基-吡咯烷-2-甲酸甲酯(H-Hyp-OMe HCl)(1.81g,10.0mmol)、HATU(5.70g,15.0mmol)和N-BOC-叔丁基-L-甘氨酸(2.42g,10.5mmol)在二氯甲烷(100mL)中的混合物内加入DIPEA(3.47g,31.0mmol)。在环境温度下搅拌12h后,将前述溶液用二氯甲烷(100mL)稀释,用冰冻的5%柠檬酸水溶液洗涤。有机层依次用5%柠檬酸、1M NaOH、盐水洗涤,经硫酸镁干燥并过滤。将滤液真空蒸发,得到3.55g(99%)所需的产物,为类白色泡沫状物。该产物未经进一步纯化,直接用于下一步反应中。
1H NMR(CD3OD)δ1.04(s,9H),1.43(s,9H),1.99-2.03(m,1H),2.20-2.30(m,1H),3.69(s,3H),3.70-3.79(m,2H),4.28(b,1H),4.46(b,1H),4.74-4.80(m,1H);LC-MS(保留时间:1.28min,方法B),MS m/z 359(M++H)。
步骤4:
将步骤3的产物(3.55g,9.9mmol)在THF(50mL)、甲醇(50mL)和一水合氢氧化锂(0.83g,19.9mmol,50mL H2O)中的混合物在环境温度下搅拌过夜。真空除去挥发份后,将剩余物溶于0.1M NaOH(100mL)中。该水溶液用乙醚(50mL)洗涤,用1M HCl酸化至pH为4。用EtOAc(100mL)萃取。有机层用5%柠檬酸和盐水洗涤,经硫酸镁干燥,蒸发至干,得到3.20g(95%)所需的产物,为白色泡沫状物。该产物未经进一步纯化,直接使用。
1H NMR(CD3OD)δ1.02(s,9H),1.43(s,9H),2.01-2.09(m,1H),2.25-2.32(m,1H),3.70-3.85(m,2H),4.264.30(m,1H),4.46-4.51(m,2H),6.37-6.41(m,1H);LC-MS(保留时间:1.14min,方法B),MS m/z 345(M++H)。
步骤5:
向步骤4的产物(1.01g,2.93mmol)的DMSO(30mL)溶液中加入叔丁醇钾(1.02g,9.08mmol)。将所形成的溶液在环境温度下搅拌1h,随后加入7-氯-3-甲基-5-苯基-异_唑并[4,5-b]吡啶(0.75g,3.08mmol)。将最终的溶液搅拌12h。随后用冰冻的水猝灭,用1M HCl酸化至pH为4,用EtOAc(两次,每次200mL)萃取。有机层用盐水洗涤,经硫酸镁干燥,过滤,蒸发。剩余物经制备型HPLC纯化(60%B-100%B,15min梯度),得到305mg(19%)所需的产物,为浅黄色固体物。
1H NMR(CD3OD)δ1.02(s,9H),1.17(s,9H),2.37-2.47(m,1H),2.64(s,3H),2.85-2.93(m,1H),4.00-4.08(m,1H),4.14(b,1H),4.49-4.55(m,1H),4.62-4.71(m,1H),5.70(m,1H),7.45-7.53(m,3H),7.56(s,1H),8.03-8.06(m,2H);
LC-MS(保留时间:1.89min,方法B),MS m/z 553(M++H)。
方案3
步骤6a
如部分A所述。
步骤6b:
向1(R)-叔丁氧基羰基氨基-2(S)-乙烯基-环丙烷甲酸乙酯、步骤6a的产物(3.28g,13.2mmol)在THF(7mL)和甲醇(7mL)中的溶液内加入LiOH(1.27g,53.0mmol)的水(14mL)悬浮液。将混合物在室温下搅拌过夜并用1N NaOH(15mL)和水(20mL)猝灭。所得的混合物用EtOAc(20mL)洗涤,有机相用20mL 0.5N NaOH萃取。合并的水相用1N HCl酸化,直至pH为4,用EtOAc(3×40mL)萃取。合并的有机萃取液用盐水洗涤并干燥(硫酸镁),得到题述化合物,为白色固体物(2.62g,87%)。
1H NMR:(DMSO-d6)δ1.22-1.26(m,1H),1.37(s,9H),1.50-1.52(m,1H),2.05(q,J=9Hz,1H),5.04(d,J=10Hz,1H),5.22(d,J=17Hz,1H),5.64-5.71(m,1H),7.18,7.53(s,NH(外消旋体),12.4(br s,1H));
LC-MS(保留时间:1.67min,方法B),MS m/z 228(M++H)。
步骤7:
在氮气气氛中,将步骤6的产物(2.62g,11.5mmol)和CDI(2.43g,15.0mmol)的THF(40mL)溶液加热回流50min。将溶液冷却至室温并经插管转移至环丙基磺酰胺(1.82g,15.0mmol)的THF(10mL)溶液中。向所得的溶液中加入DBU(2.40mL,16.1mmol),继续搅拌20h。混合物用1N HCl猝灭至pH为1,真空蒸发出THF。悬浮液用EtOAc萃取(2×50mL),将合并的有机萃取液干燥(硫酸钠)。在己烷-EtOAc(1∶1)中重结晶纯化,得到题述化合物(2.4g),为白色固体物。母液经Biotage40S柱纯化(用9%丙酮/DCM洗脱),得到第二部分的题述化合物(1.1g)。将两部分合并(总收率92%)。
1H NMR:(DMSO-d6)δ0.96-1.10(m,4H),1.22(dd,J=5.5,9.5Hz,1H),1.39(s,9H),1.70(t,J=5.5Hz,1H),2.19-2.24(m,1H),2.90(m,1H),5.08(d,J=10Hz,1H),5.23(d,J=17Hz,1H),5.45(m,1H),6.85,7.22(s,NH(外消旋体);
LC-MS(保留时间:1.70min,方法B),MS m/z 331(M++H)。
步骤8:
将步骤7的产物(3.5g,10.6mmol)在DCM(35mL)和TFA(32mL)中的溶液在室温下搅拌1.5h。真空除去挥发份,将剩余物悬浮于1NHCl的乙醚溶液(20mL)中并真空浓缩。重复一次该过程。所得的混合物在戊烷中研磨并过滤,得到题述化合物,为易潮的、灰白色固体物(2.60g,92%)。
1H NMR:(DMSO-d6)δ1.01-1.15(m,4H),1.69-1.73(m,1H),1.99-2.02(m,1H),2.38(q,J=9Hz,1H),2.92-2.97(m,1H),5.20(d,J=11Hz,1H),5.33(d,J=17Hz,1H),5.52-5.59(m,1H),9.17(br s,3H);
LC-MS(保留时间:0.24min,方法B),MS m/z 231(M++H)。
步骤9:
向冰冻的步骤5的产物(70mg,0.13mmol)、步骤8的产物(1R,2S)-N-(1-氨基-2-乙烯基-环丙烷羰基)-环丙烷磺酰胺盐酸盐(37mg,0.14mmol)和HATU(72mg,0.19mmol)在DCM(2mL)中的混合物内加入二异丙基乙胺(50mg,0.39mmol)。将所形成的溶液升温至环境温度下反应12h并真空蒸发。剩余物经制备型HPLC纯化(60%B-100%B,15min梯度),得到52mg(54%)化合物1,为浅灰色固体物。1H NMR(CD3OD)δ0.96-1.09(m,12H),1.16-1.25(m,10H),1.44-1.48(m,1H),1.87-1.91(m,1H),2.20-2.40(m,2H),2.63-2.65(m,4H),2.89-2.98(m,1H),4.08-4.20(m,2H),4.44-4.65(m,2H),5.13(d,J=11.7Hz,1H),5.32(d,J=15Hz,1H),5.72-5.85(b,2H),6.62(d,J=15.0Hz,1H),7.46-7.53(m,3H),7.58(s,1H),8.04-8.07(m,2H);
LC-MS(保留时间:1.92min,方法B),MS m/z 765(M++H)。
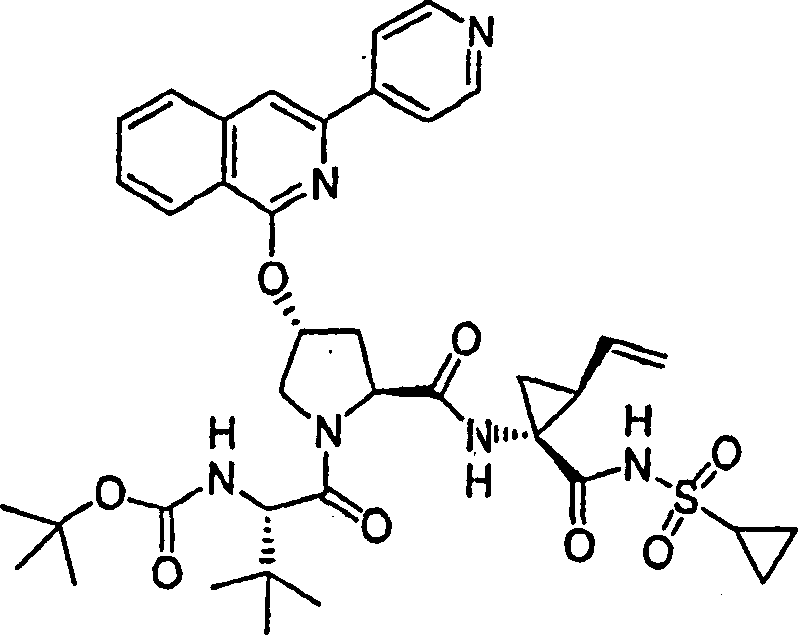
实施例2:化合物2的制备
化合物2
方案1
步骤1:
将2-氨基-6-甲基吡啶(1.08g,10.0mmol)、苯甲酰基乙酸乙酯(2.30g,12.0mmol)和聚磷酸(6.00g,61.2mmol)的混合物加热至110℃下反应5h。冷却至环境温度下,将混合物倒入冰冻的水(20mL)中,并用10M NaOH中和至pH为7。用氯仿萃取。有机层用盐水洗涤,经硫酸镁干燥,过滤,蒸发。剩余物经快速层析纯化(1∶1己烷-EtOAc),得到510mg(22%)所需的产物,为浅黄色固体物。
1H NMR(CDCl3)δ3.08(s,3H),6.64(d,J=7.0Hz,1H),6.71(s,1H),7.42-7.52(m,5H),8.04-8.06(m,2H);
LC-MS(保留时间:1.21min,方法B),MS m/z 237(M++H)。
步骤2:
将6-甲基-2-苯基-吡啶并[1,2a]嘧啶-4-酮(489mg,2.07mmol)在熔融的二苯基醚(5mL)的溶液加热至温和回流5h。冷却至环境温度下,将所形成的悬浮液用乙醚(10mL)稀释,过滤。滤饼用乙醚充分洗涤,得到450mg(92%)所需的产物,为褐色固体物。
LC-MS(保留时间:1.25min,方法B),MS m/z 237(M++H)。
步骤3:
将7-甲基-2-苯基-1H-[1,8]萘啶-4-酮(450mg,1.91mmol)的磷酰氯(10mL)悬浮液加热至温和回流3h。真空蒸发。将剩余物倒入冰冻的水(20mL)中,用10M NaOH中和至pH为10。用氯仿萃取。有机层用盐水洗涤,经硫酸镁干燥,过滤,蒸发。剩余物经快速层析纯化(2∶1己烷-EtOAc),得到450mg(92%)所需的产物,为粉红色固体物。
1H NMR(CD3OD)δ2.80(s,3H),7.54-7.56(m,3H),7.61(d,J=8.4Hz,1H),8.25-8.30(m,3H),8.58(d,J=8.4Hz,1H);
LC-MS(保留时间:1.39min,方法B),MS m/z 255,257(M++H)。
方案2
步骤4:
按照实施例1、步骤5的相同方法制备该步骤的产物,不同之处在于使用实施例2、步骤3的4-氯-7-甲基-2-苯基-[1,8]萘啶。
LC-MS(保留时间:1.55min,方法B),MS m/z 563(M++H)。
步骤5:
按照实施例1、步骤9的相同方法制备化合物2,不同之处在于使用实施例2、步骤4的产物。
1H NMR(CD3OD)δ1.01-1.10(m,12H),1.21-1.26(m,10H),1.40-1.45(m,1H),1.86-1.91(m,1H),2.20-2.29(m,1H),2.39-2.49(m,1H),2.72-2.81(m,1H),2.92-2.95(m,4H),4.10-4.16(m,2H),4.55-4.65(m,2H),5.14(d,J=12.0Hz,1H),5.30(d,J=15.0Hz,1H),5.67-5.82(m,2H),7.60-7.80(m,3H),7.78(d,J=8.6Hz,1H),7.87(s,1H),8.26-8.29(m,2H),8.95(d,J=8.4Hz,1H);
LC-MS(保留时间:1.62min,方法B),MS m/z 775(M++H)。
实施例3:化合物3的制备
化合物3
方案1
步骤1:
在0℃下,向4-甲氧基苯乙醇(1.52g,10.0mmol)的二氯甲烷(50mL)溶液中一次性加入Dess-MaRtin试剂(4.45g,10.5mmol)。将所形成的混合物升温至环境温度下反应1h。依次用饱和硫代硫酸钠水溶液、1M NaOH和盐水洗涤。经硫酸镁干燥,真空蒸发,得到1.50g(100%)所需的醛,为粘稠的油状物。该产物未经进一步纯化直接使用。
步骤2:
将3,5-二甲基-4-硝基-异_唑(142mg,1.0mmol)、实施例3步骤1的4-甲氧基-苯乙醛(180mg,1.1mmol)在哌啶(0.1mL)和乙醇(2mL)中的溶液加热回流12h。冷却至环境温度后,过滤收集沉淀出的产物。滤饼用冷乙醇充分洗涤,得到130mg(51%)所需的产物,为浅灰色固体物。
1H NMR(CDCl3)δ2.88(s,3H),3.87(s,3H),7.02(d,J=8.5Hz,2H),7.50(d,J=9.0Hz,1H),7.57(d,J=9.0Hz,1H),7.81(d,J=8.5Hz,2H);
LC-MS(保留时间:1.24min,方法B),MS m/z 257(M++H)。
步骤3:
按照实施例1、步骤2的相同方法制备该产物,不同之处在于使用实施例3、步骤2的产物。
1H NMR(CDCl3)δ2.70(s,3H),3.87(s,3H),7.00-7.03(m,2H),7.84(s,1H),7.96-7.98(m,2H);
LC-MS(保留时间:1.96min,方法B),MS m/z 275,277(M++H)。
步骤4:
按照实施例1、步骤5的相同方法制备该产物,不同之处在于使用实施例3、步骤3的产物。
1H NMR(CD3OD)δ1.02(s,9H),1.18(s,9H),2.39-2.43(m,1H),2.63(s,3H),2.75-2.80(m,1H),3.87(s,3H),4.00-4.08(m,1H),4.17(b,1H),4.49-4.55(m,1H),4.62-4.71(m,1H),5.68(b,1H),7.05(d,J=8.5Hz,2H),7.49(s,1H),8.00(d,J=8.5Hz,2H);
LC-MS(保留时间:1.89min,方法B),MS m/z 583(M++H)。
步骤5:
按照实施例1、步骤9的相同方法制备化合物3,不同之处在于使用实施例3、步骤4的产物。
1H NMR(CD3OD)δ1.01-1.09(m,12H),1.17-1.26(m,10H),1.44-1.47(m,1H),1.87-1.91(m,1H),2.20-2.40(m,2H),2.63-2.65(m,4H),2.89-2.98(m,1H),3.87(s,3H),4.08-4.20(m,2H),4.44-4.65(m,2H),5.13(d,J=11.7Hz,1H),5.32(d,J=15.0Hz,1H),5.72-5.85(m,2H),7.05(d,J=8.5Hz,2H),7.06(s,1H),8.01(d,J=8.5Hz,2H);
LC-MS(保留时间:1.96min,方法B),MS m/z 795(M++H)。
实施例4:化合物4的制备
化合物4
步骤1:
按照实施例3、步骤1和2的相同方法制备该产物,不同之处在于使用4-氟苯乙醇。
LC-MS(保留时间:1.18min,方法B),MS m/z 245(M++H)。
步骤2:
按照实施例1、步骤2的相同方法制备该产物,不同之处在于使用实施例4、步骤1的产物。
1H NMR(CDCl3)δ2.71(s,3H),7.17-7.20(m,2H),7.86(s,1H),8.00-8.02(m,2H);
LC-MS(保留时间:1.71min,方法B),MS m/z 263,265(M++H)。
步骤3:
按照实施例1、步骤5的相同方法制备该产物,不同之处在于使用实施例4、步骤2的产物。
LC-MS(保留时间:1.91min,方法B),MS m/z 571(M++H)。
步骤4:
按照实施例1、步骤9的相同方法制备化合物4,不同之处在于使用实施例4、步骤3的产物。
1H NMR(CD3OD)δ1.01-1.09(m,12H),1.17-1.26(m,10H),1.44-1.47(m,1H),1.87-1.91(m,1H),2.20-2.40(m,2H),2.63-2.65(m,4H),2.89-2.98(m,1H),4.08-4.20(m,2H),4.44-4.65(m,2H),5.13(d,J=11.7Hz,1H),5.32(d,J=15.0Hz,1H),5.72-5.85(m,2H),7.20-7.26(m,2H),7.60(s,1H),8.09-8.14(m,2H),9.26(b,1H);LC-MS(保留时间:1.91min,方法B),MS m/z 783(M++H)。
实施例5:化合物5的制备
化合物5
步骤1:
按照实施例3、步骤1和2的相同方法制备该产物,不同之处在于使用3-甲氧基-苯乙醇。
LC-MS(保留时间:1.03min,方法B),MS m/z 257(M++H)。
步骤2:
按照实施例1、步骤2的相同方法制备该产物,不同之处在于使用实施例5、步骤1的产物。
1H NMR(CDCl3)δ2.72(s,3H),3.90(s,3H),7.00-7.02(m,1H),7.41(t,J=8.0Hz,1H),7.55(d,J=7.5Hz,1H),7.59(d,J=2.0Hz,1H),7.89(s,1H);
LC-MS(保留时间:1.89min,方法B),MS m/z 275,277(M++H)。
步骤3:
按照实施例1、步骤5的相同方法制备该产物,不同之处在于使用实施例5、步骤2的产物。
1H NMR(CD3OD)δ1.02(s,9H),1.18(s,9H),2.37-2.47(m,1H),2.64(s,3H),2.85-2.93(m,1H),3.88(s,3H),4.00-4.08(m,1H),4.14(b,1H),4.49-4.55(m,1H),4.62-4.71(m,1H),5.71(b,1H),7.02-7.04(m,1H),7.40(t,J=8.0Hz,1H),7.58-7.62(m,3H);
LC-MS(保留时间:1.90min,方法B),MS m/z 583(M++H)。
步骤4:
按照实施例1、步骤9的相同方法制备化合物5,不同之处在于使用实施例5、步骤3的产物。
1H NMR(CD3OD)δ1.01-1.09(m,12H),1.17-1.29(m,10H),1.44-1.47(m,1H),1.87-1.91(m,1H),2.20-2.40(m,2H),2.63-2.65(m,4H),2.89-2.98(m,1H),3.89(s,3H),4.08-4.20(m,2H),4.44-4.65(m,2H),5.13(d,J=11.7Hz,1H),5.32(d,J=15.0Hz,1H),5.72-5.85(m,2H),7.02-7.05(m,1H),7.41(t,J=8.0Hz,1H),7.55-7.61(m,3H);
LC-MS(保留时间:1.96min,方法B),MS m/z 795(M++H)。
实施例6:化合物6的制备
化合物6
步骤1:
按照实施例3、步骤1和2的相同方法制备该产物,不同之处在于使用2-甲氧基-苯乙醇。
LC-MS(保留时间:1.10min,方法B),MS m/z 257(M++H)。
步骤2:
按照实施例1、步骤2的相同方法制备该产物,不同之处在于使用实施例6、步骤1的产物。
1H NMR(CDCl3)δ2.721(s,3H),3.88(s,3H),7.03(d,J=8.0Hz,1H),7.11(t,J=7.5Hz,1H),7.41-7.44(m,1H),7.79-7.81(m,1H),8.04(s,1H);
LC-MS(保留时间:1.92min,方法B),MS m/z 275,277(M++H)。
步骤3:
按照实施例1、步骤5的相同方法制备该产物,不同之处在于使用实施例6、步骤2的产物。
1H NMR(CD3OD)δ1.02(s,9H),1.20(s,9H),2.37-2.47(m,1H),2.63(s,3H),2.85-2.93(m,1H),3.89(s,3H),4.00-4.08(m,1H),4.14(b,1H),4.49-4.55(m,1H),4.62-4.71(m,1H),5.56(b,1H),7.09(t,J=7.5Hz,1H),7.15(d,J=8.5Hz,1H),7.41-7.44(m,1H),7.52(s,1H),7.67(d,J=8.0Hz,1H);
LC-MS(保留时间:1.76min,方法B),MS m/z 583(M++H)。
步骤4:
按照实施例1、步骤9的相同方法制备化合物6,不同之处在于使用实施例6、步骤3的产物。
1H NMR(CD3OD)δ1.01-1.08(m,12H),1.17-1.26(m,10H),1.44-1.47(m,1H),1.87-1.91(m,1H),2.20-2.40(m,2H),2.63-2.65(m,4H),2.89-2.98(m,1H),3.88(s,3H),4.08-4.12(m,1H),4.19(b,1H),4.44-4.65(m,2H),5.13(d,J=11.7Hz,1H),5.32(d,J=15.0Hz,1H),5.59(b,1H),5.72-5.80(m,1H),7.09(t,J=7.5Hz,1H),7.15(d,J=8.5Hz,1H),7.41-7.45(m,1H),7.66(s,1H),7.66-7.67(m,1H);
LC-MS(保留时间:1.93min,方法B),MS m/z 795(M++H)。
实施例7:化合物7的制备
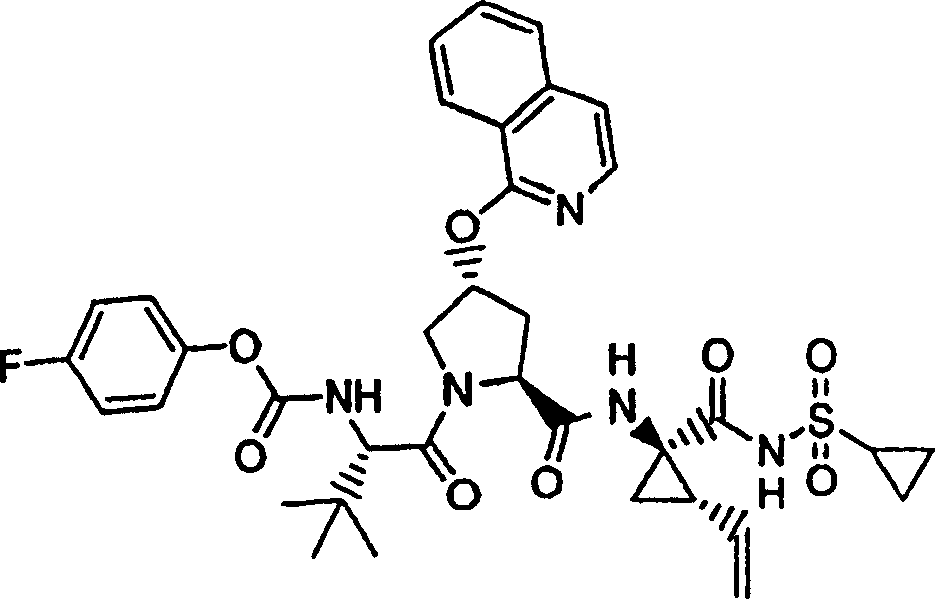
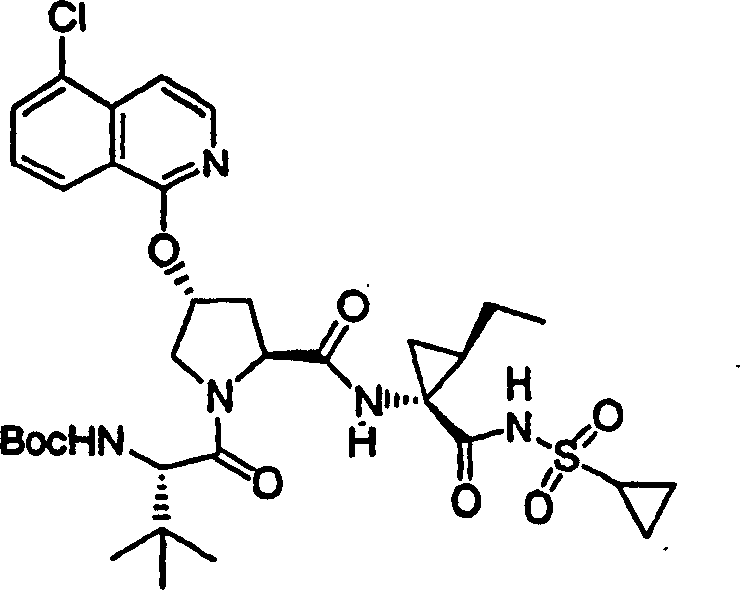
化合物7
步骤1:
按照实施例1、步骤5的相同方法制备该产物,不同之处在于使用2-氯-喹啉。
LC-MS(保留时间:1.73min,方法B),MS m/z 472(M++H)。
步骤2:
按照实施例1、步骤9的相同方法制备化合物7,不同之处在于使用实施例7、步骤1的产物。
1H NMR(CD3OD)δ1.01-1.08(m,12H),1.17-1.26(m,10H),1.44-1.47(m,1H),1.87-1.91(m,1H),2.23-2.30(m,2H),2.52-2.57(m,1H),2.89-2.98(m,1H),4.10-4.14(m,1H),4.09-4.15(m,2H),4.47-4.51(m,1H),5.13(d,J=10.0Hz,1H),5.32(d,J=17.0Hz,1H),5.73-5.78(m,1H),5.92(b,1H),6.90-6.92(m,1H),7.42(t,J=7.5Hz,1H),7.64(t,J=7.5Hz,1H),7.78-7.82(m,2H),8.13(d,J=7.5Hz,1H),9.18(d,1H);
LC-MS(保留时间:1.75min,方法B),MS m/z 684(M++H)。
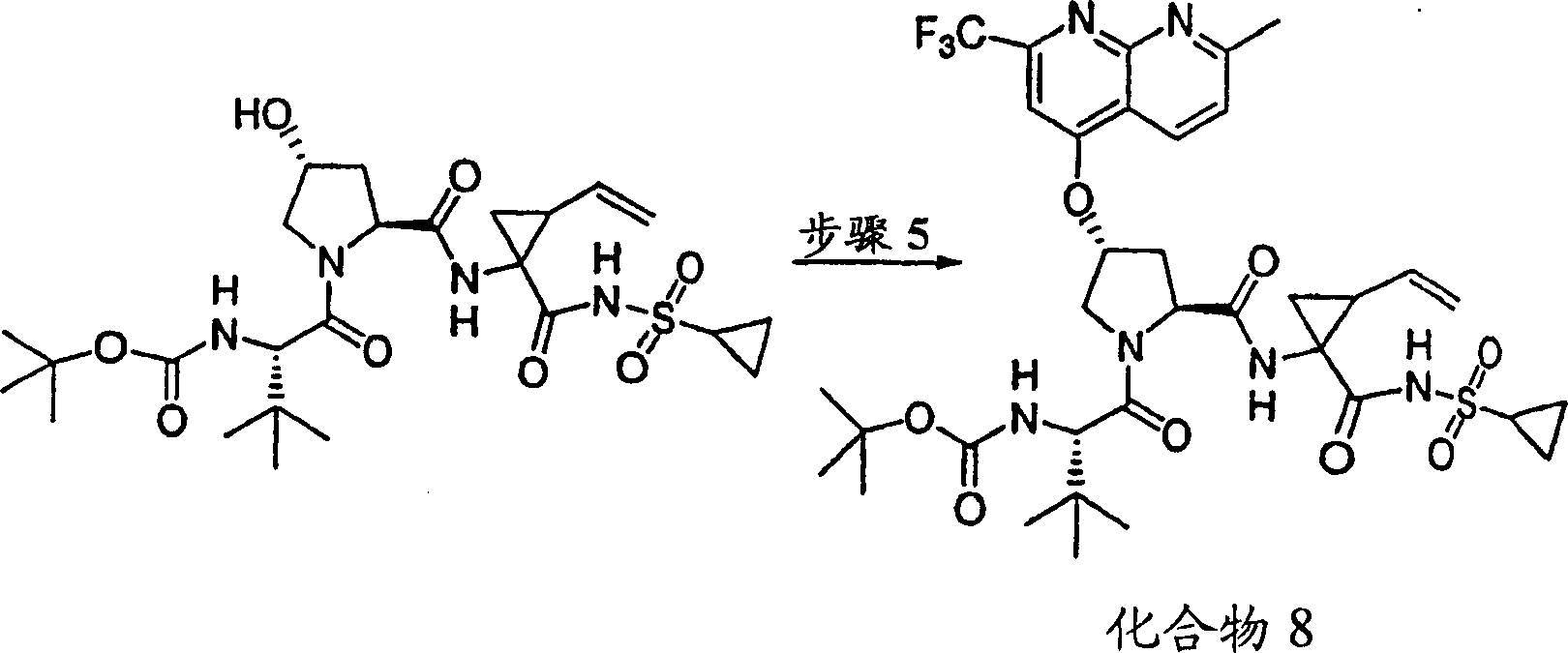
实施例8:化合物8的制备
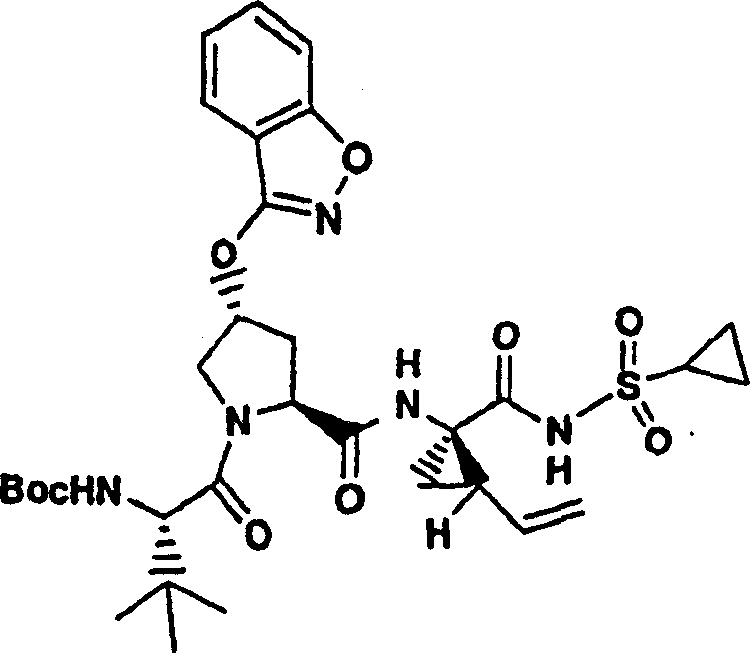
化合物8
方案1
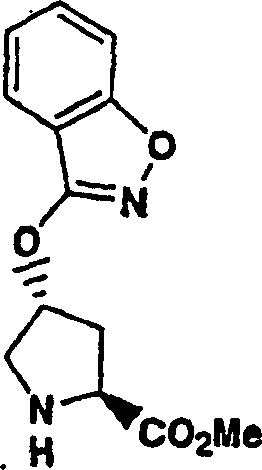
步骤1:
按照实施例1、步骤6b至8的相同方法制备该产物,没有使用步骤6a,本步骤为酶促拆分步骤。
LC-MS(保留时间:0.24min,方法B),MS m/z 231(M++H)。
方案2
步骤2:
向冰冻的N-BOC-4-反式-羟基-L-脯氨酸(1.58g,6.83mmol)、环丙烷磺酸(1-氨基-2-乙烯基-环丙烷羰基)-酰胺盐酸盐(实施例8,步骤1)(2.00g,7.52mmol)和HATU(3.89g,10.2mmol)在二氯甲烷(100mL)中的混合物内加入二异丙基乙胺(4.41g,34.2mmol)。将所形成的溶液升温至环境温度下反应12h。用EtOAc(200mL)稀释,用5%磷酸和盐水洗涤,经硫酸镁干燥,过滤,蒸发。剩余物经快速层析纯化(梯度,2∶1-1∶1 己烷-丙酮),得到1.25g(41%)所需的产物。
1H NMR(DMSO-d6)δ1.00-1.08(m,4H),1.34-1.40(m,1:2,10H),1.62-1.70(m,1H),1.76-1.87(m,1H),2.02-2.21(m,2H),2.81-2.95(m,1H),3.20-3.45(m,2H),4.04-4.09(m,1H),4.26(b,1H),5.08-5.12(m,1H),5.26(d,J=17.1Hz,1H),5.59-5.69(m,1H),8.59,8.87(外消旋物,1:2,1H),10.48-11.15(外消旋物,2:1,1H);LC-MS(保留时间:1.25min,方法B),MS m/e 444(M++H)。
步骤3:
按照实施例1、步骤8的相同方法制备该产物,不同之处在于使用实施例8、步骤2的产物。
LC-MS(保留时间:1.02min,方法B),MS m/e 344(M++H)。
步骤4:
向冰冻的N-BOC-4-反式-羟基-L-脯氨酸(1.58g,6.83mmol)、环丙烷磺酸(1-氨基-2-乙烯基-环丙烷羰基)-酰胺盐酸盐(实施例8,步骤3)(2.00g,7.52mmol)和HATU(3.89g,10.2mmol)在二氯甲烷(100mL)中的混合物内加入二异丙基乙胺(4.41g,34.2mmol)。将所形成的溶液升温至环境温度下反应12h。用EtOAc(200mL)稀释,用5%磷酸和盐水洗涤,经硫酸镁干燥,过滤,蒸发。剩余物经快速层析纯化(梯度,2∶1-1∶1己烷-丙酮),得到1.25g(41%)所需的产物。
1H NMR(CD3OD)δ0.99-1.07(m,11H),1.35-1.44(m,13H),1.75-1.87(m,1H),2.09-2.22(m,2H),2.88-2.94(m,1H),3.74-3.82(m,2H),4.28-4.30(m,1H),4.33-4.38(m,1H),4.48(b,1H),5.11-5.13(m,1H),5.30(d,J=15.0Hz,1H),5.70-5.78(m,1H),6.51-6.61(m,1H);
LC-MS(保留时间:1.26min,方法B),MS m/e 557(M++H)。
方案3
步骤5:
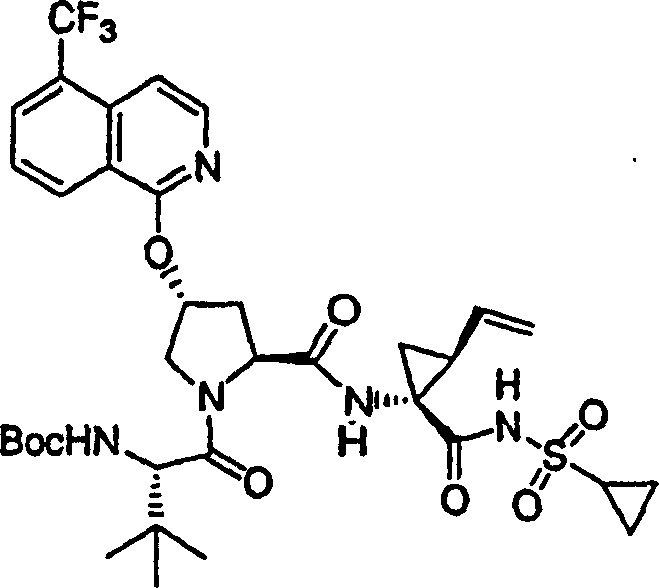
向实施例8、步骤4的产物(56mg,0.1mmol)的DMSO(2mL)溶液中加入叔丁醇钾(49mg,0.44mmol)。将所形成的溶液在环境温度下搅拌1h,随后加入4-氯-7-甲基-2-三氟甲基-[1,8]萘啶(P.Ferrarini等,J Heterocyclic Chem,1983,1053页)(30mg,0.12mmol)。将最终的溶液搅拌12h。用冰冻的水猝灭,用1M HCl酸化至pH为4,用EtOAc(20mL,×2)萃取。有机层用盐水洗涤,经硫酸镁干燥,过滤,蒸发。剩余物经制备型HPLC纯化,得到16mg(21%)化合物8,为粉红色固体物。
1H NMR(CD3OD)δ0.92-0.99(m,11H),1.01-1.04(m,11H),1.22-1.45(m,2H),1.76-1.85(m,1H),2.18-2.40(m,2H),2.76(s,3H),2.86-2.97(m,1H),4.00-4.11(m,2H),4.48-4.58(m,2H),5.09-5.12(m,1H),5.28-5.31(m,1H),5.59(b,1H),5.69-5.78(m,1H),6.39-6.48(m,1H),7.58-7.64(m,2H),8.08(s,1H),8.64-8.68(m,1H),8.85-8.91(m,1H);
LC-MS(保留时间:1.89min,方法B),MS m/e 767(M++H)。
实施例9:化合物9的制备
化合物9
按照实施例8、步骤5的相同方法制备化合物9,不同之处在于使用7-氯-5-乙基-3-甲基-异_唑并[4,5-b]吡啶(R.Nesi等,Synth Comm1992,22(16),2349)。
1H NMR(CD3OD)δ1.01-1.09(m,11H),1.21-1.25(m,11H),1.36(t,J=7.8Hz,3H),1.38-1.47(m,2H),1.80-1.90(m,1H),2.20-2.31(m,2H),2.59(s,3H),2.90-3.00(m,3H),4.01-4.18(m,2H),4.41-4.51(m,2H),5.11-5.15(m,1H),5.27-5.32(m,1H),5.58(b,1H),5.70-5.80(m,1H),7.11(s,1H),7.72,7.98(1:1,1H),9.00,9.22(1:1,1H);
LC-MS(保留时间:1.75min,方法B),MS m/e 717(M++H)。
实施例10:化合物10的制备
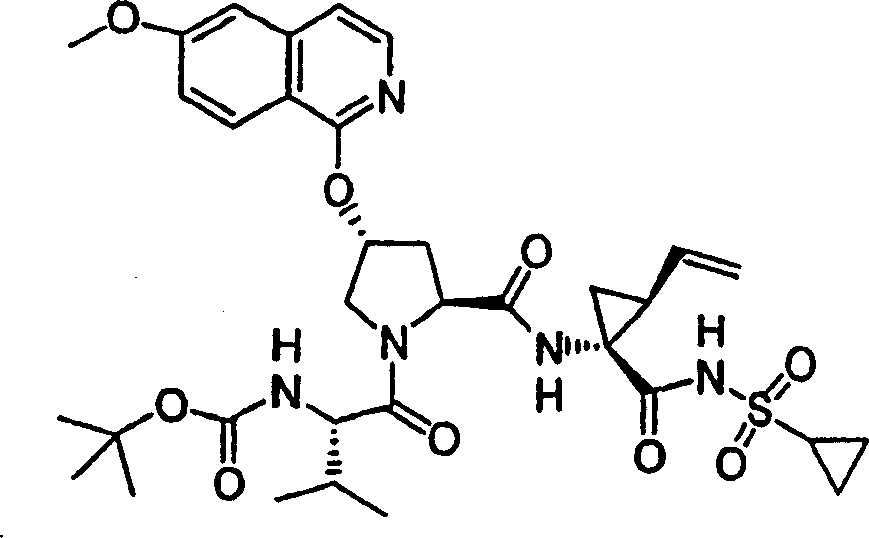
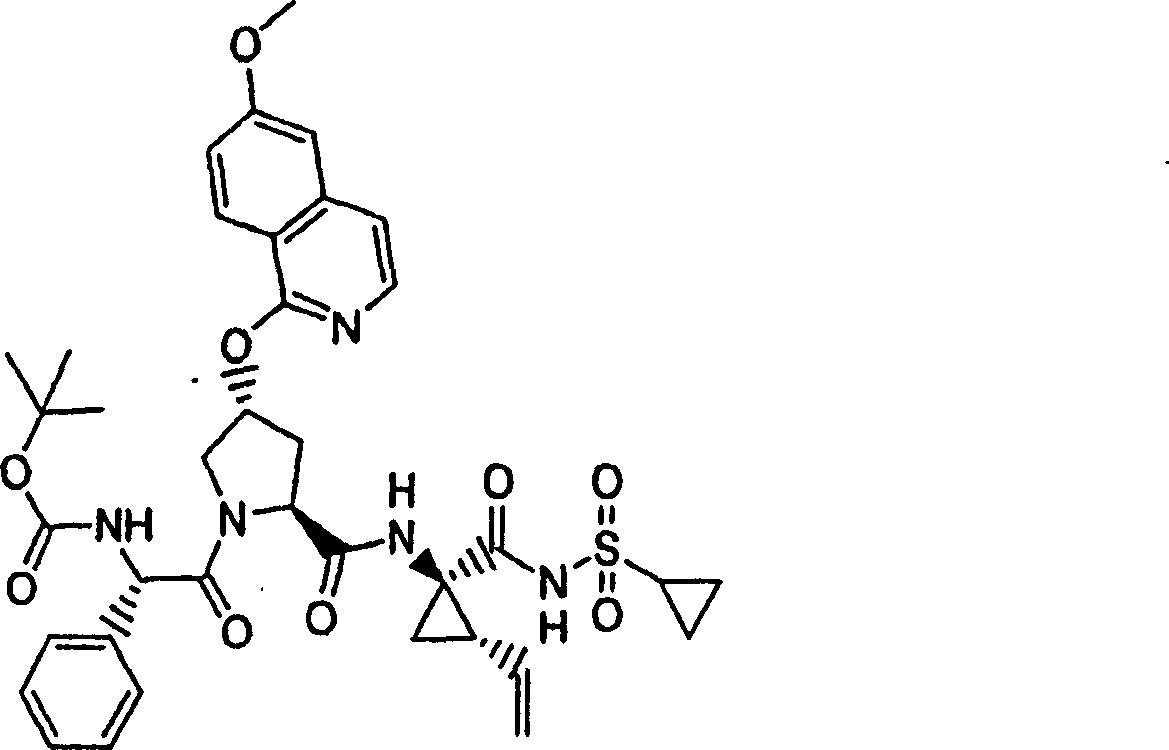
化合物10
按照实施例8、步骤5的相同方法制备化合物10,不同之处在于使用7-氯-5-苯基-3-甲基-异_唑并[4,5-b]吡啶(实施例1,步骤2)。
1H NMR(CD3OD)δ1.00-1.09(m,12H),1.16-1.25(m,10H),1.44-1.48(m,1H),1.79-1.89(m,1H),2.20-2.40(m,2H),2.64-2.66(m,4H),2.89-2.98(m,1H),4.08-4.20(m,2H),4.44-4.55(m,2H),5.11-5.16(m,1H),5.27-5.31(m,1H),5.72-5.74(m,2H),7.20-7.35(m,1H),7.46-7.51(m,2H),7.55-7.68(m,1H),8.05-8.06(m,2H);LC-MS(保留时间:1.97min,方法B),MS m/z 765(M++H)。
实施例11:化合物11的制备
化合物11
方案1
步骤1:
在0℃下,向3-甲氧基肉桂酸(11.04g,62mmol)和三乙胺(12.52g,124mmol)在丙酮(80mL)中的溶液内滴加入氯甲酸乙酯(约1.5当量)。在该温度下搅拌1h,滴加入叠氮化钠水溶液(6.40g,100mmol,35mLH2O)并将反应混合物在环境温度下搅拌16h。向混合物中加入水(100mL),真空除去挥发份。所得的浆状物用甲苯(3×50mL)萃取,并将合并的有机层经硫酸镁干燥。将该无水溶液在190℃滴加入二苯基甲烷(50mL)和三丁基胺(30mL)的热溶液中。在加入过程中蒸馏出甲苯。加入完毕,将反应温度升高至210℃下反应2h。冷却后,过滤收集沉淀出的产物,用己烷(2×50mL)洗涤,并干燥,得到所需的产物,为白色固体物(5.53g,51%)(Nicolas Briet等,Tetrahedron,2002,5761-5766)。
LC-MS(保留时间:0.82min,方法B),MS m/z 176(M++H)。
步骤2:
6-甲氧基-2H-异喹啉-1-酮(5.0g,28.4mmol)在磷酰氯(10mL)中的溶液加热至温和回流3h,真空蒸发(Nicolas Briet等,Tetrahedron,2002,5761-5766)。将剩余物倒入冰冻的水(20mL)中,用10M NaOH中和至pH为10。用氯仿萃取。有机层用盐水洗涤,经硫酸镁干燥,过滤,蒸发。剩余物经快速层析纯化(1∶1己烷-EtOAc),得到4.41g(80%)所需的产物,为白色固体物。
1H NMR(CD3OD)δ3.98(s,3H),7.34-7.38(m,2H),7.69(d,J=5.5Hz,1H),8.10(d,J=6.0Hz,1H),8.23(d,J=9.5Hz,1H);
LC-MS(保留时间:1.42min,方法B),MS m/z 194(M++H)。
方案2
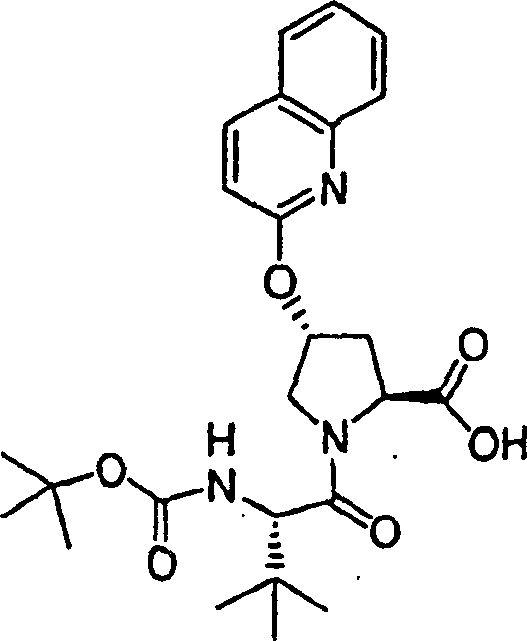
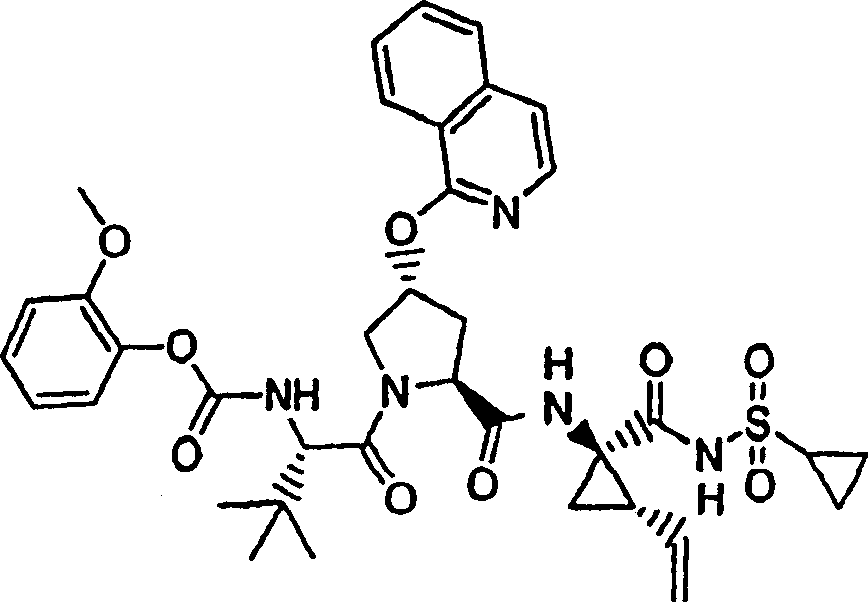
步骤3:
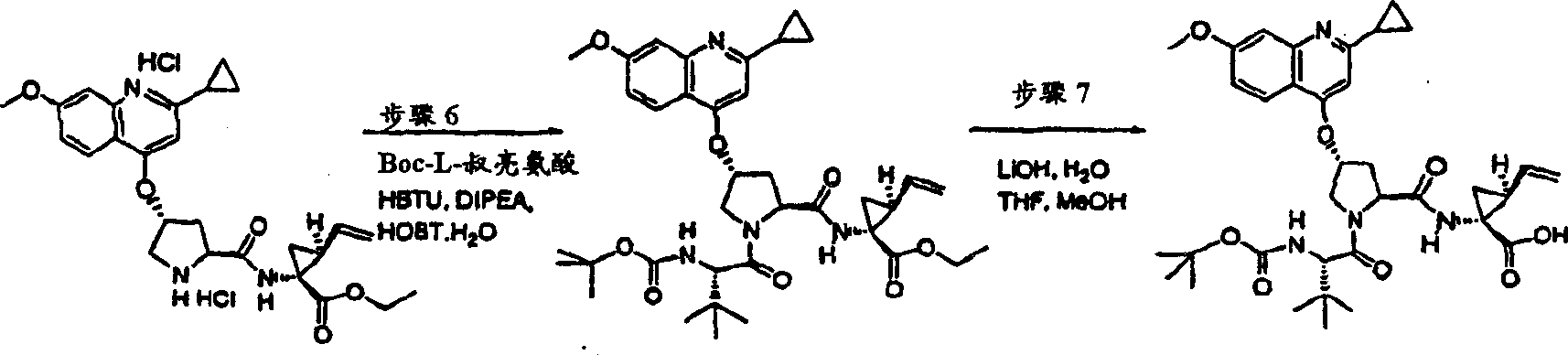
在环境温度下,向N-BOC-3-(R)-羟基-L-脯氨酸(892mg,3.89mmol)的DMSO(40mL)溶液中一次性加入叔丁醇钾(1.34g,12.0mmol)。将所形成的悬浮液在该温度下搅拌30min,随后冷却至10℃。一次性加入1-氯-6-甲氧基-异喹啉固体物(实施例11,步骤2)(785mg,4.05mmol),将最终的混合物在环境温度下搅拌12h。用冰冻的5%柠檬酸水溶液猝灭,用EtOAC(100mL)萃取。水相再次用EtOAC萃取。合并的有机层依次用5%柠檬酸水溶液和盐水洗涤,经硫酸镁干燥,过滤。将滤液真空蒸发至干,得到1.49g(99%)所需的产物,为灰白色泡沫状物。该物质未经进一步纯化直接用于下一步反应中。
1H NMR(CD3OD)δ1.42,1.44(外消旋物,9H),2.38-2.43(m,1H),2.66-2.72(m,1H),3.80-3.87(m,2H),3.92(s,3H),4.44-4.52(m,1H),5.73(b,1H),7.16-7.18(m,2H),7.24-7.25(m,1H),7.87-7.88(m,1H),8.07(d,J=8.5Hz,1H);
LC-MS(保留时间:1.62min,方法B),MS m/z 389(M++H)。
步骤4:
在0℃下,向实施例11、步骤3的产物(1.49g,3.84mmol)、HATU(2.19g,5.76mmol)和环丙烷磺酸(1-(R)-氨基-2-(S)-乙烯基-环丙烷羰基)-酰胺盐酸盐(实施例1,步骤8)(1.12g,4.22mmol)在二氯甲烷(50mL)中的混合物内加入DIPEA(1.29g,11.5mmol)。在环境温度下搅拌12h后,将形成的溶液用二氯甲烷(50mL)稀释,用冰冻的5%柠檬酸水溶液洗涤。有机层依次用5%柠檬酸水溶液和盐水洗涤,经硫酸镁干燥并过滤。将滤液真空蒸发至干。剩余物在甲醇中重结晶,得到1.60g(70%)所需的产物,为白色固体物。
1H NMR(CD3OD)δ1.05-1.08(m,2H),1.16-1.20(m,1H),1.24-1.27(m,1H),1.42-1.45(m,10H),1.88(dd,J=8.09,5.34Hz,1H),2.24-2.30(m,2H),2.53-2.57(m,1H),2.94-2.98(m,1H),3.80(d,J=12.5Hz,1H),3.86-3.89(m,1H),3.93(s,3H),4.40-4.42(m,1H),5.13(d,J=10.5Hz,1H),5.32(d,J=18.0Hz,1H),5.72-5.81(m,2H),7.17-7.20(m,2H),7.26(d,J=6.0Hz,1H),7.88(d,J=6.0Hz,1H),8.07(d,J=9.0Hz,1H);
LC-MS(保留时间:1.74min,方法B),MS m/z 601(M++H)。
步骤5:
向冰冻的实施例11、步骤4的产物(1.50g,2.50mmol)的二氯甲烷(10mL)溶液中加入TFA(10mL)。将所形成的溶液升温至环境温度下反应2h。真空除去溶剂。将剩余物在1M HCl的乙醚溶液中研磨。过滤,用乙醚洗涤,得到1.43g(99.8%)所需的产物,为易潮的白色固体物。
1H NMR(CD3OD)δ1.03-1.208(m,4H),1.26-1.31(m,1H),1.37-1.40(m,1H),1.95-1.97(m,1H),2.32-2.37(m,1H),2.42-2.48(m,1H),2.95-2.99(m,1H),3.88(d,J=12.5Hz,2H),3.98(s,3H),4.40-4.42(m,1H),5.16(d,J=10.5Hz,1H),5.33(d,J=18.0Hz,1H),5.62-5.69(m,1H),5.97(b,1H),7.30-7.34(m,2H),7.47(d,J=6.0Hz,1H),7.90(d,J=6.5Hz,1H),8.34(d,J=9.0Hz,1H),9.14(b,1H);
LC-MS(保留时间:1.12min,方法B),MS m/z 501(M++H)。
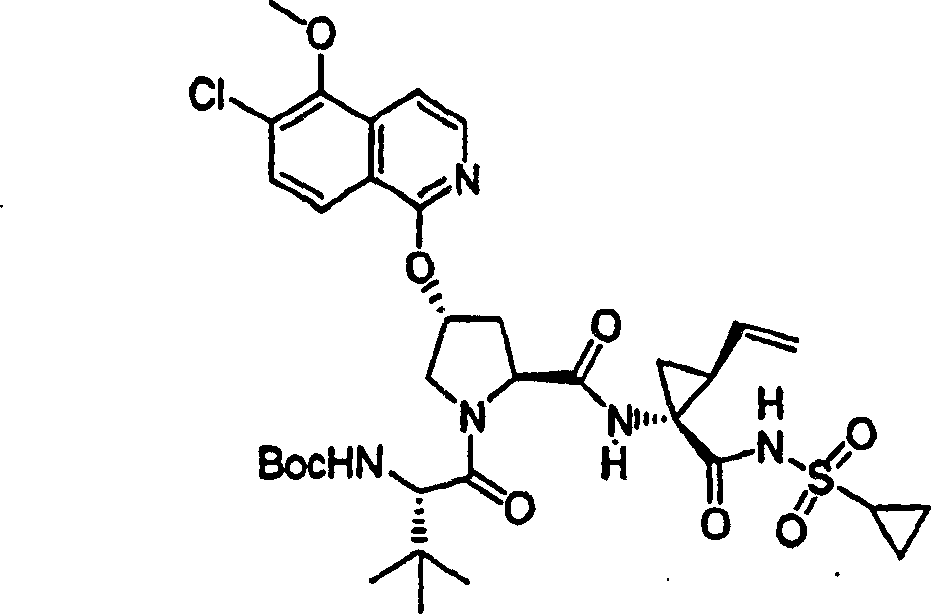
步骤6:
在0℃下,向实施例11,步骤5的产物(1.49g,3.84mmol)、HATU(2.19g,5.76mmol)和N-BOC-叔丁基-L-甘氨酸(1.12g,4.22mmol)在二氯甲烷(50mL)中的混合物内加入DIPEA(1.29g,11.5mmol)。在环境温度下搅拌12h后,将所形成的溶液用二氯甲烷(50mL)稀释,用冰冻的5%柠檬酸水溶液洗涤。有机层依次用5%柠檬酸水溶液和盐水洗涤,经硫酸镁干燥并过滤。将滤液真空蒸发至干。剩余物经制备型HPLC纯化(40%B至100%B,梯度时间15min),得到1.60g(70%)化合物11,为白色固体物。
1H NMR(CD3OD)δ1.00-1.08(m,12H),1.23-1.25(m,1H),1.27(s,9H),1.40-1.45(m,1H),1.85-1.88(m,1H),2.20-2.30(m,2H),2.55-2.61(m,1H),2.91-2.97(m,1H),3.92(s,3H),4.02-4.06(m,1H),4.21-4.24(m,1H),4.40-4.42(m,1H),4.49-4.51(m,1H),5.12(d,J=10.5Hz,1H),5.28(d,J=18.0Hz,1H),5.69-5.74(m,1H),5.81(b,1H),6.60(d,J=10.0Hz,1H),7.08-7.10(m,1H),7.18(s,1H),7.25(d,J=6.0Hz,1H),7.88(d,J=6.0Hz,1H),8.09(d,J=9.0Hz,1H);
LC-MS(保留时间:1.75min,方法B),MS m/z 714(M++H);
C35H47N5O9S·0.5H2O的计算值:C,58.16;H,6.69;N,9.69,实测值:C,58.01;H,6.46;N,9.55。
步骤7:
在-78℃下,向化合物11(71mg,0.1mmol)的二氯甲烷(5mL)溶液中加入1M HCl的乙醚溶液(0.2mL,0.2mmol)。在该温度下搅拌10min,真空除去挥发份,不使用加热浴。将剩余物在乙醚中研磨,过滤,用乙醚洗涤并干燥,得到61mg(85%)所需的化合物11的盐酸盐,为非常细的固体物。
1H NMR(CD3OD)δ1.00-1.08(m,12H),1.19(s,9H),1.23-1.25(m,1H),1.40-1.45(m,1H),1.85-1.91(m,1H),2.20-2.26(m,1H),2.31-2.42(m,1H),2.65-2.78(m,1H),2.92-2.97(m,1H),4.00(s,3H),4.10-4.16(m,2H),4.51-4.64(m,2H),5.13(d,J=10.5Hz,1H),5.30(d,J=18Hz,1H),5.69-5.79(m,1H),5.84(b,1H),7.28(d,J=9.3Hz,1H),7.40(s,1H),7.55(d,J=6.3Hz,1H),7.89-7.92(m,1H),8.29(d,J=9.0Hz,1H),9.21(b,1H);
LC-MS(保留时间:1.75min,方法B),MS m/z 714(M++H)。
C35H47N5O9S·1.0HCl的计算值:C,56.02;H,6.44;N,9.33;Cl,4.72;S,4.27,实测值:C,55.80;H,6.42;N,9.15;Cl,4.56;S,4.09。
步骤8:
25ml的2颈烧瓶中装入搅拌棒、隔膜和氮气接管。称量化合物11(99.7mg,0.140mmol)并加入反应烧瓶中。用氮气吹扫反应烧瓶并置于氮气气氛保护下。向烧瓶中加入850μl丙酮,得到澄清溶液。室温下,向该溶液中加入780μl 0.179M KOH水溶液(将固体KOH(502.8mg,8.97mmol)溶于50ml H2O中制得)。加入KOH时,溶液稍微升温,但仍保持澄清。将所述澄清溶液在室温下搅拌2h。产物从溶液中结晶析出,过滤分离。滤饼用冷丙酮洗涤,得到42mg(40%收率)所需的产物,为细白色针状物:
1H NMR(DMSO)δ0.68(m,1H),0.72(m,1H),0.88(s,1H),0.92(s,1H),1.24(s,1H),1.38(s,1H),1.50(b,1H),1.81(b,1H),2.68(b,2H),3.90(s,3H),3.95-4.10(m,3H),4.40(t,J=10Hz,1H),4.85(m,1H),5.04(m,1H),5.71(b,1H),6.01(b,1H),6.64(d,J=10Hz,1H),7.10(m,1H),7.30(m,J=5Hz,2H),7.95(d,J=10Hz,1H),8.08(d,J=15Hz,1H).
对C35H46KN5O9S·H2O进行元素分析;计算值:C,54.60;H,6.28;K,5.08;N,9.10;实测值:C,54.88;H,6.23;K,5.05;N,9.01;MS m/e714(MH+);
实施例12:化合物12的制备
化合物12
按照实施例11、步骤6的相同方法制备化合物12,不同之处在于使用N-BOC-L-缬氨酸。
1H NMR(CD3OD)δ0.94-0.98(m,6H),1.07-1.09(m,3H),1.21-1.25(m,10H),1.40-1.43(m,1H),1.88-1.89(m,1H),2.05-2.09(m,1H),2.22-2.35(m,2H),2.57-2.61(m,1H),2.94-2.97(m,1H),3.92(s,3H),4.03-4.06(m,2H),4.47-4.55(m,2H),5.12(d,J=10.5Hz,1H),5.32(d,J=18.1Hz,1H),5.74-5.81(m,1H),5.86(b,1H),7.10(d,J=9.0Hz,1H),7.18(s,1H),7.25(d,J=6.0Hz,1H),7.88(d,J=6.0Hz,1H),8.10(d,J=9.0Hz,1H);
LC-MS(保留时间:1.71min,方法B),MS m/z 700(M++H)。
实施例13:化合物13的制备
化合物13
按照实施例11、步骤6的相同方法制备化合物13,不同之处在于使用N-BOC-L-别异亮氨酸。
1H NMR(CD3OD)δ0.89-0.96(m,6H),1.07-1.18(m,5H),1.28(s,9H),1.42-1.45(m,1H),1.50-1.54(m,1H),1.87-1.89(m,2H),2.23-2.34(m,2H),2.57-2.61(m,1H),2.92-2.95(m,1H),3.92(s,3H),4.05-4.07(m,1H),4.22-4.24(m,1H),4.37-4.40(m,1H),4.54-4.56(m,1H),5.13(d,J=10.5Hz,1H),5.32(d,J=18.0Hz,1H),5.75-5.82(m,1H),5.86(b,1H),7.12(d,J=9.0Hz,1H),7.19(s,1H),7.24(d,J=6.0Hz,1H),7.88(d,J=6.0Hz,1H),8.10(d,J=9.0Hz,1H);
LC-MS(保留时间:1.77min,方法B),MS m/z 714(M++H)。
实施例14:化合物14的制备
化合物14
方案1
步骤1:
将化合物11(150mg,0.21mmol)和Pearlmann催化剂(Pd(OH)2,15mg)在EtOAc(10mL)中的混合物置于10psi H2的Parr摇荡器中20min。经celite硅藻土过滤。将滤液真空蒸发。剩余物经制备型HPLC纯化,得到67mg(45%)化合物14,为白色固体物。
1H NMR(CD3OD)δ0.96-0.99(m,4H),1.04(s,9H),1.07-1.09(m,2H),1.21-1.24(m,2H),1.27(s,9H),1.51-1.65(m,4H),2.25-2.27(m,1H),2.55-2.61(m,1H),2.94-2.98(m,1H),3.92(s,3H),4.02-4.06(m,1H),4.21-4.24(m,1H),4.40-4.42(m,1H),4.49-4.51(m,1H),5.81(b,1H),6.59(d,J=10.0Hz,1H),7.08-7.10(m,1H),7.18(d,J=1.5Hz,1H),7.24(d,J=6.0Hz,1H),7.88(d,J=6.0Hz,1H),8.08(d,J=9.0Hz,1H);
LC-MS(保留时间:1.76min,方法B),MS m/z 716(M++H)。
实施例15:化合物15的制备
化合物15
化合物15从制备化合物14的同一反应物中,作为副产物分离得到,保留时间稍长,收率15%。
1H NMR(CD3OD)δ0.92-1.10(m,17H),1.26-1.36(m,13H),1.64-1.72(m,1H),1.90-1.96(m,1H),2.30-2.40(m,1H),2.63-2.67(m,1H),2.96-3.00(m,1H),3.92(s,3H),4.03-4.07(m,1H),4.24(b,1H),4.40-4.42(m,1H),4.49-4.51(m,1H),5.83(b,1H),7.08-7.11(m,1H),7.19(d,J=2.0Hz,1H),7.25(d,J=6.0Hz,1H),7.89(d,J=6.0Hz,1H),8.10(d,J=9.0Hz,1H),8.51(b,1H);
LC-MS(保留时间:1.83min,方法B),MS m/z 718(M++H)。
实施例16:化合物16的制备
化合物16
方案1
步骤1:
在0℃下,向化合物11(420mg,0.59mmol)的DCM(5mL)溶液中加入TFA(5mL)。在该温度下搅拌2h,真空除去挥发份。将剩余物在1MHCl的乙醚(5mL)溶液中研磨,过滤,用乙醚洗涤并干燥,得到360mg(89%)所需的HCl盐,为非常细的固体物。
LC-MS(保留时间:1.28min,方法B),MS m/z 614(M++H)。
步骤2:
在0℃下,向实施例16、步骤1的产物(39mg,0.06mmol)和DIPEA(20mg,0.18mmol)在DCM(1mL)中的悬浮液内加入氯甲酸甲酯(6.8mg,0.072mmol)。在该温度下搅拌2h,真空除去挥发份。剩余物经制备型HPLC纯化,得到21mg(58%)化合物16,为白色晶体。
1H NMR(CD3OD)δ1.05-1.09(m,11H),1.22-1.25(m,2H),1.41-1.44(m,1H),1.86-1.89(m,1H),2.22-2.32(m,2H),2.59-2.63(m,1H),2.89-2.93(m,1H),3.48(s,3H),3.92(s,3H),4.06-4.10(m,1H),4.31-4.33(m,1H),4.38-4.40(m,1H),4.50-4.52(m,1H),5.12(d,J=10.5Hz,1H),5.30(d,J=18.0Hz,1H),5.71-5.80(m,1H),5.85(b,1H),6.95(d,J=10.0Hz,1H),7.13-7.16(m,1H),7.19(s,1H),7.25(d,J=6.0Hz,1H),7.88(d,J=6.0Hz,1H),8.09(d,J=9.0Hz,1H);
LC-MS(保留时间:1.54min,方法B),MS m/z 672(M++H)。
实施例17:化合物17的制备
化合物17
按照实施例16、步骤2的相同方法制备化合物17,不同之处在于使用氯甲酸异丙酯。
1H NMR(CD3OD)δ1.00-1.09(m,15H),1.13-1.16(m,2H),1.24-1.26(m,2H),1.40-1.45(m,1H),1.86-1.89(m,1H),2.21-2.31(m,2H),2.55-2.61(m,1H),2.91-2.97(m,1H),3.92(s,3H),4.04-4.08(m,1H).4.30(b,1H),4.40(d,J=10Hz,1H),4.49-4.54(m,2H),5.12(d,J=10.5Hz,1H),5.29(d,J=18.0Hz,1H),5.71-5.77(m,1H),5.84(b,1H),6.80(d,J=10.0Hz,1H),7.11(d,J=9.0Hz,1H),7.19(s,1H),7.25(d,J=6.0Hz,1H),7.88(d,J=6.0Hz,1H),8.08(d,J=9.0Hz,1H);
LC-MS(保留时间:1.74min,方法B),MS m/z 700(M++H)。
实施例18:化合物18的制备
化合物18
按照实施例16、步骤2的相同方法制备化合物18,不同之处在于使用氯甲酸新戊酯。
1H NMR(CD3OD)δ0.61(b,1H),0.84(s,8H),1.05-1.09(m,11H),1.23-1.25(m,2H),1.39-1.44(m,1H),1.85-1.88(m,1H),2.20-2.30(m,2H),2.56-2.62(m,1H),2.91-2.97(m,1H),3.38(d,J=9.0Hz,1H),3.55(d,J=9.0Hz,1H),3.92(s,3H),4.02-4.06(m,1H),4.32(d,J=9.5Hz,1H),4.41(d,J=9.0Hz,1H),4.49-4.51(m,1H),5.12(d,J=10.5Hz,1H),5.28(d,J=18.0Hz,1H),5.69-5.74(m,1H),5.81(b,1H),6.90(d,J=10.0Hz,1H),7.08-7.10(m,1H),7.19(s,1H),7.26(d,J=6.0Hz,1H),7.88(d,J=6.0Hz,1H),8.07(d,J=9.0Hz,1H);
LC-MS(保留时间:1.84min,方法B),MS m/z 728(M++H)。
实施例19:化合物19的制备
化合物19
按照实施例16、步骤2的相同方法制备化合物19,不同之处在于使用(S)-氯甲酸(3-呋喃)酯(呋喃ochlorolate)(J.Campbell,A.Good,WO 20020808)。
1H NMR(CD3OD)δ1.03-1.08(m,11H),1.23-1.26(m,2H),1.38-1.46(m,1H),1.64-1.71(m,1H),1.85-1.90(m,2H),2.20-2.30(m,2H),2.55-2.61(m,1H),2.91-2.97(m,1H),3.66-3.72(m,4H),3.93(s,3H),4.05-4.09(m,1H),4.27-4.29(m,1H),4.40-4.42(m,1H),4.55-4.59(m,1H),4.75-4.77(m,1H),5.12(d,J=10.5Hz,1H),5.28(d,J=18Hz,1H),5.73-5.80(m,1H),5.85(b,1H),7.06(d,J=10.0Hz,1H),7.13(d,J=9.0Hz,1H),7.20(s,1H),7.25(d,J=6.0Hz,1H),7.89(d,J=6.0Hz,1H),8.07(d,J=9.0Hz,1H);
LC-MS(保留时间:1.52min,方法B),MS m/z 728(M++H)。
实施例20:化合物20的制备
化合物20
步骤1:
按照实施例11、步骤2的相同方法制备该产物,不同之处在于使用6-氯-2H-异喹啉-1-酮(Nicolas Briet等,Tetrahedron,2002,5761-5766)。
LC-MS(保留时间:1.07min,方法B),MS m/z 180(M++H)。
步骤2:
按照实施例1、步骤5的相同方法制备该产物,不同之处在于使用实施例20、步骤1的产物。
1H NMR(CD3OD)δ1.04(s,9H),1.20(s,9H),2.36-2.41(m,1H),2.74-2.78(m,1H),4.01-4.04(m,1H),4.19-4.21(m,1H),4.47-4.49(m,1H),4.67-4.70(m,1H),5.84(b,1H),7.28(d,J=6.0Hz,1H),7.47(d,J=6.0Hz,1H),7.84(s,1H),8.00(d,J=6.0Hz,1H),8.20(d,J=9.0Hz,1H);
LC-MS(保留时间:1.88min,方法B),MS m/z 506(M++H)。
步骤3:
按照实施例1、步骤9的相同方法制备化合物20,不同之处在于使用实施例20、步骤2的产物。
1H NMR(CD3OD)δ0.99-1.11(m,12H),1.20-1.26(m,10H),1.43-1.46(m,1H),1.87-1.90(m,1H),2.22-2.31(m,2H),2.60-2.64(m,1H),2.92-2.97(m,1H),4.06-4.08(m,1H),4.21-4.23(m,1H),4.45-4.47(m,1H),4.53-4.56(m,1H),5.13(d,J=10.5Hz,1H),5.29(d,J=18.0Hz,1H),5.72-5.80(m,1H),5.88(b,1H),6.58(d,J=10.0Hz,1H),7.29(d,J=6.0Hz,1H),7.47(d,J=9.0Hz,1H),7.86(s,1H),8.01(d,J=6.0Hz,1H),8.18(d,J=9.0Hz,1H);
LC-MS(保留时间:1.94min,方法B),MS m/z 718(M++H)。
实施例21:化合物21的制备
化合物21
方案1
步骤1:
在0℃下,向实施例1、步骤4的产物(3.00g,8.72mmol)、HATU(4.97g,13.1mmol)和实施例1、步骤8的产物(2.55g,9.59mmol)在二氯甲烷(100mL)中的混合物内加入DIPEA(3.02g,27.0mmol)。在环境温度下搅拌后12h,将所形成的溶液用二氯甲烷(100mL)稀释,用冰冻的5%柠檬酸水溶液洗涤。有机层依次用5%柠檬酸水溶液和盐水洗涤,经硫酸镁干燥并过滤。将滤液真空蒸发至干。剩余物经快速柱层析纯化(1∶1己烷∶丙酮),得到3.64g(75%)所需的产物,为泡沫状物。
LC-MS(保留时间:1.41min,方法B),MS m/z 557(M++H)。
方案2
步骤2:
向冰冻的6-溴异喹啉(4.16g,20mmol)的二氯甲烷(100mL)溶液中一次性加入mCPBA固体物(9.38g,纯度77%,42mmol)。在环境温度下搅拌12h后,用二氯甲烷(100mL)稀释并用1M NaOH(100mL,×2)和盐水洗涤。有机层经硫酸镁干燥,过滤,蒸发至干,得到3.83g(86%)所需的产物,为白色固体物。该物质未经进一步纯化直接使用。
LC-MS(保留时间:0.77min,方法B),MS m/z 224,226(M++H)。
步骤3:
将6-溴-异喹啉2-氧化物(88mg,0.2mmol)、吡唑(68mg,1.0mmol)、CuBr(57mg,0.4mmol)和碳酸铯(130mg,0.4mmol)在DMF(2mL)中的混合物在140℃的密封管中加热4h。过滤后,滤液经制备型HPLC纯化,得到41mg(98%)所需的产物,为灰白色固体物。
1H NMR(CDCl3)δ6.58-6.59(m,1H),7.82(d,J=1.0Hz,1H),7.89(d,J=7.0Hz,1H),8.02(d,J=9.0Hz,1H),8.11(d,J=2.5Hz,1H),8.18-8.22(m,2H),8.29(d,J=7.0Hz,1H),9.07(b,1H);
LC-MS(保留时间:0.77min,方法B),MS m/z 212(M++H)。
步骤4:
按照实施例11、步骤2的相同方法制备该产物,为类白色固体物,不同之处在于使用6-吡唑-异喹啉2-氧化物。
1H NMR(CD3OD)δ7.82-7.83(m,2H),8.23-8.32(m,4H),8.44-8.49(m,2H);
LC-MS(保留时间:1.35min,方法B),MS m/z 230,(M++H)。
步骤5:
向实施例21、步骤1的产物(45mg,0.08mmol)的DMSO(2mL)溶液中加入叔丁醇钾(41mg,0.37mmol)。将形成的溶液在环境温度下搅拌30min,随后加入1-氯-6-吡唑-1-基-异喹啉(17mg,0.07mmol)。将最终的溶液搅拌12h。用冰冻的水猝灭,用1M HCl酸化至pH为4,用EtOAc(20mL,×2)萃取。有机层用盐水洗涤,经硫酸镁干燥,过滤,蒸发。剩余物经制备型HPLC纯化,得到10mg(16%)化合物21,为粉红色固体物。
1H NMR(CD3OD)δ1.04-1.10(m,12H),1.23-1.27(m,10H),1.43-1.47(m,1H),1.87-1.91(m,1H),2.22-2.29(m,2H),2.61-2.68(m,1H),2.92-2.98(m,1H),4.07-4.11(m,1H),4.24(b,1H),4.46-4.60(m,2H),5.13(d,J=10.5Hz,1H),5.29(d,J=18Hz,1H),5.70-5.83(m,1H),5.89(b,1H),6.59-6.61(m,1H),7.40(d,J=10.0Hz,1H),7.80(d,J=2.5Hz,1H),8.01(d,J=10.0Hz,2H),8.15(s,1H),8.31(d,J=15.0Hz,1H),8.42(d,J=4.5Hz,1H);
LC-MS(保留时间:1.77min,方法B),MS m/z 750(M++H)。
实施例22:化合物22的制备
化合物22
步骤1:
按照实施例11、步骤2的相同方法制备该产物,为灰白色固体物,不同之处在于使用6-溴-异喹啉2-氧化物。
1H NMR(CD3OD)δ7.73(d,J=5.5Hz,1H),7.85-7.91(m,1H),8.22-8.31(m,3H);LC-MS(保留时间:1.53min,方法B),MS m/z 241,243,245(M++H)。
步骤2:
按照实施例21、步骤5的相同方法制备化合物22,为白色固体物,不同之处在于使用1-氯-6-溴-异喹啉。
1H NMR(CD3OD)δ0.99-1.09(m,12H),1.22-1.27(m,10H),1.40-1.47(m,1H),1.86-1.91(m,1H),2.20-2.34(m,2H),2.57-2.66(m,1H),2.90-2.97(m,1H),4.05-4.09(m,1H),4.21(b 1H),4.44-4.57(m,2H),5.13(d,J=10.5Hz,1H),5.29(d,J=18.0Hz,1H),5.70-5.82(m,1H),5.88(b,1H),7.29(d,J=9.5Hz,1H),7.60-7.63(m,1H),8.00-8.12(m,3H);
LC-MS(保留时间:1.90min,方法B),MS m/z 762,764(M++H)。
实施例23:化合物23的制备
步骤1:
按照实施例11、步骤3的相同方法制备该产物,为白色固体物,不同之处在于使用1-氯-异喹啉。
1H NMR(CD3OD)δ1.42,1.44(外消旋物,9H),2.39-2.44(m,1H),2.68-2.72(m,1H),3.80-3.87(m,2H),4.44-4.52(m,1H),5.78(b,1H),7.32-7.33(m,1H),7.58(t,J=7.8Hz,1H),),7.71(t,J=7.5Hz,1H),7.81(d,J=8.0Hz,1H),7.95(d,J=6.0Hz,1H),8.19(d,J=8.0Hz,1H);
LC-MS(保留时间:1.61min,方法B),MS m/z 359(M++H)。
步骤2:
按照实施例11、步骤4的相同方法制备该产物,不同之处在于使用实施例23、步骤1的产物。
1H NMR(DMSO-d6)δ1.00-1.09(m,4H),1.35-1.38(m,10H),1.69-1.84(m,1H),2.11-2.66(m,3H),2.89-2.93(m,1H),3.62-3.89(m,2H),4.31(t,J=8.1Hz,1H),5.12(d,J=10.8Hz,1H),5.27(d,J=16.8Hz,1H),5.58-5.70(m,1H),5.76(b,1H),7.43(d,J=5.7Hz,1H),7.66(t,J=7.4Hz,1H),7.79(t,J=7.5Hz,1H),7.92(d,J=8.1Hz,1H),8.02(d,J=10.0Hz,1H),8.13(d,J=8.1Hz,1H),9.02(b,1H);
LC-MS(保留时间:1.72min,方法B),MS m/z 571(M++H)。
步骤3:
按照实施例11、步骤5的相同方法制备该产物,不同之处在于使用实施例23、步骤2的产物。
LC-MS(保留时间:1.16min,方法B),MS m/z 471(M++H)。
步骤4:
按照实施例11、步骤6的相同方法制备化合物23,为白色固体物,不同之处在于使用实施例23、步骤3的产物。
1H NMR(CD3OD)δ1.00-1.09(m,12H),1.25-1.27(m,10H),1.42-1.46(m,1H),1.86-1.90(m,1H),2.22-2.34(m,2H),2.60-2.67(m,1H),2.92-2.99(m,1H),4.06-4.11(m,1H),4.26(b,1H),4.45-4.57(m,2H),5.12(d,J=10.2Hz,1H),5.27(d,J=16.8Hz,1H),5.70-5.82(m,1H),5.88(b,1H),7.32(d,J=6.0Hz,1H),7.52(t,J=7.4Hz,1H),7.70(t,J=7.5Hz,1H),7.80(d,J=8.1Hz,1H),7.97(d,J=6Hz,1H),8.20(d,J=8.4Hz,1H),9.18(b,1H);
LC-MS(保留时间:1.80min,方法B),MS m/z 684(M++H)。
实施例24:化合物24的制备
化合物24
方案1
步骤1:
在0℃下,向N-BOC-3-(R)-羟基-L-脯氨酸(6.22g,26.9mmol)的DMF(250mL)溶液中分批加入NaH(60%,3.23g,80.8mmol)。将所形成的悬浮液在该温度下搅拌30min。一次性加入1,3-二氯异喹啉固体物(5.33g,26.9mmol)并将最终的混合物在环境温度下搅拌12h。用冰冻的5%柠檬酸水溶液猝灭,用EtOAC(300mL)萃取。水相再次用乙酸乙酯萃取。合并的有机层依次用5%柠檬酸水溶液和盐水洗涤,经硫酸镁干燥,过滤。将滤液真空蒸发至干,得到10.53g(99.8%)4-(6-甲氧基-异喹啉-1-氧基)-吡咯烷-1,2-二甲酸1-叔丁酯,为灰白色泡沫状物。该物质未经进一步纯化直接用于下一步反应中。
1H NMR(CD3OD)δ1.43,1.44(外消旋物,9H),2.39-2.44(m,1H),2.68-2.72(m,1H),3.80-3.90(m,2H),4.44-4.52(m,1H),5.77(b,1H),7.39(s,1H),7.58(t,J=7.3Hz,1H),7.71-7.78(m,2H),8.16(d,J=7.5Hz,1H);
LC-MS(保留时间:1.80min,方法B),MS m/z 392(M++H)。
步骤2:
按照实施例11、步骤4的相同方法制备该产物,不同之处在于使用实施例24、步骤1的产物。
1H NMR(CD3OD)δ1.02-1.08(m,2H),1.18-1.26(m,2H),1.44-1.48(m,10H),1.84-1.91(m,1H),2.22-2.36(m,2H),2.57-2.60(m,1H),2.95-2.99(m,1H),3.81-3.93(m,2H),4.38-4.41(m,1H),5.13(d,J=10.8Hz,1H),5.31(d,J=16.8Hz,1H),5.75-5.82(m,2H),7.41(s,1H),7.59(t,J=7.4Hz,1H),7.74-7.79(m,2H),8.16(d,J=8.0Hz,1H);
LC-MS(保留时间:1.82min,方法B),MS m/z 605(M++H)。
步骤3:
按照实施例11、步骤5的相同方法制备该产物,不同之处在于使用实施例24、步骤2的产物。
LC-MS(保留时间:1.30min,方法B),MS m/z 505(M++H)。
步骤4:
按照实施例11、步骤6的相同方法制备化合物24,为白色固体物,不同之处在于使用实施例24、步骤3的产物。
1H NMR(CD3OD)δ0.99-1.09(m,12H),1.22-1.29(m,10H),1.42-1.46(m,1H),1.86-1.90(m,1H),2.21-2.34(m,2H),2.62-2.66(m,1H),2.92-2.99(m,1H),4.06-4.11(m,1H),4.26(b,1H),4.46-4.56(m,2H),5.13(d,J=10.5Hz,1H),5.29(d,J=17.2Hz,1H),5.72-5.79(m,1H),5.89(b,1H),7.40(d,J=6.0Hz,1H),7.52(t,J=7.4Hz,1H),7.72-7.76(m,2H),8.18(d,J=8.5Hz,1H);
LC-MS(保留时间:1.95min,方法B),MS m/z 718(M++H)。
实施例25,化合物25
化合物25
方案1
步骤1:
将实施例24、步骤1(39mg,0.10mmol)、苯基硼酸(14.6mg,0.12mmol)、叔丁醇钠(38mg,0.40mmol)和((t-Bu)2POH)2PdCl2(POPd)(5mg,0.01mmol)在THF(2mL)中的混合物加热回流4h。冷却后,所形成的混合物用5%柠檬酸水溶液猝灭并用EtOAc(20mL)萃取。有机层用盐水洗涤,经硫酸镁干燥,过滤,蒸发。剩余物经制备型HPLC纯化,得到36mg(83%)所需的产物,为灰白色泡沫状物。
1H NMR(CD3OD)δ1.43,1.45(外消旋物,9H),2.51-2.56(m,1H),2.74-2.82(m,1H),3.88-3.92(m,1H),3.98-4.01(m,1H),4.50-4.57(m,1H),5.95(b,1H),7.36-7.39(m,1H),7.45-7.48(m,2H),7.55(t,J=7.3Hz,1H),7.70(t,J=7.5Hz,1H),7.84-7.89(m,2H),8.14-8.17(m,3H),9.05(b,1H);
LC-MS(保留时间:1.97min,方法B),MS m/z 435(M++H)。
步骤2:
按照实施例11、步骤4的相同方法制备该产物,不同之处在于使用实施例25、步骤1的产物。
1H NMR(DMSO-d6)δ0.98-1.10(m,4H),1.38-1.41(m,10H),1.74-1.81(m,1H),2.18-2.34(m,2H),2.47-2.49(m,1H),2.95-2.99(m,1H),3.74-3.96(m,2H),4.34-4.37(m,1H),5.12(d,J=10.5Hz,1H),5.26(d,J=17.8Hz,1H),5.75-5.82(m,1H),5.95(b,1H),7.41-7.45(m,1H),7.51-7.54(m,2H),7.61-7.64(m,1H),7.78-7.82(m,1H),7.98(d,J=9.0Hz,1H),8.06(s,1H),8.13-8.14(m,1H),8.18-8.20(m,2H),9.05(b,1H),10.34(b,1H);
LC-MS(保留时间:1.99min,方法B),MS m/z 647(M++H)。
步骤3:
按照实施例11、步骤5的相同方法制备该产物,为白色固体物,不同之处在于使用实施例25、步骤2的产物。
LC-MS(保留时间:1.55min,方法B),MS m/z 547(M++H)。
步骤4:
按照实施例11、步骤6的相同方法制备化合物25,为白色固体物,不同之处在于使用实施例25、步骤3的产物。
1H NMR(CD3OD)δ0.92-1.09(m,12H),1.26-1.30(m,10H),1.43-1.46(m,1H),1.87-1.90(m,1H),2.21-2.26(m,1H),2.36-2.41(m,1H),2.70-2.75(m,1H),2.93-2.97(m,1H),4.18-4.30(m,2H),4.46-4.48(m,1H),4.55-4.58(m,1H),5.12(d,J=10.5Hz,1H),5.29(d,J=18.0Hz,1H),5.72-5.79(m,1H),6.10(b,1H),7.37-7.40(m,1H),7.46-7.49(m,3H),7.70(t,J=7.5Hz,1H),7.85-7.89(m,2H),8.16-8.20(m,3H);
LC-MS(保留时间:2.08min,方法B),MS m/z 760(M++H)。
实施例26:化合物26的制备
化合物26
步骤1:
按照实施例25、步骤1的相同方法制备该产物,不同之处在于使用4-甲氧基苯基硼酸。
1H NMR(CD3OD)δ1.40,1.45(外消旋物,9H),2.50-2.55(m,1H),2.73-2.81(m,1H),3.81-3.89(m,4H),3.98-4.01(m,1H),4.50-4.57(m,1H),5.93(b,1H),7.02(d,J=9.0Hz,2H),7.50(t,J=7.3Hz,1H),7.67(t,J=7.5Hz,1H),7.73(s,1H),7.83(d,J=8.5Hz,1H),8.09(d,J=8.5Hz,2H),8.15(d,J=8.0Hz,1H);
LC-MS(保留时间:2.00min。方法B),MS m/z 465(M++H)。
步骤2:
按照实施例11、步骤4的相同方法制备该产物,不同之处在于使用实施例26、步骤1的产物。
1H NMR(CD3OD)δ1.06-1.09(m,2H),1.17-1.27(m,2H),1.42-1.47(m,10H),1.88-1.90(m,1H),2.21-2.26(m,1H),2.33-2.39(m,1H),2.61-2.65(m,1H),2.95-2.99(m,1H),3.85(s,3H),3.86-3.90(m,1H),3.99-4.00(m,1H),4.43-4.45(m,1H),5.13(d,J=10.8Hz,1H),5.31(d,J=18.0Hz,1H),5.77-5.80(m,1H),5.99(b,1H),7.02(d,J=9.0Hz,2H),7.51(t,J=7.3Hz,1H),7.68(t,J=7.5Hz,1H),7.76(s,1H),7.84(d,J=8.5Hz,1H),8.09(d,J=8.5Hz,2H),8.15(d,J=8.0Hz,1H);
LC-MS(保留时间:2.02min,方法B),MS m/z 677(M++H)。
步骤3:
按照实施例11、步骤5的相同方法制备该产物,为白色固体物,不同之处在于使用实施例26、步骤2的产物。
LC-MS(保留时间:1.53min,方法B),MS m/z 577(M++H)。
步骤4:
按照实施例11、步骤6的相同方法制备化合物26,不同之处在于使用实施例26、步骤3的产物。
1H NMR(CD3OD)δ0.93-1.09(m,12H),1.26-1.30(m,10H),1.44-1.46(m,1H),1.87-1.90(m,1H),2.21-2.26(m,1H),2.36-2.41(m,1H),2.70-2.75(m,1H),2.93-2.97(m,1H),3.86(s,3H),4.18-4.25(m,1H),4.30(b,1H),4.46-4.48(m,1H),4.55-4.58(m,1H),5.12(d,J=10.5Hz,1H),5.29(d,J=18.0Hz,1H),5.72-5.79(m,1H),6.08(b,1H),7.02(d,J=9.0Hz,2H),7.44(t,J=7.3Hz,1H),7.66(t,J=7.5Hz,1H),7.75(s,1H),7.83(d,J=8.5Hz,1H),8.09(d,J=8.5Hz,2H),8.15(d,J=8.0Hz,1H);LC-MS(保留时间:2.03min,方法B),MS m/z 790(M++H)。
实施例27:化合物27的制备
化合物27
步骤1:
按照实施例25、步骤1的相同方法制备该产物,不同之处在于使用4-吡啶基硼酸。
1H NMR(CD3OD)δ1.43,1.46(外消旋物,9H),2.53-2.56(m,1H),2.80-2.89(m,1H),3.90-3.93(m,1H),4.00-4.05(m,1H),4.50-4.57(m,1H),6.00,6.05(外消旋物,1H),7.80(t,J=7.3Hz,1H),7.87(t,J=7.5Hz,1H),8.08(d,J=8.5Hz,1H),8.32(d,J=8.0Hz,1H),8.49(s,1H),8.84(d,J=6.0Hz,2H),8.84(d,J=6.5Hz,2H);LC-MS(保留时间:1.39min,方法B),MS m/z 436(M++H)。
步骤2:
按照实施例11、步骤4的相同方法制备该产物,不同之处在于使用实施例27、步骤1的产物。
1H NMR(CD3OD)δ1.06-1.09(m,2H),1.17-1.27(m,2H),1.42-1.46(m,10H),1.88-1.90(m,1H),2.21-2.26(m,1H),2.33-2.39(m,1H),2.61-2.65(m,1H),2.95-2.99(m,1H),3.88-3.90(m,1H),4.01-4.08(m,1H),4.43-4.45(m,1H),5.15(d,J=10.8Hz,1H),5.32(d,J=18.0Hz,1H),5.77-5.80(m,1H),6.10(b,1H),7.79(t,J=7.3Hz,1H),7.88(t,J=7.5Hz,1H),8.08(d,J=8.5Hz,1H),8.31(d,J=8.0Hz,1H),8.47(s,1H),8.79(d,J=7.0Hz,2H),8.86(d,J=6.5Hz,2H);
LC-MS(保留时间:1.49min,方法B),MS m/z 648(M++H)。
步骤3:
按照实施例11、步骤5的相同方法制备该产物,为白色固体物,不同之处在于使用实施例27、步骤2的产物。
LC-MS(保留时间:0.96min,方法B),MS m/z 548(M++H)。
步骤4:
按照实施例11、步骤6的相同方法制备化合物27,不同之处在于使用实施例27、步骤3的产物。
1H NMR(CD3OD)δ0.94-1.09(m,12H),1.22-1.26(m,10H),1.44-1.49(m,1H),1.88-1.92(m,1H),2.22-2.25(m,1H),2.41-2.44(m,1H),2.70-2.75(m,1H),2.93-2.98(m,1H),4.18-4.21(m,1H),4.25(b,1H),4.53-4.62(m,2H),5.12(d,J=10.0Hz,1H),5.29(d,J=20.0Hz,1H),5.72-5.77(m,1H),6.12(b,1H),7.67(t,J=7.3Hz,1H),7.82(t,J=7.5Hz,1H),8.02(d,J=8.5Hz,1H),8.29(d,J=8.0Hz,1H),8.31(s,1H),8.55(d,J=7.0Hz,2H),8.76(d,J=6.5Hz,2H);
LC-MS(保留时间:1.49min,方法B),MS m/z 761(M++H)。
实施例28:化合物28的制备
化合物28
步骤1:
按照实施例25、步骤1的相同方法制备该产物,不同之处在于使用4-N,N-二甲基氨基-苯基硼酸。
LC-MS(保留时间:1.64min,方法B),MS m/z 478(M++H)。
步骤2:
按照实施例11、步骤4的相同方法制备该产物,不同之处在于使用实施例28、步骤1的产物。
LC-MS(保留时间:1.70min,方法B),MS m/z 690(M++H)。
步骤3:
按照实施例11、步骤5的相同方法制备该产物,为白色固体物,不同之处在于使用实施例28、步骤2的产物。
LC-MS(保留时间:1.20min,方法B),MS m/z 590(M++H)。
步骤4:
按照实施例11、步骤6的相同方法制备化合物28,不同之处在于使用实施例28、步骤3的产物。
1H NMR(d6-DMSO)δ0.92-1.10(m,13H),1.30(s,9H),1.35-1.38(m,1H),1.68-1.71(m,1H),2.12-3.00(m,2H),2.59-2.62(m,1H),2.91-2.95(m,1H),2.99(s,6H),3.93-4.10(m,2H),4.32-4.40(m,2H),5.09(d,J=11.5Hz,1H),5.23(d,J=19.0Hz,1H),5.54-5.64(m,1H),5.92(b,1H),6.83(d,J=9.0Hz,2H),7.42(t,J=7.3Hz,1H),7.70(t,J=7.5Hz,1H),7.81(s,1H),7.87(d,J=8.5Hz,1H),8.04(d,J=9.0Hz,2H),8.15(d,J=8.0Hz,1H);
LC-MS(保留时间:1.72min,方法B),MS m/z 803(M++H)。
实施例29:化合物29的制备
化合物29
步骤1:
按照实施例25、步骤1的相同方法制备该产物,不同之处在于使用4-氰基-苯基硼酸。
LC-MS(保留时间:1.87min,方法B),MS m/z 460(M++H)。
步骤2:
按照实施例11、步骤4的相同方法制备该产物,不同之处在于使用实施例29、步骤1的产物。
LC-MS(保留时间:1.88min,方法B),MS m/z 672(M++H)。
步骤3:
按照实施例11、步骤5的相同方法制备该产物,为白色固体物,不同之处在于使用实施例29、步骤2的产物。
LC-MS(保留时间:1.41min,方法B),MS m/z 572(M++H)。
步骤4:
按照实施例11、步骤6的相同方法制备化合物29,为白色固体物,不同之处在于使用实施例29、步骤3的产物。
1H NMR(CD3OD)δ0.92-1.09(m,12H),1.25-1.26(m,10H),1.42-1.46(m,1H),1.86-1.89(m,1H),2.20-2.22(m,1H),2.33-2.34(m,1H),2.68-2.71(m,1H),2.93-2.95(m,1H),4.13-4.28(m,2H),4.49-4.60(m,2H),5.12(d,J=10.5Hz,1H),5.28(d,J=18.0Hz,1H),5.71-5.80(m,1H),6.09(b,1H),7.56(t,J=7.3Hz,1H),7.74(t,J=7.5Hz,1H),7.83(d,J=10.5Hz,2H),7.93(d,J=7.5Hz,1H),8.01(s,1H),8.22(d,J=7.5Hz,1H),8.37(d,J=10.5Hz,2H);
LC-MS(保留时间:1.87min,方法B),MS m/z 785(M++H)。
实施例30:化合物30的制备
化合物30
步骤1:
按照实施例25、步骤1的相同方法制备该产物,不同之处在于使用3-呋喃硼酸。
LC-MS(保留时间:1.85min,方法B),MS m/z 425(M++H)。
步骤2:
按照实施例11、步骤4的相同方法制备该产物,不同之处在于使用实施例30、步骤1的产物。
LC-MS(保留时间:1.88min,方法B),MS m/z 637(M++H)。
步骤3:
按照实施例11、步骤5的相同方法制备该产物,不同之处在于使用实施例30、步骤2的产物。
LC-MS(保留时间:1.38min,方法B),MS m/z 537(M++H)。
步骤4:
按照实施例11、步骤6的相同方法制备化合物30,为白色固体物,不同之处在于使用实施例30、步骤3的产物。
1H NMR(CD3OD)δ0.95-1.09(m,12H),1.23-1.30(m,10H),1.43-1.46(m,1H),1.87-1.90(m,1H),2.21-2.23(m,1H),2.30-2.34(m,1H),2.64-2.70(m,1H),2.93-2.96(m,1H),4.11-4.29(m,2H),4.41-4.44(m,1H),4.54-4.56(m,1H),5.12(d,J=10.5Hz,1H),5.29(d,J=17.5Hz,1H),5.71-5.80(m,1H),6.02(b,1H),7.00(s,1H),7.44(t,J=7.2Hz,1H),7.52(s,1H),7.57(s,1H),7.66(t,J=7.0Hz,1H),7.79(d,J=8.0Hz,1H),8.14-8.17(m,2H);
LC-MS(保留时间:1.93min,方法B),MS m/z 750(M++H)。
实施例31:化合物31的制备
化合物31
方案1
步骤1:
按照实施例21、步骤2的相同方法制备该产物,为白色固体物,不同之处在于使用3-溴-异喹啉(Atkins等,JOC,1973,400)。
1H NMR(CDCl3)δ7.60-7.62(m,2H),7.71-7.73(m,2H),8.12(s,1H),8.99(s,1H);
LC-MS(保留时间:0.78min,方法B),MS m/z 224,226(M++H)。
步骤2:
按照实施例11、步骤2的相同方法制备该产物,为白色固体物,不同之处在于使用3-溴-异喹啉2-氧化物。
1H NMR(CDCl3)δ7.66-7.71(m,1H),7.74-7.76(m,2H),7.83(s,1H),8.29(d,J=8.5Hz,1H);
LC-MS(保留时间:1.55min,方法B),MS m/z 242,244(M++H)。
步骤3:
按照实施例11、步骤3的相同方法制备该产物,为泡沫状物,不同之处在于使用3-溴-1-氯-异喹啉。
1H NMR(CD3OD)δ1.43,1.44(外消旋物,9H),2.41-2.47(m,1H),2.69-2.72(m,1H),3.80-3.84(m,1H),3.88-3.90(m,1H),4.46-4.52(m,1H),5.76(b,1H),7.57-7.61(m,2H),7.73-7.75(m,2H),8.15(d,J=8.0Hz,1H);
LC-MS(保留时间:1.79min,方法B),MS m/z 437,439(M++H)。
步骤4:
将2-三丁基锡烷基-吡嗪(44mg,0.12mmol)、四(三苯基膦)合钯(0)(12mg,0.01mmol)和实施例31、步骤3的产物(44mg,0.1mmol)在甲苯(1mL)中的混合物加热回流3h。真空除去挥发份后,剩余物经制备型HPLC纯化,得到35mg(80%)所需的产物,为黄色固体物。
LC-MS(保留时间:1.77min,方法B),MS m/z 437(M++H)。
步骤5:
按照实施例11、步骤4的相同方法制备该产物,不同之处在于使用实施例31、步骤4的产物。
LC-MS(保留时间:1.78min,方法B),MS m/z 649(M++H)。
步骤6:
按照实施例11、步骤5的相同方法制备该产物,为白色固体物,不同之处在于使用实施例31、步骤5的产物。
LC-MS(保留时间:1.26min,方法B),MS m/z 549(M++H)。
步骤7:
按照实施例11、步骤6的相同方法制备化合物31,不同之处在于使用实施例31、步骤6的产物。
1H NMR(CD3OD)δ0.95-1.10(m,12H),1.24-1.27(m,10H),1.44-1.47(m,1H),1.87-1.90(m,1H),2.19-2.22(m,1H),2.38-2.44(m,1H),2.71-2.76(m,1H),2.93-2.96(m,1H),4.18-4.28(m,2H),4.50-4.61(m,2H),5.12(d,J=10.5Hz,1H),5.29(d,J=17.5Hz,1H),5.71-5.80(m,1H),6.12(b,1H),7.60(t,J=7.2Hz,1H),7.77(t,J=7.0Hz,1H),7.97(d,J=8.5Hz,1H),8.26(d,J=8.5Hz,1H),8.44(s,1H),8.59(s,1H),8.70(s,1H),9.61(s,1H);
LC-MS(保留时间:1.84min,方法B),MS m/z 762(M++H)。
实施例32:化合物32的制备
化合物32
步骤1:
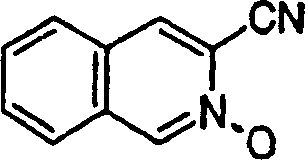
按照实施例21、步骤2的相同方法制备2-氧基-异喹啉-3-甲腈产物,为白色固体物,不同之处在于使用3-氰基-异喹啉。
1H NMR(DMSO-d6)δ7.74(t,J=8.0Hz,1H),7.84(t,J=8.2Hz,1H),7.97(d,J=8.5Hz,1H),8.03(d,J=8.5Hz,1H),8.85(s,1H),9.17(s,1H);
LC-MS(保留时间:0.48min,方法B),MS m/z 171(M++H)。
步骤2:
按照实施例11、步骤2的相同方法制备1-氯-异喹啉-3-甲腈产物,为白色固体物,不同之处在于使用3-氰基-异喹啉2-氧化物。
1H NMR(CDCl3)δ7.87-7.91(m,2H),7.92-7.94(m,1H),8.09(s,1H),8.42-8.44(m,1H);
LC-MS(保留时间:1.22min,方法B),MS m/z 189(M++H)。
步骤3:
按照实施例1、步骤5的相同方法制备该产物,不同之处在于使用1-氯-异喹啉-3-甲腈。
1H NMR(CD3OD)δ1.05(s,9H),1.17(s,9H),2.34-2.40(m,1H),2.71-2.78(m,1H),4.09-4.11(m,1H),4.21(b,1H),4.48-4.52(m,1H),4.68-4.72(m,1H),5.89(b,1H),7.74(t,J=7.5Hz,1H),7.86(t,J=7.5Hz,1H),7.94-7.97(m,2H),8.31(d,J=8.0Hz,1H);
LC-MS(保留时间:1.66min,方法B),MS m/z 497(M++H)。
步骤4:
按照实施例1、步骤9的相同方法制备化合物32,为白色固体物,不同之处在于使用实施例32、步骤3的产物。
1H NMR(CD3OD)δ1.04-1.09(m,12H),1.20-1.27(m,10H),1.39-1.45(m,1H),1.85-1.88(m,1H),2.20-2.30(m,2H),2.63-2.71(m,1H),2.91-2.97(m,1H),4.09-4.13(m,1H),4.23(d,J=9.3Hz,1H),4.49-4.58(m,2H),5.13(d,J=10.5Hz,1H),5.28(d,J=18.0Hz,1H),5.69-5.81(m,1H),5.92(b,1H),6.60(d,J=10.0Hz,1H),7.72(t,J=7.5Hz,1H),7.86(t,J=7.5Hz,1H),7.96-7.99(m,2H),8.29(d,J=8.0Hz,1H);
LC-MS(保留时间:1.75min,方法B),MS m/z 714(M++H)。
实施例33:化合物33的制备
化合物33
步骤1:
按照实施例21、步骤2的相同方法制备3-甲基-异喹啉2-氧化物产物,为白色固体物,不同之处在于使用3-甲基-异喹啉。
1H NMR(CD3OD)δ2.64(s,3H),7.64-7.72(m,2H),7.88-7.95(m,2H),9.05(s,1H);
LC-MS(保留时间:0.61min,方法B),MS m/z 160(M++H)。
步骤2:
按照实施例11、步骤2的相同方法制备1-氯-3-甲基-异喹啉产物,为白色固体物,不同之处在于使用3-甲基-异喹啉2-氧化物。
1H NMR(CDCl3)δ2.65(s,3H),7.25(s,1H),7.61(t,J=7.5Hz,1H),7.69(t,J=7.5Hz,1H),7.74(d,J=8.0Hz,1H),8.27(d,J=8.5Hz,1H);
LC-MS(保留时间:1.47min,方法B),MS m/z 178(M++H)。
步骤3:
按照实施例1、步骤5的相同方法制备该产物,为白色固体物,不同之处在于使用1-氯-3-甲基-异喹啉。
1H NMR(CD3OD)δ1.05(s,9H),1.23(s,9H),2.51(s,3H),2.34-2.40(m,1H),2.72-2.78(m,1H),4.05-4.12(m,1H),4.26(b,1H),4.41(d,J=10Hz,1H),4.62-4.67(m,1H),5.90(b,1H),7.14(s,1H),7.38(t,J=7.5Hz,1H),7.62(t,J=7.5Hz,1H),7.68(d,J=8.0Hz,1H),8.14(d,J=8.0Hz,1H);
LC-MS(保留时间:1.84min,方法B),MS m/z 486(M++H)。
步骤4:
按照实施例1、步骤9的相同方法制备化合物33,为白色固体物,不同之处在于使用实施例33、步骤3的产物。
1H NMR(CD3OD)δ0.99-1.09(m,12H),1.23-1.25(m,10H),1.41-1.45(m,1H),1.86-1.90(m,1H),2.21-2.31(m,2H),2.52(s,3H),2.58-2.61(m,1H),2.91-2.97(m,1H),4.08-4.12(m,1H),4.28(b,1H),4.40(d,J=10.0Hz,1H),4.50-4.55(m,1H),5.12(d,J=10.0Hz,1H),5.30(d,J=18.0Hz,1H),5.71-5.81(m,1H),5.93(b,1H),7.13(s,1H),7.38(t,J=7.5Hz,1H),7.62(t,J=7.5Hz,1H),7.68(d,J=8.0Hz,1H),8.12(d,J=8.0Hz,1H),9.12(b,1H);
LC-MS(保留时间:1.85min,方法B),MS m/z 698(M++H)。
实施例34:化合物34的制备
化合物34
步骤1:
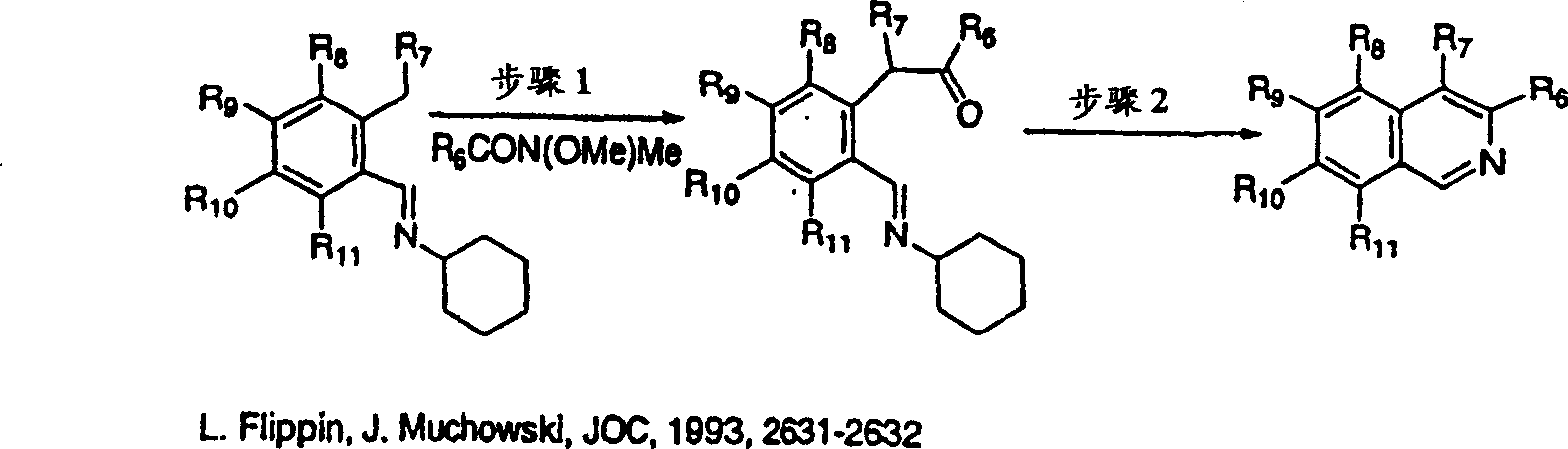
按照实施例21、步骤2的相同方法制备3-环丙基-异喹啉2-氧化物产物,为白色固体物,不同之处在于使用3-环丙基-异喹啉(L.Flippin,J.Muchowski,J.O.C,1993,2631-2632)。
LC-MS(保留时间:0.95min,方法B),MS m/z 186(M++H)。
步骤2:
按照实施例11、步骤2的相同方法制备1-氯-3-环丙基-异喹啉产物,为白色固体物,不同之处在于使用3-环丙基-异喹啉2-氧化物。
1H NMR(CD3OD)δ1.00-1.04(m,4H),2.11-2.18(m,1H),7.55(s,1H),7.61(t,J=8.0Hz,1H),7.72(t,J=8.0Hz,1H),7.83(d,J=13.5Hz,1H),8.27(d,J=14.5Hz,1H);
LC-MS(保留时间:1.70min,方法B),MS m/z 204(M++H)。
步骤3:
按照实施例1、步骤5的相同方法制备该产物,为白色固体物,不同之处在于使用1-氯-3-环丙基-异喹啉。
1H NMR(CD3OD)δ0.93-1.05(m,13H),1.29(s,9H),2.06-2.10(m,1H),2.39-2.44(m,1H),2.70-2.76(m,1H),4.05-4.12(m,1H),4.27(b,1H),4.35(d,J=10.0Hz,1H),4.62-4.67(m,1H),5.78(b,1H),7.18(s,1H),7.38(t,J=7.5Hz,1H),7.61(t,J=7.5Hz,1H),7.66(d,J=8.0Hz,1H),8.09(d,J=8.0Hz,1H);
LC-MS(保留时间:1.96min,方法B),MS m/z 512(M++H)。
步骤4:
按照实施例1、步骤9的相同方法制备化合物34,为白色固体物,不同之处在于使用实施例34、步骤3的产物。
1H NMR(CD3OD)δ0.93-1.09(m,16H),1.24-1.30(m,10H),1.42-1.46(m,1H),1.87-1.90(m,1H),2.06-2.11(m,1H),2.21-2.32(m,2H),2.56-2.61(m,1H),2.92-2.97(m,1H),4.08-4.12(m,1H),4.28(b,1H),4.32(d,J=10.0Hz,1H),4.48-4.53(m,1H),5.12(d,J=10.5Hz,1H),5.30(d,J=17.5Hz,1H),5.72-5.77(m,1H),5.82(b,1H),7.18(s,1H),7.36(t,J=7.5Hz,1H),7.60(t,J=7.5Hz,1H),7.67(d,J=8.0Hz,1H),8.07(d,J=8.0Hz,1H);
LC-MS(保留时间:2.00min,方法B),MS m/z 724(M++H)。
实施例35:化合物35的制备
化合物35
方案1
步骤1:
将3-羟基-异喹啉(725mg,5.0mmol)、碳酸铯(4.89g,15.0mmol)、MeI(781mg,5.5mmol)在DMF(50mL)中的混合物在环境温度下搅拌12h。混合物用EtOAc(200mL)稀释,过滤,依次用水(200mL,×2)、1M NaOH水溶液和盐水洗涤。有机层经硫酸镁干燥,过滤,蒸发。剩余物经制备型HPLC纯化,得到120mg(15%)所需的产物,为白色固体物。
1H NMR(CDCl3)δ4.03(s,3H),6.99(s,1H),7.36(t,J=8.0Hz,1H),7.56(t,J=8.2Hz,1H),7.68(d,J=8.5Hz,1H),7.87(J=8.5Hz,1H);
LC-MS(保留时间:0.54min,方法B),MS m/z 160(M++H)。
步骤2:
按照实施例21、步骤2的相同方法制备3-甲氧基-异喹啉2-氧化物产物,为白色固体物,不同之处在于使用3-甲氧基-异喹啉。
LC-MS(保留时间:0.83min。方法B),MS m/z 176(M++H)。
步骤3:
按照实施例11、步骤2的相同方法制备1-氯-3-甲氧基-异喹啉产物,为白色固体物,不同之处在于使用3-甲氧基-异喹啉2-氧化物。
LC-MS(保留时间:1.62min,方法B),MS m/z 194(M++H)。
步骤4:
按照实施例1、步骤5的相同方法制备该产物,为白色固体物,不同之处在于使用1-氯-3-甲氧基-异喹啉。
1H NMR(CD3OD)δ1.05(s,9H),1.23(s,9H),2.35-2.43(m,1H),2.72-2.79(m,1H),3.96(s,3H),4.01-4.11(m,1H),4.26(b,1H),4.48(d,J=10.0Hz,1H),4.62-4.67(m,1H),5.83(b,1H),6.61(s,1H),7.25(t,J=7.5Hz,1H),7.54(t,J=7.5Hz,1H),7.63(d,J=8.1Hz,1H),8.08(d,J=8.4Hz,1H);
LC-MS(保留时间:1.82min,方法B),MS m/z 502(M++H)。
步骤5:
按照实施例1、步骤9的相同方法制备化合物35,为白色固体物,不同之处在于使用实施例35、步骤4的产物。
1H NMR(CD3OD)δ1.04-1.08(m,12H),1.24-1.27(m,10H),1.43-1.45(m,1H),1.86-1.89(m,1H),2.21-2.26(m,1H),2.30-2.34(m,1H),2.62-2.66(m,1H),2.91-2.97(m,1H),3.99(s,3H),4.09-4.12(m,1H),4.27-4.28(m,1H),4.46(d,J=10.0Hz,1H),4.51-4.58(m,1H),5.12(d,J=10.5Hz,1H),5.30(d,J=18.0Hz,1H),5.72-5.76(m,1H),5.88(b,1H),6.62(s,1H),7.26(t,J=7.5Hz,1H),7.55(t,J=7.5Hz,1H),7.65(d,J=8.0Hz,1H),8.06(d,J=8.5Hz,1H);
LC-MS(保留时间:1.85min,方法B),MS m/z 714(M++H)。
实施例36:化合物36的制备
化合物36
方案1
步骤1:
将4-甲氧基-2-甲基-苯甲酸(5.00g,30.1mmol)和亚硫酰氯(20.0g,0.17mol)的混合物加热回流30min。真空除去挥发份。抽真空过夜后,所得的粘稠油状酰氯未经进一步纯化直接用于下一步反应。
在0℃下,向4-甲氧基-2-甲基-苯甲酰氯的二氯甲烷(60mL)溶液中滴加入二乙胺。将所形成的混合物在搅拌下升温至环境温度反应2h。真空除去挥发份。将剩余物在EtOAc(100mL)中研磨并过滤。将滤液用1M HCl、1M NaOH和盐水洗涤,经硫酸镁干燥。蒸发溶剂,得到6.51g(98%)所需的产物,为粘稠油状物。
LC-MS(保留时间:1.20min,方法B),MS m/z 222(M++H)。
步骤2:
在-78℃下,向N,N-二乙基-4-甲氧基-2-甲基-苯甲酰胺(221mg,1.0mmol)的THF(2mL)溶液内滴加入n-BuLi(0.84ml 2.5M的己烷溶液,2.10mmol)。所形成的橙色溶液再于该温度下保温30min,随后滴加入苄腈(103mg,1.0mmol)。将最终的溶液在搅拌下升温至环境温度过夜。用冰冻的5%柠檬酸猝灭。过滤,用水洗涤并干燥。在2∶1己烷-EtOAc(5mL)中研磨,得到205mg(82%)所需的产物,为白色固体物。
1H NMR(d6-DMSO)δ3.89(s,3H),6.84(s,1H),7.05-7.07(m,1H),7.18(d,J=2.5Hz,1H),7.44-7.51(m,3H),7.78(d,J=7.0Hz,1H),8.11(d,J=9.0Hz,1H);
LC-MS(保留时间:1.20min,方法B),MS m/z 252(M++H)。
步骤3:
按照实施例11、步骤2的相同方法制备1-氯-6-甲氧基-3-苯基-异喹啉产物,为白色固体物,不同之处在于使用6-甲氧基-3-苯基-2H-异喹啉-1-酮。
1H NMR(CDCl3)δ3.97(s,3H),7.12(d,J=2.5Hz,1H),7.23-7.26(m,1H),7.40-7.42(m,1H),7.46-7.50(m,2H),7.89(s,1H),8.08(d,J=7.0Hz,2H),8.21(d,J=9.0Hz,1H);
LC-MS(保留时间:1.90min,方法B),MS m/z 270,271(M++H)。
步骤4:
向实施例21、步骤1的产物(320mg,0.57mmol)的DMSO(5mL)溶液中加入叔丁醇钾(321mg,2.87mmol)。将所形成的溶液在环境温度下搅拌30min,随后加入1-氯-6-甲氧基-3-苯基-异喹啉(实施例36,步骤3)(155mg,0.57mmol)。将最终的溶液搅拌12h。用冰冻的水猝灭,用1M HCl酸化至pH为4,用EtOAc萃取(20mL,×2)。有机层用盐水洗涤,经硫酸镁干燥,过滤,蒸发。剩余物经制备型HPLC纯化(40%B至100%B,15min梯度),得到289mg(64%)化合物36,为白色固体物。
1H NMR(CD3OD)δ0.95-1.05(m,12H),1.24-1.32(m,10H),1.44-1.46(m,1H),1.87-1.90(m,1H),2.20-2.26(m,1H),2.30-2.36(m.1H),2.65-2.71(m,1H),2.93-2.97(m,1H),3.94(s,3H),4.12-4.28(m,2H),4.38-4.52(m,2H),5.12(d,J=10.0Hz,1H),5.28(d,J=17.0Hz,1H),5.69-5.74(m,1H),6.05(b,1H),7.06-7.07(m,1H),7.26(s,1H),7.37-7.39(m,1H),7.44-7.48(m,2H),7.77(s,1H),8.07(d,J=9.0Hz,1H),8.15(d,J=8.5Hz,2H);
LC-MS(保留时间:2.02min,方法B),MS m/z 790(M++H)。
实施例37:化合物37的制备
化合物37
方案1
步骤1:
在-78℃下,向N,N-二乙基-4-甲氧基-2-甲基-苯甲酰胺(633mg,2.9mmol)的THF(15mL)溶液中滴加入n-BuLi(2.3ml,2.5M己烷溶液,5.74mmol)。所形成的红色溶液再于该温度下保温30min,随后在-78℃下,经插管转移到噻唑-2-甲酸乙酯(A.Medici等,Tetrahedron Lett.1983,2901页)(450mg,2.9mmol)的THF(5mL)溶液中。最终的深绿色溶液在搅拌下保持在该温度下反应2h。用饱和氯化铵水溶液猝灭并用EtOAc(50mL)萃取。有机层用饱和氯化铵水溶液和盐水洗涤,干燥,经快速柱层析纯化,用2∶1 EtOAc∶己烷洗脱,得到405mg(45%)所需的产物,为灰白色粘稠油状物。
1H NMR(CDCl3)δ1.08(t,J=7.0Hz,6H),3.22(b,2H),3.44(b,2H),3.79(s,3H),4.59(s,2H),6.79-6.81(m,1H),6.86(d,J=2.5Hz,1H),7.16(d,J=8.5Hz,1H),7.66(d,J=3.0Hz,1H),8.00(d,J=3.0Hz,1H);
LC-MS(保留时间:1.30min,方法B),MS m/z 333(M++H)。
步骤2:
将N,N-二乙基-4-甲氧基-2-(2-氧代-2-噻唑-2-基-乙基)-苯甲酰胺(405mg,1.22mmol)和乙酸铵(3.0g,38.9mmol)的混合物在140℃的密封管中加热1h。将熔融的溶液倒入冰冻的水中,过滤,用水充分洗涤滤饼。经干燥的褐色固体物(240mg,76%)未经进一步纯化直接用于下一反应中。
LC-MS(保留时间:1.24min,方法B),MS m/z 259(M++H)。
步骤3:
按照实施例11、步骤2的相同方法制备1-氯-6-甲氧基-3-噻唑-2-基-异喹啉产物,为白色固体物,不同之处在于使用6-甲氧基-3-噻唑-2-基-2H-异喹啉-1-酮。
1H NMR(CDCl3)δ3.97(s,3H),7.16(d,J=4.0Hz,1H),7.27-7.31(m,1H),7.46(d,J=5.0Hz,1H),7.93(d,J=5.5Hz,1H),8.22(d,J=15.5Hz,1H),8.39(s,1H);
LC-MS(保留时间:1.66min,方法B),MS m/z 277(M++H)。
步骤4:
按照实施例36、步骤4的相同方法制备化合物37,不同之处在于使用1-氯-6-甲氧基-3-噻唑-2-基-异喹啉。
1H NMR(CD3OD)δ0.97-1.09(m,12H),1.24-1.29(m,10H),1.44-1.46(m,1H),1.87-1.90(m,1H),2.20-2.26(m,1H),2.30-2.36(m,1H),2.65-2.71(m,1H),2.93-2.96(m,1H),3.96(s,3H),4.12-4.27(m,2H),4.38-4.52(m,2H),5.12(d,J=10.5Hz,1H),5.29(d,J=17.5Hz,1H),5.69-5.74(m,1H),5.99(b,1H),7.14(d,J=9.0Hz,1H),7.33(s,1H),7.66(d,J=3.5Hz,1H),7.93(d,J=3.0Hz,1H),8.05(s,1H),8.11(d,J=9.0Hz,1H),9.14(b,1H);
LC-MS(保留时间:1.89min,方法B),MS m/z 797(M++H)。
实施例38:化合物38的制备
化合物38
方案1
步骤1:
将3-羟基-异_唑-5-甲酸甲酯(5.72g,0.04mol)、甲基碘(6.82g,0.044mol)和碳酸铯(39.1g,0.12mol)在DMF(200mL)中的混合物在环境温度下搅拌过夜。用EtOAc(1L)稀释,过滤。将滤液依次用水(IL,×2)、1M NaOH和盐水洗涤,经硫酸镁干燥,真空蒸发,得到4.80g(76%)所需的产物,为白色固体物。所得的产物未经进一步纯化直接使用。
1H NMR(CDCl3)δ3.92(s,3H),4.00(s,3H),6.51(s,1H);
LC-MS(保留时间:0.69min,方法B),MS m/z 158(M++H)。
步骤2:
按照实施例37、步骤1的相同方法制备N,N-二乙基-4-甲氧基-2-[2-(3-甲氧基-异_唑-5-基)-2-氧代-乙基]-苯甲酰胺产物,不同之处在于使用3-甲氧基-异_唑-5-甲酸甲酯。
LC-MS(保留时间:1.28min,方法B),MS m/z 347(M++H)。
步骤3:
按照实施例37、步骤2的相同方法制备6-甲氧基-3-(3-甲氧基-异_唑-5-基)-2H-异喹啉-1-酮产物,不同之处在于使用N,N-二乙基-4-甲氧基-2-[2-(3-甲氧基-异_唑-5-基)-2-氧代-乙基]-苯甲酰胺。
1H NMR(DMSO-d6)δ3.89(s,3H),3.97(s,3H),7.01(s,1H),7.14-7.16(m,2H),7.43(s,1H),8.13(d,J=8.5Hz,1H);
LC-MS(保留时间:1.31min,方法B),MS m/z 273(M++H)。
步骤4:
按照实施例11、步骤2的相同方法制备1-氯-6-甲氧基-3-(3-甲氧基-异_唑-5-基)-异喹啉产物,为白色固体物,不同之处在于使用6-甲氧基-3-(3-甲氧基-异_唑-5-基)-2H-异喹啉-1-酮。
1H NMR(CDCl3)δ3.97(s,3H),4.04(s,3H),6.60(s,1H),7.17(d,J=2.5Hz,1H),7.31-7.33(m,1H),8.02(s,1H),8.23(d,J=9.0Hz,1H);
LC-MS(保留时间:1.73min,方法B),MS m/z 291,293(M++H)。
步骤5:
按照实施例36、步骤4的相同方法制备化合物38,不同之处在于使用1-氯-6-甲氧基-3-(3-甲氧基-异_唑-5-基)-异喹啉。
1H NMR(CD3OD)δ0.99-1.09(m,12H),1.23-1.28(m,10H),1.44-1.46(m,1H),1.87-1.90(m,1H),2.20-2.26(m,1H),2.30-2.36(m,1H),2.65-2.71(m,1H),2.93-2.96(m,1H),3.95(s,3H),4.02(s,3H),4.13-4.14(m,1H),4.24-4.26(m,1H),4.41-4.42(m,1H),4.52-4.55(m,1H),5.12(d,J=10.5Hz,1H),5.29(d,J=17.0Hz,1H),5.72-5.79(m,1H),5.96(b,1H),6.60(s,1H),7.15-7.17(m,1H),7.32(s,1H),7.80(s,1H),8.10(d,J=9.0Hz,1H);
LC-MS(保留时间:1.95min,方法B),MS m/z 811(M++H)。
实施例39:化合物39的制备
化合物39
步骤1:
按照实施例37、步骤1的相同方法制备N,N-二乙基-4-甲氧基-2-[2-(5-甲氧基-_唑-2-基)-2-氧代-乙基]-苯甲酰胺产物,不同之处在于使用5-甲氧基-_唑-2-甲酸乙酯。
LC-MS(保留时间:1.24min,方法B),MS m/z 347(M++H)。
步骤2:
按照实施例37、步骤2的相同方法制备6-甲氧基-3-(5-甲氧基-_唑-2-基)-2H-异喹啉-1-酮产物,不同之处在于使用N,N-二乙基-4-甲氧基-2-[2-(5-甲氧基-_唑-2-基)-2-氧代-乙基]-苯甲酰胺。
1H NMR(DMSO-d6)δ3.94(s,3H),4.01(s,3H),6.34(s,1H),6.99(d,J=2.0Hz,1H),7.12-7.14(m,1H),7.25(s,1H),8.32(d,J=9.0Hz,1H);
LC-MS(保留时间:1.22min,方法B),MS m/z 274(M++H)。
步骤3:
按照实施例11、步骤2的相同方法制备1-氯-6-甲氧基-3-(5-甲氧基-_唑-2-基)-异喹啉产物,为白色固体物,不同之处在于使用6-甲氧基-3-(5-甲氧基-_唑-2-基)-2H-异喹啉-1-酮。
1H NMR(CDCl3)δ3.96(s,3H),4.00(s,3H),6.34(s,1H),7.12(d,J=2.5Hz,1H),7.28-7.31(m,1H),8.13(s,1H),8.23(d,J=9.0Hz,1H);
LC-MS(保留时间:1.58min,方法B),MS m/z 291,293(M++H)。
步骤4:
按照实施例36、步骤4的相同方法制备化合物39,不同之处在于使用1-氯-6-甲氧基-3-(3-甲氧基-异_唑-5-基)-异喹啉。
1H NMR(CD3OD)δ0.99-1.09(m,12H),1.23-1.28(m,10H),1.44-1.46(m,1H),1.87-1.90(m,1H),2.20-2.26(m,1H),2.30-2.36(m,1H),2.65-2.71(m,1H),2.93-2.96(m,1H),3.95(s,3H),4.02(s,3H),4.13-4.14(m,1H),4.25(b,1H),4.41-4.42(m,1H),4.52-4.55(m,1H),5.12(d,J=10.0Hz,1H),5.29(d,J=17.0Hz,1H),5.72-5.79(m,1H),6.07(b,1H),6.45(s,1H),7.15-7.16(m,1H),7.29(s,1H),7.85(s,1H),8.10(d,J=9.0Hz,1H),9.11(b,1H);
LC-MS(保留时间:1.75min,方法B),MS m/z 811(M++H)。
实施例40:化合物40的制备
化合物40
方案1
步骤1:
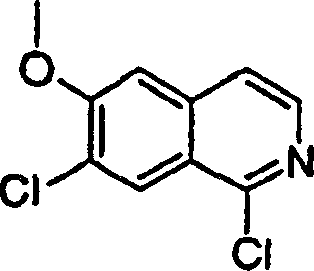
按照实施例21、步骤2的相同方法制备1-氯-6-甲氧基-异喹啉2-氧化物产物,不同之处在于使用1-氯-6-甲氧基-异喹啉(实施例11、步骤2的产物)。
1H NMR(CDCl3)δ4.00(s,3H),7.14(d,J=2.5Hz,1H),7.41-7.43(m,1H),7.62(d,J=7.0Hz,1H),8.15(d,J=9.5Hz,1H),8.36(d,J=7.0Hz,1H);
LC-MS(保留时间:0.85min,方法B),MS m/z 210(M++H)。
步骤2:
按照实施例11、步骤2的相同方法制备1,3-二氯-6-甲氧基-异喹啉产物,不同之处在于使用1-氯-6-甲氧基-异喹啉2-氧化物。
1H NMR(CDCl3)δ3.94(s,3H),6.98(s,1H),7.25-7.26(m,1H),7.529s,1H),8.16(d,J=9.5Hz,1H);
LC-MS(保留时间:1.54min,方法B),MS m/z 228,230(M++H)。
步骤3:
按照实施例24、步骤1的相同方法制备该产物,为泡沫状物,不同之处在于使用1,3-二氯-6-甲氧基-异喹啉。
1H NMR(CD3OD)δ1.43,1.44(外消旋物,9H),2.39-2.44(m,1H),2.68-2.72(m,1H),3.80-3.90(m,2H),3.91(s,3H),4.79-4.82(m,1H),5.71(b,1H),7.10-7.14(m,2H),7.26(s,1H),7.99-8.01(m,1H);
LC-MS(保留时间:1.79min,方法B),MS m/z 422(M++H)。
步骤4:
按照实施例11、步骤4的相同方法制备该产物,不同之处在于使用实施例40、步骤3的产物。
LC-MS(保留时间:1.83min,方法B),MS m/z 635(M++H)。
步骤5:
按照实施例11、步骤5的相同方法制备该产物,为白色固体物,不同之处在于使用实施例40、步骤4的产物。
LC-MS(保留时间:1.36min,方法B),MS m/z 535(M++H)。
步骤6:
按照实施例11、步骤6的相同方法制备化合物40,为白色固体物,不同之处在于使用实施例40、步骤5的产物。
1H NMR(CD3OD)δ1.07-1.11(m,12H),1.26-1.30(m,10H),1.46-1.48(m,1H),1.87-1.91(m,1H),2.21-2.34(m,2H),2.62-2.66(m,1H),2.94-2.99(m,1H),3.95(s,3H),4.06-4.11(m,1H),4.26-4.28(m,1H),4.46-4.56(m,2H),5.15(d,J=10.0Hz,1H),5.29(d,J=17.0Hz,1H),5.72-5.79(m,1H),5.89(b,1H),6.63(d,J=9.0Hz,1H),7.08-7.09(m,1H),7.18(s,1H),7.34(s,1H),8.08(d,J=9.5Hz,1H);
LC-MS(保留时间:1.99min,方法B),MS m/z 748(M++H)。
实施例41:化合物41的制备
化合物41
步骤1:
按照实施例30、步骤1的相同方法制备该产物,不同之处在于使用实施例40、步骤3的产物。
LC-MS(保留时间:1.85min,方法B),MS m/z 455(M++H)。
步骤2:
按照实施例11、步骤4的相同方法制备该产物,为泡沫状物,不同之处在于使用实施例41、步骤1的产物。
LC-MS(保留时间:1.88min,方法B),MS m/z 667(M++H)。
步骤3:
按照实施例11、步骤5的相同方法制备该产物,为白色固体物,不同之处在于使用实施例41、步骤2的产物。
LC-MS(保留时间:1.38min,方法B),MS m/z 567(M++H)。
步骤4:
按照实施例11、步骤6的相同方法制备化合物41,为白色固体物,不同之处在于使用实施例41、步骤3的产物。
1H NMR(CD3OD)δ0.99-1.04(m,12H),1.22-1.31(m,10H),1.43-1.45(m,1H),1.87-1.89(m,1H),2.22-2.24(m,1H),2.30-2.34(m,1H),2.65-2.68(m,1H),2.93-2.96(m,1H),3.92(s,3H),4.11-4.14(m,1H),4.28-4.30(m,1H),4.38-4.42(m,1H),4.53-4.55(m,1H),5.12(d,J=10.0Hz,1H),5.29(d,J=18.0Hz,1H),5.72-5.77(m,1H),5.99(b,1H),6.61(d,J=5.0Hz,1H),6.98(s,1H),6.99-7.02(m,1H),7.17(s,1H),7.44(s,1H),7.57(d,J=5.0Hz,1H),8.03(d,J=10.0Hz,1H),8.14(s,1H);
LC-MS(保留时间:1.92min,方法B),MS m/z 780(M++H)。
实施例42:化合物42的制备
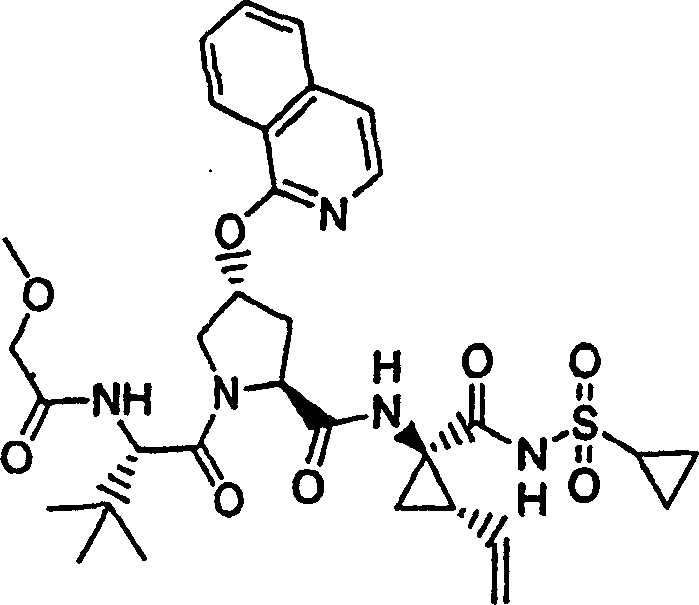
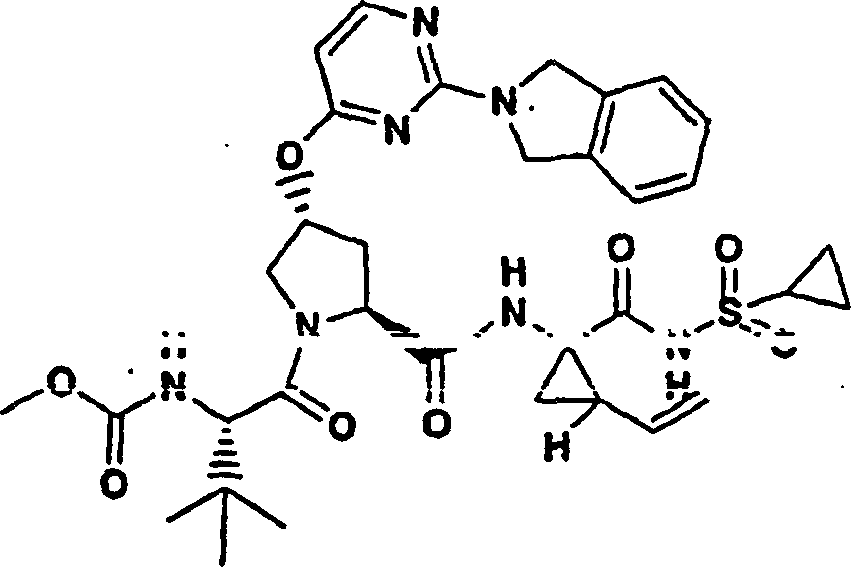
按照制备实施例11的化合物11的方法制备题述产物,不同之处在于使用6-乙氧基肉桂酸代替6-甲氧基肉桂酸作为用于P2组件的原料。
化合物42
1H NMR(500MHz,CD3OD)δppm 0.98-1.09(m,15H),1.24-1.31(m,10H),1.42-1.46(m,1H),1.85-1.90(m,1H),2.19-2.32(m,2H),2.57-2.63(m,1H),2.91-2.97(m,1H),4.03-4.09(m,1H),4.17(q,J=7.0Hz,2H),4.42(d,J=11.3Hz,1H),4.49-4.54(m,1H),5.12(d,J=17.4Hz,1H),5.72-5.78(m,1H),5.83(s,1H),7.07-7.10(M,1H),7.15(s,1H),7.22(d,J=5.8Hz,1H),7.87(d,J=5.8Hz,1H),8.08(d,J=8.8Hz,1H);MS:(M+H)+728.
部分C:
实施例45:化合物45的制备
P1为(1R,2S)和(1S,2R)1∶1的混合物
化合物45
方案1
步骤1:
向装于中压烧瓶(Chemglass)中的2-溴-5-甲氧基苯甲酸(1.68g,7.27mmol)的DMF(50mL)溶液内加入苄脒(1.25g,8.00mmol)、碳酸钾(6.0g,43.6mmol)和铜粉(336mg,1.45mmol)。将反应混合物在180℃下加热1h。真空抽滤除去铜和过量的碳酸钾,并用甲醇洗涤。浓缩滤液并将所得的粗产物经快速柱层析纯化(SiO2,5%甲醇/DCM),得到浅绿色固体物(1.55g,收率84%):
1H NMR(DMSO-d6)δ3.84(s,3H),7.26(d,J=7.8Hz,1H),7.46(br s,5H),7.57(s,1H),8.38(br s,1H);MS m/z(MH+)253.
步骤2:
向0℃的Boc-顺-羟基脯氨酸-OMe(2.0g,8.15mmol)和实施例45、步骤1的产物(2.26g,8.97mmol)在THF(82mL)中的浆状物内加入三苯基膦和偶氮二甲酸二异丙酯(1.98g,8.97mmol)。在室温下搅拌17h后,将反应混合物用EtOAc(100mL)稀释并用H2O(50mL)洗涤。分离出水层并用EtOAc(2×50mL)反萃取。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩,得到粘稠油状物。将所述油状物再次溶于尽可能少量的乙酸乙酯中,加入己烷以沉淀出大部分的Ph3PO副产物。抽滤除去Ph3PO并浓缩滤液。将所得的粘稠的油状物经快速柱层析纯化(SiO2,4∶1己烷∶乙酸乙酯),得到白色固体产物(1.76g,收率45%):
1H NMR(60/40外消旋物,CDCl3)δ1.47(s,9H),2.49-2.55(m,1H),2.73-2.83(m,1H),3.80(s,1.8H),3.81(s,1.2H),3.96(s,3H),4.03-4.09(m,1H),4.54(t,J=8.0Hz,0.6H),4.66(t,J=7.8Hz),4.96-5.06(m,1H),5.97(br s,0.6H),6.04(br s,0.4H),7.33(dd,J=6.1,2.7Hz,1H),7.46-7.51(m,4H),7.91(d,J=9.2Hz,1H),8.49(t,J=8.5Hz,2H);13C NMR(外消旋物,CDCl3)δ21.7,22.0,28.3,28.4,35.8,36.8,52.3,52.4,52.6,55.8,55.9,57.9,58.3,74.5,74.9,80.6,101.2,101.3,115.7,125.8,126.0,128.1,128.5,129.7,130.2,137.9,147.8,153.8,157.7,158.0,158.0,164.8,173.1,173.3;MS m/z(MH+)480.
方案2
步骤3:
将实施例45、步骤2的产物(760.0mg,1.59mmol)溶于50%TFA的DCM溶液中并在室温下搅拌2h。浓缩溶剂,将所得的棕色粘稠油状物真空干燥过夜。产物直接用于下一步反应。
步骤4:
向实施例45、步骤3的棕色粘稠油状产物(963mg,1.59mmol)和DIPEA(1.23g,9.54mmol)的DCM(11mL)溶液中加入N-BOC L-tBuGly(440mg,1.90mmol)、HBTU(902mg,2.38mmol)和HOBt(364mg,2.38mmol)。在室温下搅拌14h后,浓缩溶剂和过量的DIPEA,将所得的棕色粘稠油状物经快速柱层析纯化(SiO2,4∶1己烷∶乙酸乙酯),得到白色固体物(0.922mg,两步收率98%):
1HNMR(CDCl3/MeOD)δ0.94(s,9H),1.15(s,9H),2.38-2.42(m,1H),2.60-2.73(m,1H),3.61(s,3H),3.83(s,3H),4.08-4.17(m,2H),4.25(d,J=11.5Hz,1H),4.69(t,J=8.0Hz,1H),5.99(br s,1H),7.13(s,1H),7.38(s,5H),7.80(d,J=9.0Hz,1H),8.32(d,J=5.5Hz,1H);13C NMR(CDCl3/MeOD)δ29.6,31.4,31.6,33.04,38.2,39.0,55.8,56.9,59.2,61.5,62.1,78.3,83.1,105.0,119.0,129.4,131.5,131.9,132.6,133.8,141.2,151.0,161.4,161.6,168.2,175.2,175.7;MS m/z(MH+)593.
步骤5:
向实施例45、步骤4的产物(409mg,0.69mmol)的THF(10mL)溶液中加入1N NaOH(2mL)。在室温下搅拌19h后,用浓盐酸将反应物酸化至约pH约为5,用DCM萃取(3×50mL)。合并的有机层经硫酸镁干燥并浓缩,得到黄色固体产物(370mg,收率92%),真空干燥后,该产物直接用于下一步反应中:
1H NMR(CDCl3)δ1.05(s,9H),1.25(s,9H),2.76-2.83(m,2H),3.94(s,3H),4.23-4.27(m,2H),4.41(d,J=11.6Hz,1H),4.92(t,J=7.6Hz,1H),5.20(d,J=8.9Hz,1H),6.08(br s,1H),7.31(s,1H),7.46-7.50(m,5H),7.93(d,J=9.15Hz,1H),8.51(d,J=7.3Hz,2H);MS m/z(MH+)579.
方案3
步骤6:
向N-Boc-乙烯基环丙烷甲酸(1R,2S/1S,2R 1∶1混合物)(1.01g,4.46mmol)在THF(20mL)和DMSO(2mL)中的溶液内加入CDI(1.08g,6.69mmol)和DMAP(817mg,6.69mmol)。在70℃下搅拌1h后,将反应混合物冷却至室温并用异丙基磺酰胺(1.1g,8.92mmol)和DBU(1.36g,8.92mmol)处理。反应混合物在室温下搅拌16h,浓缩并经快速柱层析纯化(SiO2,5%甲醇/DCM),得到棕色粘稠油状物(1.4g,98%收率):
1H NMR(甲醇-d4)δ1.25(m,1H),1.33(d,J=6.7Hz,3H),1.36(d,J=6.7Hz,3H),1.45(s,9H),1.84(dd,J=7.6,5.2Hz,1H),2.16(d,J=7.6Hz,1H),3.58(br s,1H),5.08(d,J=11.6Hz,1H),5.27(d,J=15.6Hz,1H),5.58-5.66(m,1H);MS m/z(MH+)332.
步骤7:
将实施例45、步骤6的产物(113mg,0.34mmol)用50%三氟乙酸的DCM(10mL)溶液处理,并在室温下搅拌1.4h。真空除去溶剂和过量的三氟乙酸。将所得的棕色粘稠油状物真空干燥(1.3g,收率99%),未经进一步纯化直接使用:
1H NMR(DMSO-d6)δ1.24(d,J=6.7Hz,3H),1.26(d,J=6.7Hz,3H),1.54(dd,J=9.6,6.6Hz,1H),1.99(t,J=6.9Hz,1H),2.24(d,J=8.5Hz,1H),3.58-3.63(m,1H),5.18(d,J=10.4Hz,1H),5.33(d,J=17.1Hz,1H),5.61-5.69(m,1H),8.83(br s,3H);13C NMR(DMSO-d6)δ15.2,15.9,16.5,29.9,41.6,52.1,116.0,118.9,132.0,158.2,167.3;MS m/z(MH+)233.
步骤8:
向实施例45、步骤5的产物(117mg,0.338mmol)和DIPEA(174mg,1.35mmol)在DCM(5mL)中的混合物内加入HBTU(128mg,0.338mmol)、HOBt(52mg,0.338mmol)和实施例45步骤7的产物(130mg,0.225mmol)。在室温下搅拌16h后,将该混合物浓缩并将所得的棕色粘稠油状物经快速柱层析纯化(SiO2,1∶3己烷∶乙酸乙酯,接着用95∶5 DCM∶甲醇),得到类白色固体产物(150mg,收率84%)。终产物化合物45为各种异构体的混合物;不同之处在于分子的P1乙烯基环丙基部分(1R,2S/1S,2R 1∶1混合物):
1H NMR(甲醇-d4)δ0.92(br s,2H),1.03(s,9H),1.17(s,9H),1.27-1.38(m,9H),1.42-1.46(m,1H),1.83(dd,J=8.1,5.3Hz,0.4H),1.90(dd,J=7.9,5.5Hz,0.6H),2.24-2.31(m,1H),2.37-2.45(m,1H),2.67-2.75(m,1H),3.73-3.79(m,1H),3.90(s,3H),4.21(dd,J=9.3,6.0Hz,2H),4.48(d,J=11.3Hz,1H),4.61(q,J=8.9Hz,1H),5.14(t,J=9.0Hz,1H),5.33(t,J=17.9Hz,1H),5.70-5.76(m,1H),6.06(d,J=11.9Hz,1H),6.61(d,J=8.9Hz,1H),7.34(d,J=2.8Hz,1H),7.49(br s,5H),7.87(d,J=8.9Hz,1H),8.46(d,J=4.3Hz,2H);13C NMR(甲醇-d4)δ15.7,16.1,16.5,16.8,23.9,27.1,28.6,35.8,36.0,36.2,36.3,36.4,42.6,42.8,54.7,54.8,55.5,56.4,61.1,61.2,80.5,102.9,117.0,118.8,118.9,126.8,129.4,129.6,130.2,131.5,134.4,139.2,148.8,158.0,159.3,159.8,166.3,171.1,175.1,184.3;MS m/z(MH+)793.
实施例46:化合物46的制备
P1为(1R,2S)和(1S,2R)的1∶1混合物
化合物46
按照实施例45的步骤1至5和步骤8的方法制备化合物46,不同之处在于进行如下修改:
步骤1:
修改:使用2-溴-4,5-二甲氧基苯甲酸和环丙基胍盐酸盐作为原料。
产物:
数据:
1H NMR(DMSO-d6)δ0.97-1.01(m,2H),1.03-1.06(m,2H),1.90-1.94(m,1H),3.84(s,3H),3.87(s,3H),6.93(s,1H),7.37(s,3H),12.28(s,1H);13C NMR(DMSO-d6)δ9.03,13.17,55.47,55.73,104.81,107.27,113.26,145.16,147.48,154.44,157.21,160.89;MS m/z(MH+)247.
步骤2:
修改:使用实施例46、步骤1的产物作为原料代替实施例45、步骤1的产物。
产物:
数据:
1H NMR(CDCl3)δ1.00-1.04(m,2H),1.07-1.11(m,2H),1.43(s,5.4H),1.46(s,3.6H),2.17-2.21(m,1H),2.37-2.43(m,1H),2.62-2.69(m,1H),3.75(s,1.8H),3.78(s,1.2H),3.92(d,J=2.8Hz,1H),4.00(s,3.6H),4.01(s,2.4H),4.48(t,J=8.0Hz,0.6H),4.59(t,J=7.6Hz,0.4H),5.7(br s,0.6H),5.74(br s,0.4H),7.18(s,1H),7.20(s,1H);13C NMR(CDCl3)δ9.6,9.7,18.1,28.3,28.4,35.8,36.7,52.2,52.4,56.3,57.8,58.2,74.0,74.5,80.5,80.6,101.0,101.1,106.3,108.6,148.8,149.1,153.8,155.4,164.4,165.9,172.9,173.2;LC-MS m/z(MH+)474.
步骤3和4:
使用实施例46、步骤2的产物作为原料代替实施例45、步骤2的产物。
产物:
数据:
1H NMR(甲醇-d4)δ1.04(s,9H),1.08-1.21(m,4H),1.14(s,9H),2.17-2.21(m,1H),2.39-2.41(m,1H),2.74-2.77(m,1H),3.77(s,3H),3.92(s,3H),3.98(m,3H),4.09(dd,J=11.4,3.8Hz,1H),4.17(d,J=8.9Hz,1H),4.42(d,J=11.3Hz,1H),4.76(t,J=8.2Hz,1H),5.81(br s,1H),6.43(d,J=8.6Hz,1H),7.14(d,J=6.1Hz,1H),7.27(d,J=5.8Hz,1H);13C NMR(甲醇-d4)δ10.0,10.3,18.6,26.9,28.5,28.8,35.8,36.1,38.9,52.8,54.9,56.7,59.6,60.5,76.6,80.4,102.7,106.2,109.9,149.8,150.7,157.6,166.0,167.3,173.5,173.6;MS m/z(MH+)587.
步骤5:
使用实施例46、步骤4的产物作为原料代替实施例45、步骤4的产物。
产物:
数据:
1H NMR(甲醇-d4)δ1.03(s,9H),1.13(s,9H),1.20-1.23(m,4H),2.15-2.19(m,1H),2.40-2.45(m,1H),2.70-2.76(m,1H),3.90(s,3H),3.96(s,3H),4.08(dd,J=11.4,3.8Hz,1H),4.17(d,J=5.8Hz,1H),4.37(d,J=11.3Hz,1H),4.71(t,J=8.1Hz,1H),5.77(br s,1H),7.09(s,1H),7.20(s,1H);13C NMR(甲醇-d4)δ10.2,10.5,18.6,26.9,28.5,28.8,36.0,36.3,54.9,56.8,59.7,60.4,76.8,80.4,102.6,105.9,109.9,126.9,127.9,149.3,150.8,157.65,157.8,166.1,167.3,173.3,175.1;MS m/z(MH+)573.
步骤8:
使用实施例46、步骤5的产物作为原料代替实施例45、步骤5的产物。终产物化合物46为各种异构体的混合物;不同之处在于分子的P1乙烯基环丙基部分(1R,2S/1S,2R 1∶1混合物)。
产物:
P1为(1R,2S)和(1S,2R)的1∶1混合物
化合物46
数据:
1H NMR(甲醇-d4)δ1.03(s,9H),1.05-1.09(m,4H),1.16(s,4.5H),1.17(s,4.5H),1.19-1.22(m,1H),1.31(d,J=6.7Hz,2H),1.33-1.38(m,7H),1.18-1.89(m,1H),2.15-2.20(m,2H),2.35-2.44(m,1H),3.23(q,J=7.4Hz,1H),3.70-3.75(m,1H),3.91(s,3H),3.98(s,3H),4.08-4.13(m,2H),4.16(dd,J=8.9,3.1Hz,1H),4.38(t,J=13.1Hz,1H),4.58-4.62(m,1H),4.06(m,1H),5.29(t,J=15.2Hz,1H),5.83(br s,1H),7.15(s,1H),7.27(d,J=4.3Hz,1H);MS m/z(MH+)787.
实施例47:化合物47的制备
化合物47
按照制备实施例45的化合物45的类似步骤制备化合物47,代替原料邻-溴苯甲酸的是N-(1R-氨基-2S-乙烯基-环丙烷羰基)环丙烷磺酰胺盐酸盐。化合物47:MH+=761。
实施例48:化合物48的制备
化合物48
按照实施例45的步骤1至5和步骤8的方法制备化合物48,不同之处在于进行如下修改:
步骤1:
修改:使用盐酸乙脒和2-溴-5-甲氧基苯甲酸作为原料。
产物:
数据:
1H NMR(DMSO)δ2.31(s,3H),3.85(s,3H),7.36(d,J=6.2Hz,1H),7.37(s,1H),7.51(d,J=7.8Hz,1H),12.15(s,1H);13C NMR(DMSO)δ21.11,55.41,105.57,121.22,123.59,128.12,143.34,151.68,157.00,161.45;LC-MS m/e(MH+)191.
步骤2:
修改:使用实施例48、步骤1的产物作为原料代替实施例45、步骤1的产物。
产物:
数据:
1H NMR(CDCl3)δ1.43(s,5.4H),1.45(s,3.6H),2.38-2.45(m,1H),2.62-2.71(m,1H),2.66(s,1.8H),2.68(s,1.2H),3.77(1.8H),3.79(s,1.2H),3.92(s,3H),3.93-3.98(m,2H),4.49(t,J=8.0Hz,0.6H),4.61(t,J=7.8Hz,0.4H),5.82(t,J=2.1Hz,0.6H),5.89(t,J=2.3Hz,0.4H),7.26(dd,J=4.7,3.2Hz,1H),7.42(dd,J=6.3,2.8Hz,1H),7.75(d,J=9.15Hz,1H);13C NMR(CDCl3)δ26.1,28.3,28.4,35.8,36.7,52.2,52.2,52.4,52.5,55.755.8,57.9,58.2,74.1,74.7,80.6,101.0,101.2,114.9,125.6,125.9,128.6,147.3,153.8,154.5,157.6,157.6,161.2,164.6,173.0,173.3;LC-MS m/e(MH+)418.
步骤3和4:
修改:使用实施例48、步骤2的产物作为原料代替实施例45、步骤2的产物。
产物:
数据:1H NMR(MeOD)δ1.03(s,9H),1.07(s,9H),2.38-2.42(m,1H),2.68(s,3H),2.80(q,J=7.8Hz,1H),3.76(s,3H),3.89(s,3H),4.07(dd,J=11.9,3.4Hz,1H),4.13(br s,1H),4.55(d,J=12.2Hz,1H),4.78(t,J=8.7Hz,1H),5.93(s,1H),7.37(d,J=2.75Hz,1H),7.48-7.51(m,2H),7.70(d,J=5.7Hz,1H);13C NMR(MeOD)δ25.6,26.9,28.4,28.8,35.9,52.8,55.0,56.4,59.7,60.6,77.2,80.4,102.9,111.6,116.5,127.0,128.4,147.5,162.7,166.4,173.6;LC-MS m/e(MH+)531。
步骤5:
修改:使用实施例48、步骤4的产物作为原料代替实施例45、步骤4的产物。
产物:
数据:
1H NMR(MeOD)δ1.03(s,9H),1.08(s,9H),2.41-2.46(m,1H),2.68(s,3H),2.81(q,J=8.1Hz,1H),3.89(s,3H),4.07(dd,J=11.8,3.2Hz,1H),4.18(d,J=5.5Hz,1H),4.52(d,J=11.9Hz,1H),4.74(t,J=8.7Hz,1H),5.93(br s,1H),7.37(d,J=2.81Hz,1H),7.49(dd,J=9.2,2.4Hz,1H),7.71(d,J=9.2Hz,1H);13C NMR(MeOD)δ25.7,26.9,28.5,36.1,55.0,56.4,59.7,60.5,77.1,80.4,103.0,116.5,127.0,128.5,147.7,157.8,159.6,162.7,166.4,173.5,174.9;LC-MS m/e(MH+)517.
实施例48:化合物48的制备
化合物48
步骤8:
向实施例48、步骤5的产物(45.8mg,0.089mmol)、N-(1R-氨基-2S-乙烯基-环丙烷羰基)-环丙烷磺酰胺盐酸盐(21.0mg,0.089mmol)和DIEA(34.5mg,0.267mmol)的DCM(1mL)溶液中加入HATU(44.0mg,0.116mmol)。室温下搅拌过夜后,将反应混合物用5%碳酸氢钠水溶液(1mL)洗涤。水层用2×2mL DCM萃取。有机层用5%柠檬酸水溶液(1mL)和盐水洗涤,经硫酸镁干燥,浓缩并经反相制备型HPLC纯化。该纯化步骤脱去了分子的P3叔-亮氨酸部分的N-BOC保护基:
1H NMR(MeOD)δ1.07-1.12(m,2H)1.14(s,2H)1.14-1.16(m,2H)1.17(s,9H)1.20-1.30(m,3H)1.45(dd,J=9.46,5.49.Hz,1H)1.56(s,1H)1.92(dd,J=8.20,5.60Hz,1H)2.25-2.31(m,1H)2.39-2.45(m,1H)2.73(m,1H)2.76(s,3H)2.93-2.97(m,1H)3.94(s,1H)3.96(s,3H)4.07(s,1H)4.21(d,J=3.97Hz,0.4H)4.23(d,J=3.97Hz,0.6H)4.31(m,1H)4.73(dd,J=10.38,7.02Hz,1H)5.15(dd,J=10.38,1.52Hz,1H)5.32(dd,J=17.1,1.52Hz,1H),5.71-5.78(m,1H)6.11(t,J=3.51Hz,1H)7.46(d,J=2.75Hz,1H)7.67(d,J=3.06Hz,0.4H)7.69(d,J=3.05Hz,0.6H)7.82(s,0.6H)7.84(s,0.4H).
实施例49:化合物49的制备
化合物49
按照制备化合物48的相同方法制备化合物49,不同之处在于使用实施例46、步骤5的产物和N-(1R-氨基-2S-乙烯基-环丙烷羰基)-环丙烷磺酰胺盐酸盐作为原料。制备型HPLC纯化步骤脱去了分子的P3叔-亮氨酸部分的N-BOC保护基:
1H NMR(MeOD)δ1.09(m,2H)1.14(d,J=3.97Hz,2H)1.17(s,9H)1.25(m,3H)1.37(m,3H)1.44(dd,J=9.31,5.65Hz,2H)1.57(s,1H)1.92(dd,J=8.09,5.65Hz,1H)2.28(dd,J=17.70,8.55Hz,1H)2.32(m,1H)2.68(dd,J=14.19,7.78Hz,1H)2.95(m,1H)3.98(s,3H)4.06(s,3H)4.08(s,1H)4.22(d,J=2.75Hz,1H)4.70(dd,J=9.77,7.32Hz,1H)5.15(dd,J=10.38,1.53Hz,1H)5.32(dd,J=17.40,1.22Hz,1H)5.74(m,1H)6.04(m,1H)7.24(s,1H)7.37(s,1H)
实施例50:化合物50的制备
化合物50
按照制备化合物48的相同方法制备化合物50,不同之处在于使用实施例45、步骤5的产物和N-(1R-氨基-2S-乙烯基-环丙烷羰基)-环丙烷磺酰胺盐酸盐作为原料。制备型HPLC纯化步骤脱去了分子的P3叔-亮氨酸部分的N-BOC保护基:
1H NMR(MeOD)δ1.10(m,2H)1.14(s,1H)1.15(d,J=3.36Hz,1H)1.17(d,J=3.05Hz,9H)1.22(m,1H)1.27(m,2H)1.46(dd,J=9.46,5.49Hz,1H)1.56(s,1H)1.93(dd,J=8.24,5.49Hz,1H)2.29(q,J=8.55Hz,1H)2.48(m,1H)2.78(dd,J=13.89,8.09Hz,1H)2.97(m,1H)3.96(s,2H)4.07(s,1H)4.32(d,J=2.14Hz,2H)4.76(d,J=7.02Hz,1H)4.78(m,1H)4.86(d,J=3.05Hz,1H)5.32(dd,J=17.09,1.22Hz,1H)5.75(m,1H)6.24(d,J=2.44Hz,1H)7.45(d,J=2.75Hz,1H)7.52(m,3H)7.61(dd,J=9.16,2.75Hz,1H)7.96(d,J=9.16Hz,1H).
实施例51:化合物51的制备
P1为(1R,2S)和(1S,2R)的1∶1混合物
化合物51
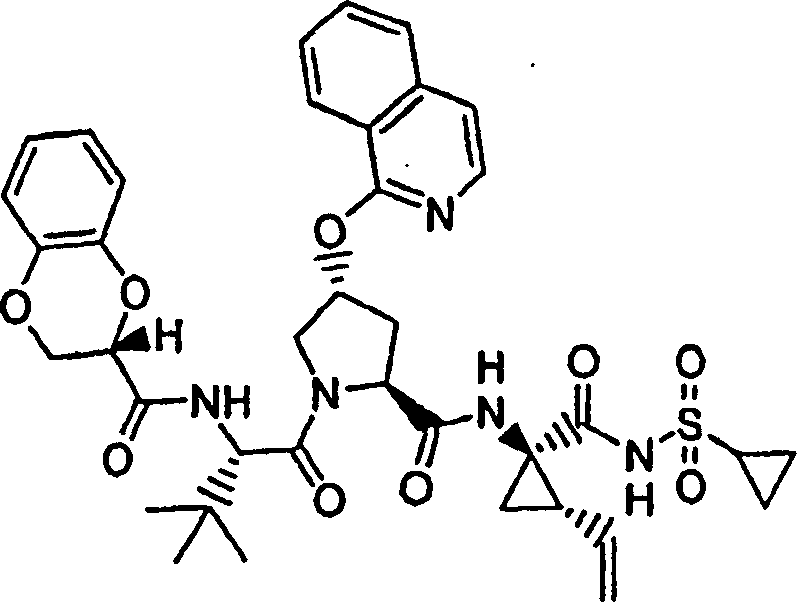
按照实施例45的步骤1至5和步骤8的方法制备化合物51,不同之处在于进行如下修改:
步骤1:
修改:使用2-溴-4,5-二甲氧基苯甲酸和三氟脒作为原料。
产物:
数据:
1H NMR(DMSO)δ3.92(s,3H),3.94(s,3H),7.33(s,1H),7.50(s,1H),13.40(br s,1H);13C NMR(DMSO)δ55.8,56.1,104.9,108.7,150.2,155.0;LC-MS m/e(MH+)275.
步骤2:
修改:使用实施例51、步骤1的产物作为原料代替实施例45、步骤1的产物。
产物:
数据:
1H NMR(CDCl3)δ1.42(s,3.6H),1.44(s,5.4H),2.42-2.49(m,1H),2.67-2.73(m,1H),3.37(s,1.2H),3.78(s,1.8H),3.97(t,J=6.5Hz,1H),4.02(s,2.4H),4.04(s,3.6H),4.48(t,J=7.9Hz,0.6H),4.60(t,J=7.7Hz,0.4H),5.86(br s,0.6H),5.90(br s,0.4H),7.27-7.29(m,1H),7.38-7.44(m,1H);13C NMR(CDCl3)δ8.2,28.3,35.7,36.7,52.1,52.2,52.4,56.5,57.8,58.2,75.5,76.0,80.7,100.8,107.6,111.0,119.7,148.2,150.2,151.4,153.8,154.5,156.4,165.1,172.7,173.0;LC-MSm/e(MH+)502.
步骤3和4:
修改:使用实施例51、步骤2的产物作为原料代替实施例45、步骤2的产物。
产物:
数据:
1H NMR(MeOD)δ1.03(s,9H),1.08(s,9H),2.41-2.45(m,1H),2.80-2.84(m,1H),3.76(s,3H),3.96(s,3H),4.00(s,3H),4.10-4.14(m,2H),4.52(d,J=11.6Hz,1H),4.80(t,J=8.7Hz,1H),5.92(br s,1H),7.35(br s,2H);13C NMR(MeOD)□26.9,28.4,28.8,35.7,36.0,52.8,54.8,56.9,59.6,60.7,77.9,80.3,102.2,107.9,112.4,120.3,149.3,153.2,157.8,158.3,173.5;LC-MS m/e(MH+)615.
步骤5:
修改:使用实施例51、步骤4的产物作为原料代替实施例45、步骤4的产物。
产物:
数据:
1H NMR(MeOD)δ1.03(s,9H),1.09(s,9H),2.44-2.49(m,1H),2.80-2.84(m,1H),3.97(s,3H),4.01(s,3H),4.10-4.24(m,3H),4.50(d,J=11.9Hz,1H),4.76(t,J=7.9Hz,1H),5.93(br s,1H),7.36(br s,2H);13C NMR(MeOD)δ26.9,28.4,28.8,36.0,36.1,54.8,56.9,57.0,60.6,77.9,80.3,102.3,108.0,112.5,120.3,149.3,151.3,153.2,158.2,158.3,166.7,173.5;LC-MS m/e(MH+)601.
步骤8:
修改:使用实施例51、步骤5的产物作为原料代替实施例45、步骤5的产物。终产物化合物51为各种异构体的混合物;不同之处在于分子的P1乙烯基环丙基部分(1R,2S/1S,2R 1∶1混合物)。
产物:
P1为(1R,2S)和(1S,2R)的1∶1混合物
化合物51
数据:
1H NMR(DMSO)δ0.23(s,4.5H),0.23(s,4.5H),0.35(s,4.5H),0.36(s,4.5H),0.45-0.59(m,8H),0.63-0.66(m,1H),1.04(dd,J=8.2,5.2Hz,1H),1.10(dd,J=8.2,5.5Hz,1H),1.47-1.53(m,1H),1.58-1.61(m,1H),1.87-1.90(m,1H),2.95-3.01(m,1H),3.17(s,1.5H),3.18(s,1.5H),3.22(s,3H),3.37(br s,2H),3.68(q,J=5.9Hz,1H),3.82(q,J=8.6Hz,1H),4.33-4.37(m,1H),4.54(t,J=16.5Hz,1H),4.93(q,J=8.9Hz,1H),5.17(d,J=15.9Hz,1H),6.53(s,1H),6.58(s,1H);13CNMR(DMSO)δ12.8,13.2,13.7,13.9,19.5,20.6,21.1,24.3,25.6,32.9,33.1,33.4,33.6,36.1,39.7,39.9,51.8,51.9,52.4,54.0,54.2,57.7,57.7,58.1,58.3,75.1,75.3,77.5,84.1,99.2,105.2,107.9,109.5,116.0,116.1,118.7,123.9,127.4,131.5,146.5,148.6,150.3,155.1,155.5,163.7,164.7,168.2,168.3,170.7,172.2;LC-MS m/e(MH+)815.
实施例52:化合物52的制备
化合物52
按照制备化合物48的相同方法制备化合物52,不同之处在于使用实施例51、步骤5的产物和N-(1R-氨基-2S-乙烯基-环丙烷羰基)-环丙烷磺酰胺盐酸盐作为原料。制备型HPLC纯化步骤脱去了分子的P3叔-亮氨酸部分的N-BOC保护基:
1H NMR(MeOD)δ1.11(m,3H)1.17(s,9H)1.25(m,3H)1.46(dd,J=9.46,5.49Hz,1H)1.92(dd,J=8.24,5.49Hz,1H)2.28(q,J=8.95Hz,1H)2.42(m,1H)2.72(dd,J=14.19,7.17Hz,1H)2.96(m,1H)4.01(s,3H)4.04(m,5H)4.24(m,2H)4.73(dd,J=10.22,7.17Hz,1H)5.15(dd,J=10.53,1.37Hz,1H)5.32(d,J=17.09Hz,1H)5.75(m,1H)6.07(s,1H)7.41(s,1H)7.47(s,1H).
实施例53:化合物53的制备
化合物53
方案1
步骤1:
向(1R,2S/1S,2R 1∶1的混合物)N-(1-氨基-2-乙烯基-环丙烷羰基)-环丙烷磺酰胺三氟乙酸盐(626mg,1.82mmol)的DCM(17mL)溶液中加入DIEA(555mg,4.29mmol)的DCM(17mL)溶液、HATU(754mg,1.98mmol)和(2S,4R)Fmoc-4-氨基-1-boc-吡咯烷-2-甲酸(747mg,1.65mmol)。在室温下搅拌24h后,将混合物用1N HCl(10mL)、5%碳酸氢钠水溶液(4mL)洗涤。各水层用DCM(25mL)萃取。合并的DCM经硫酸镁干燥并浓缩。所得的棕色粘稠油状物经快速柱层析纯化(SiO2,95∶5DCM∶甲醇),得到黄色固体物(822mg,收率75%):
1H NMR(DMSO-d6)δ1.04-1.09(m,3H),1.15-1.27(m,4H),1.38-1.44(m,7H),1.47(s,9H),1.84(dd,J=8.2,5.2Hz,1H),2.01-2.30(m,4H),2.90-2.98(m,1H),3.64-3.71(m,1H),4.16-4.22(m,4H),4.39(bs,2H),5.13(dd,J=10.7,0.9Hz,1H),3.31(d,J=17.1Hz,1H),5.72-5.79(m,1H),7.31(t,J=7.3Hz,3H),7.38(t,J=7.5Hz,3H),7.64(d,J=7.02Hz,3H),7.79(d,J=7.63Hz,3H);LC-MSm/e(Na+MH+)687.
步骤2:
将实施例53、步骤1的产物(500mg,0.752mmol)用50%TFA的DCM(10mL)溶液处理。在室温下搅拌0.5h后,将所得的棕色反应混合物浓缩,得到棕色固体物(489mg,收率84%):
1H NMR(DMSO-d6)δ1.03-1.19(m,4H),1.24-1.26(m,1H),1.35(dd,J=9.5,5.5Hz,1H),1.91-1.96(m,1H),2.22-2.30(m,1H),2.40(bs,1H),2.93-2.98(m,1H),3.60(bs,1H),4.21(t,J=5.6Hz,2H),4.47(bs,3H),5.17(d,J=9.2Hz,1H),5.32(d,J=17.1Hz,1H),5.64-5.67(m,1H),7.31(t,J=7.3Hz,3H),7.39(t,J=7.5Hz,3H),7.63(d,J=7.3Hz,2H),7.80(d,J=7.3Hz,2H);(LC-MS m/e(MH+)565.
方案2
步骤3:
向实施例53、步骤2的产物(260mg,0.383mmol)的DCM(4mL)溶液中加入DIPEA(218mg,1.69mmol)、HATU(165mg,0.422mmol)和N-BOC L-tBuGly(100mg,0.422mmol)。在室温下搅拌16h后,将反应混合物用H2O(3mL)稀释,用1N HCl酸化至pH=1。水层用DCM(2×15mL)萃取。将合并的有机层用5%碳酸氢钠(3mL)和盐水(5mL)洗涤,经硫酸镁干燥并浓缩。所得的棕色粘稠油状物经快速柱层析纯化(SiO2,95∶5 DCM∶甲醇),得到棕色泡沫状固体物(281mg,94%收率):
1H NMR(DMSO-d6)δ0.96-1.08(m,4H),1.05(s,9H),1.15-1.26(m,2H),1.35-1.38(m,5H),1.42(s,9H),1.85(dd,J=9.5,5.5Hz,1H),2.07(bs,1H),2.22(q,J=8.7Hz,1H),2.92-2.95(m,1H),3.90(bs,1H),4.20(d,J=6.4Hz,3H),4.29-4.39(m,5H),5.13(d,J=10.7,1H),5.31(dd,J=18.0,5.8Hz,1H),5.70-5.77(m,1H),7.30(t,J=7.3Hz,3H),7.39(t,J=7.3Hz,4H),7.63(dd,J=6.7,2.8Hz,3H),8.80(d,J=7.63Hz,3H);LC-MS m/z(MH+)678.
步骤4:
将实施例53、步骤3的产物用10%哌啶的DMF(3.3mL)溶液处理。在室温下搅拌14h后,除去溶剂并将所得的棕色粘稠油状物经快速柱层析纯化(SiO2,95∶5 DCM∶甲醇)来分离出高纯度的Rf 1R,2S P1非对映异构体,为浅黄色固体物(31mg)。其它的异构体以混合物形式分离出,不使用:LC-MS m/z(MH+)556。
方案3
步骤5和6:
向实施例53、步骤4的产物的DMF(2mL)溶液中加入聚乙烯基吡啶(13mg)和Fmoc-异硫氰酸酯。在室温下搅拌14h后,将反应混合物用哌啶(172mg,2.02mmol)处理。将反应物再于室温下搅拌6h,随后浓缩并真空干燥过夜。再次将粗品剩余物溶于DMF(2mL)中,并用2-溴苯乙酮处理,在室温下再搅拌14h。将反应混合物浓缩并将所得的剩余物经快速柱层析纯化(SiO2,95∶5 DCM∶甲醇),得到化合物53,为浅黄色固体物(21.9mg,收率50%):
1H NMR(DMSO-d6)δ0.87-0.92(m,1H),1.05(bs,13H),1.16-1.25(m,4H),1.34-1.38(m,2H),1.42(s,9H),1.87(t,J=6.6Hz,1H),2.22-2.25(m,2H),2.48(t,J=10.7Hz,1H),2.93(bs,1H),3.04(q,J=7.3Hz,1H)3.30-3.31(m,2H),3.43-3.49(m,1H),4.01(d,J=10.4Hz,1H),4.07-4.12(m,1H),4.27(t,J=9.5Hz,1H),4.44(t,J=7.0Hz,1H),4.58(bs,1H),5.11(d,J=10.1Hz,1H),5.30(dd,J=16.8,9.6Hz,1H),5.73-5.78(m,1H),6.69(d,J=8.2Hz,1H),6.86(s,1H),7.25(t,J=7.3Hz,1H),7.35(t,J=7.63Hz,2H),7.82(d,J=8.2Hz,2H);LC-MS m/z(MH+)715.
用于化合物55至155的反相制备型-HPLC条件如下:
Waters Xterra Prep MS C18柱,5mm(这表示粒径为5μm),30mm×100mm
溶剂A:10%甲醇,90%H2O,10mM乙酸铵
溶剂B:90%甲醇,10%H2O,10mM乙酸铵
流速50mL/min
梯度:10min内0%B至100%B,保持100%B 4min。
实施例55:化合物55的制备
方案1
步骤1:
向化合物11(1.5g,2.10mmol)的DCE(25mL)溶液中加入TFA(25mL)。在室温下搅拌15min后,将反应混合物浓缩。所得的红色粘稠油状物再次溶于DCE(50mL)中并再次浓缩。随后再将其溶于DCM(15mL)中,用1N HCl/乙醚(25mL)处理。将所得的悬浮液冷却至0℃下,抽滤,用乙醚洗涤并在真空烘箱中干燥,得到步骤1的产物,为白色固体物(1.4g,收率97%):
1H NMR(CD3OD,500MHz)δ1.07-11.2(m,3H)1.14(t,J=4.12Hz,1H)1.17(s,9H)1.22(dd,J=10.53,4.43Hz,1H)1.21-1.27(m,2H)1.42(dd,J=9.61,5.34Hz,1H)1.91(dd,J=7.93,5.49Hz,1H)2.27(q,J=8.85Hz,1H)2.32-2.38(m,1H)2.70(dd,J=13.43,6.71Hz,1H),2.93-2.98(m,1H)3.96(s,3H)4.09(s,1H)4.14(dd,J=12.21,3.66Hz,1H)4.32-4.35(m,1H)4.69(dd,J=10.53,6.87Hz,1H)5.14(dd,J=10.38,1.53Hz,1H)5.31(dd,J=17.40,1.22Hz,1H)5.70-5.77(m,1H)5.90(t,J=3.51Hz,1H)7.24-7.27(m,1H)7.29(d,J=4.27Hz,1H)7.39(t,J=4.88Hz,1H)7.90(d,J=6.10Hz,1H)8.19(m,1H)9.22(s,1H).
步骤2:
向实施例55、步骤1的产物(70.0mg,0.108mmol)和DIEA(41.8mg,0.323mmol)在DCM(2mL)中的溶液混合物内加入乙酸酐(33.0mg,0.323mmol)。在室温下搅拌14h后,除去溶剂,产物经反相制备型-HPLC纯化,得到化合物55(39.1mg,收率14%):
1HNMR(CD3OD,500MHz)δ1.00-1.03(m,1H),1.06(s,9H),1.07-1.10(m,1H),1.21-1.28(m,2H),1.43(dd,J=9.46,5.19Hz,1H),1.88(dd,J=8.55,5.49Hz,1H),2.23(q,J=8.85Hz,1H),2.27-2.32(m,1H),2.59(dd,J=13.73,7.02Hz,1H),2.92-2.97(m,1H),3.93(s,3H),4.12(dd,J=11.90,3.97Hz,1H),4.35(d,J=11.60Hz,1H),4.51(dd,J=10.38,7.02Hz,1H),4.61(dd,J=5.80,3.05Hz,1H),4.80(d,J=4.27Hz,1H),4.88(d,J=3.96Hz,1H),5.12(dd,J=10.38,1.83Hz,1H),5.29(dd,J=17.24,1.37Hz,1H),5.73-5.78(m,1H),5.84(t,J=3.66Hz,1H),7.15(dd,J=8.85,2.44Hz,1H),7.19(d,J=2.44Hz,1H),7.25(d,J=6.10Hz,1H),7.88(d,J=6.10Hz,1H),8.06(d,J=9.16Hz,1H);
LC-MS(保留时间:1.49min),MS m/z 656(MH+)。
实施例56:化合物56的制备
化合物56
按照化合物55的相同方法制备化合物56,但作如下修改:
修改:使用环戊烷甲酰氯作为原料,得到化合物56(18.0mg,24%收率):
1H NMR(CD3OD,500MHz)δ1.00-1.03(m,1H),1.05(s,9H),1.06-1.10(m,2H),1.24-1.27(m,2H),1.25-1.61(m,9H),1.80-1.83(m,1H),1.88(dd,J=8.24,5.49Hz,1H),2.22-2.31(m,2H),2.58-2.65(m,2H),2.93-2.98(m,1H),3.92(s,3H),4.10(dd,J=11.90,3.66Hz,1H),4.35(d,J=11.91Hz,1H),4.52(dd,J=10.38,7.02Hz,1H),4.65(d,J=9.46Hz,1H),4.80(d,J=5.49Hz,1H),4.88(d,J=5.19Hz,1H),5.13(dd,J=10.37,1.83Hz,1H),5.30(dd,J=16.80,1.22Hz,1H),5.73-5.78(m,1H),5.84(t,J=4.27Hz,1H),7.11(dd,J=9.16,2.44Hz,1H),7.19(d,J=2.44Hz,1H),7.25(d,J=5.80Hz,1H),7.88(d,J=6.10Hz,1H),8.05(d,J=9.16Hz,1H));
LC-MS(保留时间:1.71min),MS m/z 710(MH+)。
实施例57:化合物57的制备
化合物57
按照化合物55的相同方法制备化合物57,但作如下修改:
修改:使用2-乙基丁酰氯作为原料,得到化合物57(20.7mg,27%收率):
1H NMR(CD3OD,500MHz)δ0.66(t,J=7.32Hz,3H),0.85(t,J=7.32Hz,3H),1.02-1.05(m,1H),1.07(s,9H),1.10-1.12(m,1H),1.24-1.33(m,4H),1.36-1.39(m,1H),1.43(dd,J=9.46,5.19Hz,1H),1.48-1.51(m,1H),1.88(dd,J=8.24,5.19Hz,1H),2.12-2.14(m,1H),2.22(q,J=8.85Hz,1H),2.26-2.30(m,1H),2.59(dd,J=13.73,6.71Hz,1H),2.94-2.97(m,1H),3.92(s,3H),4.11(dd,J=11.90,3.66Hz,1H),4.40(d,J=11.90Hz,1H),4.50(dd,J=10.68,7.02Hz,1H),4.75(d,J=9.46Hz,1H),4.81(d,J=9.16Hz,1H),4.89(d,J=9.16Hz,1H),5.12(dd,J=10.38,1.53Hz,1H),5.29(dd,J=17.09,1.22Hz,1H),5.72-5.79(m,1H),5.85(t,J=3.66Hz,1H),7.08(dd,J=9.16,2.44Hz,1H),7.19(d,J=2.44Hz,1H),7.25(d,J=5.80Hz,1H),7.88(d,J=6.10Hz,1H),7.98(d,J=9.16Hz,1H),8.02(d,J=9.16Hz,1H);
LC-MS(保留时间:1.73min),MS m/z 712(MH+)。
实施例58:化合物58的制备
向实施例55、步骤1的产物(70.0mg,0.108mmol)、DIEA(41.8mg,0.323mmol)和环丙烷乙酸(16.2mg,0.162mmol)在DCM(2mL)中的溶液混合物内加入HATU(61.6mg,0.162mmol)。室温下搅拌反应混合物过夜后,用5%碳酸氢钠水溶液(1mL)洗涤。水层用2×2mLDCM萃取。将合并的有机层用5%柠檬酸水溶液(2mL)和盐水洗涤,经硫酸镁干燥并浓缩。产物经反相制备型-HPLC纯化,得到化合物58(21.9mg,29%收率):
1H NMR(CD3OD,500MHz)δ0.11-0.14(m,2H),0.43-0.47(m,2H),0.87-0.09(m,1H),1.01-1.04(m,1H),1.07(s,9H),1.09-1.12(m,1H),1.23-1.27(m,2H),1.45(dd,J=9.46,5.49Hz,1H),1.88(dd,J=8.24,5.49Hz,1H),2.03(d,J=7.32Hz,2H),2.23(q,J=8.75Hz,1H),2.27-2.31(m,1H),2.59(dd,J=13.73,7.02Hz,1H),2.92-2.96(m,1H),3.93(m,3H),4.13(dd,J=11.90,3.97Hz,1H),4.34(d,J=11.90Hz,1H),4.53(dd,J=10.38,7.02Hz,1H),4.66(d,J=9.46Hz,1H),4.81(d,J=6.10Hz,1H),4.89(d,J=6.10Hz,1H),5.12(dd,J=10.37,1.52Hz,1H),5.30(dd,J=17.09,1.22Hz,1H),5.75-5.81(m,1H),5.86(s,1H),7.12(dd,J=9.16,2.44Hz,1H),7.19(d,J=2.44Hz,1H),7.81(d,J=9.46Hz,1H),7.89(d,J=5.80Hz,1H),8.06(d,J=9.16Hz,1H);
LC-MS(保留时间:1.63min),MS m/z 696(MH+)。
实施例59:化合物59的制备
化合物59
按照化合物58的相同方法制备化合物59,但作如下修改:
修改:使用甲氧基乙酸作为原料,得到化合物59(23.5mg,32%收率):
1H NMR(CD3OD,500MHz)δ10.99-1.04(m,2H),1.06(s,9H),1.09-1.12(m,1H),1.22-1.27(m,2H),1.45(dd,J=9.46,5.49Hz,1H),1.88(dd,J=8.24,5.49Hz,1H),2.22(q,J=8.85Hz,1H),2.29-2.32(m,1H),2.60(dd,J=13.89,6.87Hz,1H),2.92-2.97(m,1H),3.35(s,3H),3.70(d,J=15.26Hz,1H),3.84(d,J=15.26Hz,1H),3.93(s,3H),4.13(dd,J=11.90,3.97Hz,1H),4.32(d,J=11.60Hz,1H),4.54(dd,J=10.38,7.02Hz,1H),4.65(s,1H),4.81(d,J=7.32Hz,1H),4.89(d,J=7.32Hz,1H),5.12(d,J=10.38Hz,1H),5.30(d,J=16.79Hz,1H),5.74-5.81(m,1H),5.86(t,J=3.36Hz,1H),7.14(dd,J=9.00,2.59Hz,1H),7.19(d,J=2.44Hz,1H),7.26(d,J=6.10Hz,1H),7.89(d,J=6.10Hz,1H),8.04(d,J=9.16Hz,1H);
LC-MS(保留时间:1.54min),MS m/z 686(MH+)。
实施例60:化合物60的制备
化合物60
按照化合物58的相同方法制备化合物60,但作如下修改:
修改:使用(+)-甲氧基乙酸作为原料,得到化合物60(23.8mg,27%收率):
1H NMR(CD3OD,500MHz)δ0.78(d,J=7.02Hz,3H),0.83-0.88(m,2H),0.92(t,J=7.17Hz,6H),0.94-0.98(m,1H),1.00-1.03(m,2H),1.06(s,9H),1.07-1.11(m,1H),1.22-1.26(m,2H),1.31-1.36(m,2H),1.45(dd,J=9.46,5.49Hz,1H),1.63-1.68(m,2H),1.89(dd,J=8.09,5.34Hz,1H),2.01-2.04(m,1H),2.17-2.21(m,1H),2.24(q,J=9.00Hz,2H),2.28-2.33(m,1H),2.60(dd,J=13.73,7.02Hz,1H),2.93-2.98(m,1H),3.15-3.20(m,1H),3.77(d,J=15.26Hz,1H),3.87(d,J=15.26Hz,1H),3.93(s,3H),4.12(dd,J=11.90,3.66Hz,1H),4.32(d,J=11.90Hz,1H),4.56(dd,J=10.38,7.02Hz,1H),4.65(d,J=9.77Hz,1H),4.81(d,J=5.80Hz,1H),4.89(d,J=5.80Hz,1H),5.13(dd,J=10.22,1.68Hz,1H),5.30(dd,J=17.09,1.53Hz,1H),5.75-5.79(m,1H),5.85(t,J=3.66Hz,1H),7.14(dd,J=9.16,2.44Hz,1H),7.19(d,J=2.44Hz,1H),7.26(d,J=5.80Hz,1H),7.52(d,J=9.77Hz,1H),7.89(d,J=5.80Hz,1H),8.03(d,J=9.16Hz,1H);
LC-MS(保留时间:2.043min),MS m/z 810(MH+)。
实施例61:化合物61的制备
化合物61
按照化合物58的相同方法制备化合物61,但作如下修改:
修改:使用(-)-甲氧基乙酸作为原料,得到化合物61(26.4mg,30%收率):
1H NMR(CD3OD,500MHz)δ0.78(d,J=7.02Hz,3H),0.82-084(m,1H),0.88(dd,J=8.39,3.81Hz,1H),0.91(d,J=7.01Hz,3H),0.92(d,J=6.41Hz,3H),0.94-0.99(m,2H),1.00-1.03(m,2H),1.06(s,9H),1.08-1.10(m,1H),1.23-1.26(m,2H),1.30-1.37(m,2H),1.44(dd,J=9.61,5.34Hz,1H),1.62-1.68(m,2H),1.89(dd,J=8.24,5.49Hz,1H),1.98-2.02(m,1H),2.13-2.16(m,1H),2.24(q,J=8.85Hz,1H),2.28-2.32(m,1H),2.60(dd,J=13.73,7.02Hz,1H),2.94-2.98(m,1H),3.08-3.13(m,1H),3.63(d,J=15.56Hz,1H),3.93(S,3H),4.11(dd,J=12.05,3.81Hz,1H),4.32(d,J=11.90Hz,1H),4.56(dd,J=10.38,7.02Hz,1H),4.62(d,J=9.46Hz,1H),4.81(d,J=6.41Hz,1H),4.89(d,J=6.72Hz,1H),5.13(dd,J=10.38,1.83Hz,1H),5.30(dd,J=17.09,1.23Hz,1H),5.76-5.80(m,1H),5.85(t,J=3.51Hz,1H),7.14(dd,J=9.00,2.59Hz,1H),7.20(d,J=2.44Hz,1H),7.26(d,J=5.80Hz,1H),7.52(d,J=9.77Hz,1H),7.89(d,J=6.10Hz,1H),8.04(d,J=9.16Hz,1H);
LC-MS(保留时间:2.05min),MS m/z 810(MH+)。
实施例62:化合物62的制备
化合物62
按照化合物58的相同方法制备化合物62,但作如下修改:
修改:使用双环[1.1.1]戊烷-2-甲酸作为原料,得到化合物62(35.1,45%收率):
1H NMR(CD3OD,500MHz)δ1.03(d,J=5.49Hz,1H),1.06(s,9H),1.08-1.11(m,3H),1.23-1.29(m,3H),1.37(dd,J=7.02,3.36Hz,13H),1.46(dd,J=9.46,5.19Hz,1H),1.65(dd,J=9.77,2.14Hz,1H),1.69(d,J=2.14Hz,1H),1.72(dd,J=7.32,3.05Hz,1H),1.88(dd,J=8.09,5.34Hz,1H),2.12(dd,J=9.77,3.05Hz,1H),2.23(d,J=8.85Hz,1H),2.27-2.31(m,1H),2.60(t,J=6.87Hz,1H),2.63(d,J=1.83Hz,2H),2.68(d,J=7.63Hz,1H),2.93-2.96(m,1H),3.23(q,J=7.43Hz,2H),3.70-3.75(m,2H),3.93(s,3H),4.11(dd,J=11.90,3.66Hz,1H),4.35(d,J=11.90Hz,1H),4.53(dd,J=10.53,6.87Hz,1H),4.71(d,J=9.46Hz,1H),4.79(d,J=5.80Hz,2H),4.87(d,J=5.49Hz,2H),5.13(dd,J=10.38,1.83Hz,1H),5.30(dd,J=17.24,1.37Hz,1H),5.74-5.79(m,1H),5.86(t,J=3.20Hz,1H),7.12(dd,J=9.16,2.44Hz,1H),7.19(d,J=2.44Hz,1H),7.25(d,J=5.80Hz,1H),7.72(d,J=9.46Hz,1H),7.88(d,J=5.80Hz,1H),8.05(d,J=9.16Hz,1H);
LC-MS(保留时间:1.16min),MS m/z 708(MH+)。
实施例63:化合物63的制备
化合物63
按照化合物58的相同方法制备化合物63,但作如下修改:
修改:使用吡嗪-2-甲酸作为原料,得到化合物63(42.3,54%收率):
1H NMR(CD3OD,500MHz)δ1.00-1.04(m,3H),1.05-1.09(m,1H),1.10(s,9H),1.15(s,1H),1.18-1.22(m,2H),1.43(dd,J=9.46,5.49Hz,1H),1.88(dd,J=7.93,5.49Hz,1H),2.17(q,J=8.65Hz,1H),2.37-2.42(m,1H),2.64(dd,J=13.73,7.32Hz,1H),2.91-2.95(m,1H),3.88(s,3H),3.93(d,J=3.35Hz,1H),4.13(dd,J=11.90,3.36Hz,1H),4.46(d,J=11.90Hz,1H),4.61(dd,J=10.07,7.32Hz,1H),4.76(s,1H),4.80(d,J=7.63Hz,1H),4.88(d,J=7.93Hz,1H),5.09(d,J=10.38Hz,1H),5.27(d,J=17.09Hz,1H),5.77-5.83(m,1H),5.85(s,1H),6.85(dd,J=9.16,2.44Hz,1H),7.08(d,J=2.14Hz,1H),7.23(d,J=5.80Hz,1H),7.87(d,J=8.85Hz,1H),7.90(d,J=6.10Hz,1H),8.57(d,J=1.53Hz,1H),8.73(d,J=2.44Hz,1H),8.81(s,1H).
实施例64:化合物64的制备
向实施例55、步骤1的产物(70.0mg,0.108mmol)和DIEA(41.8mg,0.323mmol)在DCM(2mL)中的溶液混合物内加入氯甲酸苄酯(55.1mg,0.323mmol)。在室温下搅拌14h后,除去溶剂,产物经反相制备型-HPLC纯化,得到化合物64(26.9mg,31%收率):
1H NMR(CD3OD,500MHz)δ1.00(d,J=2.14Hz,1H),1.02(d,J=5.80Hz,1H),1.04(s,9H),1.08-1.14(m,1H),1.16(d,J=6.71Hz,1H),1.18-1.22(m,2H),1.43(dd,J=9.46,5.19Hz,1H),1.87(dd,J=8.09,5.34Hz,1H),2.17-2.22(m,1H),2.30-2.35(m,1H),2.62(dd,J=13.73,7.02Hz,1H),2.90-2.95(m,1H),3.88(s,3H),4.08(dd,J=11.90,3.66Hz,1H),4.31(s,1H),4.43(d,J=11.60Hz,1H),4.55(dd,J=10.07,7.32Hz,1H),4.74(d,J=12.21Hz,1H),4.81(d,J=6.10Hz,1H),4.89(d,J=5.79Hz,1H),5.10(d,J=9.16Hz,1H),5.16(s,1H),5.28(d,J=17.09Hz,1H),5.75-5.81(m,1H),5.83(s,1H),7.07(dd,J=9.16,2.44Hz,1H),7.17(d,J=2.44Hz,1H),7.20(d,J=7.32Hz,2H),7.25(t,J=5.65Hz,3H),7.30-7.33(m,1H),7.34-7.37(m,2H),7.89(d,J=5.80Hz,1H),8.07(d,J=9.16Hz,1H);
LC-MS(保留时间:1.79min),MS m/z 748(MH+)。
实施例65:化合物65的制备
化合物65
按照化合物64的相同方法制备化合物65,但作如下修改:
修改:使用(+)-氯甲酸甲酯作为原料,得到化合物65(28.8mg,36%收率):
1H NMR(CD3OD,500MHz)δ0.72(d,J=6.71Hz,3H),0.80(t,J=5.80Hz,6H),0.87(d,J=7.02Hz,4H),0.90-0.95(m,6H),0.98-1.02(m,5H),1.05(s,9H),1.07-1.12(m,2H),1.18-1.23(m,2H),1.32-1.38(m,3H),1.41(dd,J=9.46,5.19Hz,1H),1.46-1.48(m,1H),1.63-1.71(m,5H),1.85(dd,J=7.93,5.49Hz,1H),1.89-1.93(m,1H),2.00-2.03(m,1H),2.15(q,J=8.70Hz,1H),2.34-2.38(m,1H),2.61(dd,J=13.73,7.33Hz,1H),2.89-2.93(m,1H),3.73(s,2H),3.92(s,3H),4.10(dd,J=11.60,3.36Hz,1H),4.33(s,1H),4.41(d,J=11.29Hz,1H),4.46-4.52(m,1H),4.54(dd,J=9.76,7.90Hz,1H),4.81(d,J=5.80Hz,1H),4.89(m,1H),5.08(d,J=11.60Hz,1H),5.26(d,J=17.09Hz,1H),5.77-5.81(m,1H),5.83(d,J=3.97Hz,1H),7.11(dd,J=11.29,1.83Hz,1H),7.18(d,J=1.83Hz,1H),7.24(d,J=5.80Hz,1H),7.88(d,J=6.10Hz,1H),8.08(d,J=8.85Hz,1H);
LC-MS(保留时间:2.06min),MS m/z 796(MH+)。
实施例66:化合物66的制备
化合物66
按照化合物64的相同方法制备化合物66,但作如下修改:
修改:使用(-)-氯甲酸甲酯作为原料,得到化合物66(26.9mg,31%收率):
1H NMR(CD3OD,500MHz)δ0.35(d,J=6.41Hz,1H),0.51(d,J=6.71Hz,2H),0.68(d,J=6.71Hz,1H),0.73(d,J=7.02Hz,2H),0.77-0.82(m,4H),0.88-0.98(m,10H),1.0-1.03(m,2H),1.05(s,9H),1.09-1.18(m,3H),1.25-1.29(m,1H),1.3-1.41(m,3H),1.60-1.71(m,3H),1.82-1.89(m,3H),2.00-2.04(m,J=2.14Hz,1H),2.10(q,J=8.24Hz,,1H),2.39-2.43(m,1H),2.61(dd,J=14.04,7.32Hz,1H),2.87-2.91(m,1H),3.73(s,1H),3.92(s,3H),4.13(dd,J=11.75,3.51Hz,1H),4.22-4.27(m,2H),4.30(s,1H),4.39(d,J=11.90Hz,1H),4.48-4.55(m,1H),4.79(d,J=5.19Hz,1H),4.87(d,J=4.27Hz,1H),5.05(d,J=10.07Hz,1H),5.22(d,J=16.79Hz,1H),5.78-5.85(m,2H),7.09(dd,J=9.16,1.83Hz,1H),7.17(d,J=1.83Hz,1H),7.23(d,J=5.80Hz,1H),7.88(d,J=5.80Hz,1H)8.09(d,J=9.16Hz,1H);
LC-MS(保留时间:2.05min),MS m/z 796(MH+)。
实施例67:化合物67的制备
化合物67
按照化合物64的相同方法制备化合物67,但作如下修改:
修改:使用二碳酸二叔戊酯作为原料,得到化合物67(35.3mg,41%收率):
1H NMR(CD3OD,500MHz)δ0.78(t,J=7.17Hz,3H),0.89-0.95(m,6H),1.04(s,9H),1.005-1.09(m,3H),1.15(s,1H),1.22(d,J=12.51Hz,6H),1.40(s,2H),1.42-1.46(m,1H),1.48(s,3H),1.56-1.66(m,2H),1.78(q,J=7.63Hz,1H),1.84(q,J=7.53Hz,1H),1.88(d,J=5.80Hz,1H),2.22(d,J=8.55Hz,1H),2.27-2.31(m,1H),2.61(dd,J=13.73,7.02Hz,1H),2.91-2.96(m,1H),3.92(s,3H),4.08(d,J=12.21Hz,1H),4.26(d,J=9.16Hz,1H),4.42(d,J=11.29Hz,1H),4.52(t,J=7.93Hz,1H),5.12(d,J=10.07Hz,1H),5.29(d,J=17.09Hz,1H),5.73-5.80(m,1H),5.84(s,1H),6.57(d,J=8.85Hz,1H),7.09(d,J=8.54Hz,1H),7.18(s,1H),7.25(d,J=5.80Hz,1H),7.89(d,J=5.80Hz,1H),8.08(d,J=9.16Hz,1H);
LC-MS(保留时间:1.82min),MS m/z 728(MH+)。
实施例68:化合物68的制备
化合物68
按照化合物64的相同方法制备化合物68,但作如下修改:
修改:使用氯甲酸(2,2,2-三氯-1,1-二甲基)酯作为原料,得到化合物68(30.5mg,37%收率):
1H NMR(CD3OD,500MHz)δ0.99(s,9H),1.04(s,6H),1.08-1.09(m,3H),1.23-1.26(m,3H),1.44(s,2H),1.46(d,J=5.80Hz,1H),1.71(s,2H),2.23-2.33(m,2H),2.60-2.64(m,1H),2.93-2.96(m,1H),3.70(m,1H),3.71(s,3H),3.93(s,3H),4.04-4.06(m,2H),4.27(d,J=9.16Hz,1H),4.41(d,J=11.60Hz,1H),4.57(d,J=10.98,6.11Hz,1H),5.14(d,J=12.21Hz,1H),5.32(d,J=17.70Hz,1H),5.75-5.80(m,1H),5.84(s,1H),7.10(dd,J=9.16,2.44Hz,1H),7.19(d,J=2.44Hz,1H),7.26(d,J=6.10Hz,1H),7.90(d,J=5.80Hz,1H),8.07(d,J=9.16Hz,1H));
LC-MS(保留时间:1.95min),MS m/z 816(MH+)。
实施例69:化合物69的制备
向实施例55、步骤1的产物(102mg,0.149mmol)和DIEA(48.2,0.373mmol)在THF(2mL)中的溶液混合物内加入碳酸N,N’-二琥珀酰亚胺酯(57.1mg,0.223mmol)。所得的悬浮液经微波辐射加热至80℃下反应15min。随后加入1-甲基环戊醇钠的浆状液,该浆状液通过在室温下用NaH(60%装于油中,59.6mg,1.49mmol)处理0℃的1-甲基环戊醇(149.2mg,1.49mmol)的THF(1mL)溶液15min得到。在室温下搅拌15min后,反应物用饱和氯化铵水溶液(3mL)猝灭并用EtOAc(10mL)萃取。随后将有机层通过celite hydromatrix柱,浓缩并经反相制备型-HPLC纯化,得到化合物69(49.0mg,44%):
1H NMR(CD3OD,500MHz)δ0.95-0.98(m,3H),0.99-1.01(m,J=12.51Hz,1H),1.03(s,9H),1.14-1.18(m,2H),1.30(s,3H),1.40-1.47(m,3H),1.50-1.56(m,3H),1.60-1.64(m,1H),1.76-1.81(m,1H),1.83-1.85(m,1H),2.10-2.19(m,1H),2.36-2.43(m,1H),2.63(dd,J=14.50,7.17Hz,1H),2.86-2.90(m,1H),3.92(s,3H),4.09(d,J=12.51Hz,1H),4.25(d,J=1.53Hz,1H),4.43(d,J=10.99Hz,1H),4.51-4.55(m,1H),5.06(d,J=11.60Hz,1H),5.23(d,J=16.78Hz,1H),5.80-5.85(m,J=12.67,12.67Hz,2H),7.09(d,J=8.55Hz,1H),7.17(s,1H),7.24(d,J=5.49Hz,1H),8.07(d,J=9.16Hz,1H));
LC-MS(保留时间:1.87min),MS m/z 740(MH+)。
实施例70:化合物70的制备
化合物70
按照化合物69的相同方法制备化合物70,但作如下修改:
修改:使用环戊醇作为原料,得到化合物70(85.1mg,收率40%):
1H NMR(CD3OD,500MHz)δ0.98(s,1H),1.00(d,J=4.88Hz,1H),1.03(s,9H),1.06-110(m,2H),1.24-1.29(m,3H),1.36-1.40(m,2H),1.44(dd,J=9.31,5.04Hz,2H),1.57-1.62(m,5H),1.69-1.73(m,2H),1.88(dd,J=8.09,5.65Hz,1H),2.22-29(m,2H),2.59-2.62(m,1H),2.92-2.96(m,1H),3.93(s,3H),4.07(dd,J=10.99,2.44Hz,1H),4.29(s,1H),4.42(dd,J=12.51,1.53Hz,1H),4.55(dd,J=9.77,7.93Hz,1H),4.68-4.71(m,1H),4.81(d,J=8.55Hz,1H),4.89(d,J=9.46Hz,1H),5.13(d,J=10.68Hz,1H),5.30(d,J=16.48Hz,1H),5.73-5.78(m,1H),5.84(s,1H),7.12(dd,J=9.15,1.83Hz,1H),7.20(d,J=2.14Hz,1H),7.27(d,J=5.80Hz,1H),7.89(d,J=5.80Hz,1H),8.09(d,J=8.85Hz,1H);
LC-MS(保留时间:1.81min),MS m/z 726(MH+)。
实施例71:化合物71的制备
化合物71
按照化合物69的相同方法制备化合物71,但作如下修改:修改:使用环丁醇作为原料,得到化合物71(16.2mg,收率39%):
1H NMR(CD3OD,500MHz)δ0.97(s,3H),1.03(s,9H),1.06-1.08(m,2H),1.17-1.22(m,3H),1.37-1.44(m,1H),1.82-1.85(m,1H),2.02-2.08(m,1H),2.15-2.21(m,1H),2.30-2.36(m,1H),2.49-2.54(m,0.4H),2.59-2.67(m,0.6H),2.90-2.94(m,1H),3.93(s,3H),4.09(dd,J=8.09,4.73Hz,1H),4.20(d,J=10.68Hz,0.4H),4.38(d,J=10.68Hz,0.6H),4.46(dd,J=10.22,7.17Hz,0.4H),4.48-4.56(m,1H),5.07-5.11(m,1H),5.26(d,J=17.24Hz,0.4H),5.28(d,J=17.24Hz,0.6H),5.71-5.79(m,1H),5.84(s,1H),7.07(dd,J=8.39,2.90Hz,1H),7.14(d,J=2.14Hz,1H),7.20(d,J=2.44Hz,1H),7.25(d,J=5.80Hz,1H),7.59(d,J=5.80Hz,1H),7.88(m,1H),8.00(d,J=9.16Hz,0.4H),8.07(d,J=8.85Hz,0.6H));
LC-MS(保留时间:1.25min),MS m/z 712(MH+)。
实施例72:化合物72的制备
化合物72
按照化合物69的相同方法制备化合物72,但作如下修改:
修改:使用2-苯基-2-丙醇作为原料,得到化合物72(19.0mg,收率42%):
1H NMR(CD3OD,500MHz)δ0.97(m,1H),1.03(s,9H),1.06-1.09(m,3H),1.16-1.22(m,4H),1.41-1.44(m,1H),1.57(s,3H),1.86(t,J=7.80Hz,1H),2.14-2.18(m,1H),2.30-2.35(m,1H),2.57-2.61(m,1H),2.90-2.94(m,1H),3.92(d,J=4.27Hz,1H),3.94(s,3H),4.04(dd,J=10.99,3.66Hz,1H),4.18(s,1H),4.24(d,J=10.99Hz,1H),4.52(s,1H),5.09(d,J=10.07Hz,1H),5.26(d,J=14.95Hz,1H),5.78-5.82(m,2H),7.07-7.12(m,2H),7.16-7.20(m,3H),7.23(d,J=5.19Hz,1H),7.29(d,J=7.02Hz,2H),7.84(d,J=5.80Hz,1H),8.03(d,J=9.46Hz,1H));
LC-MS(保留时间:1.84min),MS m/z 776(MH+)。
实施例73:化合物73的制备
化合物73
按照化合物69的相同方法制备化合物73,但作如下修改:
修改:使用4-(三氟甲基)苯基二甲基甲醇作为原料,得到化合物73(22.1mg,收率45%):
1H NMR(CD3OD,500MHz)δ0.91(s,1H),0.97-1.00(m,J=15.56Hz,4H),1.04(s,9H),1.07-1.10(m,2H),1.16-1.20(m,3H),1.30-1.31(m,1H),1.41(dd,J=9.61,5.34Hz,1H),1.55(d,J=7.32Hz,6H),1.83-1.87(m,1H),2.11-2.14(m,1H),2.34-2.39(m,1H),2.57-2.62(m,1H),2.89-2.92(m,J=11.60,4.27Hz,1H),3.92(s,2H),3.94(s,3H),4.02-4.05(m,1H),4.17(s,1H),4.26(d,J=11.90Hz,1H),4.53(t,J=8.85Hz,1H),5.07(d,J=10.07Hz,1H),5.24(d,J=18.01Hz,1H),5.78-5.83(m,2H),7.08(d,J=7.02Hz,1H),7.19(s,1H),7.22(d,J=5.80Hz,1H),7.45(dd,J=13.74,7.63Hz,3H),7.60(d,J=6.41Hz,1H),7.67(d,J=7.63Hz,1H),7.84(d,J=5.80Hz,1H),8.02(d,J=8.54Hz,1H));
LC-MS(保留时间:1.92min),MS m/z 844(MH+)。
实施例74:化合物74的制备
向实施例55、步骤1的产物(70.0mg,0.108mmol)和DIEA(41.8mg,0.323mmol)在DCM(2mL)中的溶液混合物内加入异氰酸叔丁酯(32.0,0.323mmol)。室温下搅拌过夜后,将反应物浓缩并经反相制备型HPLC纯化,得到化合物74(42.3mg,收率55%):
1H NMR(CD3OD,500MHz)δ0.96-1.00(m,1H)1.04(s,9H)1.08-1.10(m,3H)1.19(s,9H)1.22-1.31(m,2H)1.30(m,1H)1.41(dd,J=9.46,5.49Hz,1H)1.87(dd,J=8.24,5.49Hz,1H)2.20-2.29(m,2H)2.61(dd,J=14.04,6.72Hz,1H)2.92-2.97(m,1H)3.92(s,3H)4.08(dd,J=11.60,3.97Hz,1H)4.36(s,1H)4.47-4.52(m,2H)4.81(d,J=3.36Hz,1H)4.88(d,J=8.85Hz,1H)5.11(dd,J=10.22,1.68Hz,1H)5.28(dd,J=17.09,1.53Hz,1H)5.72-5.76(m,1H)5.85(s,1H)7.08(dd,J=9.16,2.44Hz,1H)7.18(d,J=2.14Hz,1H)7.24(d,J=5.80Hz,1H)7.88(d,J=6.10Hz,1H)8.12(d,J=9.16Hz,1H);
LC-MS(保留时间:1.70min),MS m/z 713(MH+)。
实施例75:化合物75的制备
化合物75
按照化合物74的相同方法制备化合物75,但作如下修改:
修改:使用异氰酸环戊酯作为原料,得到化合物75(38.5mg,49%):
1H NMR(CD3OD,500MHz)δ0.92(d,J=7.63Hz,1H)0.96(s,9H)0.98-1.02(m,1H)1.05(s,9H)1.07-1.10(m,2H)1.21-1.25(m,3H)1.28-1.34(m,1H)1.36-1.55(m,8H)1.58-1.65(m,13H)1.81(m,1H)1.88(m,6H)2.23(dd,J=18.01,8.85Hz,1H)2.29(m,1H)2.59(dd,J=13.73,7.02Hz,1H)2.94(m,1H)3.27(d,J=1.83Hz,1H)3.35(d,J=1.53Hz,1H)3.75(m,1H)3.92(s,3H)3.95(d,J=6.41Hz,1H)3.97(s,1H)4.09(m,2H)4.40(s,1H)4.45(d,J=11.90Hz,1H)4.52(dd,J=10.07,7.02Hz,1H)4.81(d,J=7.02Hz,1H)4.89(d,J=7.02Hz,1H)5.11(m,1H)5.29(d,J=17.40Hz,1H)5.75(m,1H)5.85(s,1H)7.11(dd,J=9.16,2.44Hz,1H)7.18(d,J=2.44Hz,1H)7.25(d,J=5.80Hz,1H)7.88(d,J=6.10Hz,1H)7.95(m,1H)8.12(d,J=9.16Hz,1H));
LC-MS(保留时间:1.67min),MS m/z 725(MH+)。
实施例76:化合物76的制备
向实施例55、步骤1的产物(70mg,0.102mmol)和DIEA(33.0mg,0.255mmol)在THF(2mL)中的溶液混合物内加入碳酸N,N’-二琥珀酰亚胺酯(39.2mg,0.153mmol)。所得的悬浮液经微波辐射加热至80℃下反应15min。随后将其用叔戊胺(88.9mg,1.02mmol)处理。在室温下搅拌15min后,将反应物浓缩并经反相制备型HPLC纯化,得到化合物76(51mg,69%):
1H NMR(CD3OD,500MHz)δ0.76(t,J=7.48Hz,4H),0.97(s,1H),1.04(s,9H),1.13(s,6H),1.22-1.25(m,2H),1.41(dd,J=9.61,5.34Hz,1H),1.53(dd,J=13.89,7.48Hz,1H),1.58-1.62(m,1H),1.87(dd,J=7.93,5.49Hz,1H),2.20(q,J=8.65Hz,1H),2.27-2.31(m,1H),2.60(dd,J=13.73,7.32Hz,1H),2.92-2.96(m,1H),3.92(s,3H),4.08(dd,J=11.75,3.81Hz,1H),4.36(s,1H),4.46(d,J=11.90Hz,1H),4.50(dd,J=10.22,7.17Hz,1H),5.10(dd,J=10.22,1.37Hz,1H),5.27(dd,J=16.94,1.07Hz,1H),5.73-5.77(m,1H),5.84(t,J=3.51Hz,1H),7.09(dd,J=9.16,2.44Hz,1H),7.18(d,J=2.44Hz,1H),7.24(d,J=5.80Hz,1H),7.88(d,J=6.10Hz,1H),8.11(d,J=9.16Hz,1H);
LC-MS(保留时间:1.753min),MS m/z 727(MH+)。
实施例77:化合物77的制备
化合物77
按照化合物76的相同方法制备化合物77,但作如下修改:
修改:使用叔丁基甲胺作为原料,得到化合物77(160.7mg,收率74%):
1H NMR(CD3OD,500MHz)δ0.96-1.10(m,1H),1.06(s,9H),1.08-1.12(m,J=5.80Hz,3H),1.26(s,9H),1.46(dd,J=9.46,5.19Hz,1H),1.87(dd,J=7.93,5.49Hz,1H),2.21(q,J=8.75Hz,1H),2.26-2.31(m,1H),2.57-2.62(m,1H),2.86(s,3H),2.91-2.95(m,1H),3.92(s,3H),4.09(dd,J=11.90,3.66Hz,1H),4.43(s,1H),4.46(d,J=11.90Hz,1H),4.52(dd,J=10.68,7.02Hz,1H),5.11(dd,J=10.22,1.37Hz,1H),5.29(d,J=17.09Hz,1H),5.75-5.82(m,1H),5.86(s,1H),7.09(dd,J=9.16,2.14Hz,1H),7.18(d,J=2.44Hz,1H),7.24(d,J=5.80Hz,1H),7.88(d,J=5.80Hz,1H),8.09(d,J=8.85Hz,1H));
LC-MS(保留时间:1.76min),MS m/z 727(MH+)。
实施例78:化合物78的制备
化合物78
按照化合物76的相同方法制备化合物78,但作如下修改:
修改:使用N,O-二甲基羟胺盐酸盐作为原料,得到化合物78(62.1mg,收率60%):
1H NMR(CD3OD,500MHz)δ0.99(t,J=6.10Hz,1H),1.07(s,11H),1.22-1.26(m,J=3.97Hz,2H),1.47(dd,J=9.46,5.49Hz,1H),1.88(dd,J=8.24,5.49Hz,1H),2.22(d,J=8.54Hz,1H),2.3-30-2.33(m,1H),2.60(dd,J=13.43,7.02Hz,1H),2.92(s,3H),2.93-2.96(m,1H),3.66(s,3H),3.92(s,3H),4.12(dd,J=11.90,3.66Hz,1H),4.34(d,J=12.21Hz,1H),4.44(d,J=9.46Hz,1H),4.54(dd,J=10.53,6.87Hz,1H),5.12(d,J=10.38Hz,1H),5.30(d,J=17.09Hz,1H),5.75-5.83(m,1H),5.86(t,J=3.97Hz,1H),6.70(d,J=9.77Hz,1H),7.13(dd,J=9.16,2.44Hz,1H),7.19(d,J=2.44Hz,1H),7.25(d,J=6.10Hz,1H),7.88(d,J=5.80Hz,1H),8.07(d,J=9.16Hz,1H));
LC-MS(保留时间:1.59min),MS m/z 701(MH+)。
实施例79:化合物79的制备
化合物79
按照化合物76的相同方法制备化合物79,但作如下修改:
修改:使用二乙胺作为原料,得到化合物79(56.5mg,收率54%):
1H NMR(CD3OD,500MHz)δ1.03(q,J=15.6Hz,4H),1.06(d,J=1.53Hz,9H),1.05-1.10(m,3H),1.13-1.23(m,4H),1.46(dd,J=9.46,5.19Hz,1H),1.86(dd,J=7.93,5.30Hz,1H),2.17(q,J=8.85Hz,1H),2.32-2.36(m,1H),2.60(dd,J=14.04,7.32Hz,1H),2.89-2.93(m,1H),3.16-3.24(m,4H),3.92(s,3H),4.14(dd,J=11.90,3.66Hz,1H),4.37(d,J=11.60Hz,1H),4.51-4.55(m,2H),5.09(d,J=10.07Hz,1H),5.27(d,J=17.09Hz,1H),5.55(d,J=9.46Hz,1H),5.79-5.84(m,1H),5.86(s,1H),7.11(dd,J=8.85,2.44Hz,1H),7.18(d,J=2.44Hz,1H),7.24(d,J=5.80Hz,1H),7.88(d,J=5.80Hz,1H),8.08(d,J=9.16Hz,1H));
LC-MS(保留时间:1.68min),MS m/z 713(MH+)。
实施例80:化合物80的制备
化合物80
按照化合物76的相同方法制备化合物80,但作如下修改:
修改:使用饱和氯化铵水溶液作为原料,得到化合物76(12.2mg,收率32%):
1H NMR(CD3OD,500MHz)δ1.00-1.03(m,3H),1.06(s,9H),1.20-1.25(m,2H),1.42(dd,J=9.31,5.34Hz,1H),2.22(d,J=9.77Hz,1H),2.29-2.35(m,1H),2.59(dd,J=13.28,6.87Hz,1H),2.92-2.96(m,1H),3.92(s,3H),4.14(dd,J=11.75,4.12Hz,1H),4.38-4.43(m,1H),4.51(dd,J=9.92,6.87Hz,1H),5.11(d,J=11.90Hz,1H),5.28(d,J=17.70Hz,1H),5.72-5.79(m,1H),5.84(s,1H),7.15(d,J=2.44Hz,1H),7.17(d,J=2.75Hz,1H),7.23(d,J=5.80Hz,1H),7.87(d,J=7.87Hz,1H),8.10(d,J=8.85Hz,1H));
LC-MS(保留时间:1.43min),MS m/z 657(MH+)。
实施例81:化合物81的制备
化合物81
按照化合物76的相同方法制备化合物81,但作如下修改:
修改:使用叔辛胺作为原料,得到化合物81(16.1mg,收率48%):
1H NMR(CD3OD,500MHz)δ0.88(s,9H),1.00(d,J=9.77Hz,5H),1.04(s,9H),1.17(s,3H),1.18-1.20(m,1H),1.21(s,3H),1.35(d,J=2.44Hz,1H),1.40-1.43(m,1H),1.57(d,J=14.95Hz,1H),1.67(d,J=14.65Hz,1H),1.85(dd,J=8.09,5.34Hz,1H),2.15(d,J=8.24Hz,1H),2.34-2.43(m,1H),2.60(dd,J=13.73,7.02Hz,1H),2.89-2.93(m,1H),3.92(s,3H),4.13(dd,J=11.60,3.97Hz,1H),4.38(s,1H),4.43(d,J=11.90Hz,1H),4.50(dd,J=9.77,7.32Hz,1H),5.07(d,J=10.38Hz,1H),5.24(d,J=17.09Hz,1H),5.75-5.81(m,1H),5.84(s,1H),7.09(dd,J=9.16,2.44Hz,1H),7.17(d,J=2.44Hz,1H),7.23(d,J=5.80Hz,1H),7.88(d,J=5.80Hz,1H),8.10(d,J=9.16Hz,1H));
LC-MS(保留时间:1.92min),MS m/z 769(MH+)。
实施例82:化合物82的制备
化合物82
按照化合物76的相同方法制备化合物82,但作如下修改:
修改:使用1-(4-氟苯基)-2-甲基-2-丙基胺作为原料,得到化合物82(14.8mg,收率42%):
1H NMR(CD3OD,500MHz)δ0.88(s,9H),1.00(d,J=9.46Hz,6H),1.04(s,9H),1.17(s,3H),1.21(s,3H),1.32-1.37(m,2H),1.39-1.43(m,1H),1.57(d,J=14.65Hz,1H),1.67(d,J=14.96Hz,1H),1.82-1.86(m,1H),2.15(t,J=9.46Hz,1H),2.33-2.43(m,2H),2.58-2.62(dd,J=14.50,7.78Hz,1H),2.89-2.93(m,1H),3.92(s,3H),4.12(dd,J=11.90,3.97Hz,1H),4.38(s,1H),4.43(d,J=12.82Hz,1H),4.49-4.52(m,1H),5.24(d,J=16.48Hz,1H),5.76-5.82(m,1H),5.83-5.85(m,1H),7.09(dd,J=9.00,2.59Hz,1H),7.17(d,J=2.14Hz,1H),7.23(d,J=5.80Hz,1H),7.88(d,J=5.80Hz,1H),8.10(d,J=9.16Hz,1H));
LC-MS(保留时间:1.40min),MS m/z 807(MH+)。
实施例83:化合物83的制备
化合物83
按照化合物76的相同方法制备化合物83,但作如下修改:
修改:使用枯基胺作为原料,得到化合物83(64.6mg,收率57%):
1H NMR(CD3OD,500MHz)δ0.87-0.91(m,1H),0.98(d,J=9.46Hz,2H),1.01(s,9H),1.02-1.05(m,1H),1.17-1.21(m,3H),1.29(s,2H),1.40(dd,J=9.46,5.19Hz,1H),1.51(d,J=3.05Hz,5H),1.85(dd,J=8.09,5.34Hz,1H),2.17(q,J=8.85Hz,1H),2.30-2.33(m,1H),2.58(dd,J=13.58,7.48Hz,1H),2.91-2.94(m,1H),3.92(d,J=2.14Hz,1H),3.93(s,3H),4.05(dd,J=11.60,3.66Hz,1H),4.32(d,J=9.77Hz,1H),4.51(dd,J=9.92,7.17Hz,1H),5.09(dd,J=1.59,1.52Hz,1H),5.27(dd,J=16.71,1.22Hz,1H),5.74-5.78(m,1H),5.82(s,1H),7.05-7.09(m,1H),7.18(d,J=2.44Hz,1H),7.19(d,J=7.33Hz,1H),7.22(d,J=5.80Hz,1H),7.33(d,J=7.63Hz,1H),7.84(d,J=5.80Hz,1H);
LC-MS(保留时间:1.76min),MS m/z 775(MH+)。
实施例84:化合物84的制备
向实施例11、步骤5的产物(77.0mg,0.136mmol)、DIEA(70.4mg,0.544mmol)和HATU(77.5mg,0.204mmol)的溶液中加入Boc-MeIle-OH(43.4mg,0.177mmol)。在室温下搅拌14hr后,将反应混合物用5%碳酸氢钠水溶液(1mL)洗涤。水层用2×2mL DCM萃取。合并的有机层用5%柠檬酸水溶液(2mL)和盐水洗涤,经硫酸镁干燥,浓缩并经快速柱层析纯化(SiO2,97∶3 DCM∶甲醇),得到化合物84(68.4mg,收率69%):
1H NMR(CD3OD,500MHz)δ0.89(t,J=7.32Hz,3H)0.94(dd,J=5.95,4.43Hz,3H)1.07(d,J=7,63Hz,3H)1.13(s,5H)1.16-1.20(m,J=4.88Hz,2H)1.23(s,3H)1.28(m,1H)1.34-1.38(m,1H)1.41-1.47(m,1H)1.55-1.60(m,J=7.63Hz,1H)1.87-1.91(m,1H)2.22-2.26(m,2H)2.36-2.38(m,1H)2.56-2.62(m,1H)2.81(d,J=11.30Hz,2H)2.94-2.99(m,1H)3.92(s,3H)4.05-4.12(m,2H)4.48-4.57(m,2H)5.12(d,J=10.07Hz,1H)5.32(m,1H)5.75-5.82(m,1H)5.84-5.88(m,1H)7.09-7.13(m,1H)7.16-7.20(m,1H)7.23-7.27(m,1H)7.88(dd,J=5.95,2.29Hz,1H)8.04(d,J=9.16Hz,0.6H)8.09(d,J=9.46Hz,0.4H);
LC-MS(保留时间:1.83min),MS m/z 728(MH+)。
实施例85:化合物85的制备
化合物85
按照化合物84的相同方法制备化合物85,但作如下修改:
修改:使用Boc-MeVal-OH作为原料,得到化合物84(72.1mg,收率74%):
1H NMR(CD3OD,500MHz)δ0.84(t,J=5.80Hz,3H),0.96(d,J=6.41Hz,3H),1.08(d,J=7.32Hz,2H),1.13(s,6H),1.16(s,4H),1.18-1.21(m,1H),1.23-1.29(m,1H),1.44(dd,J=9.61,5.34Hz,1H),1.88-1.92(m,1H),2.24(d,J=10.07Hz,1H),2.32-2.39(m,2H),2.58(dd,J=13.89,6.26Hz,1H),2.80(s,3H),2.93-2.98(m,1H),3.93(s,3H),4.01(dd,J=11.90,3.36Hz,0.6H),4.12(dd,J=11.90,3.66Hz,0.4H),4.16(d,J=11.29Hz,0.6H),4.38(d,J=10.99Hz,0.4H),4.45(d,J=10.68Hz,1H),4.47(d,J=10.69Hz,0.6H),4.53(dd,J=10.38,7.02Hz,0.4H),4.58(dd,J=10.07,7.02Hz,1H),5.12(d,J=4.28,0.6H),5.14(d,J=4.27Hz,0.4H),5.30(d,J=7.32Hz,0.6H),5.34(d,J=7.32Hz,0.4H),5.78-5.85(m,1H),5.88(t,J=3.05Hz,0.6H),5.96(t,J=3.97Hz,0.4H),7.13(dd,J=9.00,2.29Hz,0.6H),7.16(dd,J=9.46,2.44Hz,0.4H),7.19(m,1H),7.24(d,J=6.10Hz,0.6H),7.26(d,J=6.10Hz,0.4H),7.88(d,J=5.80Hz,1H),8.02(d,J=9.16Hz,0.6H)8.05(d,J=9.16Hz,0.4H).
实施例86:化合物86的制备
化合物86
按照化合物84的相同方法制备化合物86,但作如下修改:
修改:使用Boc-MeLeu-OH作为原料,得到化合物85(56.5mg,收率57%):
1H NMR(CD3OD,500MHz)δ0.94-0.96(m,6H),1.04-1.13(m,2H),1.17(s,4.5H),1.18(s,4.5H),1.26-1.31(m,1H),1.42(dd,J=9.46,5.49Hz,1H),1.46-1.51(m,2H),1.56-1.60(m,0.5H),1.69-1.72(m,0.5H),1.75-1.81(m,0.5H),1.90(q,J=7.50Hz,1H),2.27(dd,J=13.89,7.78Hz,1H),2.32-2.38(m,1H),2.58(dd,J=14.80,7.48Hz,1H),2.75(s,3H),2.95-2.99(m,1H),3.93(s,3H),4.03(d,J=12.21Hz,1H),4.11-15(m,0.5H),4.28(d,J=12.21Hz,1H),4.53(t,J=8.50Hz,0.5H),4.59(t,J=8.55Hz,0.5H),4.83-4.87(m,J=6.41Hz,0.5H),4.96(m,0.5H),5.14(dd,J=11.14,4.73Hz,1H),5.32(dd,J=17.70,6.41Hz,1H),5.75-5.82(m,1H),5.90(s,0.5H),5.92(s,0.5H),7.13-7.18m,1H),7.20(s,1H),7.25-7.27(m,1H),7.87(t,J=4.40Hz,1H)8.05(d,J=8.85Hz,1H).
实施例87:化合物87的制备
化合物87
按照化合物84的相同方法制备化合物87,但作如下修改:
修改:使用Boc-MeNle-OH作为原料,得到化合物87(82.3mg,收率83%):
1H NMR(CD3OD,500MHz)δ0.90-0.96(q,J=7.63Hz,3H)1.05-1.10(m,2H)1.18(s,4.5H)1.20(s,4.5H)1.24-1.30(m,3H)1.31-1.38(m,1H)1.42(dd,J=9.46,5.19Hz,2H)1.72-1.81(m,2H)1.88-1.92(m,1H)2.22-2.29(m,1H)2.32-2.38(m,1H)2.58(dd,J=13.89,7.17Hz,1H)2.72(s,3H)2.94-2.99(m,1H)3.93(s,3H)4.02(dd,J=9.77,4.27Hz,1H)4.12(dd,J=11.90,3.35Hz,0.5H)4.24(dd,J=11.90,0.6Hz,0.5H)4.51-4.60(m,1H)5.14(d,J=10.38Hz,1H)5.33(dd,J=17.24,4.73Hz,1H)5.75-5.82(m,1H)5.91(s,1H)7.15(dd,J=14.34,7.63Hz,1H)7.20(d,J=2.44Hz,1H)7.25(d,J=5.80Hz,1H)7.87(t,J=4.37Hz,1H)8.05(d,J=8.85Hz,1H).
实施例88:化合物88的制备
化合物88
按照化合物84的相同方法制备化合物88,但作如下修改:
修改:使用Boc-N-Me-NVa-OH作为原料,得到化合物88(70.5mg,收率73%):
1H NMR(CD3OD,500MHz)δ0.96(d,J=6.41Hz,3H)1.06-1.10(m,2H)1.18(s,9H)1.27-1.30(m,4H)1.42(dd,J=9.46,5.49Hz,1H)1.66-1.80(m,2H)1.88-1.92(m,1H)2.23-2.29(m,1H)2.30-2.37(m,1H)2.58(dd,J=13.58,7.17Hz,1H)2.73(s,3H)2.94-2.98(m,1H)3.93(s,3H)4.00-4.04(m,1H)4.12(d,J=12.82Hz,0.5H)4.25(d,J=12.21Hz,0.5H)4.51-4.60(m,1H)5.13(d,J=10.68Hz,1H)5.32(d,J=17.09Hz,1H)5.75-5.81(m,1H)5.90(s,1H)7.13-7.18(m,1H)7.20(d,J=2.14Hz,1H)7.25(d,J=5.80Hz,1H)7.87(s,1H)8.05(d,J=9.16Hz,1H).
实施例89:化合物89的制备
向实施例11、步骤5的产物(66.0mg,0.123mmol)、DIEA(63.7mg,0.492mmol)和HATU(70.0,0.184mmol)的溶液中加入2S叔丁氧基羰基氨基-3-羟基-3-甲基-丁酸(34.0mg,0.147mmol)。在室温下搅拌14hr后,将反应混合物用5%碳酸氢钠水溶液(1mL)洗涤。水层用2×2mL DCM萃取。合并的有机层用5%柠檬酸水溶液(2mL)和盐水洗涤,经硫酸镁干燥,浓缩并经反相制备型-HPLC纯化,得到化合物89(36.1mg,收率41%):
1H NMR(CD3OD,500MHz)δ1.07(d,J=7.93Hz,2H),1.18(s,1H),1.20(s,9H),1.24-1.27(m,J=11.60Hz,3H),1.30(s,3H),1.43-1.48(m,10H),1.59(s,1H),1.65(s,1H),1.87(dd,J=8.24,5.19Hz,1H),2.24(q,J=9.16Hz,1H),2.33-2.36(m,1H),2.63(dd,J=12.97,6.56Hz,1H),2.94-2.99(m,1H),3.92(s,3H),3.93(s,1H),4.12(dd,J=11.60,3.05Hz,1H),4.27-4.31(m,1H),4.54(t,J=9.77Hz,1H),5.12(dd,J=10.53,1.37Hz,1H),5.30(d,J=17.09Hz,1H),5.79-5.83(m,1H),5.85(s,1H),7.11(dd,J=8.55,1.83Hz,1H),7.18(d,J=2.24Hz,1H),7.24(d,J=5.49Hz,1H),7.88(m,1H),8.10(d,J=8.85Hz,1H));
LC-MS(保留时间:1.637min),MS m/z 716(MH+)。
实施例91:化合物91的制备
化合物91
按照化合物89的相同方法制备化合物91,但作如下修改:
修改:使用Boc-L-Thr-OH作为原料,得到化合物91(80.5mg,收率66%):
1H NMR(CD3OD,500MHz)δ0.93(dd,J=8.24,2.14Hz,2H),1.08-1.18(m,4H),1.20(d,J=6.10Hz,3H),1.29(s,9H),1.32(dd,J=9.61,5.04Hz,1H),1.45(d,J=4.27Hz,1H),1.84(dd,J=7.63,5.19Hz,1H),2.15(q,J=8.85Hz,1H),2.42-2.48(m,1H),2.64(dd,J=14.04,7.63Hz,1H),2.85-2.89(m,1H),3.92(s,3H),4.1-4.14(m,2H),4.30(d,J=4.88Hz,1H),4.38(d,J=11.60Hz,1H),4.60(t,J=8.55Hz,1H),5.04(dd,J=10.22,1.68Hz,1H),5.80-5.84(m,2H),7.11(d,J=9.16Hz,1H),7.17(d,J=1.83Hz,1H),7.23(d,J=5.80Hz,1H),7.87(d,J=5.80Hz,1H),8.10(d,J=8.85Hz,1H);
LC-MS(保留时间:1.560min),MS m/z 702(MH+)。
实施例92:化合物92的制备
化合物92
按照化合物89的相同方法制备化合物92,但作如下修改:
修改:使用Boc-L-Thr(Me)-OH作为原料,得到化合物92(47.1mg,收率69%):
1H NMR(CD3OD,500MHz)δ0.95(d,J=4.27Hz;2H),1.11-1.16(m,3H),1.18(d,J=6.10Hz,6H),132(s,9H),1.38(dd,J=9.31,5.04Hz,1H),1.45(s,1H),1.85(dd,J=7.78,5.04Hz,1H),2.13(d,J=9.15Hz,1H),2.46-2.51(m,1H),2.63(dd,J=14.19,7.78Hz,1H),2.81-2.91(m,1H),3.68-3.73(m,1H),3.92(s,4H),4.14(d,J=12.21Hz,1H),4.35(d,J=5.80Hz,1H),4.42(d,J=11.29Hz,1H),4.60(t,J=8.70Hz,1H),5.05(d,J=10.68Hz,1H),5.24(dd,J=16.79,0.92Hz,1H),5.81-5.85(m,2H),7.11(dd,J=9.16,0.92Hz,1H),7.17(s,1H),7.24(d,J=5.80Hz,1H),7.88(d,J=5.80Hz,1H),8.11(d,J=8.55Hz,1H);
LC-MS(保留时间:1.660min),MS m/z 716(MH+)。
实施例93:化合物93的制备
化合物93
按照化合物89的相同方法制备化合物93,但作如下修改:
修改:使用Boc-L-Thr(tBu)-OH作为原料,得到化合物93(52.7mg,收率53%):
1H NMR(CD3OD,500MHz)δ1.09(dd,J=8.24,2.44Hz,2H),1.201.24.(m,3H),1.28(s,9H),1.45(s,9H),1.89(dd,J=7.93,5.19Hz,1H),2.25(q,J=8.34Hz,1H),2.32-2.36(m,1H),2.59(dd,J=12.82,6.41Hz,1H),2.95-3.00(m,1H),3.71(s,2H),3.92(s,3H),3.93-3.99(m,1H),4.10(d,J=7.02Hz,1H),4.15(d,J=11.90Hz,1H),4.22(dd,J=6.10,2.44Hz,2H),4.40(d,J=11.60Hz,1H),4.54(dd,J=10.01,6.71Hz,1H),5.13(d,J=10.38Hz,1H),5.31(d,J=17.09Hz,1H),5.76-5.83(m,1H),5.87(s,1H),6.06(d,J=9.46Hz,1H),6.36(d,J=7.02Hz,1H),7.11(d,J=8.85Hz,1H),7.18(s,1H),7.24(d,J=5.49Hz,1H),7.88(m,1H),8.08(d,J=9.16Hz,1H).
实施例94:化合物94的制备
化合物94
按照化合物89的相同方法制备化合物94,但作如下修改:
修改:使用Boc-(2S,3S)-2-氨基-3-甲氧基丁酸作为原料,得到化合物94(150.2mg,收率80%):
1H NMR(CD3OD,500MHz)δ1.04-1.13(m,3H),1.17(d,J=6.10Hz,3H),1.20-1.24(m,2H),1.27(s,9H),1.44-1.48(m,2H),1.86(dd,J=7.93,5.49Hz,1H),2.24(q,J=8.65Hz,1H),2.34-2.37(m,1H),2.61(dd,J=14.19,7.17Hz,1H),2.94-2.99(m,1H),3.66(m,1H),3.92(s,3H),4.13(dd,J=12.36,3.81Hz,1H),4.37(dd,J=22.58,10.99Hz,2H),4.54(dd,J=10.38,7.63Hz,1H),5.12(d,J=10.68Hz,1H),5.31(d,J=17.40Hz,1H),5.77-5.82(m,1H),5.85(s,1H),7.12(d,J=9.16Hz,1H),7.18(s,1H),7.25(d,J=6.10Hz,1H),7.89(d,J=5.80Hz,1H),8.10(d,J=8.85Hz,1H);
LC-MS(保留时间:1.673min),MS m/z 716(MH+)。
实施例95:化合物95的制备
方案1
步骤1
向H-allo-THr-OH(5.0g,41.98mmol)和DIEA(10.9g,83.96mmol)在DCM(150mL)中的混合物内加入二碳酸二叔丁酯(13.7g,62.97mmol)。在室温下搅拌14h后,将反应混合物用3×100mL DCM洗涤。合并的有机层经硫酸镁干燥并浓缩。LC/MS表明大部分产物在水层中。因此,将水层浓缩。产物经快速柱层析纯化(SiO2,90∶10 DCM∶甲醇),得到Boc-allo-THr-OH;LC-MS(保留时间:0.727min),MS m/z242(MNa+)。
步骤2
向实施例11、步骤5的产物(100.0mg,0.174mmol)、DIEA(67.6mg,0.522mmol)和HATU(106.0mg,0.278mmol)的溶液中加入上述步骤1的产物(57.3mg,0.262mmol)。在室温下搅拌3hr后,将反应混合物用5%碳酸氢钠水溶液(1mL)洗涤。水层用2×2mL DCM萃取。合并的有机层用5%柠檬酸水溶液(2mL)和盐水洗涤,经硫酸镁干燥,浓缩并经反相制备型-HPLC纯化,得到化合物95(39.1mg,收率32%):
1HNMR(CD3OD,500MHz)δ1.02(d,J=8.55Hz,1H),1.18-1.23(m,3H),1.25(s,9H),1.38(dd,J=9.15,6.30Hz 1H),1.84(dd,J=7.93,5.19Hz,1H),2.19-2.24(m,1H),2.38-2.43(m,1H),2.65(dd,J=14.19,6.87Hz,1H),2.92-2.96(m,1H),3.92(s,3H),3.93(s,2H),4.16-4.19(m,1H),4.23(d,J=8.24Hz,1H),4.44(d,J=12.21Hz,1H),4.57-5.81(m,1H),5.09(d,J=10.68Hz,1H),5.29(d,J=17.09Hz,1H),5.75-5.81(m,1H),5.83-5.85(m,1H),7.11(d,J=10.38Hz,1H),7.18(d,J=1.83Hz,1H),7.24(d,J=6.41Hz,1H),7.88(d,J=5.80Hz,1H),8.11(d,J=9.16Hz,1H);
LC-MS(保留时间:1.583min),MS m/z 702(MH+)。
实施例96:化合物96的制备
方案1
按照化合物95的相同方法制备化合物96,但作如下修改:
修改:
步骤1
使用(2S,3S)-2-氨基-3-乙氧基丁酸盐酸盐作为步骤1的原料,得到Boc-(2S,3S)-2-氨基-3-乙氧基丁酸;LC-MS(保留时间:1.067min),MS m/z 270(M+Na+)。
步骤2
随后按照相同的方法,使步骤1的产物与实施例11、步骤5的产物偶合,得到化合物96(55.3mg,收率44%):
1HNMR(CD3OD,500MHz)δ0.94(t,J=6.87Hz,1H),0.97-1.03(m,2H),1.08-1.11(m,2H),1.13-1.15(m,2H),1.17(d,J=6.10Hz,6H),1.29(s,9H),1.41-1.45(m,3H),1.85(dd,J=7.48,5.34Hz,1H),2.12-2.19(m,1H),2.43-2.49(m,1H),2.60(dd,J=13.73,6.80Hz,1H),2.89-2.93(m,1H),3.50-3.57(m,2H),3.73-3.78(m,1H),3.92(s,3H),4.18(d,J=8.85Hz,1H),4.35(d,J=12.21Hz,1H),4.39(d,J=8.55Hz,1H),4.53(t,J=7.78Hz,1H),5.07(d,J=9.16Hz,1H),5.25(d,J=18.01Hz,1H),5.82(t,J=9.85Hz,1H),5.88(t,J=9.80Hz,1H),7.11(d,J=5.19Hz,1H),7.18(d,J=2.14Hz,1H),7.24(d,J=5.49Hz,1H),7.88(d,J=6.10Hz,1H),8.10(d,J=8.85Hz,1H),
LC-MS(保留时间:1.743min),MS m/z 730(MH+)。
实施例97:化合物97的制备
方案1
按照化合物95的相同方法制备化合物97,但作如下修改:
修改:
步骤1
使用H-allo-Thr(t-Bu)-OH作为步骤1的原料,得到Boc-(2S,3S)-2-氨基-3-乙氧基丁酸;LC-MS(保留时间:1.363min),MS m/z298(M+Na+)。
步骤2
随后按照相同的方法,使步骤1的产物与实施例11、步骤5的产物偶合,得到化合物97(48.2mg,收率37%):LC-MS(保留时间:1.820min),MS m/z 758(MH+)。
实施例99:化合物99的制备
化合物99
按照实施例55、步骤1的相同方法制备化合物99,但作如下修改:
修改:使用化合物84作为原料,得到化合物99(60.3mg,收率98%):
1H NMR(CD3OD,500MHz)δ1.00(q,J=7.12Hz,3H)1.10-113(m,5H)1.20-1.31(m,3H)1.41(dd,J=9.46,5.49Hz,1H)1.61-1.68(m,1H)1.92(dd,J=8.24,5.49Hz,1H)2.04-2.09(m,1H)2.28(q,J=8.55Hz,1H)2.34-2.39(m,1H)2.57(s,3H)2.64-2.70(m,1H)2.94-2.97(m,1H)3.93(s,3H)4.07-4.14(dd,J=12.05,3.81Hz,1H)4.13(d,J=6.10Hz,1H)4.18(d,J=5.80Hz,1H)4.25(d,J=12.21Hz,1H)4.66-4.73(m,1H)5.15(d,J=10.68Hz,1H)5.32(d,J=17.09Hz,1H)5.70-5.79(m,1H)5.92(t,J=3.66Hz,0.4H)5.95(t,J=3.66Hz,0.6H)7.17(dd,J=9.16,2.44Hz,1H)7.22(d,J=2.14Hz,1H)7.28(dd,J=5.80,3.36Hz,1H)7.91(dd,J=5.80,4.27Hz,1H)8.03(d,J=8.85Hz,0.6H)8.07(d,J=9.16Hz,0.4H);
LC-MS(保留时间:1.33min),MS m/z 6.28(XH+)。
实施例100:化合物100的制备
向化合物94(0.600g,0.838mmol)的DCE(3mL)溶液中加入TFA(3mL)。在室温下搅拌15min后,将反应混合物浓缩,所得的粘稠油状物再次溶于DCE(5mL)并再次浓缩。随后再次溶于DCM(2mL)中,用1N HCl/乙醚(10mL)溶液处理。将所得的悬浮液冷却至0℃下,抽滤,用乙醚洗涤并在真空烘箱中干燥,得到双盐酸盐产物,为白色固体物(527.1g,收率91%):
1H NMR(CD3OD,500MHz)δ1.08-1.15(m,2H),1.21(d,J=6.71Hz,4H),1.28-1.33(m,1H),1.41(dd,J=9.46,5.49Hz,1H),1.91(dd,J=8.24,5.49Hz,1H),2.28(q,J=8.65Hz,1H),2.34-2.37(m,1H),2.68(dd,J=13.12,7.02Hz,1H),2.81(s,3H),2.93-2.98(m,1H),3.45(s,3H),3.94(s,3H),3.96-4.00(m,1H),4.16(dd,J=11.90,3.66Hz,1H),4.27(d,J=11.60Hz,1H),4.59(d,J=4.58Hz,1H),4.69(dd,J=10.07,7.02Hz,1H),5.14(dd,J=10.53,1.37Hz,1H),5.32(d,J=17.09Hz,1H),5.70-5.77(m,1H),5.94(t,J=3.66Hz,1H),7.19(d,J=9.16Hz,1H),7.24(s,1H),7.32(s,1H),7.91(d,J=5.80Hz,1H),8.09(d,J=9.16Hz,1H);
LC-MS(保留时间:1.213min),MS m/z 616(MH+)。
实施例101:化合物101的制备
向化合物100(80mg,0.116mmol)和DIEA(31.5mg,0.244mmo1)在THF(2mL)中的溶液混合物内加入碳酸N,N’-二琥珀酰亚胺酯(44.6mg,0.174mmo1)。所得的悬浮液经微波辐射加热至80℃下反应15min。随后将其用叔戊胺(84.8mg,1.16mmo1)处理。在室温下搅拌15min后,将反应物浓缩并经反相制备型HPLC纯化,得到化合物101(65.1mg,79%):
1H NMR(CD3OD,500MHz)δ1.03-1.08(m,3H),1.16(d,J=6.41Hz,3H),1.19(s,9H),1.21-1.25(m,2H),1.27(d,J=6.10Hz,1H),1.45(dd,J=9.46,5.19Hz,1H),1.85(dd,J=8.24,5.19Hz,1H),2.23(q,J=9.46Hz,1H),2.34-2.41.(m,1H),2.61(dd,J=14.34,7.32Hz,1H),2.94-2.97(m,1H),3.58-3.63(m,1H),3.92(s,3H),4.15(dd,J=12.05,3.81Hz,1H),4.39(d,J=11.60Hz,1H),4.55(dd,J=9.92,7.48Hz,1H),5.11(d,J=10.68Hz,1H),5.29(d,J=17.40Hz,1H),5.77-5.83(m,1H),5.86(s,1H),7.12(dd,J=9.00,2.29Hz,1H),7.18(d,J=2.14Hz,1H),7.24(d,J=5.80Hz,1H),7.88(d,J=5.80Hz,1H),8.10(d,J=9.16Hz,1H);
LC-MS(保留时间:1.617min),MS m/z 715(MH+)。
实施例102:化合物102的制备
化合物102
按照化合物101的相同方法制备化合物102,但作如下修改:
修改:使用叔戊胺作为原料,得到化合物102(62.5mg,收率74%):
1H NMR(CD3OD,500MHz)δ0.77(t,J=7.48Hz,2H),0.84(t,J=7.48Hz,1H),1.04-1.08(m,2H),1.13(d,J=1.22Hz,9H),1.16(d,J=6.41Hz,3H),1.21(s,1H),1.22-1.28(m,2H),1.44(dd,J=9.46,5.19Hz,1H),1.52-1.57(m,1H),1.58-1.62(m,1H),1.85(dd,J=7.93,5.19Hz,1H),2.21-2.25(m,1H),2.34-2.39(m,1H),2.61(dd,J=13.58,7.17Hz,1H),2.93-2.98(m,1H),3.59-3.64(m,1H),4.15(dd,J=11.75,3.81Hz,1H),4.38(d,J=12.51Hz,1H),4.50(d,J=7.63Hz,1H),4.55(dd,J=9.92,7.78Hz,1H),5.11(d,J=9.77Hz,1H),5.29(d,J=16.79Hz,1H),5.77-5.83(m,1H),5.86(t,J=4.73Hz,1H),7.12(dd,J=8.85,2.44Hz,1H),7.18(d,J=2.44Hz,1H),7.24(d,J=6.10Hz,1H),7.88(d,J=5.80Hz,1H),8.10(d,J=9.16Hz,1H);LC-MS(保留时间:1.690min),MS m/z 729(MH+)。
实施例103:化合物103的制备
化合物103
按照化合物101的相同方法制备化合物103,但作如下修改:
修改:使用环戊基胺作为原料,得到化合物103(56.4mg,收率67%):
1H NMR(CD3OD,500MHz)δ1.01-1.08(m,2H),1.07(d,J=6.10Hz,1H),1.16(d,J=6.10Hz,3H),1.21-1.25(m,3H),1.30-1.33(m,1H),1.44(dd,J=9.77,5.19Hz,1H),1.51-1.56(m,2H),1.60-1.65(m,2H),1.71-1.75(m,1H),1.80-1.84(m,1H),1.86(dd,J=8.09,5.34Hz,1H),2.20-2.25(m,1H),2.37-2.41(m,1H),2.61(dd,J=14.04,7.32Hz,1H),2.93-2.98(m,1H),3.60-3.65(m,1H),3.75-3.80(m,1H),3.92(s,3H),4.17(dd,J=12.05,3.81Hz,1H),4.37(d,J=11.90Hz,1H),4.55-4.59(m,2H),5.10(d,J=11.60Hz,1H),5.29(d,J=16.48Hz,1H),5.78-5.83(m,1H),5.85(d,J=2.44Hz,1H),7.13(dd,J=9.16,2.44Hz,1H),7.18(d,J=2.44Hz,1H),7.24(d,J=5.80Hz,1H),7.88(d,J=5.80Hz,1H),8.09(d,J=9.16Hz,1H);
LC-MS(保留时间:1.607min),MS m/z 727(MH+)。
实施例104:化合物104的制备
向化合物100(80mg,0.116mmol)和DIEA(31.5mg,0.244mmol)在THF(2mL)中的溶液混合物内加入碳酸N,N’-二琥珀酰亚胺酯(44.6mg,0.174mmol)。将所得的悬浮液经微波辐射加热至80℃下反应15min。随后加入环戊醇钠的浆状液,该浆状液通过在室温下用NaH(60%装于油中,46.4mg,1.16mmol)处理0℃的环戊醇(110mg,1.28mmol)的THF(1mL)溶液15min得到。在室温下搅拌15min后,该反应物用饱和氯化铵水溶液(1mL)猝灭并用EtOAc(5mL)萃取。随后将有机层通过celite hydromatrix柱,浓缩并经反相制备型-HPLC纯化,得到化合物104(38.2mg,45%):
1H NMR(CD3OD,500MHz)δ1.03-1.09(m,3H),1.16(d,J=6.10Hz,3H),1.201.25(m,1H),1.25-1.30(m,J=10.22,5.34Hz,1H),1.40-1.45(m,J=10.83,3.81Hz,1H),1.46(dd,J=9.61,5.34Hz,1H),1.58-1.63(m,3H),1.70-1.75(m,2H),1.86(dd,J=7.63,5.49Hz,1H),2.22-2.26(m,1H),2.34-2.39(m,1H),2.59-2.64(m,1H),2.94-2.98(m,1H),3.67(dd,J=7.78,6.56Hz,1H),3.92(s,3H),4.13(dd,J=10.83,4.12Hz,1H),4.37-4.42(m,1H),4.56(dd,J=10.07,7.32Hz,1H),4.71-4.76(m,1H),5.12(d,J=10.68Hz,1H),5.31(d,J=16.79Hz,1H),5.80(m,1H),5.85(s,1H),7.13(d,J=10.68Hz,1H),7.19(d,J=1.83Hz,1H),7.25(d,J=6.10Hz,1H),7.89(d,J=5.80Hz,1H),8.09(d,J=9.16Hz,1H),;
LC-MS(保留时间:1.697min),MS m/z 728(MH+)。
实施例105:化合物105的制备
向化合物100(80.0mg,0.116mmol)和DIEA(31.5mg,0.244mmol)在DCM(2mL)中的溶液混合物内加入二碳酸二叔戊酯(57.1mg,0.232mmol)。在室温下搅拌14h后,除去溶剂,产物经反相制备型-HPLC纯化,得到化合物105(62.5mg,收率74%):
1H NMR(CD3OD,500MHz)δ0.79(t,J=7.48Hz,3H),1.04-1.08(m,3H),1.17(d,J=6.10Hz,3H),1.19-1.23(s,3H),1.24(s,3H),1.39-1.43(m,1H),1.46(dd,J=9.61,5.34Hz,1H),1.60-1.65(m,2H),1.86(dd,J=7.93,5.49Hz,1H),2.22(q,J=8.85Hz,1H),2.35-2.40(m,1H),2.61(dd,J=14.04,7.15Hz,1H),2.94-3.00(m,1H),3.64-4.00(m,1H),3.92(s,4H),4.14(dd,J=11.90,3.05Hz,1H),4.35(d,J=7.93Hz,1H),4.40(d,J=11.90Hz,1H),4.55(dd,J=9.31,7.78Hz,1H),5.11(d,J=10.68Hz,1H),5.30(d,J=16.79Hz,1H),5.79-5.83(m,1H),5.85(s,1H),7.12(d,J=9.16Hz,1H),7.18(s,1H),7.25(d,J=5.80Hz,1H),7.86-7.90(m,1H),8.09(d,J=9.16Hz,1H);
LC-MS(保留时间:1.740min),MS m/z 730(MH+)。
实施例106:化合物106的制备
化合物106
按照化合物105的相同方法制备化合物106,但作如下修改:
修改:使用碳酸吡啶-2-基酯·2,2,2-三氟-1,1-二甲基-乙酯作为原料,得到化合物106(58.1mg,收率65%):
1HNMR(CD3OD,500MHz)δ1.04-1.08(m,3H),1.17(d,J=6.10Hz,3H),1.19.1.23(m,1H),1.23-1.27(m,1H),1.28(s,3H),1.46(dd,J=9.46,5.19Hz,2H),1.49(s,2H),1.86(dd,J=8.09,5.34Hz,1H),2.21(q,J=8.85Hz,1H),2.36-2.40(m,1H),2.62(dd,J=13.74,7.32Hz,1H),2.93-2.98(m,1H),3.65-3.70(m,1H),3.92(s,3H),4.12(dd,J=11.90,3.66Hz,1H),4.31(d,J=8.24Hz,1H),4.42(d,J=11.90Hz,1H),4.57(dd,J=10.07,7.32Hz,1H),5.11(d,J=10.38Hz,1H),5.30(d,J=16.79Hz,1H),5.78-5.83(m,1H),5.84(s,1H),7.12(dd,J=9.00,2.29Hz,1H),7.19(d,J=2.14Hz,1H),7.25(d,J=5.80Hz,1H),7.89(d,J=6.10Hz,1H),8.09(d,J=8.85Hz,1H);
LC-MS(保留时间:1.770min),MS m/z 770(MH+)。
实施例107:化合物107的制备
向实施例25、步骤3的产物(100.0mg,0.116mmol)、DIEA(62.5mg,0.483mmol)和HATU(92.0mg,0.242mmol)的溶液中加入Boc-allo-Thr-OH(43.5mg,0.177mmol)。在室温下搅拌3hr后,将反应混合物用5%碳酸氢钠水溶液(1mL)洗涤。水层用2×2mL DCM萃取。合并的有机层用5%柠檬酸水溶液(1mL)和盐水洗涤,经硫酸镁干燥,浓缩并经反相制备型-HPLC纯化,得到化合物107(62.5mg,收率52%):
1H NMR(CD3OD,500MHz)δ0.8-1.02(m,1H),1.04-1.08(m,2H),1.23-1.27(m,12H),1.42(dd,J=9.46,5.19Hz,1H),1.86(t,J=6.26Hz,1H),2.23-2.27(m,1H),2.46-2.50(m,1H),2.76(dd,J=14.04,6.71Hz,1H),2.95-2.99(m,1H),3.94-3.98(m,1H),4.28(d,J=7.32Hz,2H),4.52(d,J=12.51Hz,1H),4.63(t,J=9.00Hz,1H),5.12(d,J=10.07Hz,1H),5.31(d,J=16.79Hz,1H),5.77-5.83(m,1H),6.09(s,1H),7.36-7-41(m,1H),7.47(t,J=7.17Hz,3H),7.52(d,J=7.63Hz,1H),7.70(t,J=7.17Hz,1H),7.85(s,1H),7.88(d,J=8.24Hz,1H),8.17(d,J=7.93Hz,2H),8.22(d,J=7.63Hz,1H);
LC-MS(保留时间:1.937min),MS m/z 748(MH+)。
实施例108:化合物108的制备
化合物108
按照化合物107的相同方法制备化合物108,但作如下修改:
修改:使用Boc-(2S,3S)-氨基-3-甲氧基丁酸作为原料,得到化合物108(75.1mg,收率51%):
1H NMR(CD3OD,500MHz)δ0.80-1.02(m,4H),1.18(d,J=6.10Hz,3H),1.28(s,9H),1.44(dd,J=9.77,1.30Hz,1H),1.45-1.50(m,1H),1.85-1.90(m,1H),2.14-2.18(m,1H),2.55-2.59(m,1H),2.72-2.76(m,1H),2.91-2.95(m,1H),3.34(s,3H),3.65-3.69(m,1H),4.32(d,J=10.68Hz,1H),4.40(d,J=7.93Hz,1H),4.46(d,J=13.12Hz,1H),4.60(t,J=8.24Hz,1H),5.07(d,J=9.46Hz,1H),5.26(d,J=17.40Hz,1H),5.82-5.86(m,1H),6.08(s,1H),7.38(dd,J=7.32,6.10Hz,1H),7.47(t,J=7.02Hz,3H),7.51(d,J=5.80Hz,1H),7.69(t,J=6.56Hz,1H),7.69(t,J=6.56Hz,1H),7.85(s,1H),7.88(d,J=7.63Hz,1H),8.18(d,J=8.24Hz,3H),8.21(d,J=9.16Hz,1H);
LC-MS(保留时间:1.973min),MS m/z 762(MH+)。
实施例109:化合物109的制备
化合物109
按照化合物107的相同方法制备化合物109,但作如下修改:
修改:使用Boc-(2S,3S)-氨基-3-乙氧基丁酸作为原料,得到化合物109(57.2mg,收率47%):
1H NMR(CD3OD,500MHz)δ1.02-1.08(m,4H),1.17(d,J=6.10Hz,6H),1.19(s,1H),1.19-1.24(m,1H),1.23-1.27(m,J=3.97Hz,1H),1.30(s,9H),1.44(dd,J=9.77,5.50Hz,1H),1.46(s,1H),1.88(dd,J=7.78,5.95Hz,1H),2.20-1.25(m,1H),2.49-2.54(m,1H),2.69-2.73(m,1H),2.93-2.97(m,1H),3.53-3.57(m,2H),3.75-3.80(m,1H),4.34(dd,J=11.75,3.20Hz,1H),4.42(t,J=8.30Hz,2H),4.57(t,J=8.09Hz,1H),5.11(d,J=10.38Hz,1H),5.29(d,J=17.40Hz,1H),5.78-5.83(m,1H),6.09(s,1H),7.38(t,J=7.32Hz,1H),7.47(t,J=7.63Hz,2H),7.52(d,J=7.02Hz,1H),7.70(t,J=7.93Hz,1H),7.86(s,1H),7.88(d,J=7.93Hz,1H),8.18(d,J=7.32Hz,2H),8.22(d,J=8.24Hz,1H);
LC-MS(保留时间:2.030min),MS m/z 776(MH+)。
实施例110:化合物110的制备
化合物110
按照化合物89的相同方法制备化合物110,但作如下修改:
修改:使用Boc-L-Thr(Bn)-OH作为原料,得到化合物110(49.8mg,收率48%);LC-MS(保留时间:1.857min),MS m/z 792(MH+)。
部分D:
实施例120:化合物120的制备
化合物120
方案1
步骤1:
将化合物23(参见实施例23)(1.50g,2.19mmol)在DCM(50mL)和三氟乙酸(50mL)中的溶液在室温下搅拌3h。将混合物真空浓缩至得到粘稠剩余物,随后溶于1,2-二氯乙烷并再真空浓缩,得到所需的双-三氟乙酸盐产物,为灰白色玻璃状固体物(定量)。该物质未经进一步纯化直接用于下一步骤中。
步骤2:
向实施例120、步骤1的产物(118mg,0.146mmol)的1,2-二氯乙烷(3mL)溶液中加入氯甲酸对甲苯酯(32.4mg,0.190mm0l)和N,N-二异丙基乙胺(94.5mg,0.731mmol)。将混合物在室温下搅拌72h。将反应混合物用pH=4的缓冲溶液(3×3mL)洗涤,将洗液用1,2-二氯乙烷(3mL)反萃取。合并有机相并真空浓缩。随后将粗产物溶于甲醇中并经反相制备型HPLC纯化,得到题述化合物(化合物120),为黄色玻璃状固体物(64.2mg,收率61.1%):
1H NMR(CD3OD)δ1.06-110(m,3H),1.12(s,9H),1.24-1.28(m,2H),1.44(dd,J=9.31,5.34Hz,1H),1.89(dd,J=7.93,5.49Hz,1H),2.21-2.28(m,2H),2.31(s,3H),2.62-2.66(m,1H),2.93-2.99(m,1H),4.12(dd,J=11.90,3.66Hz,1H),4.42(d,J=11.60Hz,1H),4.57dd,J=10.22,7.17Hz,1H),5.13(d,J=10.38Hz,1H),5.30(d,J=17.09Hz,1H),5.76(ddd,J=17.09,9.77,9.46Hz,1H),5.87(s,1H),6.79(d,J=8.24Hz,2H),7.07(d,J=8.24Hz,2H),7.30(d,J=6.10Hz,1H),7.40(t,J=7.63Hz,1H),7.68(t,J=7.63Hz,1H),7.79(d,J=8.24Hz,1H),7.93(d,J=5.80Hz,1H),8.17(d,J=8.24Hz,1H);MS m/z 718(MH+).
实施例121:化合物121的制备
化合物121
按照实施例120、方案1的方法制备化合物121,不同之处在于在步骤2中使用氯甲酸苯酯代替氯甲酸对甲苯酯。
步骤2:
修改:使用30mg(0.19mmol)氯甲酸苯酯,得到89.0mg产物,为黄色玻璃状固体物(收率50%):MS m/z 704(MH+)。
实施例122:化合物122的制备
化合物122
按照实施例120、方案1的方法制备化合物122,不同之处在于在步骤2中使用氯甲酸4-氟苯酯代替氯甲酸对甲苯酯。
步骤2:
修改:使用33mg(0.19mmol)氯甲酸4-氟苯酯,得到83.1mg产物,为黄色粘稠油状物(收率78.8%):MS m/z 722(MH+)。
实施例123:化合物123的制备
化合物123
按照实施例120、方案1的方法制备化合物123,不同之处在于在步骤2中使用氯甲酸4-甲氧基苯酯代替氯甲酸对甲苯酯。
步骤2:
修改:使用35mg(0.19mmol)氯甲酸4-甲氧基苯酯,得到70.2mg产物,为黄色玻璃状固体物(收率65.4%):
1H NMR(CD3OD)δ1.06-1.10(m,3H),1.11(s,9H),1.24-1.28(m,2H),1.44(dd,J=9.46,5.49Hz,1H),1.89(dd,J=7.93,5.49Hz,1H),2.24(q,J=8.85Hz,1H),2.31(ddd,J=13.81,10.30,3.97Hz,1H),2.62-2.66(m,1H),2.94-2.98(m,1H),3.77(s,3H),4.12(dd,J=11.60,3.66Hz,1H),4.42(d,J=11.60Hz,1H),4.57(dd,J=10.07,7.32Hz,1H),5.13(d,J=10.68Hz,1H),5.30(d,J=16.79Hz,1H),5.72-5.80(m,1H),5.87(s,1H),6.80(d,J=2.44Hz,4H),7.30(d,J=5.80Hz,1H),7.42(t,J=7.48Hz,1H),7.69(t,J=7.63Hz,1H),7.80(d,J=7.93Hz,1H),7.93(d,J=5.80Hz,1H),8.18(d,J=8.24Hz,1H);MS m/z734(MH+).
实施例124:化合物124的制备
化合物124
按照实施例120、方案1的方法制备化合物124,不同之处在于在步骤2中使用氯甲酸2-甲氧基乙酯代替氯甲酸对甲苯酯。
步骤2:
修改:使用26mg(0.19mmol)氯甲酸2-甲氧基乙酯,得到87.4mg产物,为黄色粘稠油状物(收率87.2%):
1H NMR(CD3OD)δ0.96-1.02(m,3H),1.05(s,9H),1.16-1.18(m,2H),1.40(dd,J=9.46,5.19Hz,1H),1.85(dd,J=7.93,5.19Hz,1H),2.15(q,J=8.75Hz,1H),2.40(ddd,J=13.89,10.07,4.12Hz,1H),2.65(dd,J=13.58,7.17Hz,1H),2.90(ddd,J=12.89,8.16,4.88Hz,1H),3.27(s,3H),3.36-3.44(m,2H),3.81-3.84(m,1H),3.92-3.96(m,1H),4.12(dd,J=11.60,3.36Hz,1H),4.44(d,J=11.60Hz,1H),4.57(dd,J=9.46,7.93Hz,1H),5.07(d,J=10.38Hz,1H),5.25(d,J=17.09Hz,1H),5.80(ddd,J=17.32,9.77,9.54Hz,1H),5.87(s,1H),7.32(d,J=5.80Hz,1H),7.55(t,J=7.32Hz,1H),7.70(t,J=7.48Hz,1H),7.80(d,J=7.93Hz,1H),7.96(d,J=5.80Hz,1H),8.19(d,J=8.24Hz,1H);MS m/z686(MH+).
实施例125:化合物125的制备
化合物125
按照实施例120、方案1的方法制备化合物125,不同之处在于在步骤2中使用氯甲酸新戊酯代替氯甲酸对甲苯酯。
步骤2:
修改:使用29mg(0.19mmol)氯甲酸新戊酯,得到57.4mg产物,为黄色玻璃状固体物(收率56.2%):
1H NMR(CD3OD)δ0.83(s,9H),1.05(d,J=2.44Hz,9H),1.07-1.09(m,2H),1.23-1.27(m,2H),1.43-1.46(m,1H),1.87-1.90(m,1H),2.21-2.25(m,1H),2.29-2.33(m,1H),2.61-2.65(m,1H),2.92-2.96(m,1H),3.42(d,J=10.07Hz,1H),3.56(d,J=10.07Hz,1H),4.09-4.11(m,1H),4.33(d,J=9.16Hz,1H),4.43(d,J=11.29Hz,1H),4.54-4.57(m,1H),5.12(d,J=10.07Hz,1H),5.30(d,J=17.40Hz,1H),5.73-5.80(m,1H),5.88(s,1H),7.33(d,J=5.49Hz,1H),7.53(m,1H),7.71(t,J=6.87Hz,1H),7.81(d,J=7.93Hz,1H),7.97(d,J=5.80Hz,1H),8.19(d,J=7.63Hz,1H);MS m/z 698(MH+).
实施例126:化合物126的制备
化合物126
按照实施例120、方案1的方法制备化合物126,不同之处在于在步骤2中使用氯甲酸2-氟乙酯代替氯甲酸对甲苯酯。
步骤2:
修改:使用24mg(0.19mmol)氯甲酸2-氟乙酯,得到58.9mg产物,为黄色玻璃状固体物(收率59.8%):
1H NMR(CD3OD)δ1.05(d,J=2.14Hz,9H),1.07-1.09(m,2H),1.22-1.27(m,2H),1.42-1.45(m,1H),1.87-1.90(m,1H),2.24(q,J=8.75Hz,1H),2.28-2.33(m,1H),2.63(dd,J=13.43,6.41Hz,1H),2.92-2.96(m,1H),3.92-4.10(m,3H),4.31-4.37(m,2H),4.42-4.46(m,2H),4.54-4.57(m,1H),5.12(d,J=10.38Hz,1H),5.29(d,J=17.09Hz,1H),5.71-5.79(m,1H),5.88(s,1H),7.33(d,J=5.80Hz,1H),7.55(t,J=7.17Hz,1H),7.71(m,1H),7.81(d,J=7.93Hz,1H),7.96(d,J=5.80Hz,1H),8.19(d,J=7.63Hz,1H);MS m/z 674(MH+).
实施例127:化合物127的制备
化合物127
按照实施例120、方案1的方法制备化合物127,不同之处在于在步骤2中使用氯甲酸2-甲氧基苯酯代替氯甲酸对甲苯酯。
步骤2:
修改:使用35mg(0.19mmol)氯甲酸2-甲氧基苯酯,得到97.6mg产物,为黄色粘稠油状物(收率91.0%):MS m/z 734(MH+)。
实施例128:化合物128的制备
化合物128
按照实施例120、方案1的方法制备化合物128,不同之处在于在步骤2中使用2-(-)-(1R)-氯甲酸薄荷基酯代替氯甲酸对甲苯酯。
步骤2:
修改:使用42mg(0.19mmol)(-)-(1R)-氯甲酸薄荷基酯,得到69.1mg产物,为白色玻璃状固体物(收率61.7%):MS m/z 766(MH+)。
实施例129:化合物129的制备
化合物129
按照实施例120、方案1的方法制备化合物129,不同之处在于在步骤2中使用氯甲酸己酯代替氯甲酸对甲苯酯。
步骤2:
修改:使用31mg(0.19mmol)氯甲酸己酯,得到66.7mg产物,为黄色玻璃状固体物(收率64.1%):
1H NMR(CD3OD)δ0.87-0.99(m,5H),1.05(s,9H),1.07-1.09(m,2H),1.22-1.28(m,6H),1.43-1.48(m,3H),1.88(dd,J=8.24,5.49Hz,1H),2.24(q,J=8.85Hz,1H),2.28-2.33(m,1H),2.63(dd,J=14.34,7.63Hz,1H),2.92-2.97(m,1H),3.72(dt,J=10.61,6.60Hz,1H),3.81-3.86(m,1H),4.10(dd,J=11.60,3.36Hz,1H),4.32(d,J=8.85Hz,1H),4.43(d,J=11.90Hz,1H),4.55(dd,J=9.77,7.32Hz,1H),5.13(d,J=10.38Hz,1H),5.30(d,J=17.09Hz,1H),5.76(ddd,J=17.09,10.07,9.16Hz,1H),5.89(s,1H),7.33(d,J=5.80Hz,1H),7.54(t,J=7.48Hz,1H),7.69-7.72(m,1H),7.81(d,J=8.24Hz,1H),7.97(d,J=6.10Hz,1H),8.20(d,J=8.24Hz,1H);MS m/z 712(Hz+).
实施例130:化合物130的制备
化合物130
方案1
步骤1:
将化合物23(参见实施例23)(1.50g,2.19mmol)在DCM(50mL)和三氟乙酸(50mL)中的溶液在室温下搅拌3h。将混合物真空浓缩至得到粘稠剩余物,随后溶于1,2-二氯乙烷并再真空浓缩,得到所需的双-三氟乙酸盐产物,为灰白色玻璃状固体物(定量)。该物质未经进一步纯化直接用于下一步骤中。
步骤2:
将步骤1的产物(118mg,0.146mmol)、叔丁基乙酸(22mg,0.19mmol)、HATU(72mg,0.19mmol)和N-甲基吗啉(59mg,0.58mmol)在1,2-二氯乙烷中的混合物在室温下搅拌24h。将反应混合物用pH=4的缓冲溶液(3×3mL)洗涤,将洗液用1,2-二氯乙烷(3mL)反萃取。合并有机相并真空浓缩。随后将粗产物溶于甲醇并经反相制备型HPLC纯化,得到题述化合物(化合物130),为浅黄色玻璃状固体物(43.4mg,43.5%收率):
1H NMR(CD3OD)δ0.82(d,J=1.83Hz,9H),1.06(d,J=2.14Hz,9H),1.07-1.10(m,2H),1.22-1.28(m,2H),1.43-1.46(m,1H),1.87-1.90(m,1H),1.99(d,J=1.83Hz,2H),2.20-2.26(m,1H),2.27-2.33(m,1H),2.59-2.64(m,1H),2.93-2.97(m,1H),4.12-4.14(m,1H),4.42(d,J=11.60Hz,1H),4.51-4.55(m,1H),4.67(dd,J=9.31,1.98Hz,1H),5.11-5.14(m,1H),5.29(d,J=17.40Hz,1H),5.72-5.80(m,1H),5.89(d,J=1.83Hz,1H),7.32(dd,J=5.80,2.14Hz,1H),7.52-7.55(m,1H),7.69-7.72(m,1H),7.81(d,J=8.24Hz,1H),7.96(dd,J=5.80,1.83Hz,1H),8.17(d,J=8.24Hz,1H);MS m/z682(MH+).
实施例131:化合物131的制备
化合物131
按照实施例130、方案1的方法制备化合物131,不同之处在于在步骤2中使用甲氧基乙酸代替叔丁基乙酸。
步骤2:
修改:使用17mg(0.19mmol)甲氧基乙酸,得到49.9mg产物,为浅黄色玻璃状固体物(收率52.0%):1H NMR(CD3OD)δ1.05-1.08(m,11H),1.24-1.26(m,2H),1.45(ddd,J=9.31,5.34,3.66Hz,1H),1.88(ddd,J=8.39,5.19,3.81Hz,1H),2.21-2.27(m,1H),2.29-2.35(m,1H),2.60-2.64(m,1H),2.91-2.97(m,1H),3.34(d,J=3.66Hz,314),3.69(dd,J=15.26,3.66Hz,1H),3.81-3.85(m,1H),4.15(dt,J=11.67,3.62Hz,1H),4.35(d,J=11.90Hz,1H),4.55(ddd,J=10.30,6.94,3.20Hz,1H),4.66(dd,J=9.61,3.51Hz,1H),5.11-5.14(m,1H),5.28-5.32(m,1H),5.73-5.81(m,1H),5.90(d,J=3.36Hz,1H),7.33(dd,J=5.65,3.20Hz,1H),7.54-7.58(m,1H),7.69-7.73(m,1H),7.80-7.82(m,1H),7.95-7.97(m,1H),8.15(dd,J=8.39,2.59Hz,1H);MS m/z656(MH+)。
实施例132:化合物132的制备
化合物132
按照实施例130、方案1的方法制备化合物132,不同之处在于在步骤2中使用甲氧基丙酸代替叔丁基乙酸。
步骤2:
修改:使用20mg(0.19mmol)甲氧基丙酸,得到50.0mg产物,为黄色玻璃状固体物(收率51.1%):
1H NMR(CD3OD)δ1.06(d,J=1.83Hz,9H),1.07-1.09(m,2H),1.23-1.27(m,2H),1.44(ddd,J=9.38,5.26,1.83Hz,1H),1.87-1.90(m,1H),2.21-2.27(m,1H),2.29-2.33(m,2H),2.40-2.46(m,1H),2.59-2.64(m,1H),2.92-2.97(m,1H),3.25(d,J=1.83Hz,3H),3.45-3.54(m,2H),4.12-4.16(m,1H),4.37(d,J=11.60Hz,1H),4.52-4.55(m,1H),4.65(dd,J=9.16,1.83Hz,1H),5.12(d,J=10.38Hz,1H),5.30(d,J=17.40Hz,1H),5.72-5.80(m,1H),5.89(s,1H),7.33(dd,J=5.65,1.98Hz,1H),7.54-7.58(m,1H),7.69-7.73(m,1H),7.81(d,J=8.24Hz,1H),7.96(dd,J=6.10,1.83Hz,1H),8.18(d,J=8.24Hz,1H);MS m/z 670(MH+).
实施例133:化合物133的制备
化合物133
按照实施例130、方案1的方法制备化合物133,不同之处在于在步骤2中使用(S)-1,4-苯并二_烷-2-甲酸代替叔丁基乙酸。
步骤2:
修改:使用35mg(0.19mmol)(S)-1,4-苯并二_烷-2-甲酸,得到54.0mg产物,为浅黄色玻璃状固体物(49.5%收率):
1H NMR(CD3OD)δ0.83(d,J=3.36Hz,9H),1.05-1.08(m,2H),1.20-1.25(m,1H),1.26-1.31(m,1H),1.45-1.49(m,1H),1.86-1.90(m,1H),2.21-2.25(m,1H),2.28-2.34(m,1H),2.59-2.65(m,1H),2.90-2.94(m,1H),4.12-4.17(m,2H),4.32(d,J=11.90Hz,1H),4.35-4.39(m,1H),4.55-4.61(m,3H),5.11-5.14(m,1H),5.28-5.32(m,1H),5.75-5.83(m,1H),5.90(d,J=3.66Hz,1H),6.80-6.89(m,3H),7.03-7.07(m,1H),7.32-7.34(m,1H),7.55-7.58(m,1H),7.68-7.72(m,1H),7.80-7.82(m,1H),7.96-7.98(m,1H),8.15-8.18(m,1H);MS m/z746(MH+)。
实施例134:化合物134的制备
化合物134
方案1
步骤1:
将实施例11步骤5的产物(100mg,0.172mmol)、N-α-叔丁氧基羰基-L-苯基甘氨酸(45.3mg,0.180mmol)、HATU(84.9mg,0.223mmol)和N-甲基吗啉(87.0mg,0.859mmol)在DMF(1.0mL)中的混合物在室温下搅拌18h。混合物直接经反相制备型HPLC纯化,得到29.7mg(收率23.6%)化合物134,为白色粉末:
1HNMR(CD3OD)δ0.97-1.07(m,2H),1.12-1.17(m,1H),1.22-1.32(m,2H),1.38(s,9H),1.90(dd,J=8.09,5.34Hz,1H),2.20-2.28(m,2H),2.54(dd,J=13.58,6.56Hz,1H),2.85-2.89(m,J=8.24Hz,1H),3.50(d,J=10.99Hz,1H),3.93(s,3H),4.11(d,J=11.60Hz,1H),4.63(dd,J=9.46,7.32Hz,1H),5.13(dd,J=10.38,1.53Hz,1H),5.32(d,J=17.09Hz,1H),5.47(s,1H),5.74-5.84(m,2H),7.16-7.19(m,1H),7.25(d,J=5.80Hz,1H),7.32-7.43(m,6H),7.86(d,J=5.80Hz,1H),8.13(d,J=9.16Hz,1H);MS m/z 734(MH+).
实施例135:化合物135的制备
化合物135
按照实施例134、方案1的方法制备化合物135,不同之处在于在步骤1中使用N-α-叔丁氧基羰基-赤型-DL-β-甲基苯丙氨酸代替N-α-叔丁氧基羰基-L-苯甘氨酸。由N-α-叔丁氧基羰基-赤型DLβ-甲基苯丙氨酸的混合物制备化合物135,将所得的两种非对映异构体经反相制备型HPLC分离。该化合物为先从制备型HPLC柱中洗脱出的单一异构体。分子的β-甲基苯丙氨酸部分的确切的立体化学构型未知。
步骤1:
修改:使用50.4mg(0.180mmol)N-α-叔丁氧基羰基-赤型-DL-β-甲基苯丙氨酸,得到29.7mg产物,为白色粉末(收率22.7%):
1H NMR(CD3OD)δ1.11(d,J=7.93Hz,2H),1.15(d,J=6.10Hz,3H),1.24-1.32(m,11H),1.44(dd,J=9.16,5.19Hz,1H),1.90-1.94(m,1H),2.25-2.29(m,1H),2.36(t,J=13.28Hz,1H),2.62(dd,J=13.58,7.17Hz,1H),2.98-3.02(m,1H),3.20-3.24(m,1H),3.91(s,3H),4.11(dd,J=11.60,3.05Hz,1H),4.51(d,J=10.68Hz,1H),4.57(dd,J=10.07,7.32Hz,1H),4.63(d,J=12.21Hz,1H),5.14(d,J=10.07Hz,1H),5.32(d,J=16.79Hz,1H),5.76-5.84(m,1H),5.88(s,1H),7.08(dd,J=8.70,1.68Hz,1H),7.16-7.18(m,2H),7.23-7.27(m,5H),7.89(d,J=5.80Hz,1H),8.09(d,J=9.46Hz,1H);MS m/z 762(MH+).
实施例136:化合物136的制备
化合物136
按照实施例134、方案1的方法制备化合物136,不同之处在于在步骤1中使用N-α-叔丁氧基羰基-赤型-DL-β-甲基苯丙氨酸代替N-α-叔丁氧基羰基-L-苯甘氨酸。由N-叔丁氧基羰基-赤型DL[β]-甲基苯丙氨酸的混合物制备化合物136,所得的两种非对映异构体经反相制备型HPLC分离。该化合物为从制备型HPLC柱中较后洗脱出的单一异构体。分子的β-甲基苯丙氨酸部分的确切立体化学构型未知。
步骤1:
修改:使用50.4mg(0.180mmol)N-α-叔丁氧基羰基-赤型-DL-β-甲基苯丙氨酸,得到26.3mg产物,为白色粉末(收率20.1%):
1H NMR(CD3OD)δ1.04(s,1H),1.13(d,J=6.71Hz,3H),1.12-1.17(m,2H),1.30(s,9H),1.33-1.36(m,1H),1.41(dd,J=9.46,5.19Hz,1H),1.87(dd,J=7.78,5.34Hz,1H),2.29(q,J=8.85Hz,1H),2.36(ddd,J=13.81,9.99,4.27Hz,1H),2.54(dd,J=13.58,7.17Hz,1H),3.00-3.04(m,1H),3.05-3.08(m,1H),3.80(d,J=11.90Hz,1H),3.94(s,3H),4.10(dd,J=12.05,3.81Hz,1H),4.53-4.57(m,1H),4.59(d,J=8.24Hz,1H),5.14(d,J=10.38Hz,1H),5.34(d,J=17.09Hz,1H),5.78-5.85(m,2H),6.75(t,J=7.32Hz,1H),7.03(t,J=7.48Hz,2H),7.12(s,1H),7.14(s,1H),7.19(dd,J=9.31,1.68Hz,1H),7.22(s,1H),7.28(d,J=6.10Hz,1H),7.89(d,J=5.80Hz,1H),8.03(d,J=8.85Hz,1H);MS m/z 762(MH+).
实施例137:化合物137的制备
化合物137
方案1
步骤1:
将实施例11、步骤5的产物(100mg,0.172mmol)、N-α-叔丁氧基羰基-L-天冬氨酸4-苄酯(59.5mg,0.180mmol)、HATU(84.9mg,0.223mmol)和N-甲基吗啉(87.0mg,0.859mmol)在DCM(3.0mL)中的混合物在室温下搅拌18h。将反应混合物用pH=4的缓冲溶液(3×3mL)洗涤,将洗液用DCM(3mL)反萃取。合并有机相并真空浓缩。随后将粗产物溶于甲醇中并经反相制备型HPLC纯化,得到化合物137,为浅灰白色玻璃状固体物(26.0mg,收率18.8%):
1H NMR(CD3OD)δ0.95-1.01(m,2H),1.16(s,9H),1.22-1.29(m,2H),1.44(dd,J=9.46,5.19Hz,1H),1.86dd,J=7.93,5.19Hz,1H),2.26(q,J=8.85Hz,1H),2.32-2.37(m,1H),2.61(dd,J=13.73,7.32Hz,1H),2.66(dd,J=16.48,6.10Hz,1H),2.89(ddd,J=12.67,8.09,4.88Hz,1H),3.05(dd,J=16.63,839Hz,1H),3.92(s,3H),4.04-4.07(m,1H),4.47(d,J=11.90Hz,1H),4.52-4.56(m,1H),4.75(dd,J=8.24,6.41Hz,1H),5.12-5.14(m,1H),5.14(s,2H),5.31(d,J=17.09Hz,1H),5.75-5.82(m,2H),7.12(d,J=9.16Hz,1H),7.18(d,J=2.14Hz,1H),7.24(d,J=5.80Hz,1H),7.31-7.33(m,1H),7.36(t,J=7.32Hz,2H),7.39(s,1H),7.41(s,1H),7.88(d,J=6.10Hz,1H),8.10(d,J=9.16Hz,1H);MS m/z 806(MH+).
实施例138:化合物138的制备
化合物138
按照实施例137、方案1的方法制备化合物138,不同之处在于在步骤1中使用N-叔丁氧基羰基-L-天冬氨酸4-甲基酯代替N-α-叔丁氧基羰基-L-天冬氨酸4-苄酯。
步骤1:
修改:使用45.5mg(0.180mmol)N-叔丁氧基羰基-L-天冬氨酸4-甲酯,得到93.5mg产物,为灰白色玻璃状固体物(收率74.6%):
1H NMR(CD3OD)δ1.07-1.09(m,2H),1.17(s,9H),1.20-1.29(m,2H),1.41-1.44(m,1H),1.84-1.86(m,1H),2.26(q,J=8.85Hz,1H),2.33-2.38(m,1H),2.58-2.64(m,2H),2.92-3.02(m,2H),3.69(s,3H),3.92(s,3H),4.15(dd,J=11.44,2.29Hz,1H),4.49-4.56(m,2H),4.72--4.76(m,1H),5.13(d,J=10.38Hz,1H),5.32(d,J=17.09Hz,1H),5.74-5.82(m,1H),5.87(s,1H),7.12(d,J=9.16Hz,1H),7.18(s,1H),7.24(d,J=5.80Hz,1H),7.88(dd,J=5.80,0.92Hz,1H),8.10(d,J=8.85Hz,1H);MS m/z 730(MH+).
实施例139:化合物139的制备
化合物139
按照实施例137、方案1的方法制备化合物139,不同之处在于在步骤1中使用N-叔丁氧基羰基-L-天冬氨酸4-叔丁酯代替N-α-叔丁氧基羰基-L-天冬氨酸4-苄酯。
步骤1:
修改:使用52.2mg(0.180mmol)N-叔丁氧基羰基-L-天冬氨酸4-叔丁酯,得到125mg产物,为灰白色玻璃状固体物(收率99.8%):
1H NMR(CD3OD)δ1.08-1.10(m,2H),1.17(s,9H),1.21-1.29(m,2H),1.46(s,10H),1.82(dd,J=7.78,5.34Hz,1H),2.26(q,J=8.75Hz,1H),2.32-2.38(m,1H),2.51(dd,J=16.33,7.17Hz,1H),2.63(dd,J=14.04,7.02Hz,1H),2.89(dd,J=16.48,7.63Hz,1H),2.92-2.98(m,1H),3.92(s,3H),4.15(dd,J=11.60,3.05Hz,1H),4.50-4.57(m,2H),4.70-4.75(m,1H),5.13(dd,J=10.38,1.53Hz,1H),5.32(d,J=17.40Hz,1H),5.76-5.83(m,1H),5.87(s,1H),7.13(dd,J=8.85,1.53Hz,1H),7.18(d,J=2.14Hz,1H),7.25(d,J=5.80Hz,1H),7.88(d,J=5.80Hz,1H),8.10(d,J=9.16Hz,1H);MS m/z 772(MH+).
实施例140:化合物140的制备
化合物140
方案1
将化合物138(50.0mg,0.0685mmol)溶于THF(1mL)、甲醇(1mL)和1.0M氢氧化钠水溶液(0.137mL,0.137mmol)的混合物中。3小时后,加入1.0M盐酸(0.137mL,0.137mmol)中和反应混合物。将粗品混合物真空浓缩,随后加入pH=4的缓冲溶液(3mL)和DCM(3mL)并将混合物摇荡。分离各层,水层再次用DCM(2×1mL)萃取。将有机相合并,用无水硫酸镁干燥,过滤并真空浓缩,得到48.0mg(收率97.9%)化合物140,为类白色玻璃状固体物:
1H NMR(CD3OD)δ1.04-1.07(m,2H),1.17(s,9H),1.22-1.25(m,2H),1.38(dd,J=9.31,5.34Hz,1H),1.78(dd,J=7.78,5.34Hz,1H),2.30(q,J=8.75Hz,1H),2.37-2.42(m,1H),2.48(dd,J=15.87,4.58Hz,1H),2.72(dd,J=13.12,7.63Hz,1H),2.88(dd,J=15.72,10.53Hz,1H),2.90-2.95(m,1H),3.92(s,3H),4.21(dd,J=11.44,2.90Hz,1H),4.57(t,J=8.70Hz,1H),4.62-4.65(m,2H),5.10(d,J=10.68Hz,1H),5.34(d,J=17.09Hz,1H),5.69-5.76(m,1H),5.83(s,1H),7.10(d,J=9.16Hz,1H),7.17(s,1H),7.23(d,J=6.10Hz,1H),7.88(d,J=6.10Hz,1H),8.12(d,J=8.85Hz,1H);MS m/z716(MH+).
实施例141:化合物141的制备
化合物141
方案1
步骤1:
将实施例55、步骤1的产物(65mg,0.0947mmol)、碳酸N,N’-二琥珀酰亚胺酯(41.0mg,0.142mmol)和N,N-二异丙基乙胺(30.6mg,0.237mmol)与无水THF(1mL)混合,将所得的悬浮液在微波反应器中加热至80℃下反应15min。冷却至室温后,将该粗品混合物直接用于下一步骤。
步骤2:
将步骤1的粗品反应混合物用L-缬氨酸甲酯盐酸盐(159mg,0.947mmol)和N,N-二异丙基乙胺(122mg,0.947mmol)在无水THF(2mL)中的混合物处理。将所得的混合物在室温下搅拌18h。真空除去溶剂,将剩余物溶于DCM(2mL)中,用pH=4的缓冲溶液(3×2mL)洗涤。将缓冲液洗液合并并用DCM(2mL)反萃取。将合并的DCM相真空浓缩,将所得的剩余物溶于甲醇中并经反相制备型HPLC纯化,得到38.1mg(收率52.2%)化合物141,为白色粉末:
1H NMR(CD3OD)δ0.83(dd,J=6.87,3.81Hz,6H),1.06(s,11H),1.21-1.26(m,2H),1.41(dd,J=9.46,5.49Hz,1H),1.87(dd,J=8.09,5.34Hz,1H),1.95-2.02(m,1H),2.21(q,J=8.85Hz,1H),2.29(ddd,J=13.89,9.92,4.27Hz,1H),2.60(dd,J=13.73,7.02Hz,1H),2.91-2.97(m,1H),3.67(s,3H),3.93(s,3H),4.00(d,J=5.49Hz,1H),4.09(dd,J=11.90,3.97Hz,1H),4.40-4.43(m,2H),4.52(dd,J=10.07,7.02Hz,1H),5.11(dd,J=10.38,1.53Hz,H)),5.28(dd,J=17.09,1.22Hz,1H),5.75(ddd,J=17.17,10.15,9.00Hz,1H),5.83(s,1H),7.11(dd,J=9.16,2.44Hz,1H),7.18(d,J=2.44Hz,1H),7.24(d,J=5.80Hz,1H),7.87(d,J=6.10Hz,1H),8.10(d,J=8.85Hz,1H);MS m/z 771(MH+).
实施例142:化合物142的制备
化合物142
按照实施例141、方案1的方法制备化合物142,不同之处在于在步骤2中使用D-缬氨酸甲酯盐酸盐代替L-缬氨酸甲酯盐酸盐。
步骤2:
修改:使用159mg(0.947mmol)D-缬氨酸甲酯盐酸盐,得到23.0mg产物,为白色粉末(收率31.5%):
1H NMR(CD3OD)δ0.88(dd,J=13.89,6.87Hz,6H),1.06(s,9H),1.07-1.09(m,2H),1.23-1.27(m,2H),1.41(dd,J=9.46,5.49Hz,1H),1.88(dd,J=7.93,5.49Hz,1H),2.01-2.07(m,1H),2.23(q,J=9.05Hz,1H),2.31(ddd,J=14.11,9.99,4.27Hz,1H),2.63(dd,J=13.89,7.17Hz,1H),2.93-2.98(m,1H),3.62(s,3H),3.96(s,3H),4.03(d,J=5.19Hz,1H),4.09(dd,J=11.75,3.81Hz,1H),4.38(s,1H),4.48-4.54(m,2H),5.12(dd,J=10.38,1.22Hz,1H),5.29(dd,J=17.24,1.07Hz,1H),5.70-5.77(m,1H),5.83(s,1H),7.22(dd,J=9.00,2.59Hz,1H),7.25(d,J=2.44Hz,1H),7.35(d,J=6.10Hz,1H),7.87(d,J=6.10Hz,1H),8.16(d,J=9.16Hz,1H);MS m/z 771(MH+).
实施例143:化合物143的制备
化合物143
按照实施例137、方案1的方法制备化合物143,不同之处在于在步骤1中使用N-叔丁氧基羰基-L-环己基甘氨酸代替N-α-叔丁氧基羰基-L-天冬氨酸4-苄酯。
步骤1:
修改:使用46.2mg(0.180mmol)N-叔丁氧基羰基-L-环己基甘氨酸,得到93.9mg产物,为白色粉末(收率73.8%):
1H NMR(CD3OD)δ1.04-1.08(dd,J=7.78,2.29Hz,4H),1.19-1.26(m,4H),1.25(s,9H),1.41(dd,J=9.46,5.19Hz,1H),1.63-1.82(m,7H),1.88(dd,J=7.93,5.49Hz,1H),2.22(q,J=9.05Hz,1H),2.32-2.37(m,1H),2.59(dd,J=13.58,6.87Hz,1H),2.91-2.96(m,1H),3.92(s,3H),4.05(dd,J=11.75,3.20Hz,1H),4.09(d,J=8.85Hz,1H),4.47(d,J=11.90Hz,1H),4.53(dd,J=10.22,7.17Hz,1H),5.11(d,J=10.38Hz,1H),5.29(d,J=16.79Hz,1H),5.79(ddd,J=16.86,9.92,9.54Hz,1H),5.84(s,1H),7.10(d,J=8.85Hz,1H),7.17(d,J=1.53Hz,1H),7.24(d,J=5.80Hz,1H),7.88(d,J=6.10Hz,1H),8.09(d,J=8.85Hz,1H);MS m/z 740(MH+).
实施例144:化合物144的制备
化合物144
按照实施例141、方案1的方法制备化合物144,不同之处在于在步骤2中扩大规模并使用甘氨酸甲酯盐酸盐代替L-缬氨酸甲酯盐酸盐。
步骤1:
修改:使用100mg(0.146mmol)实施例55、步骤1的产物;62.2mg(0.219mmol)碳酸N,N’-二琥珀酰亚胺酯;47.0mg(0.364mmol)N,N-二异丙基乙胺。
步骤2:
修改:使用183mg(1.46mmol)甘氨酸甲酯盐酸盐、188mg(1.46mmol)N,N-二异丙基乙胺,得到56.3mg产物,为白色粉末(收率52.9%):
1H NMR(CD3OD)δ1.01-1.04(m,2H),1.05(s,9H),1.17-1.21(m,2H),1.40(dd,J=9.46,5.49Hz,1H),1.85(dd,J=7.78,5.34Hz,1H),2.18(q,J=8.55Hz,1H),2.33(ddd,J=13.89,9.92,4.27Hz,1H),2.61(dd,J=13.73,7.32Hz,1H),2.89-2.94(m,1H),3.65(s,3H),3.69-3.77(m,2H),3.92(s,3H),4.10(dd,J=11.75,3.81Hz,1H),4.40-4.42(m,2H),4.53(dd,J=9.92,7.17Hz,1H),5.08(d,J=10.38Hz,1H),5.26(d,J=17.09Hz,1H),5.77(ddd,J=17.09,10.22,9.00Hz,1H),5.83(s,1H),7.13(dd,J=8.85,2.44Hz,1H),7.17(s,1H),7.23(d,J=5.80Hz,1H),7.87(d,J=5.80Hz,1H),8.09(d,J=8.85Hz,1H);MS m/z 729(MH+).
实施例145:化合物145的制备
化合物145
按照实施例141、方案1的方法制备化合物145,不同之处在于扩大规模并在步骤2中使用L-丙氨酸甲酯盐酸盐代替L-缬氨酸甲酯盐酸盐。
步骤1:
修改:使用100mg(0.146mmol)实施例55、步骤1的产物;62.2mg(0.219mmol)碳酸N,N’-二琥珀酰亚胺酯;47.0mg(0.364mmol)N,N-二异丙基乙胺。
步骤2:
修改:使用203mg(1.46mmol)L-丙氨酸甲酯盐酸盐,188mg(1.46mmol)N,N-二异丙基乙胺,得到64.3mg产物,为白色粉末(收率59.3%):
1H NMR(CD3OD)δ0.97-1.02(m,2H),1.05(s,9H),1.19(d,J=7.02Hz,3H),1.18-1.22(m,2H),1.41(dd,J=9.46,5.19Hz,1H),1.86(dd,J=8.09,5.34Hz,1H),2.19(q,J=8.85Hz,1H),2.32(ddd,J=13.81,9.84,4.43Hz,1H),2.61(dd,J=13.73,7.02Hz,1H),2.93(ddd,J=12.82,8.09,4.73Hz,1H),3.65(s,3H),3.93(s,3H),3.99(q,J=7.22Hz,1H),4.08(dd,J=11.75,3.81Hz,1H),4.38(s,1H),4.42(d,J=11.60Hz,1H),4.53(dd,J=10.07,7.32Hz,1H),5.09(dd,J=10.38,1.53Hz,1H),5.27(dd,J=17.09,1.22Hz,1H),5.77(ddd,J=17.09,10.07,9.16Hz,1H),5.82(s,1H),7.13(dd,J=9.16,2.44Hz,1H),7.18(d,J=2.44Hz,1H),7.24(d,J=5.80Hz,1H),7.87(d,J=5.80Hz,1H),8.10(d,J=9.16Hz,1H);MSm/z 743(MH+).
实施例146:化合物146的制备
化合物146
按照实施例141、方案1的方法制备化合物147,不同之处在于扩大规模并在步骤2中使用L-叔-亮氨酸甲酯盐酸盐代替L-缬氨酸甲酯盐酸盐。
步骤1:
修改:使用100mg(0.146mmol)实施例55、步骤1的产物;62.2mg(0.219mmol)碳酸N,N’-二琥珀酰亚胺酯;47.0mg(0.364mmol)N,N-二异丙基乙胺。
步骤2:
修改:使用265mg(1.46mmol)L-叔-亮氨酸甲酯盐酸盐、188mg(1.46mmol)N,N-二异丙基乙胺,得到68.3mg产物,为白色粉末(收率59.6%):
1H NMR(CD3OD)δ0.88(s,9H),0.97-1.03(m,2H),1.06(s,9H),1.18-1.24(m,2H),1.41(dd,J=9.46,5.49Hz,1H),1.86(dd,J=7.93,5.49Hz,1H),2.19(q,J=8.75Hz,1H),2.30(ddd,J=13.89,10.07,4.43Hz,1H),2.60(dd,J=13.73,7.32Hz,1H),2.91-2.96(m,1H),3.65(s,3H),3.91(s,1H),3.93(s,3H),4.09(dd,J=11.60,3.97Hz,1H),4.38(s,1H),4.43(d,J=11.60Hz,1H),4.51(dd,J=10.07,7.32Hz,1H),5.10(dd,J=10.38,1.53Hz,1H),5.27(dd,J=17.24,1.37Hz,1H),5.76(ddd,J=17.09,10.07,9.16Hz,1H),5.82(s,1H),7.12(dd,J=9.16,2.44Hz,1H),7.18(d,J=2.14Hz,1H),7.24(d,J=6.10Hz,1H),7.87(d,J=5.80Hz,1H),8.11(d,J=9.16Hz,1H);MS m/z 785(MH+).
实施例147:化合物147的制备
化合物147
按照实施例141、方案1的方法制备化合物147,不同之处在于扩大规模并在步骤2中使用L-组氨酸甲酯盐酸盐代替L-缬氨酸甲酯盐酸盐,并在步骤2中使用2倍量的N,N-二异丙基乙胺。
步骤1:
修改:使用100mg(0.146mmol)实施例55、步骤1的产物;62.2mg(0.219mmol)碳酸N,N’-二琥珀酰亚胺酯;47.0mg(0.364mmol)N,N-二异丙基乙胺。
步骤2:
修改:使用352mg(1.46mmol)L-组氨酸甲酯盐酸盐、377mg(2.91mmol)N,N-二异丙基乙胺,得到51.0mg产物,为白色粉末(收率43.2%):
1H NMR(CD3OD)δ1.04(s,11H),1.20-1.22(m,2H),1.41(ddd,J=9.46,5.34,1.07Hz,1H),1.86-1.88(m,1H),2.23(q,J=8.75Hz,1H),2.28-2.33(m,1H),2.60(dd,J=13.73,7.02Hz,1H),2.92(d,J=6.41Hz,2H),2.92-2.96(m,1H),3.64(s,3H),3.91(d,J=1.53Hz,3H),4.04(dd,J=11.90,3.66Hz,1H),4.35(s,1H),4.36-4.41(m,2H),4.53(dd,J=9.77,7.63Hz,1H),5.10(d,J=10.38Hz,1H),5.28(d,J=17.40Hz,1H),5.73-5.78(m,1H),5.81(s,1H),6.82(s,1H),7.06-7.09(m,1H),7.15(s,1H),7.23(d,J=5.80Hz,1H),7.63(s,1H),7.87(dd,J=5.80,1.22Hz,1H),8.08(d,J=9.16Hz,1H);MS m/z 809(MH+).
实施例148:化合物148的制备
化合物148
按照实施例141、方案1的方法制备化合物148,不同之处在于在步骤2中扩大规模并使用L-缬氨酸乙酯盐酸盐代替L-缬氨酸甲酯盐酸盐。
步骤1:
修改:使用100mg(0.146mmol)实施例55、步骤1的产物;62.2mg(0.219mmol)碳酸N,N’-二琥珀酰亚胺酯;47.0mg(0.364mmol)N,N-二异丙基乙胺。
步骤2:
修改:使用265mg(1.46mmol)L-缬氨酸乙酯盐酸盐、188mg(1.46mmol)N,N-二异丙基乙胺,得到69.2mg产物,为白色粉末(收率60.4%):
1H NMR(CD3OD)δ0.83(dd,J=6.71,5.19Hz,6H),1.01-1.03(m,2H),1.06(s,9H),1.17-1.22(m,2H),1.23(t,J=7.17Hz,3H),1.40(dd,J=9.46,5.19Hz,1H),1.86(dd,J=8.09,5.34Hz,1H),1.95-2.02(m,1H),2.18(q,J=9.05Hz,1H),2.33(ddd,J=13.89,9.92,4.27Hz,1H),2.60(dd,J=13.89,7.17Hz,1H),2.92(ddd,J=12.82,8.09,4.73Hz,1H),3.93(s,3H),3.98(d,J=5.19Hz,1H),4.08-4.17(m,3H),4.41(s,1H),4.41-4.43(m,1H),4.52(dd,J=10.07,7.32Hz,1H),5.09(dd,J=10.38,1.53Hz,1H),5.26(dd,J=17.09,1.22Hz,1H),5.77(ddd,J=17.09,10.07,9.16Hz,1H),5.82(s,1H),7.12(dd,J=9.16,2.44Hz,1H),7.18(d,J=2.44Hz,1H),7.23(d,J=6.10Hz,1H),7.87(d,J=6.10Hz,1H),8.10(d,J=9.16Hz,1H);MS m/z 785(MH+).
实施例149:化合物149的制备
化合物149
按照实施例137、方案1的方法制备化合物149,不同之处在于在步骤1中使用N-叔丁氧基羰基-L-环戊基甘氨酸二环己基胺盐代替N-α-叔丁氧基羰基-L-天冬氨酸4-苄酯。
步骤1:
修改:使用76.2mg(0.180mmol)N-叔丁氧基羰基-L-环戊基甘氨酸二环己基胺盐,得到111mg产物,为白色粉末(收率89.3%):
1H NMR(CD3OD)δ0.98(d,J=8.24Hz,2H),1.15-1.18(m,2H),1.24(s,9H),1.29-1.32(m,J=18.01Hz,2H),1.38-1.40(m,1H),1.44(dd,J=4.88,1.53Hz,1H),1.49-1.55(m,2H),1.62-1.67(m,2H),1.74-1.80(m,1H),1.85-1.88(m,1H),2.16(q,J=8.75Hz,1H),2.21-2.26(m,1H),2.42(t,J=11.90Hz,1H),2.60-2.64(m,1H),2.89-2.93(m,1H),3.92(d,J=1.53Hz,3H),4.06-4.11(m,2H),4.51-4.57(m,2H),5.07(d,J=10.38Hz,1H),5.25(d,J=17.09Hz,1H),5.78-5.85(m,2H),7.10(d,J=8.85Hz,1H),7.17(s,1H),7.23(d,J=4.27Hz,1H),7.88(dd,J=5.95,1.68Hz,1H),8.10(d,J=9.16Hz,1H);MS m/z 726(MH+).
实施例150:化合物150的制备
化合物150
按照实施例141、方案1的方法制备化合物150,不同之处在于在步骤2中扩大规模并使用L-缬氨酸苄酯盐酸盐代替L-缬氨酸甲酯盐酸盐。
步骤1:
修改:使用100mg(0.146mmol)实施例55、步骤1的产物;62.2mg(0.219mmol)碳酸N,N’-二琥珀酰亚胺酯;47.0mg(0.364mmol)N,N-二异丙基乙胺。
步骤2:
修改:使用356mg(1.46mmol)L-缬氨酸苄酯盐酸盐、188mg(1.46mmol)N,N-二异丙基乙胺,得到41.0mg产物,为白色粉末(收率33.2%):MS m/z 848(MH+)。
实施例151:化合物151的制备
化合物151
按照实施例141、方案1的方法制备化合物151,不同之处在于在步骤2中扩大规模并使用L-异亮氨酸甲酯盐酸盐代替L-缬氨酸甲酯盐酸盐。
步骤1:
修改:使用100mg(0.146mmol)实施例55、步骤1的产物;62.2mg(0.219mmol)碳酸N,N’-二琥珀酰亚胺酯;47.0mg(0.364mmol)N,N-二异丙基乙胺。
步骤2:
修改:使用265mg(1.46mmol)L-异亮氨酸甲酯盐酸盐、188mg(1.46mmol)N,N-二异丙基乙胺,得到75.5mg产物,为白色粉末(收率65.9%):
1H NMR(CD3OD)δ0.78-0.80(m,3H),0.83-0.84(m,3H),0.90-0.95(m,1H),1.01-1.03(m,2H),1.06(d,J=3.05Hz,9H),1.17-1.21(m,2H),1.32-1.42(m,2H),1.68-1.72(m,1H),1.84-1.87(m,1H),2.14-2.20(m,1H),2.30-2.36(m,1H),2.57-2.62(m,1H),2.90-2.95(m,1H),3.66(d,J=2.75Hz,3H),3.92(d,J=2.75Hz,3H),4.05-4.12(m,2H),4.39-4.42(m,2H),4.50-4.53(m,1H),5.07-5.10(m,1H),5.23-5.28(m,1H),5.73-5.79(m,1H),5.81-5.83(m,1H),7.10-7.13(m,1H),7.17(t,J=2.44Hz,1H),7.22-7.24(m,1H),7.85-7.87(m,1H),8.10(dd,J=9.16,2.75Hz,1H);MS m/z785(MH+).
实施例152:化合物152的制备
化合物152
按照实施例141、方案1的方法制备化合物152,不同之处在于在步骤2中扩大规模并使用L-缬氨酸叔丁酯盐酸盐代替L-缬氨酸甲酯盐酸盐。
步骤1:
修改:使用100mg(0.146mmol)实施例55、步骤1的产物;62.2mg(0.219mmol)碳酸N,N’-二琥珀酰亚胺酯,47.0mg(0.364mmol)N,N-二异丙基乙胺。
步骤2:
修改:使用306mg(1.46mmol)L-缬氨酸叔丁酯盐酸盐、188mg(1.46mmol)N,N-二异丙基乙胺,得到93.5mg产物,为白色粉末(收率78.8%):MS m/z 814(MH+)。
实施例153:化合物153的制备
化合物153
按照实施例141、方案1的方法制备化合物153,不同之处在于在步骤2中扩大规模并使用(S)-(+)-1-甲氧基-2-丙基胺代替L-缬氨酸甲酯盐酸盐。
步骤1:
修改:使用100mg(0.146mmol)实施例55、步骤1的产物;62.2mg(0.219mmol)碳酸N,N’-二琥珀酰亚胺酯;47.0mg(0.364mmol)N,N-二异丙基乙胺。
步骤2:
修改:使用130mg(1.46mmol)(S)-(+)-1-甲氧基-2-丙胺、188mg(1.46mmol)N,N-二异丙基乙胺,得到50.3mg产物,为白色粉末(收率47.3%):
1H NMR(CD3OD)δ0.96(d,J=7.02Hz,3H),0.99-1.01(m,2H),1.04(s,9H),1.15-1.18(m,2H),1.39(dd,J=9.61,5.34Hz,1H),1.84(dd,J=7.93,5.19Hz,1H),1.93(s,3H),2.15(q,J=9.05Hz,1H),2.37(ddd,J=13.96,9.84,4.58Hz,1H),2.61(dd,J=14.04,7.32Hz,1H),2.90(ddd,J=12.89,8.16,4.88Hz,1H),3.19-3.28(m,2H),3.64-3.67(m,1H),3.92(s,3H),4.12(dd,J=11.60,3.97Hz,1H),4.40(s,1H),4.44(d,J=11.90Hz,1H),4.53(dd,J=9.77,7.32Hz,1H),5.07(dd,J=10.22,1.68Hz,1H),5.24(dd,J=17.24,1.37Hz,1H),5.76-5.81(m,1H),5.83(s,1H),7.10(dd,J=9.16,2.44Hz,1H),7.17(d,J=2.44Hz,1H),7.23(d,J=5.80Hz,1H),7.87(d,J=5.80Hz,1H),8.10(d,J=9.16Hz,1H);MS m/z 729(MH+).
实施例154:化合物154的制备
化合物154
按照实施例141、方案1的方法制备化合物154,不同之处在于在步骤2中扩大规模并使用N-甲基L-缬氨酸甲酯盐酸盐代替L-缬氨酸甲酯盐酸盐。
步骤1:
修改:使用100mg(0.146mmol)实施例55、步骤1的产物;62.2mg(0.219mmol)碳酸N,N’-二琥珀酰亚胺酯;47.0mg(0.364mmol)N,N-二异丙基乙胺。
步骤2:
修改:使用265mg(1.46mmol)N-甲基L-缬氨酸甲酯盐酸盐、188mg(1.46mmol)N,N-二异丙基乙胺,得到68.2mg产物,为白色粉末(收率59.5%):
1H NMR(CD3OD)δ0.70(dd,J=6.71,2.14Hz,3H),0.89(dd,J=6.41,2.44Hz,3H),0.96-0.98(m,1H),1.02-1.04(m,2H),1.07(d,J=2.14Hz,9H),1.18-1.22(m,2H),1.43-1.47(m,1H),1.84-1.87(m,1H),2.11-2.19(m,2H),2.31-2.37(m,1H),2.58-2.63(m,1H),2.87(d,J=2.44Hz,3H),2.90-2.94(m,1H),3.65(d,J=2.14Hz,3H),3.92(d,J=2.14Hz,3H),4.10-4.14(m,1H),4.25(dd,J=10.07,1.22Hz,1H),4.48(d,J=2.44Hz,1H),4.50-4.54(m,1H),5.08-5.10(m,1H),5.25-5.28(dd,J=17.09,1.53Hz,1H),5.78-5.85(m,2H),7.10-7.13(m,1H),7.18-7.19(m,1H),7.24(dd,J=5.95,2.59Hz,1H),7.87-7.89(m,1H),8.10(dd,J=9.00,2.59Hz,1H);MS m/z 785(MH+).
实施例155:化合物155的制备
化合物155
方案1
步骤1:
向实施例55、步骤1的产物(100mg,0.146mmol)的无水THF(2mL)溶液中加入碳酸吡啶-2-基酯·2,2,2-三氟-1,1-二甲基-乙酯(44.0mg,0.175mmol)和N-甲基吗啉(59mg,0.58mmol)。将混合物在室温下搅拌24h。将反应混合物洗涤、真空浓缩并将剩余物溶于DCM(2mL)中。将溶液用pH=4的缓冲溶液(3×3mL)洗涤,将洗液用DCM(3mL)反萃取。合并有机相并真空浓缩。随后将粗产物溶于甲醇并经反相制备型HPLC纯化,得到化合物155,为白色粉末(38.5mg,收率34.3%):
1H NMR(CD3OD)δ1.04(s,11H),1.19-1.22(m,2H),1.23(s,3H),1.43(dd,J=9.31,5.34Hz,1H),1.46(s,3H),1.87(dd,J=7.93,5.49Hz,1H),2.19(q,J=8.85Hz,1H),2.34(m,1H),2.62(dd,J=13.73,7.02Hz,1H),2.92(ddd,J=12.67,8.09,4.88Hz,1H),3.92(s,3H),4.06(dd,J=11.90,3.36Hz,1H),4.23(s,1H),4.43(d,J=11.60Hz,1H),4.56(dd,J=10.38,7.32Hz,1H),5.10(d,J=10.38Hz,1H),5.27(d,J=17.09Hz,1H),5.75-5.80(m,1H),5.82(s,1H),7.10(dd,J=9.16,2.44Hz,1H),7.18(d,J=2.44Hz,1H),7.25(d,J=6.10Hz,1H),7.89(d,J=5.80Hz,1H),8.07(d,J=9.16Hz,1H);MS m/z 768(MH+).
部分E:
部分E的LC-MS条件
″方法A″为3.0×50mm Xterra@4min梯度和流速4mL/min
″方法B″为3.0×50mm Xterra@3min梯度和流速4mL/min
″方法C″为4.6×50mm Xterra@4min梯度和流速4mL/min
″方法D″为4.6×50mm Xterra@3min梯度和流速4mL/min
实施例180:化合物180的制备
通用合成流程
按照上述通用合成方案制备化合物180-183。其中各反应将在其它地方详细描述。除了在第一步烷基化步骤中为叔丁醇钾(以THF为溶剂,由Aldrich Chemicals提供)的DMF溶液提供了更方便的后处理方法外,一旦烷基化完成后,大部分的DMF溶剂均用水除去。
由此得到化合物180:BOCNH-P3(L-叔-BuGly)-P2[(4R)-(2-三氟甲基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷,为白色泡沫状物,收率61%。LC/MS Rt-min(MNa+)[方法A]:3.35(774)。
1H NMR(400MHz,CD3OD)δppm 1.05(m,13H)1.21(s,9H)1.42(dd,J=9.17,5.26Hz,1H)1.86(dd,J=8.07,5.38Hz,1H)2.21(m,1H)2.33(m,1H)2.64(dd,J=13.94,6.60Hz,1H)2.93(m,1H)4.09(dd,J=11.49,2.69Hz,1H)4.21(s,1H)4.52(m,1H)4.56(d,J=12.23Hz,1H)5.10(dd,J=10.39,1.59Hz,1H)5.27(d,J=16.87Hz,1H)5.58(s,1H)5.72(m,1H)7.36(s,1H)7.61(t,J=7.70Hz,1H)7.83(t,J=7.34Hz,1H)8.07(d,J=8.56Hz,1H)8.26(d,J=8.56Hz,1H).
实施例181:化合物181的制备
得到BOCNH-P3(L-叔-BuGly)-P2[(4R)-(2,8-双(三氟甲基)喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷,为白色泡沫状物,收率52%。LC/MS Rt-min(MNa+)[方法A]:3.60(843)。
1HNMR(400MHz,CD3OD)δppm 1.05(m,11H)1.16(s,9H)1.22(m,2H)1.42(dd,J=9.29,5.38Hz,1H)1.86(dd,J=8.07,5.38Hz,1H)2.21(q,J=8.97Hz,1H)2.33(m,1H)2.65(dd,J=13.94,6.85Hz,1H)2.93(m,1H)4.07(dd,J=11.98,2.69Hz,1H)4.17(s,1H)4.52(dd,J=10.52,6.85Hz,1H)4.58(d,J=11.98Hz,1H)5.10(d,J=10.27Hz,1H)5.27(d,J=17.12Hz,1H)5.60(s,1H)5.72(m,1H)7.46(s,1H)7.69(t,J=7.83Hz,1H)8.18(d,J=7.34Hz,1H)8.50(d,J=8.31Hz,1H).
实施例182:化合物182的制备
得到BOCNH-P3(L-叔-BuGly)-P2[(4R)-(2-三氟甲基-8-三氟甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷,为白色泡沫状物,收率99%。LC/MS Rt-min(MNa+)[方法A]:3.62(858)。
1H NMR(400MHz,CD3OD)δppm 1.05(m,11H)1.21(m,11H)1.42(dd,J=9.05,5.14Hz,1H)1.86(dd,J=8.07,5.38Hz,1H)2.21(q,J=8.64Hz,1H)2.33(m,1H)2.64(dd,J=13.94,6.60Hz,1H)2.93(m,1H)4.08(dd,J=11.98,2.69Hz,1H)4.18(s,1H)4.51(dd,J=10.52,6.85Hz,1H)4.57(d,J=12.23Hz,1H)5.10(dd,J=10.52,1.22Hz,1H)5.27(d,J=17.12Hz,1H)5.59(s,1H)5.72(m,1H)7.44(s,1H)7.63(t,J=8.07Hz,1H)7.78(d,J=7.58Hz,1H)8.24(d,J=8.56Hz,1H).
实施例183:化合物183的制备
得到BOCNH-P3(L-叔-BuGly)-P2[(4R)-(2-三氟甲基,8-氯喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷,为白色泡沫状物,收率64%。LC/MS Rt-min(MNa+)[方法A]:3.52(808)。
1HNMR(400MHz,CD3OD)δppm 1.04(m,11H)1.21(m,11H)1.41(dd,J=9.41,5.50Hz,1H)1.86(dd,J=8.07,5.62Hz,1H)2.20(q,J=8.80Hz,1H)2.32(m,1H)2.63(dd,J=13.82,6.72Hz,1H)2.92(m,1H)4.07(dd,J=12.10,2.81Hz,1H)4.19(s,1H)4.50(dd,J=10.52,6.85Hz,1H)4.56(d,J=11.98Hz,1H)5.10(dd.J=10.27,1.47Hz,1H)5.26(d,J=17.12Hz,1H)5.57(s,1H)5.72(m,1H)7.41(s,1H)7.52(t,J=8.07Hz,1H)7.93(d,J=7.58Hz,1H)8.19(d,J=8.56Hz,1H).
实施例184:用三肽(化合物184)和P2*烷基化的通用方法
通用方案-实施例184的化合物(化合物184)的制备
三肽组分的制备,以HATU为偶合剂,通过连续的酰胺偶合得到实施例184的化合物。应理解许多标准偶合剂可用于以下方案。
中间体实施例184a的制备:
室温下,向装于火焰干燥的烧瓶中的HATU(820mg,2.2mmol)、实施例180a的化合物(Boc-4R-羟基脯氨酸,417mg,1.8mmol)和实施例180c的化合物(环丙烷磺酸(1(R)-氨基-2(S)-乙烯基-环丙烷羰基)-酰胺盐酸盐,490mg,1.8mmol)的混合物内加入无水二氯甲烷(8mL)。将混合物用干燥氮气保护,随后将其冷却至-78℃。在5分钟内缓慢加入Hunig氏碱(二异丙基乙胺,625μL,3.6mmol),将混合物变为浅橙色悬浮液。继续搅拌1小时,同时使温度升高至环境温度。LC/MS显示完全转化为所需的产物184a。粗品反应物经常规后处理,用水洗涤三次(每次5mL),有机剩余物用乙酸乙酯(3×5mL)萃取。真空除去有机溶剂得到粗产物。该物质未经进一步纯化直接用于下一步骤。
中间体实施例184b的制备:
室温下,将得自前述步骤的干燥固体物溶于9ml二氯甲烷中。向该溶液中加入3ml三氟乙酸,形成浅黄色溶液。室温下,继续搅拌2小时。LC/MS显示不存在原料184a,而主要的信号对应于所需的产物184b,还存在对应于先前步骤中HATU所带的副产物的信号。蒸发溶剂,固体剩余物未经进一步纯化立即用于下一步骤。
三肽化合物184的制备:
BOCNH-P3(L-叔-BuGly)-P2[(4R)-羟基-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷[编号46877-128]
在室温下,将先前步骤的粗产物(实施例184b,1.8mmol)与HATU(700mg,1.8mmol)和BOC-L-叔-亮氨酸(实施例180f,FlukaChemicals,420mg,1.8mmol)在二氯甲烷(10mL)中混合。将该悬浮液用Hünig氏碱(1mL,过量)处理,形成稍稀、橙色悬浮液。LC/MS显示有一些转化为化合物184。室温下继续搅拌两天后观察到完全转化为所需的产物184。将粗品反应混合物蒸发至干。将剩余物溶于乙酸乙酯中。用半饱和、新鲜制备的碳酸氢钠溶液萃取,除去大部分的HATU剩余物。用去离子水洗涤除去最终痕量的HATU剩余物(1-羟基7-氮杂苯并三唑)。蒸发溶剂,得到990mg(98%)所需的产物,为白色泡沫状物。该物质可未经进一步纯化,直接用于随后的用亲电体(如喹啉和异喹啉)进行的烷基化。LC/MS Rt-min(MNa+)[方法A]:2.65(579)。
1H NMR(400MHz,CD3OD)δppm 0.95(s,2H)0.99(s,9H)1.05(m,2H)1.21(m,1H)1.39(m,9H)1.84(dd,J=8.31,5.38Hz,1H)1.96(m,1H)2.11(m,1H)2.20(m,1H)2.91(m,1H)3.80(m,2H)4.28(d,J=9.78Hz,1H)4.35(dd,J=9.90,6.97Hz,1H)4.47(s,1H)5.11(m,1H)5.29(d,J=17.12Hz,1H)5.75(m,1H).
亲电体对三肽(化合物184)进行烷基化:
向火焰干燥的25mL圆底烧瓶中装入化合物184(0.5-1.0mmol)、取代的4-氯喹啉(1.0当量)和氯化镧(LaCl3无水珠粒,由Aldrich供应,购来即用,M.W.245g/mol;1.0当量。注:使用这种添加剂在某些情况下有用,采用那些低活性亲电体时更是如此。当亲电体对阴离子烷基化作用具有足够的活性时可不使用这种试剂)在2mL无水DMF中的溶液。所述无机盐在室温下在DMF中仅为微溶。在氮气气氛中,将混合物在搅拌下冷却至-78℃(干冰/丙酮浴)。向该冷却的混合物中加入叔丁醇钾的THF溶液(1.0M,由Aldrich提供,购来即用,5.5当量),混合物的颜色由无色转变成浅黄色或绿色。将混合物在-78℃下搅拌,搅拌时间取决于4-氯喹啉的活性(在-78℃下数小时至在室温下过夜)。还发现最终所述无机盐转变成细乳液。用半饱和的氯化铵水溶液(2mL)猝灭。有机物质用乙酸乙酯(10mL×3)萃取。将有机层合并,用去离子水(10mL×2)回洗。蒸发有机部分,经LC/MS确定,得到富含所需产物的粗制混合物。采用制备型HPLC,使用标准分离参数,分离所需产物(典型条件:3.0×50mm Xterra柱,梯度4min和流速4mL/min),得到分析纯的所需产物。按照完全相同的方式进行1-卤代异喹啉系列的烷基化。
实施例185:化合物185的制备
按照实施例184的通用三肽烷基化方法,得到BOCNH-P3(L-叔-BuGly)-P2[(4R)-(7-三氟甲基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷,为白色泡沫状物,收率50%。LC/MS Rt-min(MH+)[方法B]:2.32(752)。
1H NMR(400MHz,CD3OD)δppm 1.02(s,9H)1.06(m,11H)1.22(m,2H)1.43(dd,J=9.41,5.26Hz,1H)1.88(dd,J=8.19,5.50Hz,1H)2.23(q,J=8.80Hz,1H)2.42(m,1H)2.75(dd,J=14.06,6.48Hz,1H)2.93(m,1H)4.10(m,2H)4.61(m,2H)5.12(dd,J=10.39,1.59Hz,1H)5.29(d,J=17.12Hz,1H)5.72(m,2H)7.61(d,J=6.36Hz,1H)7.96(d,J=8.80Hz,1H)8.38(s,1H)8.59(d,J=8.56Hz,1H)9.14(d,J=6.36Hz,1H),
实施例186:化合物186的制备
按照实施例184的通用三肽烷基化方法,得到所需的产物BOCNH-P3(L-叔-BuGly)-P2[(4R)-(8-三氟甲基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷,为白色泡沫状物,收率50%。LC/MS Rt-min(MH+)[方法B]:2.48(752)。
1H NMR(400MHz,CD3OD)δppm 1.02(s,9H)1.05(m,2H)1.13(s,9H)1.23(m,2H)1.42(dd,J=8.68,5.50Hz,1H)1.87(dd,J=8.07,5.38Hz,1H)2.21(q,J=8.80Hz,1H)2.36(m,1H)2.69(dd,J=14.06,6.97Hz,1H)2.93(m,1H)4.08(dd,J=11.98,2.93Hz,1H)4.15(s,1H)4.54(dd,J=10.52,7.09Hz,1H)4.60(d,J=12.47Hz,1H)5.11(dd,J=10.52,1.71Hz,1H)5.28(d,J=15.90Hz,1H)5.58(s,1H)5.72(m,1H)7.32(d,J=5.87Hz,1H)7.69(t,J=7.95Hz,1H)8.22(d,J=7.09Hz,1H)8.55(d,J=8.07Hz,1H)8.88(d,J=5.62Hz,1H).
用于6-F、6-乙基、6-异丙基和6-叔丁基异喹啉P2*组件的异喹啉中间体的制备。
总而言之,如下所示,用于以下实验的6-氟和6-烷基异喹啉通过Pomeranz-Fritsch合成法(典型方法:光学活性8,8-二取代的1,1-联异喹啉的制备,K.Hirao,R.Tsuchiya,Y.Yano,H.Tsue,Hetero cycles42(1)1996,415-422)制备。通过本文其它地方描述的N-氧化物中间体将所述产物转化为1-氯衍生物。
通用合成方案
试剂和反应条件:(a)在苯中回流,共沸除去水;(b)第一步:氯甲酸乙酯,亚磷酸三甲酯的THF溶液,第二步:四氯化钛的氯仿溶液;(c)MCPBA的二氯甲烷溶液;(d)磷酰氯的苯溶液
|
R |
异喹啉,收率 |
1-氯化物,总收率 |
|
F |
20 |
43 |
|
Et |
76 |
65 |
|
i-Pr |
14 |
18 |
|
t-Bu |
47 |
55 |
实施例187:化合物187的制备
得到BOCNH-P3(L-叔-BuGly)-P2[(4R)-(6-氟异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷,为白色泡沫状物,收率12%。LC/MS Rt-min(MNa+)[方法C]:3.81(724)。
1H NMR(400MHz,CD3OD)δppm 1.05(m,13H)1.22(s,9H)1.42(m,1H)1.86(m,1H)2.21(m,2H)2.61(dd,J=13.69,6.60Hz,1H)2.93(m,1H)4.05(d,J=13.69Hz,1H)4.21(s,1H)4.49(m,2H)5.11(d,J=10.03Hz,1H)5.28(d,J=17.61Hz,1H)5.72(m,1H)5.86(d,J=4.40Hz,1H)7.31(m,2H)7.48(d,J=8.31Hz,1H)7.97(d,J=6.36Hz,1H)8.26(d,J=6.11Hz,1H).
实施例188:化合物188的制备
上述烷基化得到1-氯异喹啉主产物:BOCNH-P3(L-叔-BuGly)-P2[(4R)-(1-氯异喹啉-6-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷,为白色泡沫状物,收率40.2%。LC/MS Rt-min(MH+)[方法C]:3.81(718)。
1H NMR(400MHz,CD3OD)δppm 1.00(s,9H)1.06(m,2H)1.25(s,11H)1.41(m,1H)1.86(dd,J=8.07,5.38Hz,1H)2.25(m,2H)2.54(dd,J=12.96,6.60Hz,1H)2.92(m,1H)4.06(dd,J=11.98,2.69Hz,1H)4.20(s,1H)4.31(d,J=11.74Hz,1H)4.45(dd,J=9.78,7.58Hz,1H)5.11(dd,J=10.27,1.71Hz,1H)5.28(dd,J=17.36,1.47Hz,1H)5.36(s,1H)5.74(m,1H)7.35(d,J=9.29Hz,1H)7.40(s,1H)7.70(d,J=5.87Hz,1H)8.13(d,J=5.87Hz,1H)8.25(d,J=9.29Hz,1H).
实施例189:化合物189的制备
得到BOCNH-P3(L-叔-BuG1y)-P2[(4R)-(6-乙基异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷,为白色泡沫状物,得到4.6mg黄色固体物(4.2%)。LC/MS Rt-min(MH+)[方法B]:2.70(712)。
1H NMR(400MHz,CD3OD)δppm 1.01(s,9H)1.07(m,2H)1.22(m,11H)1.28(t,J=7.91Hz,3H)1.41(m,1H)1.85(m,1H)2.25(m,2H)2.60(dd,J=13.69,6.85Hz,1H)2.80(q,J=7.66Hz,2H)2.93(m,1H)4.02(d,J=31.06Hz,1H)4.23(s,1H)4.42(m,1H)4.54(m,1H)5.10(d,J=10.27Hz,1H)5.28(d,J=17.12Hz,1H)5.74(m,1H)5.84(s,1H)7.25(d,J=5.87Hz,1H)7.38(d,J=8.56Hz,1H)7.59(s,1H)7.90(d,J=6.24Hz,1H)8.09(d,J=8.56Hz,1H).
实施例190:化合物190的制备
得到BOCNH-P3(L-叔-BuGly)-P2[(4R)-(6-异丙基异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷,为白色泡沫状物,收率69%。LC/MS Rt-min(MNa+)[方法B]:2.76(749)。
1H NMR(400MHz,CD3OD)δppm 1.05(m,13H)1.20(m,9H)1.31(d,J=6.85Hz,6H)1.42(m,1H)1.86(dd,J=8.07,5.62Hz,1H)2.26(m,2H)2.62(dd,J=13.69,6.85Hz,1H)2.93(m,1H)3.07(m,1H)4.06(m,1H)4.21(s,1H)4.52(m,2H)5.11(d,J=10.27Hz,1H)5.28(d,J=17.12Hz,1H)5.74(m,1H)5.84(s,1H)7.32(d,J=6.11Hz,1H)7.46(d,J=8.56Hz,1H)7.64(s,1H)7.90(d,J=6.11Hz,1H)8.13(d,J=8.56Hz,1H).
实施例191:化合物191的制备
得到BOCNH-P3(L-叔-BuGly)-P2[(4R)-(6-叔丁基异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷,为白色泡沫状物,收率81%。LC/MS Rt-min(MH+)[方法B]:2.84(740)。
1H NMR(400MHz,CD3OD)δppm 1.01(s,9H)1.06(m,2H)1.18(s,9H)1.22(m,2H)1.39(s,9H)1.43(m,1H)1.87(dd,J=8.19,5.50Hz,1H)2.27(m,2H)2.63(dd,J=13.57,6.97Hz,1H)2.93(m,1H)4.06(m,1H)4.20(s,1H)4.52(m,2H)5.10(d,J=11.49Hz,1H)5.28(d,J=17.12Hz,1H)5.73(m,1H)5.84(s,1H)7.36(d,J=6.11Hz,1H)7.66(dd,J=8.80,1.22Hz,1H)7.78(s,1H)7.91(d,J=5.87Hz,1H)8.15(d,J=8.80Hz,1H).
6-异丙氧基和6-叔丁氧基异喹啉中间体的制备:
一些6-烷氧基-1-氯异喹啉直接通过相应的烷氧基金属离子如叔丁醇钾(53%)和异丙醇钠(54%)对6-氟-1-氯异喹啉进行同侧(ipso)置换得到。
通用合成方案
R=烷氧基阴离子,如叔丁基、异丙基
用异丙醇钠和叔丁醇钾的DMF溶液对6-氟-1-氯异喹啉进行芳族亲核置换,分别得到相应的6-异丙氧基主产物(54%):
1H NMR(400MHz,CHLOROFORM-d)δppm 1.43(d,J=6.11Hz,6H)4.76(m,J=6.11Hz,1H)7.08(d,J=2.45Hz,1H)7.29(dd,J=9.29,2.45Hz,1H)7.50(d,J=5.62Hz,1H)8.18(d,J=5.87Hz,1H)8.24(d,J=9.29Hz,1H)
和6-叔丁氧基-1-氯异喹啉主产物(55%):
1H NMR(400MHz,CHLOROFORM-d)δppm 1.48(s,9H)7.31(m,2H)7.47(d,J=5.62Hz,1H)8.18(d,J=5.62Hz,1H)8.21(d,J=9.78Hz,1H)
如实施例184所述,用三肽将这些6-烷氧基-1-氯异喹啉烷基化,得到如下所示的所需产物。
实施例192:化合物192的制备
得到BOCNH-P3(L-叔-BuGly)-P2[(4R)-(6-异丙氧基异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷,为白色泡沫状物,收率59%。LC/MS Rt-min(MH+)[方法C]:3.87(742)。
1H NMR(400MHz,CD3OD)δppm 0.99(s,11H)1.06(m,2H)1.21(s,9H)1.37(d,J=5.87Hz,6H)1.42(m,1H)1.86(dd,J=8.07,5.38Hz,1H)2.25(m,2H)2.61(dd,J=13.69,6.85Hz,1H)2.92(m,1H)4.04(dd,J=11.86,3.30Hz,1H)4.21(br s,1H)4.49(m,2H)4.78(h,J=5.87Hz,1H)5.10(dd,J=10.39,1.35Hz,1H)5.28(d,J=16.87Hz,1H)5.73(m,1H)5.79(d,J=11.25Hz,1H)7.07(dd,J=9.05,1.96Hz,1H)7.18(d,J=2.20Hz,1H)7.26(d,J=6.11Hz,1H)7.85(d,J=6.11Hz,1H)8.10(d,J=9.05Hz,1H).
实施例193:化合物193的制备
得到BOCNH-P3(L-叔-BuGly)-P2[(4R)-(6-叔丁氧基异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷,为白色泡沫状物,收率39%。LC/MS Rt-min(MH+)[方法C]:3.99(756)。
1H NMR(400MHz,CD3OD)δppm 1.03(br s,13H)1.19(s,9H)1.42(m,10H)1.86(dd,J=7.83,5.62Hz,1H)2.24(m,2H)2.60(dd,J=13.69,6.85Hz,1H)2.93(m,1H)4.04(dd,J=11.49,2.93Hz,1H)4.23(s,1H)4.49(m,2H)5.10(d,J=11.25Hz,1H)5.27(d,J=16.87Hz,1H)5.73(m,1H)5.81(s,1H)7.13(dd,J=8.80,1.47Hz,1H)7.25(d,J=5.87Hz,1H)7.33(d,J=2.20Hz,1)7.87(d,J=6.11Hz,1H)8.10(d,J=9.05Hz,1H).
酞嗪P2*衍生物的制备:
总而言之,1-氯酞嗪和1,4-二氯酞嗪可顺利地被烷基化,得到所需的产物。而商品1-氯酞嗪和1,4-二氯酞嗪经常含有一些水解杂质。用磷酰氯预处理,接着立即烷基化得到更一致的结果。
通用合成方案
反应条件:(a)磷酰氯的DCE溶液;(b)用三肽烷基化;(c)咪唑(R-CH)、三唑(R=N)的钠衍生物。
实施例194:化合物194的制备
得到BOCNH-P3(L-叔-BuGly)-P2[(4R)-(酞嗪-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷,为白色泡沫状物,收率41%。LC/MS Rt-min(MNa+)[方法B]:2.07(707)。
1H NMR(400MHz,CD3OD)δppm 1.03(m,9H)1.06(m,4H)1.14(s,9H)1.20(m,1H)1.43(m,1H)1.87(dd,J=8.07,5.62Hz,1H)2.24(q,J=8.80Hz,1H)2.38(m,1H)2.76(dd,J=14.18,7.09Hz,1H)2.92(m,1H)4.11(m,2H)4.62(m,1H)5.11(dd,J=10.27,1.71Hz,1H)5.29(dd,J=17.12,1.22Hz,1H)5.72(m,1H)5.96(s,1H)8.26(m,2H)8.46(m,2H)9.84(s,1H).
实施例195:化合物195的制备
得到BOCNH-P3(L-叔-BuGly)-P2[(4R)-(4-氯酞嗪-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷,为白色泡沫状物,收率23%。LC/MS Rt-min(MNa+)[方法C]:3.52(742)。
1H NMR(400MHz,CD3OD)δppm 1.01(s,11H)1.06(m,2H)1.14(s,9H)1.22(m,1H)1.43(m,1H)1.87(m,1H)2.21(m,1H)2.35(m,J=10.27Hz,1H)2.70(m,1H)2.93(m,1H)4.05(d,J=3.42Hz,1H)4.58(m,2H)5.11(dd,J=10.39,1.10Hz,1H)5.28(d,J=17.36Hz,1H)5.73(m,1H)5.93(s,1H)7.99(m,1H)8.07(t,J=7.70Hz,1H)8.26(dd,J=8.19,2.32Hz,2H).
4-(咪唑-1-基)酞嗪和4-(1,2,4-三唑-1-基)酞嗪P2*衍生物的制备:
用典型的唑类(如咪唑和三唑)阴离子代替上述步骤的产物化合物195:BOCNH-P3(L-叔-BuGly)-P2[(4R)-(4-氯酞嗪-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷,得到下述4-唑取代的酞嗪衍生物:
实施例196:化合物196的制备
在55-65℃,用咪唑钠盐的DMF溶液代替4-氯酞嗪(化合物195)制备题述化合物。
得到BOCNH-P3(L-叔-BuGly)-P2[(4R)-(4-(咪唑-1-基)酞嗪-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷,为白色泡沫状物,收率23%。LC/MS Rt-min(MNa+)[方法B]:1.94(773)。
1H NMR(400MHz,CD3OD)δppm 1.03(s,9H)1.07(m,2H)1.20(m,9H)1.24(m,1H)1.41(m,2H)1.88(dd,J=8.19,5.50Hz,1H)2.23(m,1H)2.41(m,1H)2.75(m,J=14.92Hz,1H)2.93(m,1H)4.14(m,2H)4.60(dd,J=10.15,6.97Hz,2H)5.12(dd,J=10.27,1.47Hz,1H)5.29(dd,J=17.24,1.35Hz,1H)5.74(m,1H)6.09(s,1H)7.85(s,1H)7.94(m,1H)8.10(m,2H)8.43(m,1H)9.17(s,1H)9.46(s,1H).
在所述置换反应中,还分离出少量的脱BOC副产物(化合物197):
得到NH2-P3(L-叔-BuGly)-P2[(4R)-(4-(咪唑-1-基)酞嗪-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷,为白色泡沫状物,收率17%。LC/MS Rt-min(MH+)[方法B]:1.22(651)。
1H NMR(400MHz,CD3OD)δppm 1.16(s,9H)1.23(m,2H)1.42(dd,J=9.41,5.50Hz,1H)1.89(m,1H)2.26(q,J=8.97Hz,1H)2.43(m,1H)2.78(dd,J=14.06,7.21Hz,1H)2.93(m,1H)3.74(m,1H)4.11(s,1H)4.22(dd,J=12.23,3.91Hz,1H)4.47(m,2H)4.71(dd,J=10.27,7.09Hz,1H)5.12(m,1H)5.29(d,J=17.36Hz,1H)5.71(m,1H)6.13(t,J=3.67Hz,1H)7.87(s,1H)7.97(m,1H)8.14(m,3H)8.41(m,1H)9.47(s,1H).
实施例198:化合物198的制备
在55-65℃,用1,2,4-三唑钠盐的DMF溶液代替4-氯酞嗪(化合物195)制备题述化合物。
得到BOCNH-P3(L-叔-BuGly)-P2[(4R)-(4-(1,2,4-三唑-1-基)酞嗪-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷,为白色泡沫状物,收率62%。LC/MS Rt-min(MNa+)[方法C]:3.35(774)。
1HNMR(500MHz,CD3OD)δppm 0.97(s,9H)1.01(m,2H)1.10(s,9H)1.16(m,1H)1.37(m,2H)1.82(m,1H)2.18(d,J=8.55Hz,1H)2.32(m,1H)2.69(dd,J=13.58,6.87Hz,1H)2.88(br s,1H)4.06(d,J=11.60Hz,1H)4.13(s,1H)4.53(m,J=9.16Hz,1H)4.61(d,J=11.90Hz,1H)5.06(d,J=10.07Hz,1H)5.23(d,J=17.40Hz,1H)5.68(m,1H)5.98(s,1H)7.97(m,2H)8.30(m,J=11.90Hz,2H)8.44(d,J=7.63Hz,1H)9.14(s,1H).
4-羟基和4-烷氧基酞嗪P2*衍生物的制备
反应条件:(a)醇钠,如甲醇钠、乙醇钠和异丙醇钠。
实施例199:化合物199的制备
室温下将4-氯酞嗪(实施例195)化合物溶于无水异丙醇中,加入1.0eq异丙醇钠,将所得的悬浮液回流。得到4.5mg所需的产物,为黄色固体物(20.0%)。LC/MS Rt-min(MH+):2.68(743)[方法B]。
1H NMR(400MHz,CD3OD)δppm 1.02(m,11H)1.20(m,11H)1.42(m,1H)1.49(d,J=6Hz,6H)1.87(dd,J=7.95,5.50Hz,1H)2.21(m,1H)2.23(m,1H)2.66(m,1H)2.93(m,1H)4.08(q,J=7.09Hz,1H)4.19(s,1H)4.55(m,2H)5.11(d,J=10.27Hz,1H)5.28(d,J=17.61Hz,1H)5.48(m,1H)5.70(d,J=10.03Hz,1H)5.81(m,1H)7.90(m,2H)8.16(m,2H).
实施例200:化合物200的制备
同样,制备得到4-乙氧基衍生物:BOCNH-P3(L-t-BuGly)-P2[(4R)-(4-乙氧基酞嗪-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷。
得到4.0mg黄色固体物(16.0%)。LC/MS Rt-min(MH+):
2.52(729)[方法B]。
1H NMR(400MHz,CD3OD)δppm 1.01(s,9H)1.06(m,2H)1.16(s,9H)1.24(m,2H)1.43(dd,J=9.78,5.14Hz,1H)1.53(t,J=6.97Hz,3H)1.87(dd,J=8.19,5.50Hz,1H)2.22(q,J=8.97Hz,1H)2.32(m,1H)2.67(m,1H)2.93(m,1H)4.06(d,J=8.56Hz,1H)4.19(s,1H)4.56(m,4H)5.10(m,1H)5.30(m,1H)5.74(m,1H)5.82(s,1H)7.94(m,2H)8.16(d,J=7.83Hz,1H)8.21(m,1H).
实施例201:化合物201的制备
制得BOCNH-P3(L-t-BuGly)-P2[(4R)-(4-甲氧基酞嗪-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷,收率30.2%。LC/MSRt-min(MH+):2.42(715)[方法B]。
1H NMR(400MHz,CD3OD)δppm 0.96(s,9H)1.07(m,2H)1.20(m,11H)1.43(dd,J=9.29,5.38Hz,1H)1.87(dd,J=8.07,5.62Hz,1H)2.22(q,J=8.80Hz,1H)2.31(m,1H)2.66(d,J=8.07Hz,1H)2.93(m,1H)4.06(dd,J=11.98,3.18Hz,1H)4.19(d,J=3.42Hz,4H)4.54(m,2H)5.11(m,1H)5.28(d,J=17.36Hz,1H)5.73(m,1H)5.83(s,1H)7.95(m,2H)8.19(m,2H).
实施例202:化合物202的制备
尝试用四唑钠盐代替BOCNH-P3(L-叔-BuGly)-P2[(4R)-(4-氯酞嗪-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷(实施例195)化合物,得到大部分的4-羟基水解物质。
得到BOCNH-P3(L-t-BuGly)-P2[(4R)-(4-羟基酞嗪-1-氧代)-S-脯氨酸]P1(1R,2S乙烯基Acca)-CONHSO2环丙烷(44.2%),为浅乳脂状固体。LC/MS Rt-min(MH+):2.18(701)[方法B]。
1H NMR(400MHz,CD3OD)δppm 1.01(s,9H)1.05(m,2H)1.23(m,11H)1.42(dd,J=9.29,5.38Hz,1H)1.87(dd,J=8.07,5.38Hz,1H)2.21(m,2H)2.63(m,1H)2.93(m,1H)4.00(s,1H)4.20(s,1H)4.50(m,2H)5.11(dd,J=10.27,1.47Hz,1H)5.29(d,J=16.87Hz,1H)5.59(s,1H)5.73(m,1H)7.86(dd,J=5.75,3.30Hz,2H)8.01(dd,J=5.87,3.42Hz,1H)8.29(dd,J=5.87,3.42Hz,1H).
按照烷基化方案制备5,6-二取代的异喹啉P2*衍生物
通用合成方案
反应条件:(a)LDA的THF溶液;(b)二烷基二硫,如(n-PrS)2;(c)醇钠,如MeONa;(d)噻吩-2-甲醛;(e)MnO2的苯溶液
实施例203:1-氯-5-丙硫基-6-氟异喹啉的制备:
向冷却的(-78℃)1-氯-6-氟异喹啉(59mg,0.32mmol)在2ml THF中的溶液内加入LDA的环己烷溶液(1.5mol,0.23mL,0.35mmol)。将该橙色溶液搅拌2小时,随后用二正丙基二硫(60μL,纯净物,过量)处理。在30分钟内将反应物升温至室温。用半饱和的氯化铵溶液猝灭,将有机剩余物用乙酸乙酯萃取。LC-MS分析表明约50%转化为所需的产物,并还含有大量的原料。所需的产物经短柱纯化(4cm×2cm,硅胶H-型),洗脱液:5%乙醚/己烷,得到29mg(收率36%)所需的产物。LC/MS Rt-min(MH+)[方法C]:3.79(256)。
1H NMR(400MHz,CHLOROFORM-D)δppm 0.96(t,J=7.34Hz,3H)1.52(m,2H)2.86(m,2H)7.45(dd,J=9.29,8.56Hz,1H)8.34(d,J=0.73Hz,2H)8.37(m,1H).
按照实施例184的方法,采用三肽将该化合物烷基化,得到以下化合物:
实施例204:化合物204的制备
下述的BOCNH-P3(L-t-BuGly)-P2[(4R)-(1-氯-5-丙硫基-异喹啉-6-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷
按照通用方法,得到4.6mg黄色固体物(3.2%)。LC/MS Rt-min(MH+):2.73(792)[方法B]。
1H NMR(400MHz,CD3OD)δppm 0.93(t,J=7.34Hz,3H)0.97(s,9H)1.08(m,2H)1.24(m,11H)1.43(m,3H)1.86(m,1H)2.24(m,2H)2.56(m,1H)2.78(q,J=7.09Hz,2H)2.92(m,1H)4.01(d,J=9.29Hz,1H)4.22(s,1H)4.29(s,1H)4.59(d,J=6.85Hz,1H)5.11(d,J=10.76Hz,1H)5.28(d,J=17.36Hz,1H)5.49(s,1H)5.74(m,1H)7.66(d,J=9.29Hz,1H)8.18(d,J=6.11Hz,1H)8.41(m,2H).
实施例205:5-丙硫基-6-乙氧基异喹啉P2*衍生物的制备
改变其中所用的反应试剂,可将以下方法用于其它5-烷硫基-6-烷氧基异喹啉。在氮气气氛、-78℃下,向1-氯-6-氟异喹啉(88mg,0.48mmol)在2.0mL THF中的溶液内加入LDA(1.5mol的环己烷溶液,0.42mL,0.63mmol),形成深棕色溶液。在-78℃下搅拌30min后,加入纯二正丙基二硫(85μL,过量)。在30min内,将反应物升温至室温。用半饱和的氯化铵溶液猝灭,有机剩余物用乙酸乙酯萃取。合并各有机层并在50微米(Hg)真空下干燥。将粗产物溶于2ml THF中,冷却至-78℃,加入过量的乙醇钾(60mg)。异喹啉中间体最后经硅胶柱层析纯化(H型,Merck),洗脱液:乙醚/己烷混合物,得到32.2mg(24%)纯化合物。LC-MS显示为1-氯-5-丙硫基-6-乙氧基异喹啉,Rt-min(MH+)[方法C]:3.77(282)。
1H NMR(400MHz,氯仿-D)δppm 0.94(t,J=7.34Hz,3H)1.46(M,2H)1.55(t,J=6.97Hz,3H)2.83(t,J=7.21Hz,2H)4.32(q,J=6.85Hz,2H)7.36(d,J=9.29Hz,1H)8.22(d,J=6.11Hz,1H)8.32(d,J=9.29Hz,1H)8.35(d,J=6.11Hz,1H)。
按照通用的三肽烷基化方法(实施例184),采用三肽(化合物184)化合物将1-氯-5-丙硫基-6-乙氧基异喹啉烷基化,得到40.7mg(44.8%)如下所示所需的产物。
实施例206:化合物206的制备
BOCNH-P3(L-t-BuGly)-P2[(4R)-(6-乙氧基-5-丙硫基-异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷。LC/MS Rt-min(MH+):2.93(803)[方法B]。
1H NMR(400MHz,CD3OD)δppm 0.93(t,J=7.34Hz,3H)1.01(s,9H)1.07(m,2H)1.21(m,11H)1.41(m,3H)1.48(t,J=6.85Hz,3H)1.86(dd,J=8.07,5.62Hz,1H)2.25(m,2H)2.60(dd,J=13.69,6.85Hz,1H)2.81(q,J=6.97Hz,2H)2.93(m,1H)4.05(m,1H)4.21(s,1H)4.27(q,J=7.09Hz,2H)4.43(d,J=11.74Hz,1H)4.52(m,1H)5.10(d,J=10.76Hz,1H)5.28(d,J=17.12Hz,1H)5.74(m,1H)5.82(s,1H)7.30(d,J=9.05Hz,1H)7.92(d,J=6.36Hz,1H)7.97(m,1H)8.22(d,J=9.05Hz,1H).
实施例207:化合物207的制备
采用同样的方法制备BOCNH-P3(L-t-BuGly)-P2[(4R)-(6-甲氧基-5-甲硫基-异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷。
在-78℃下,向100mg 1-氯-6-氟-异喹啉(0.55mmol)在2ml无水THF中的溶液内加入LDA的THF(1.3eq)溶液。形成深棕色溶液,随后加入二硫化物,溶液的颜色转变为绿色,接着变为浅棕色。反应物用2ml水和2ml NH4Cl猝灭,用乙酸乙酯萃取,经硫酸钠干燥。将溶剂真空蒸发,并直接使用所得的粗剩余物。LC/MS Rt-min(MH+):2.23(228)[方法B]。在-78℃下,将粗产物再次溶于2ml无水THF中,加入1.3eq KOMe,接着将反应混合物升温至室温,搅拌过夜。反应混合物用乙酸乙酯稀释并用盐水洗涤,硫酸钠干燥。得到104mg(79%)。LC/MS Rt-min(MH+):2.04(240)[方法B]。使中间体1-氯-5-甲硫基-6-甲氧基异喹啉进行前述三肽烷基化。按照通用方法,得到70.0mg黄色固体物(42.7%)。LC/MS Rt-min(MH+):2.65(760)[方法B]。1H NMR(400MHz,氯仿-D)δppm 0.94(m,11H)1.17(s,9H)1.26(m,2H)1.39(m,1H)1.83(dd,J=8.07,5.62Hz,1H)2.01(m,2H)2.23(s,3H)2.45(m,1H)2.79(m,1H)3.94(s,3H)3.97(d,J=3.91Hz,1H)4.15(s,1H)4.25(d,J=11.74Hz,1H)4.36(dd,J=9.66,7.21Hz,1H)4.99(d,J=10.27Hz,1H)5.12(d,J=16.87Hz,1H)5.69(m,1H)5.74(s,1H)7.08(d,J=9.05Hz,1H)7.83(m,2H)8.06(d,J=9.05Hz,1H)。
实施例208:1-氯-6-甲氧基-异喹啉-5-基-噻吩-2-基-甲酮的制备
按照前述1-氯-6-氟异喹啉的相同LDA脱质子方法(实施例203化合物的制备),但用2-噻吩甲醛猝灭初始阴离子,得到1-氯-6-氟异喹啉-5-基-噻吩-2-基-甲醇。使用MnO2的苯溶液将所述物质氧化成1-氯-6-氟-异喹啉-5-基-噻吩-2-甲酮,经层析纯化后总收率为49.6%。LC/MS Rt-min(MH+)[方法C]:2.98(292)。1H NMR(400MHz,氯仿-D)δppm 7.12(dd,J=4.89,3.91Hz,1H)7.40(m,1H)7.53(m,1H)7.56(dd,J=5.87,0.73Hz,1H)7.82(dd,J=5.01,1.10Hz,1H)8.27(d,J=5.87Hz,1H)8.54(ddd,J=9.29,5.38,0.73Hz,1H)。在过量甲醇钾溶液中进行氟原子的Ipso亲核芳族置换,得到主产物1-氯-6-甲氧基-异喹啉-5-基-噻吩-2-甲酮,以及25-33%的1,6-二甲氧基-异喹啉-5-基-噻吩-2-甲酮。所述粗产物(77mg)未经进一步纯化,直接用三肽进行烷基化步骤。
实施例209:化合物209的制备
按照三肽烷基化的通用方法(实施例184),得到35.3mgBOCNH-P3(L-t-BuGly)-P2[(4R)-6-甲氧基-5-(噻吩-2-羰基)-异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷,为浅色固体物(26.5%)。LC/MS Rt-min(MH+):2.54(825)[方法B]。
1H NMR(400MHz,CD3OD)δppm 1.02(s,9H)1.06(m,2H)1.22(m,2H)1.26(s,9H)1.43(m,1H)1.87(dd,J=7.95,5.50Hz,1H)2.28(m,2H)2.62(dd,J=13.82,6.97Hz,1H)2.93(m,1H)3.90(s,3H)4.07(dd,J=11.62,3.06Hz,1H)4.23(s,1H)4.43(m,1H)4.55(dd,J=9.78,7.34Hz,1H)5.09(m,1H)5.29(d,J=17.12Hz,1H)5.74(m,1H)5.86(s,1H)6.93(d,J=6.11Hz,1H)7.11(m,1H)7.32(dd,J=3.91,0.98Hz,1H)7.46(d,J=9.29Hz,1H)7.86(t,J=6.72Hz,1H)7.91(dd,J=4.89,1.22Hz,1H)8.39(d,J=9.29Hz,1H).
采用肉桂酸衍生物制备P2*。下述通用方法已在其它地方进行了详细描述。
通用合成方案
实施例210:化合物210的制备
将10.0g间甲苯基丙烯酸(61.7mmol)悬浮在50ml苯中,依次加入12.6ml DPPA(0.95eq)和10.3ml三乙胺(1.2eq)。所得的溶液在室温下搅拌1hr。真空除去挥发份,将所得的间甲苯基丙烯酰叠氮化物经快速层析纯化,得到11.5g纯化合物(定量)。将所得物质溶于100mL二苯基甲烷中,随后在1小时内滴加入100ml预加热至200℃的二苯基甲烷中。将所得的溶液于该温度下再保温4小时,随后冷却至室温。形成白色沉淀物,过滤。将固体物用己烷洗涤三次并干燥。滤液用200ml己烷稀释,将溶液静置过夜,以分离出第二批产物。将两批产物合并,得到4.2g 6-甲基-异喹啉-1-醇(50%)。LC/MS Rt-min(MH+):1.31(160)[方法B]。1H NMR(400MHz,CD3OD)δppm2.49(s,3H)6.61(d,J=7.32Hz,1H)7.13(d,J=7.02Hz,1H)7.36(d,J=8.24Hz,1H)7.45(s,1H)8.18(d,J=8.24Hz,1H)。将所述产物悬浮在15ml磷酰氯中并回流3h。真空除去磷酰氯后,将剩余物在EtOAc(1L)和冷氢氧化钠水溶液(由1.0N 200mL NaOH和20mL 10.0N NaOH制得)间分配并搅拌15min。有机层用水(2×200mL)和盐水(200mL)洗涤,干燥(硫酸镁)并真空浓缩,得到1-氯-6-甲基-异喹啉(67.4%)。LC/MSRt-min(MH+):1.92(178)[方法B]。1H NMR(400MHz,氯仿-D)δppm2.53(s,3H)7.47(d,J=6.11Hz,2H)7.56(s,1H)8.18(m,2H)。最后使用前述方法(实施例184),用三肽进行1-氯-6-甲基-异喹啉的烷基化。
得到BOCNH-P3(L-叔-BuGly)-P2[(4R)-(6-甲基异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2-环丙烷,为白色泡沫状物,收率18%。LC/MS Rt-min(MNa+)[方法B]:2.64(720)。
1H NMR(400MHz,CD3OD)δppm 1.05(m,13H)1.23(m,9H)1.42(m,1H)1.86(dd,J=7.95,5.50Hz,1H)2.25(m,2H)2.49(s,3H)2.61(dd,J=13.82,6.48Hz,1H)2.93(m,1H)4.05(dd,J=11.86,3.30Hz,1H)4.23(s,1H)4.43(d,J=11.49Hz,1H)4.52(m,1H)5.10(d,J=11.49Hz,1H)5.28(d,J=17.12Hz,1H)5.74(m,1H)5.83(s,1H)7.24(d,J=5.87Hz,1H)7.35(d,J=8.07Hz,1H)7.58(s,1H)7.89(d,J=5.87Hz,1H)8.07(d,J=8.56Hz,1H).
实施例211:化合物211的制备
按照前述通用方法,得到BOCNH-P3(L-t-BuGly)-P2[(4R)-(1,3-二氧杂-7-氮杂-环戊烯并[α]萘-6-醇)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷,51.0mg,为浅色固体物(64.9%)。LC/MSRt-min(MH+):2.57(728)[方法B]。
1H NMR(400MHz,CD3OD)δppm 0.99(s,9H)1.07(m,2H)1.18(m,11H)1.42(m,1H)1.86(dd,J=8.07,5.62Hz,1H)2.23(m,2H)2.59(dd,J=13.69,6.85Hz,1H)2.93(m,1H)4.04(dd,J=11.74,2.20Hz,1H)4.22(s,1H)4.43(d,J=11.74Hz,1H)4.51(m,1H)5.10(d,J=10.27Hz,1H)5.28(d,J=17.12Hz,1H)5.73(m,1H)5.82(s,1H)6.18(s,2H)7.13(d,J=8.56Hz,1H)7.19(d,J=6.11Hz,1H)7.81(d,J=8.56Hz,1H)7.85(d,J=6.11Hz,1H).
5,6,7-三取代异喹啉P2*衍生物的制备:
反应条件:(a)LDA的THF溶液;(b)对于R=F,用N-氟苯磺酰亚胺(NFSI),或对于R=SMe,用二甲二硫(MeS)2。
在强碱(如LDA)存在下,将如上制得的5,6-亚甲二氧基-1-氯异喹啉直接脱质子,得到相应的7-阴离子,没有破坏1-氯官能团。将7-阴离子用亲电试剂(如NFSI(N-氟苯磺酰亚胺)和二甲二硫)猝灭,得到相应的7-取代异喹啉环体系。
实施例212:化合物212的制备
步骤1:
5,6-亚甲二氧基-7-氟-1-氯异喹啉的制备。
在氮气气氛、-78℃下,向5,6-亚甲二氧基-1-氯异喹啉(126mg,0.61mmol)在4ml THF中的溶液内加入LDA的环己烷溶液(1.5mol,0.65mL,0.98mmol)。将所得的浅棕色溶液搅拌15min,接着用N-氟苯磺酰亚胺(NFSI,0.3g,1.5当量)处理。TLC分析表明除未反应的原料外,还形成了新的斑点。水处理,接着用乙酸乙酯萃取,得到油状粗产物,该粗产物经制备型HPLC纯化,得到52mg(38%)。LC/MS Rt-min(MH+)[方法C]:3.09(226)。1H NMR(400MHz,氯仿-D)δppm 6.33(s,2H)7.52(d,J=5.87Hz,1H)7.71(d,J=10.51Hz,1H)8.16(d,J=5.87Hz,1H)。步骤2:如上所述(实施例184),用三肽进行5,6-亚甲二氧基-7-氟-1-氯异喹啉的烷基化,主要得到氟置换的产物。
下述的BOCNH-P3(L-t-BuGly)-P2[(4R)-(6-氯-1,3-二氧杂-7-氮杂-环戊烯并[α]萘-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷
按照通用方法,得到24.3mg(24.3%)黄色固体物。LC/MS Rt-min(MH+):2.54(763)[方法B]。
1H NMR(400MHz,CD3OD)δppm 1.00(s,9H)1.06(m,2H)1.20(m,2H)1.29(s,9H)1.42(dd,J=9.41,5.26Hz,1H)1.86(dd,J=8.07,5.38Hz,1H)2.24(m,2H)2.54(dd,J=13.57,6.48Hz,1H)2.92(m,1H)4.04(dd,J=12.10,2.81Hz,1H)4.20(d,J=7.34Hz,1H)4.33(d,J=12.23Hz,1H)4.47(dd,J=10.52,6.85Hz,1H)5.10(dd,J=10.39,1.59Hz,1H)5.28(dd,J=17.12,1.22Hz,1H)5.46(d,J=5.87Hz,1H)5.74(m,1H)6.29(m,2H)7.40(s,1H)7.56(m,1H)8.01(d,J=5.62Hz,1H).
实施例213:化合物213的制备
在氮气气氛、-78℃下,向5,6-亚甲二氧基-1-氯异喹啉(84mg,0.41mmol)在4ml THF中的溶液内加入LDA的环己烷溶液(1.5mol,0.60mL,0.9mmol)。在-78℃下,将浅棕色溶液搅拌15min,随后用二甲二硫(50μL)纯试剂,1.4当量)处理。TLC分析表明除未反应的原料外,还形成了新的斑点。水处理,接着用乙酸乙酯萃取,得到油状粗产物,该粗产物经制备型HPLC纯化,得到51mg(49%)。LC/MS Rt-min(MH+)[方法C]:3.39(254)。1H NMR(400MHz,氯仿-D)δppm2.64(s,3H)6.29(s,2H)7.49(d,J=4.89Hz,1H)7.71(s,1H)8.11(d,J=5.87Hz,1H)。如上所述(实施例184),用三肽进行5,6-亚甲二氧基-7-甲硫基-1-氯异喹啉的烷基化,得到如下所示所需的产物:
BOCNH-P3(L-t-BuGly)-P2[(4R)-(4-甲硫基-1,3-二氧杂-7-氮杂环戊烯并[α]萘-6-氧基)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷
按照通用方法,得到59.6mg黄色固体物(42.2%)。LC/MS Rt-min(MH+):2.70(774)[方法B]。1H NMR(400MHz,氯仿-D)δppm1.02(m,11H)1.16(s,9H)1.32(s,2H)1.45(m,1H)1.94(m,1H)2.12(d,J=8.56Hz,1H)2.56(s,3H)2.62(m,2H)2.90(d,J=4.40Hz,1H)4.15(d,J=7.83Hz,2H)4.48(d,J=12.47Hz,1H)4.62(t,J=7.83Hz,1H)5.13(d,J=10.52Hz,1H)5.26(d,J=17.12Hz,1H)5.74(d,J=16.38Hz,1H)5.95(s,1H)6.28(s,2H)7.41(s,1H)7.59(s,1H)7.85(d,J=6.11Hz,1H)。
3,4-二取代的异喹啉P2*衍生物的制备
实施例215:化合物215的制备
化合物215
按照下述方案,制备下述BOCNH-P3(L-t-BuGly)-P2[(1R)-(2,3-二氢-1H-4-氮杂-环戊烯并[α]萘-5-氧基)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷:
异喹啉组分化合物214的通用合成方案
注:
按照以下引用的技术可成功合成新的1-氟代P2*:
(1)Rigby,James H.;Holswo rth,Daniel D.;James,Kelly.VinylIsocyanates In Synthesis.[4+2]Cycloaddition Reactions With BenzyneAddends.Journal Of Organic Chemistry(1989),54(17),4019-20
(2)Uchibori,Y.;Umeno,M.;Yoshiokai,H.;Heterocycles,1992,34(8),1507-1510
实施例214:化合物214,5-氯-2,3-二氢-1H-4-氮杂-环戊烯并[α]萘和实施例215的化合物215的制备
按照上述引用的Rigby的文献(参考文献1)的方法,制备2,3-二氢-1H-4-氮杂-环戊烯并[α]萘-5-醇。使用本文其它地方描述的磷酰氯,合成化合物214,收率59.8%(430mg)。LC/MS Rt-min(MH+):2.29(204)[方法B]。1H NMR(400MHz,氯仿-D)δppm 2.28(m,2H)3.19(q,J=7.74Hz,4H)7.58(m,1H)7.71(m,2H)8.32(d,J=8.56Hz,1H)。按照实施例184的方法,使所述氯化物与三肽充分反应进行烷基化,得到所需的产物化合物215。如按照上述Uchibori的方法(参考文献2),用氟化物代替氯化物,则可得到双倍的收率。由此分离出17.0mg化合物215,为浅黄色固体物(23.6%)。LC/MS Rt-min(MH+):2.80(724)[方法B]。
1H NMR(500MHz,CD3OD)δppm 1.03(s,9H)1.09(m,2H)1.24(m,11H)1.44(dd,J=8.24,5.49Hz,1H)1.88(dd,J=7.93,5.49Hz,1H)2.25(m,4H)2.63(dd,J=13.73,7.02Hz,1H)2.94(m,1H)3.05(m,2H)3.10(m,2H)4.08(dd,J=11.60,2.75Hz,1H)4.24(d,J=20.45Hz,1H)4.45(d,J=11.90Hz,1H)4.54(dd,J=9.46,7.63Hz,1H)5.11(m,1H)5.30(d,J=17.09Hz,1H)5.75(m,1H)5.87(s,1H)7.44(t,J=7.02Hz,1H)7.69(m,2H)8.18(d,J=8.24Hz,1H).
实施例217和218的3,4-二氢呋喃基和呋喃基异喹啉P2*组分的制备,
通用合成方案
实施例217:化合物217,5-氯-2,3-二氢-1-氧杂-4-氮杂-环戊烯并[α]萘和化合物218,5-氯-1-氧杂-4-氮杂-环戊烯并[α]萘的制备。
该合成方法部分按照以下参考文献中描述的技术:
(1)Hojo,Masaru;Masuda,Ryoichi;Sakaguchi,Syuhei;Takagawa,Makoto,Synthesis(1986),(12),1016-17
(2)Rigby,James H.;Holswo rth,Daniel D.;James,Kelly.VinylIsocyanates In Synthesis.[4+2]Cycloaddition Reactions With BenzyneAddends.Journal Of Organic Chemistry(1989),54(17),4019-20
(3)Uchibori,Y.;Umeno,M.;Yoshiokai,H.;Heterocycles,1992,34(8),1507-1510
当按照上述引用文献(参考文献1和2)的方法时,同时得到2,3-二氢-1-氧杂-4-氮杂-环戊烯并[α]萘-5-醇和1-氧杂-4-氮杂-环戊烯并[α]萘-5-醇。按照通用方法,采用磷酰氯将该对化合物转化成其氯代衍生物:粗羟基产物(约2g,浅黄色油状物)用15ml磷酰氯处理,并将混合物回流3h。真空除去磷酰氯后,将剩余物与EtOAc(1L)和冷氢氧化钠水溶液(220mL,1.0N)一起搅拌15min。分离出有机层,用水(2×200mL)和盐水(200mL)洗涤,经硫酸镁干燥并真空浓缩,经硅胶层析分离后,得到300mg实施例217的产物5-氯-2,3-二氢-1-氧杂-4-氮杂-环戊烯并[α]萘(13.2%)和100mg实施例218的产物5-氯-1-氧杂-4-氮杂-环戊烯并[α]萘(4.4%),均为浅棕色固体物。化合物217:LC/MS Rt-min(MH+):2.05(206)[方法B]。1H NMR(400MHz,氯仿-D)δppm 3.46(t,J=9.05Hz,2H)4.82(t,J=9.17Hz,2H)7.58(m,1H)7.66(m,1H)7.85(d,J=8.31Hz,1H)8.21(d,J=8.56Hz,1H)。化合物218:LC/MSRt-min(MH+):2.16(204)[方法B]。1H NMR(400MHz,氯仿-D)δppm7.15(d,J=2.20Hz,1H)7.70(m,1H)7.89(m,2H)8.27(d,J=8.31Hz,1H)8.44(d,J=8.80Hz,1H)。
5-氟-2,3-二氢-1-氧杂-4-氮杂-环戊烯并[α]萘和最终的P2*偶合产物的制备。
按照上述引用的方法(参考文献3)进行氯化物/氟化物的交换。为此,将90mg 5-氯-2,3-二氢-1-氧杂-4-氮杂-环戊烯并[α]萘(实施例217化合物)悬浮在1.5ml Bu4PHF2中,随后在微波下辐射(Smith反应器)至约120℃下反应2h。经水处理和柱层析纯化后,得到22mg氟化物产物(26.9%)。LC/MS Rt-min(MH+):1.91(190)[方法B]。所述呋喃衍生物(实施例218化合物)5-氯-1-氧杂-4-氮杂-环戊烯并[α]萘未经氟化物活化,直接与三肽充分反应,进行烷基化。
实施例219:化合物219的制备
BOCNH-P3(L-t-BuGly)-P2[(4R)-(2,3-二氢-1-氧杂-4-氮杂-环戊烯并[α]萘-5-氧基)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷。按照通用烷基化方法,得到22.0mg黄色固体物(26.3%)。LC/MS Rt-min(MH+):2.65(726)[方法B]。
1H NMR(400MHz,CD3OD)δppm 0.99(s,9H)1.07(m,2H)1.20(m,11H)1.40(m,1H)1.86dd,J=8.07,5.62Hz,1H)2.21(dd,J=17.48,8.93Hz,2H)2.60(dd,J=13.45,6.85Hz,1H)2.93(m,1H)3.34(m,2H)4.04(dd,J=11.74,3.18Hz,1H)4.24(s,1H)4.41(d,J=11.49Hz,1H)4.51(m,1H)4.74(t,J=9.05Hz,2H)5.11(d,J=10.27Hz,1H)5.28(d,J=17.36Hz,1H)5.73(m,1H)5.78(s,1H)7.43(m,1H)7.65(t,J=7.46Hz,1H)7.74(d,J=8.31Hz,1H)8.12(d,J=8.56Hz,1H).
实施例220:化合物220的制备
BOCNH-P3(L-t-BuGly)-P2[(4R)-(1-氧杂-4-氮杂-环戊烯并[α]萘-5-氧基)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷。按照通用烷基化方法,得到13.0mg黄色固体物(20%)。LC/MS Rt-min(MH+):2.70(724)[方法B]。
1H NMR(500MHz,CD3OD)δppm 1.01(S,,9H)1.09(m,2H)1.22(s,9H)1.27(m,2H)1.46(m,1H)1.89(dd,J=7.78,5.65Hz,1H)2.24(d,J=8.55Hz,1H)2.33(t,J=9.92Hz,1H)2.68(dd,J=13.73,7.02Hz,1H)2.95(m,1H)4.14(m,1H)4.26(s,1H)4.50(d,J=11.90Hz,1H)4.57(d,J=17.09Hz,1H)5.12(d,J=10.07Hz,1H)5.30(d,J=17.40Hz,1H)5.75(m,1H)5.93(s,1H)6.97(d,J=2.14Hz,1H)7.51(t,J=7.32Hz,1H)7.81(t,J=7.48Hz,1H)7.92(s,1H)8.13(d,J=7.94Hz,1H)8.28(d,J=8.24Hz,1H).
3-卤代和3-杂芳基4-烷氧基和4-羟基异喹啉P*衍生物的制备通用合成方案
反应条件:(1)MeOK的DMPU溶液;(2)NBS的二氯乙烷;(3)MCPBA的二氯甲烷溶液;(4)磷酰氯的二氯乙烷溶液;(5)BBr3的二氯甲烷溶液;(6)SEM-氯化物和Hunig氏碱的二氯甲烷溶液
按照新且方便的方法,使用普通实验室设备和试剂,由4-溴异喹啉制备4-甲氧基异喹啉(实施例222a化合物)。采用区域选择性NBS溴化,以良好收率得到3-溴-4-甲氧基异喹啉(实施例222b化合物)。平缓地进行MCPBA氧化,得到相应的N-氧化物(实施例222c化合物),使用常规的磷酰氯方法,将该化合物经异构化得到1-氯-3-溴-4-甲氧基异喹啉(实施例222d)。采用三肽将4-甲氧基异喹啉烷基化,得到相应的3-溴-4-甲氧基P2*衍生物,适用于Stille和Suzuki偶合。或者将所述4-甲氧基异喹啉用BBr3脱甲基,得到4-羟基-3-溴-1-氯异喹啉(实施例222e化合物)。再次用SEM-氯化物保护4-羟基,得到4-SEM保护的中间体实施例222化合物。按照酸诱导或氟诱导的脱保护方法进行偶合,再次得到4-羟基化合物。
实施例222d:1-氯-3-溴-4-甲氧基异喹啉的制备
步骤1:
向4-溴异喹啉(15g,73mmol,商品)在200mL二甲基-3,4,5,6-四氢-2(1H)-嘧啶酮(DMPU,Aldrich)中的溶液内加入固体甲醇钾(5.6gm,80mmol)。将反应容器浸入105℃的油浴中20min。在受热下,混合物的颜色由初始时的非常浅颜色很快变为深绿棕色。将反应容器从油浴中移出,并用水稀释,用乙醚多次萃取,将有机剩余物分配至乙醚中。TLC分析表明存在两个新的斑点(1∶1体积比,己烷和乙酸乙酯的混合物作为洗脱液),大小大约相同。将它们经硅胶柱层析(Merck,H型)分离。洗脱液:直链己烷,接着在流动相中逐渐加入乙醚。蒸发溶剂后,分离出所需的产物4-甲氧基异喹啉(4.1gm,35.3%)。还分离出其它产物,为还原反应副产物异喹啉。副产物经NMR确证,并与已知物质对比确认。实施例222a:LC/MS Rt-min(MH+)[方法C]:1.16(160)。1H NMR(400MHz,氯仿-D)δ4.07(s,3H)7.61(m,1H)7.69(m,1H)7.93(d,J=8.07Hz,1H)8.08(s,1H)8.19(d,J=8.56Hz,1H)8.89(s,1H)。[注:该化合物先后由Zoltewicz,John A.;Oestreich,Terence M.;Sale,Alan A,Journal of the American ChemicalSociety(1975),97(20),5889-96,″Monel Bomb″和″focusedmicrowave″,Cherng,Yie-Jia,Tetrahedron(2002),58(6),1125-1129制得。本发明的方法既不需要特殊的高压装置,也不需要制备级的微波设备。
步骤2:
将4-甲氧基异喹啉(实施例222a的产物)进行NBS溴化,为此将所述物质(实施例222a的产物,2.1gm,13.2mmol)在1,2-二氯乙烷(DCE,150mL)中的溶液在70℃下用N-溴代琥珀酰亚胺(NBS,1.5gm,8.4mmol,0.6×)处理1小时,接着加入第二部分1.5gm NBS。再将所得深棕色混合物搅拌1小时,接着加入第三部分1.0gm NBS。采用LC-MS监控该溴化反应,直至没有原料存在。将粗品混合物蒸发至干,经短硅胶柱(H型,Merck,直径3cm×高1.5cm)过滤所需的产物,洗脱液:先为直链己烷,接着逐渐增加乙醚的量。分离出所需的产物(实施例222b化合物),为油状物质(1.7gm,54%)。LC/MSRt-min(MH+)[方法C]:2.65(238)。1H NMR(400MHz,氯仿-D)δppm4.04(s,3H)7.64(t,J=7.58Hz,1H)7.76(t,J=7.09Hz,1H)7.99(d,J=8.31Hz,1H)8.11(d,J=8.31Hz,1H)8.85(s,1H)。[3-溴-4-甲氧基异喹啉预先采用不同的方法制得:Finkentey,Christel;Langhals,Elke;Langhals,Heinz.Chemische Berichte(1983),116(6),2394-7。产物的NMR位移值与报导的一致]。
步骤3:
室温下,在二氯甲烷中,将NBS溴化反应的产物进行MCPBA氧化反应。为此,将MCPBA(1.80g,纯度77%,8.0mmol)加入3-溴-4-甲氧基异喹啉(实施例222b化合物,1.65gm,6.9mmol)在35ml二氯甲烷中的溶液内。将溶液搅拌4h形成白色悬浮液。将碳酸氢钠溶液(5%,新鲜制备,20mL)加入混合物中,有机剩余物用二氯甲烷(10×25mL)萃取。需要对有机溶剂进行多次萃取,以回收一些N-氧化物产物水溶液。将蒸发溶剂得到的粗产物经硅胶过滤进一步纯化,得到1.36mg(5.4mmol,78%)所述N-氧化物(实施例222c化合物),为蜡状固体物。
LC/MS Rt-min(MH+)[方法C]:1.79(254)。1H NMR(400MHz,氯仿-D)δppm 4.07(s,3H)7.63(m,2H)7.72(m,1H)8.00(m,1H)8.86(s,1H)。
步骤4:
使用其它地方描述的方法,通常在磷酰氯中进行最终N-氧化物的重排反应。实施例222d产物的收率基本为定量。LC/MS Rt-min(MH+)[方法D]:2.69(272)。1H NMR(盐酸盐)(400MHz,氯仿-D)δppm 4.07(s,3H)7.81(m,1H)7.92(m,1H)8.17(d,J=8.31Hz,1H)8.34(d,J=8.31Hz,1H)。1H NMR(游离碱)(400MHz,氯仿-D)δppm 4.03(s,3H)7.72(m,1H)7.81(m,1H)8.12(d,J=8.56Hz,1H)8.28(d,J=8.56Hz,1H)。
实施例223:化合物223的制备
按照其它地方描述的烷基化方法(实施例184),采用三肽片断将前述步骤得到的游离碱(实施例222d的产物)烷基化,得到79%所需的产物,为纸白色固体物。LC/MS Rt-min(MNa+)[方法C]:3.91(814)。
1H NMR(400MHz,CD3OD)δppm 1.02(s,9H)1.06(dd,J=8.07,1.47Hz,2H)1.22(m,11H)1.42(dd,J=9.78,5.14Hz,1H)1.86(dd,J=8.07,5.38Hz,1H)2.22(dd,J=18.10,9.29Hz,1H)2.28(m,1H)2.61(dd,J=13.57,6.97Hz,1H)2.93(m,1H)3.92(s,3H)4.06(dd,J=11.86,2.81Hz,1H)4.22(s,1H)4.43(d,J=11.49Hz,1H)4.51(m,1H)5.10(d,J=10.52Hz,1H)5.28(d,J=17.12Hz,1H)5.74(m,1H)5.81(s,1H)7.56(t,J=7.58Hz,1H)7.78(t,J=7.58Hz,1H)8.00(d,J=8.31Hz,1H)8.16(d,J=8.56Hz,1H).
实施例224和225:化合物224和化合物225的制备
按照以下方法,将前述1-氯-3-溴-4-甲氧基异喹啉(实施例222d的产物)的4-甲氧基转化为α-三甲基甲硅烷基乙氧基甲基(SEM)部分。室温下,采用BBr3将1-氯-3-溴-4-甲氧基异喹啉(实施例222d的产物)脱甲基(最终调节反应物浓度,使得BBr3为0.2-0.3mol)12h。发现在这种脱甲基反应中使用高BBr3浓度是必需且有效的。在蒸发至干前,将粗反应混合物用50体积无水甲醇稀释。脱甲基反应基本定量完成。实施例222e的产物:LC/MS Rt-min(MH+)[方法D]:2.32(258)。1H NMR(游离盐酸盐)(400MHz,氯仿-D)δppm 5.83(br.s,1H)7.73(t,J=7.70Hz,1H)7.79(t,J=7.58Hz,1H)8.22(m,2H)。用2-(三甲基甲硅烷基)乙氧基甲基氯(SEM-Cl)再次保护所述4-羟基-3-溴-1-氯异喹啉(实施例222e的产物)。室温下,将前述制备得到的粗游离碱在40微米(Hg)下干燥,接着再用SEM-氯化物保护。在0℃下,向4-羟基化合物(实施例222e的产物,1.33gm,5.2mmol)的二氯甲烷(50ml)溶液中依次加入二异丙基乙胺(2mL,11.5mmol)和SEM-氯化物(1.8mL,10mmol)。将混合物搅拌10min,随后用新鲜制备碳酸氢钠溶液(5%,100mL)洗涤。有机剩余物用二氯甲烷萃取数次,合并的有机层用20mL去离子水回洗,接着将其真空浓缩。SEM保护基本定量进行。实施例222的产物:LC/MS Rt-min(MH+)[方法D]:3.40(410)。
1H NMR(400MHz氯仿-D)δppm 0.03(s,9H),0.99(m,2H),3.98(m,2H),5.33(s,2H),7.72(m,1H),7.80(m,1H),8.17(d,J=8.56Hz,1H),8.27(d,J=8.07Hz,1H)。用三肽将4-SEM保护的异喹啉烷基化:按照相同三肽烷基化反应得到化合物224和化合物225。4-羟基化合物(化合物224)最有可能是在制备型HPLC纯化过程中存在TFA产生。
实施例224化合物(15.4%):LC/MS Rt-min(MNa+)[方法D]:2.87(800)。
1H NMR(400MHz,CD3OD)δppm 1.02(s,9H)1.06(d,J=8.31Hz,2H)1.25(s,9H)1.42(s,2H)1.86(m,1H)2.23(m,2H)2.60(dd,J=13.21,7.34Hz,1H)2.93(m,1H)4.06(d,J=11.00Hz,1H)4.24(m,2H)4.38(d,J=11.98Hz,1H)4.49(dd,J=9.78,7.09Hz,1H)5.10(d,J=10.03Hz,1H)5.28(d,J=17.36Hz,1H)5.72(m,1H)5.76(s,1H)7.52(t,J=7.46Hz,1H)7.71(t,J=7.09Hz,1H)8.09(d,J=4.40Hz,1H)8.11(d,J=4.16Hz,1H).
实施例225化合物(8.0%):LC/MS Rt-min([M-BOC]+)[方法D]:3.46(808)。
1H NMR(400MHz,CD3OD)δppm 0.01(s,9H)0.96(m,2H)1.02(s,11H)1.06(d,J=6.60Hz,2H)1.24(s,9H)1.42(m,1H)1.86(dd,J=7.83,5.38Hz,1H)2.25(m,2H)2.62(dd,J=13.69,7.34Hz,1H)2.93(m,1H)3.97(m,2H)4.07(dd,J=10.88,3.55Hz,1H)4.23(s,1H)4.43(d,J=11.25Hz,1H)4.50(m,1H)5.10(d,J=10.76Hz,1H)5.25(m,3H)5.74(m,1H)5.82(s,1H)7.57(m,1H)7.77(t,J=7.83Hz,1H)8.06(d,J=8.56Hz,1H)8.16(d,J=8.31Hz,1H).
4H-[1,3]二_烯并[5,4-c]异喹啉P2*衍生物的制备
通用合成方案
反应条件:(1)MeOK/DMPU;(2)MCPBA的二氯甲烷溶液;(3)磷酰氯的DCE溶液;(4)BBr3的二氯甲烷溶液;(5)HCHO/40%H2SO4,按照1,3-_嗪并[5,6-c]异喹啉和相关化合物的合成方法,MiyokoToyama和Hirotaka Otomasu,Chem.Pharm.Bull.33(12),5543-5546,1985;(6)按照Uchibori,Y.;Umeno,M.;Yoshiokai,H.;Heterocycles,1992,34(8),1507-1510的氟化反应方法。
实施例227:化合物227的制备
按照Miyoko Toyama和Hirotaka Otomasu的方法,以1-氯-4-羟基异喹啉为原料,制备6-氯-1,3-_嗪并[5,6-c]异喹啉。原料1-氯-4-羟基异喹啉(实施例226c的产物)按上述合成方法制备。按通用方法进行4-甲氧基异喹啉(实施例222a的产物)的MCPBA氧化,得到79.1%相应的N-氧化物(实施例226a的产物)。随后立即在磷酰氯中将所述物质转化为1-氯衍生物,基本定量得到氯化物(实施例226b的产物)。室温下,将粗1-氯-4-甲氧基异喹啉用BBr3脱甲基,室温下用无水甲醇处理所得粗BBr3混合物,接着蒸发除去过量的硼酸盐剩余物,得到相应的1-氯-4-羟基异喹啉(实施例226c的产物)。进行MiyokoToyama和Hirotaka Otomasu反应,得到266mg 6-氯-1,3-_嗪并[5,6-c]异喹啉(实施例226d的产物,62.3%),4步反应总共得到300mg 4-甲氧基异喹啉。LC/MS Rt-min([M-HCHO]H+)[方法D]:2.45(192)。1HNMR(400MHz,氯仿-D)δppm 5.02(s,2H)5.41(s,2H)7.68(m,1H)7.77(ddd,J=8.25,6.91,1.22Hz,1H)8.10(d,J=8.31Hz,1H)8.26(d,J=8.56Hz,1H)。
发现按照实施例184的烷基化方法,所述氯化物仍未反应。按照前面引用文献[Uchibori,Y.;Umeno,M.;Yoshiokai,H.;Heterocycles,1992,34(8),1507-1510]的方法制备相应的6-氟-1,3-_嗪并[5,6-c]异喹啉(实施例226的产物)。反应没有进行完全,回收的粗反应混合物为1∶2.4(Cl∶F)的混合物。按照实施例184的方法,所述氯/氟混合物未经进一步纯化直接采用三肽烷基化,经制备型HPLC纯化后,得到66mg(50.0%)BOCNH-P3(L-t-BuGly)-P2[(4R)-(1,3-_嗪并[5,6-c]异喹啉-6-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷。LC/MS Rt-min(MNa+)[方法D]:3.03(764)。
1H NMR(400MHz,CD3OD)δppm 1.01(s,9H)1.06(dd,J=8.07,1.96Hz,2H)1.22(s,10H)1.34(d,J=6.11Hz,1H)1.42(m,1H)1.86(dd,J=8.07,5.38Hz,1H)2.23(m,2H)2.59(dd,J=13.82,6.97Hz,1H)2.93(m,1H)4.03(dd,J=11.86,3.06Hz,1H)4.23(s,1H)4.41(d,J=11.98Hz,1H)4.50(dd,J=9.66,6.97Hz,1H)4.87(m,2H)5.11(d,J=10.52Hz,1H)5.28(d,J=17.12Hz,1H)5.34(s,2H)5.74(m,2H)7.51(t,J=7.46Hz,1H)7.70(t,J=7.58Hz,1H)7.95(d,J=8.31Hz,1H)8.12(d,J=8.31Hz,1H).
通过Suzuki和Stille偶合反应制备4-甲氧基-3-杂芳基和3-偶氮基异喹啉P2*衍生物
以下所示的偶合技术说明实施例223的溴代衍生物的一般应用。应理解可采用其它偶合剂和除硼和锡外的其它催化剂的组合实施相同的方法。
实施例229:化合物229的制备
通过如下所示的Suzuki偶合制备BOCNH-P3(L-t-BuGly)-P2[(4R)-(3-呋喃-3-基-4-甲氧基-异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷:
将22mg(0.028mmol)实施例223的产物溶于1ml DMF中,加入9.4mg商品硼酸(3eq)、3mg催化剂(10%mmol)和18mg碳酸铯。将混合物脱气两次,随后在110℃下加热3h。终产物经制备型HPLC纯化,得到13.6mg黄色固体物(64.0%)。LC/MS Rt-min(MH+):2.85(780)[方法B]。
1H NMR(500MHz,CD3OD)δppm 1.09(m,11H)1.26(m,12H)1.68(m,1H)2.27(s,1H)2.64(m,2H)2.97(m,1H)3.86(s,3H)4.15(d,J=10.38Hz,1H)4.28(s,1H)4.43(d,J=10.99Hz,1H)4.56(m,1H)5.11(m,2H)5.63(m,1H)5.99(s,1H)7.20(s,1H)7.51(m,1H)7.61(m,1H)7.75(t,J=7.17Hz,1H)8.03(d,J=8.24Hz,1H)8.16(d,J=8.24Hz,1H)8.27(s,1H).
实施例230:化合物230的制备
通过如下所示的Stille偶合反应合成BOCNH-P3(L-t-BuGly)-P2[(4R)-(3-呋喃-2-基-4-甲氧基-异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷:
将40mg(0.05mmol)实施例223的产物、4mg催化剂(5%mmol)和100μl(4eq)商品锡试剂溶于1ml甲苯中,将混合物脱气两次,随后在90℃下加热过夜。经制备型HPLC分离后,得到19.6mg绿色固体物(50.0%)。LC/MS Rt-min(MH+):2.76(780)[方法B]。
1H NMR(400MHz,CD3OD)δppm 0.94(m,2H)0.98(s,9H)1.09(m,2H)1.25(s,9H)1.39(m,1H)1.60(m,1H)2.35(m,1H)2.48(m,1H)2.74(m,1H)2.95(m,1H)3.87(s,3H)4.14(m,1H)4.22(d,J=4.16Hz,1H)4.41(s,1H)4.69(m,1H)5.26(m,1H)5.35(m,1H)5.93(s,1H)6.03(m,1H)6.61(m,1H)7.16(d,J=3.18Hz,1H)7.50(d,J=7.58Hz,1H)7.67(s,1H)7.73(t,J=7.34Hz,1H)8.04(m,1H)8.17(d,J=8.31Hz,1H).
实施例231:化合物231的制备
按照类似于Stille偶合反应方法,制备BOCNH-P3(L-t-BuGly)-P2[(4(R)-(3-吡嗪-2-基-4-甲氧基-异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷,收率7.1%。LC/MS Rt-min(MH+):2.51(792)[方法B]。
1H NMR(400MHz,CD3OD)δppm0.98(m,9H)1.11(m,2H)1.19(s,9H)1.27(m,2H)1.42(m,1H)2.37(m,1H)2.48(m,2H)2.81(m,1H)2.97(m,1H)3.83(s,3H)4.07(s,1H)4.20(d,J=4.16Hz,1H)4.54(d,J=11.49Hz,1H)4.72(m,1H)5.27(m,1H)5.39(m,1H)5.96(s,1H)6.04(m,1H)7.63(s,1H)7.83(s,1H)8.17(s,1H)8.26(s,1H)8.60(d,J=2.20Hz,1H)8.76(d,J=2.20Hz,1H)9.33(s,1H).
实施例232:化合物232的制备
按照类似于Stille偶合反应方法,制备BOCNH-P3(L-t-BuGly)-P2[(4R)-(4-甲氧基-3-噻唑-2-基-异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷,收率32.2%。LC/MS Rt-min(MH+):2.42(797)[方法B]。
1H NMR(400MHz,CD3OD)δppm 1.03(S,9H)1.07(m,2H)1.13(S,9H)1.22(m,2H)1.43(dd,J=9.78,5.14Hz,1H)1.88(dd,J=8.07,5.38Hz,1H)2.23(q,J=8.97Hz,1H)2.36(m,1H)2.67(m,1H)2.94(m,1H)4.10(s,3H)4.15(m,1H)4.18(s,1H)4.53(d,J=25.92Hz,1H)4.59(dd,J=10.27,7.09Hz,1H)5.12(m,1H)5.29(d,J=17.36Hz,1H).73(m,1H)6.09(s,1H)7.74(t,J=7.58Hz,1H)7.91(t,J=7.70Hz,1H)8.00(d,J=3.42Hz,1H)8.18(d,J=3.18Hz,1H)8.22(d,J=8.31Hz,1H)8.29(d,J=8.31Hz,1H).
实施例233:化合物233的制备
按照通用的三肽烷基化方法,采用商品4-氯呋喃并[3,2-c]吡啶,得到5.7mg(8.2%)黄色固体物。LC/MS Rt-min(MH+):2.32(674)[方法B]。
1H NMR(400MHz,CD3OD)δppm 1.00(s,9H)1.07(m,2H)1.21(m,11H)1.41(m,1H)1.86(dd,J=8.07,5.38Hz,1H)2.22(dd,J=17.61,9.05Hz,2H)2.54(dd,J=13.69,7.09Hz,1H)2.92(m,1H)4.06(m,1H)4.21(m,1H)4.32(s,1H)4.49(m,1H)5.11(dd,J=10.27,1.47Hz,1H)5.29(dd,J=17.36,1.22Hz,1H)5.74(m,1H)5.81(s,1H)6.83(d,J=1.22Hz,1H)7.19(d,J=5.87Hz,1H)7.76(d,J=1.22Hz,1H)7.97(d,J=5.87Hz,1H).
实施例235:化合物235的制备
按照通用的三肽烷基化方法,采用商品4-氯噻吩并[3,2-c]吡啶,得到20.0mg(28.1%)黄色固体物。LC/MS Rt-min(MH+):2.50(690)[方法B]。
1H NMR(400MHz,CD3OD)δppm 1.01(s,9H)1.06(m,2H)1.21(m,11H)1.42(m,1H)1.86(dd,J=8.19,5.50Hz,1H)2.24(m,2H)2.57(dd,J=13.69,6.85Hz,1H)2.93(m,1H)4.05(dd,J=11.98,3.18Hz,1H)4.22(s,1H)4.39(d,J=11.74Hz,1H)4.50(dd,J=9.90,7.21Hz,1H)5.10(dd,J=10.39,1.34Hz,1H)5.28(d,J=17.12Hz,1H)5.73(m,1H)5.81(s,1H)7.45(d,J=5.62Hz,1H)7.53(m,2H)7.94(d,J=5.87Hz,1H).
实施例236:化合物236的制备
按照通用的三肽烷基化方法,采用商品3,5-二氯-1,2,4-噻二唑,得到8.0mg(11.9%)黄色固体物。LC/MS Rt-min(MNa+):2.37(697)[方法B]。
1H NMR(400MHz,CD3OD)δppm 1.00(s,9H)1.06(m,2H)1.22(m,2H)1.36(s,9H)1.42(m,1H)1.86(dd,J=8.07,5.38Hz,1H)2.25(m,2H)2.60(dd,J=14.18,6.85Hz,1H)2.92(m,1H)4.03(dd,J=12.47,3.18Hz,1H)4.17(s,1H)4.42(m,2H)5.11(dd,J=10.27,1.71Hz,1H)5.29(dd,J=17.12,1.47Hz,1H)5.68(s,1H)5.74(m,1H).
实施例237:化合物237的制备
如下所示的BOCNH-P3(L-t-BuGly)-P2[(4R)-(喹喔啉-2-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷
按照通用的三肽烷基化方法,采用商品2-氯喹喔啉,得到113.0mg(19.2%)黄色固体物。LC/MS Rt-min(MNa+):2.48(707)[方法B]。
1H NMR(400MHz,CD3OD)δppm 1.01(s,9H)1.06(m,2H)1.22(m,11H)1.42(m,1H)1.87(dd,J=8.19,5.50Hz,1H)2.24(m,1H)2.31(m,1H)2.57(dd,J=13.57,6.97Hz,1H)2.93(m,1H)4.09(dd,J=11.98,3.18Hz,1H)4.17(s,1H)4.38(d,J=11.74Hz,1H)4.50(dd,J=10.27,7.09Hz,1H)5.11(dd,J=10.27,1.71Hz,1H)5.29(dd,J=17.12,1.47Hz,1H)5.74(m,1H)5.87(s,1H)7.62(t,J=7.46Hz,1H)7.73(t,J=7.70Hz,1H)7.87(m,1H)7.96(d,J=8.31Hz,1H)8.42(s,1H).
实施例238:化合物238的制备
如下所示的BOCNH-P3(L-t-BuGly)-P2[(4R)-(2-三氟-6-氟喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷:
按照通用的三肽烷基化方法,采用商品2-三氟甲基-4-氯-6-氟喹啉,得到17.0mg(23.2%)黄色固体物。LC/MS Rt-min(MNa+):2.66(792)[方法B]。
1H NMR(400MHz,CD3OD)δppm 1.02(s,9H)1.06(m,2H)1.17(s,9H)1.23(m,2H)1.42(m,1H)1.86(dd,J=8.07,5.38Hz,1H)2.21(q,J=8.64Hz,1H)2.32(m,1H)2.63(dd,J=13.94,6.85Hz,1H)2.93(m,1H)4.08(m,1H)4.18(s,1H)4.53(m,2H)5.10(m,1H)5.27(d,J=17.12Hz,1H)5.58(s,1H)5.72(m,1H)7.39(s,1H)7.65(m,1H)7.83(dd,J=9.29,2.69Hz,1H)8.12(dd,J=9.29,5.14Hz,1H).
实施例239:化合物239的制备
如下所示的BOCNH-P3(L-t-BuGly)-P2[(4R)-(6-氟喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷:
按照通用的三肽烷基化方法,采用商品4-氯-6-氟喹啉,得到26.0mg(39.0%)黄色固体物。LC/MS Rt-min(MH+):1.98(702)[方法B]。
1H NMR(400MHz,CD3OD)δppm 1.04(m,11H)1.14(s,9H)1.23(m,2H)1.42(m,1H)1.87(dd,J=8.07,5.38Hz,1H)2.23(q,J=8.80Hz,1H)2.41(m,1H)2.75(dd,J=14.43,6.85Hz,1H)2.93(m,1H)4.09(s,1H)4.12(d,J=2.69Hz,1H)4.61(m,2H)5.11(dd,J=10.39,1.59Hz,1H)5.28(dd,J=17.24,1.34Hz,1H)5.70(m,1H)5.75(s,1H)7.62(d,J=6.60Hz,1H)7.93(m,1H)8.06(dd,J=8.68,2.57Hz,1H)8.20(dd,J=9.29,4.40Hz,1H)9.06(d,J=6.60Hz,1H).
由于F置换,在该反应中还存在少量的副产物,经制备型HPLC分离。
实施例240:化合物240的分离
如下所示的BOCNH-P3(L-t-BuGly)-P2[(4R)-(4-氯喹啉-6-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷:
得到8.0mg黄色固体物副产物(11.7%)。LC/MS Rt-min(MNa+):2.240(740)[方法B]。
1H NMR(400MHz,CD3OD)δppm 1.00(s,9H)1.06(m,2H)1.23(m,11H)1.42(m,1H)1.86(dd,J=8.19,5.50Hz,1H)2.22(m,1H)2.29(m,1H)2.55(m,1H)2.92(m,1H)4.09(m,1H)4.21(s,1H)4.30(m,1H)4.46(dd,J=10.27,6.85Hz,1H)5.11(dd,J=10.27,1.47Hz,1H)5.28(dd,J=17.36,1.47Hz,1H)5.43(s,1H)5.74(m,1H)7.60(dd,J=9.29,2.45Hz,1H)7.64(d,J=2.45Hz,1H)7.81(d,J=4.89Hz,1H)8.06(d,J=9.29Hz,1H)8.72(d,J=5.14Hz,1H).
实施例241:化合物241的制备
如下所示的BOCNH-P3(L-t-BuGly)-P2[(4R)-(8-氟喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷:
按照通用的三肽烷基化方法,采用商品4-氯-8-氟喹啉,得到10.3mg(14.7%)黄色固体物。LC/MS Rt-min(MH+):1.95(702)[方法B]。
1H NMR(400MHz,CD3OD)δppm 0.98(s,9H)1.06(m,2H)1.14(s,9H)1.22(m,2H)1.42(m,1H)1.87(dd,J=8.07,5.62Hz,1H)2.22(q,J=8.72Hz,1H)2.41(m,1H)2.74(dd,J=14.06,6.97Hz,1H)2.93(m,1H)4.11(m,2H)4.57(dd,J=10.39,6.97Hz,1H)4.66(d,J=12.23Hz,1H)5.11(dd,J=10.27,1.22Hz,1H)5.28(d,J=17.12Hz,1H)5.71(m,2H)7.59(d,J=6.36Hz,1H) 7.75(m,1H)7.86(m,1H) 8.23(d,J=8.56Hz,1H)9.02(d,J=6.36Hz,1H).
在制备型HPLC纯化过程中,也分离出副产物。由于氟原子置换代替了氯离去基团,形成了4-氯喹啉-8-氧代-喹啉衍生物。
实施例242:化合物242的分离
如下所示的BOCNH-P3(L-t-BuGly)-P2[(4R)-(4-氯喹啉-8-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷:
由此得到9.0mg黄色固体物副产物(13.2%)。LC/MS Rt-min(MH+):2.37(718)[方法B]。
1H NMR(400MHz,CD3OD)δppm 1.00(s,9H)1.06(m,2H)1.12(s,9H)1.23(m,2H)1.43(dd,J=9.41,5.50Hz,1H)1.87(dd,J=8.19,5.50Hz,1H)2.25(m,1H)2.35(m,1H)2.67(dd,J=13.94,7.09Hz,1H)2.93(m,1H)4.10(m,1H)4.13(s,1H)4.43(d,J=11.98Hz,1H)4.65(dd,J=10.03,7.09Hz,1H)5.12(dd,J=10.27,1.47Hz,1H)5.30(dd,J=17.12,1.22Hz,1H)5.51(s,1H)5.75(m,1H)7.61(d,J=7.83Hz,1H)7.88(t,J=8.19Hz,1H)8.04(m,2H)8.91(d,J=5.38Hz,1H).
实施例243:化合物243的制备
如下所示的BOCNH-P3(L-t-BuGly)-P2[(4R)-(3-羟基喹喔啉-2-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷:
采用商品2,3-二氯喹喔啉实施通用的三肽烷基化方法,所得的单烷基化产物自发水解,得到8.0mg浅黄色固体物(11.4%)。LC/MSRt-min(MNa+):2.42(723)[方法B]。
1H NMR(400MHz,CD3OD)δppm 0.99(s,9H)1.05(m,2H)1.24(s,9H)1.40(m,3H)1.86(m,1H)2.24(m,2H)2.53(m,1H)2.92(m,1H)4.06(m,1H)4.16(s,1H)4.40(m,1H)4.55(dd,J=10.39,6.97Hz,1H)5.11(m,1H)5.29(m,1H)5.73(m,1H)5.78(s,1H)7.15(s,1H)7.26(m,2H)7.36(t,J=7.83Hz,1H)7.61(d,J=8.07Hz,1H).
实施例244:化合物244的制备
使用Pd0偶合方案并以6-溴-1-氯异喹啉为原料进行逐步反应,制备BOCNH-P3(L-t-BuGly)-P2[(4R)-(6-甲酸二甲基酰胺异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷。
LC/MS rt-min(MNa+):2.34(777)[方法B].1H NMR(400MHz,CD3OD)δppm0.98(m,11H)1.23(m,11H)1.35(m,1H)1.91(m,1H)2.29(m,,2H)2.47(m,1H)2.58(m,1H)2.97(s,3H)3.11(s,3H)4.09(m,1H)4.24(s,1H)4.44(m,1H)4.61(m,1H)5.16(m,2H)5.57(m,1H)5.90(s,1H)7.38(d,J=5.87Hz,1H)7.50(d,J=8.07Hz,1H)7.86(s,1H)8.03(d,J=5.87Hz,1H)8.27(d,J=8.56Hz,1H),
实施例245
化合物245的制备
在一个Pd0催化的Stille偶合制备过程中(实施例230),还分离出次产物,经确证为:如下所示的BOCNH-P3(L-t-BuGly)-P2[(4R)-(3-氯-4-甲氧基异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷:
LC/MS rt-min(MNa+):2.62(770)[方法B].1H NMR(400MHz,CD3OD)δppm1.01(s,9H)1.08(m,2H)1.18(s,9H)1.27(m,2H)1.37(m,1H)1.62(m,1H)2.36(m,2H)2.73(m,1H)2.97(m,1H)3.92(s,3H)4.02(m,1H)4.18(s,1H)4.48(m,1H)4.66(m,1H)5.30(m,2H)5.78(s,1H)6.04(m,1H)7.53(t,J=7.70Hz,1H)7.77(t,J=7.58Hz,1H)8.00(d,J=8.56Hz,1H)8.19(d,J=8.07Hz,1H).
部分F:
实施例250:化合物250的制备
化合物250
方案1
步骤1:
将3-苯基-丁-2-烯酸(16.2g)、二苯基磷酰叠氮(27.5g)和三乙胺(10.1g)在苯(100mL)中的溶液搅拌1h。经硅胶柱过滤(用苯洗涤)并浓缩,将剩余物溶于二苯基甲烷(80mL)中并回流3h。冷却至室温后,经柱收集固体物,用苯洗涤并干燥,得到10g(63%)所需的产物,为固体物。1H NMR(400MHz,CD3OD)δppm 2.30(s,3H),7.00(s,1H),7.54(m,1H),7.77(m,2H),8.33(d,J=7.34Hz,1H)。
步骤2
将4-甲基-2H-异喹啉-1-酮(4.8g)的磷酰氯(50mL)溶液回流3h。冷却并浓缩后,将剩余物用5N NaOH碱化并用二氯甲烷萃取。有机层用盐水洗涤并经硫酸镁干燥。浓缩后,经Biotage快速层析纯化,用5%乙酸乙酯/己烷洗脱,得到4.8g(90%)所需的产物,为固体物。1HNMR(400MHz,CDCl3)δppm 2.59(s,3H),7.68(t,J=7.70Hz,1H),7.78(m,1H),7.94(d,J=8.31Hz,1H),8.11(s,1H),8.35(d,J=8.31Hz,1H)。
步骤3:
将Boc-Hyp-OH(231mg)和叔丁醇钾(336mg)在DMSO(10mL)中的溶液搅拌0.5h。向该溶液中加入1-氯-4-甲基-异喹啉(178mg)并将所得的混合物搅拌1天。反应物用5%柠檬酸猝灭并用乙酸乙酯萃取。有机层用盐水洗涤并经硫酸镁干燥。浓缩得到350mg(94%)所需的产物,为固体物,该产物未经进一步纯化直接用于下一步骤中。1HNMR(400MHz,CD3OD)δppm 1.39,1.43(2s,9H,外消旋体),2.40(dd,J=17.97,4.52Hz,1H),2.48(s,3H),2.68(m,1H),3.84(m,2H),4.46(m,1H),5.71(s,1H),7.58(t,J=7.70Hz,1H),7.75(m,2H),7.91(d,J=8.31Hz,1H),8.19(m,1H);MS:(M+Na)+396。
步骤4:
将4-(4-甲基-异喹啉-1-氧基)-吡咯烷-1,2-二甲酸1-叔丁酯)(74mg)、环丙烷磺酸(1(R)-氨基-2(S)-乙烯基-环丙烷羰基)-酰胺盐酸盐(59mg)、PyBOP(114mg)和i-Pr2NEt(0.2mL)在二氯甲烷(2mL)中的溶液搅拌2h。经Biotage快速层析纯化,用5%甲醇/乙酸乙酯洗脱,得到105mg(90%)所需的产物。
1H NMR(400MHz,甲醇-D4)δppm 1.18(m,5H),1.39(s,9H),1.87(dd,J=8.2,5.3Hz,1H),2.28(m,2H),2.54(m,4H),2.95(m,1H),3.86(m,2H),4.40(dd,J=9.8,6.9Hz,1H),5.12(d,J=10.5Hz,1H),5.31(d,J=17.6Hz,1H),5.79(m,2H),7.60(t,J=7.5Hz,1H),7.78(m,2H),7.93(d,J=8.3Hz,1H),8.20(d,J=8.1Hz,1H);MS:(M+Na)+607.
步骤5:
将2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-(4-甲基-异喹啉-1-氧基)-吡咯烷-1-甲酸叔丁酯(100mg)和TFA(3mL)在二氯甲烷(3mL)中的溶液搅拌1h。浓缩后,将剩余物溶于二氯甲烷(2mL)中,加入Boc-L-叔-亮氨酸(40mg)、PyBOP(104mg)和i-Pr2NEt(0.2mL)。将混合物搅拌1h。处理后,经制备型HPLC纯化,得到60mg(52%)所需的产物化合物250,为固体物。
1H NMR(400MHz,CD3OD)δppm 1.04(m,12H),1.26(m,10H),1.44(dd,J=9.5,5.1Hz,1H),1.88(dd,J=8.1,5.4Hz,1H),2.26(m,2H),2.49(s,3H),2.62(dd,J=13.7,7.1Hz,1H),2.94(m,1H),4.06(dd,J=12.0,3.4Hz,1H),4.25(m,1H),4.45(d,J=11.3Hz,1H),4.53(dd,J=10.3,6.6Hz,1H),5.12(d,J=10.0Hz,1H),5.29(d,J=17.1Hz,1H),5.77(m,2H),6.63(d,J=8.6Hz,1H),7.53(t,J=7.8Hz,1H),7.76(t,J=8.1Hz,1H),7.80(s,1H),7.91(d,J=8.1Hz,1H),8.22(d,J=8.3Hz,1H);MS:(M+Na)+720.
实施例251:化合物251的制备
化合物251
按照实施例250、方案1的方法制备化合物251,不同之处在于在步骤1中使用3-甲氧基-3-苯基-丙烯酸代替3-苯基-丁-2-烯酸。
步骤1:
修改:使用15g 3-甲氧基-3-苯基-丙烯酸,得到250mg产物(收率2%)。
产物:
1H NMR(400MHz,CD3COCD3)δppm 3.85(s,3H),6.96(s,1H),7.54(m,1H),7.71(m,1H),7.86(d,J=8.07Hz,1H),8.31(d,J=8.07Hz,1H).
步骤2:
修改:使用200mg 4-甲氧基-2H-异喹啉-1-酮,得到150mg产物(收率68%)。
产物:
1H NMR(400MHz,CDCl3)δppm 4.05(s,2H),7.71(m,1H),7.72(m,2H),7.80(s,1H),8.23(dd,J=18.71,7.70Hz,2H).
步骤3:
修改:使用122mg 1-氯-4-甲氧基-异喹啉,得到218mg产物(89%收率)。
产物:
MS:(M+Na)+411.
步骤4:
修改:使用194mg 4-(4-甲氧基-异喹啉-1-氧基)-吡咯烷-1,2-二甲酸1-叔丁酯,得到298mg产物(收率99%)。
产物:
1H NMR(400MHz,CD3OD)δppm 1.17(m,5H),1.42(s,9H),1.87(dd,J=8.2,5.5Hz,1H),2.27(m,2H),2.54(dd,J=13.3,6.2Hz,1H),2.95(m,1H),3.85(m,2H),4.00(s,3H),4.39(dd,J=9.8,6.9Hz,1H),5.12(d,J=10.5Hz,1H),5.31(d,J=17.1Hz,1H),5.76(m,2H),7.52(s,1H),7.62(t,J=7.6Hz,1H),7.74(t,J=7.2Hz,1H),8.12(t,J=8.3Hz,2H).
步骤5:
修改:使用190mg 2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-(4-甲氧基-异喹啉-1-氧基)-吡咯烷-1-甲酸叔丁酯,得到270mg产物(收率51%)。
产物:
化合物251
数据:
1H NMR(500MHz,CD3OD)δppm 1.06(m,12H),1.26(m,10H),1.43(dd,J=8.6,4.6Hz,1H),1.88(dd,J=7.9,5.5Hz,1H),2.24(m,2H),2.61(dd,J=13.6,6.9Hz,1H),2.94(m,1H),4.00(s,3H),4.06(dd,J=11.3,3.1Hz,1H),4.25(d,J=8.9Hz,1H),4.43(d,J=11.3Hz,1H),4.52(m,1H),5.12(d,J=10.1Hz,1H),5.29(d,J=17.1Hz,1H),5.75(m,2H),6.60(d,J=8.6Hz,1H),7.55(m,2H),7.71(t,J=7.3Hz,1H),8.09(d,J=8.2Hz,1H),8.14(d,J=8.2Hz,1H);MS:(M+Na)+736.
实施例252:化合物252的制备
化合物252
按照实施例250、方案1的方法制备化合物252,不同之处在于在步骤1中使用2-甲基肉桂酸代替3-苯基-丁-2-烯酸。
步骤1:
修改:使用20g 2-甲基肉桂酸,得到14.3g产物(收率72%)。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 2.54(s,1H),6.69(d,J=7.3Hz,1H),7.23(d,J=7.3Hz,1H),7.39(t,J=7.8Hz,1H),7.50(d,J=7.1Hz,1H),8.30(d,J=8.1Hz,1H),11.62(s,1H);MS:(M+H)+160.
步骤2:
修改:使用14.4g 5-甲基-2H-异喹啉-1-酮,得到10.6g产物(收率66%)。
产物:
数据:
1H NMR(400MHz,CDCl3)δppm 2.67(s,3H),7.55(m,2H),7.70(dd,J=5.9,1.0Hz,1H),8.19(m,1H),8.28(d,J=5.9Hz,1H);MS:(M+H)+178.
步骤3:
修改:使用533mg 1-氯-5-甲基-异喹啉,得到1116mg产物(收率100%)。
产物:
数据:MS:(M+H)+373。
步骤4:
修改:使用372mg 4-(5-甲基-异喹啉-1-氧基)-吡咯烷-1,2-二甲酸1-叔丁酯,得到551mg产物(收率94%)。
产物:
数据:MS:(M+Na)<+>607。
步骤5:
修改:使用551mg 2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-(5-甲基-异喹啉-1-氧基)-吡咯烷-1-甲酸叔丁酯,得到274mg产物(收率44%)。
产物:
化合物252
数据:
1H NMR(400MHz,CD3OD)δppm 1.00(m,12H,)1.23(m,10H),1.44(m,1H),1.87(dd,J=8.1,5.4Hz,1H),2.26(m,2H),2.62(m,4H),2.94(m,1H),4.07(dd,J=11.9,3.3Hz,1H),4.25(d,J=9.5Hz,1H),4.46(d,J=11.5Hz,1H),4.53(dd,J=10.3,7.1Hz,1H),5.12(d,J=10.5Hz,1H),5.29(d,J=16.9Hz,1H),5.75(m,1H),5.86(s,1H),6.62(d,J=9.3Hz,1H),7.39(t,J=7.7Hz,1H),7.44(d,J=5.9Hz,1H),7.53(d,J=7.1Hz,1H),8.00(d,J=6.1Hz,1H),8.06(d,J=8.3Hz,1H);MS:(M+H)+698.
实施例253:化合物253的制备
化合物253
按照实施例250、方案1的方法制备化合物253,不同之处在于在步骤1中使用2-甲氧基肉桂酸代替3-苯基-丁-2-烯酸。
步骤1:
修改:使用10g 2-甲氧基肉桂酸,得到5.3g产物(收率53%)。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 3.95(s,3H),6.94(d,J=7.3Hz,1H),7.08(d,J=8.1Hz,1H),7.14(d,J=7.3Hz,1H),7.43(t,J=8.1Hz,1H),7.99(d,J=8.1Hz,1H),10.92(s,1H);MS:(M+H)+176.
步骤2:
修改:使用5.3g 5-甲氧基-2H-异喹啉-1-酮,得到5.38g产物(收率92%)。
产物:
数据:
1H NMR(400MHz,CDCl3)δppm 4.01(s,3H),7.04(d,J=7.8Hz,1H),7.57(t,J=8.1Hz,1H),7.88(d,J=8.6Hz,1H),7.97(d,J=5.9Hz,1H),8.25(d,J=5.9Hz,1H);MS:(M+H)+194.
步骤3:
修改:使用581mg 1-氯-5-甲氧基-异喹啉,得到1163mg产物(收率100%)。
产物:
数据:MS:(M+H)+389。
步骤4:
修改:使用117mg 4-(5-甲氧基-异喹啉-1-氧基)-吡咯烷-1,2-二甲酸1-叔丁酯,得到180mg产物(收率100%)。
产物:
数据:MS:(M+H)+601。
步骤5:
修改:使用177mg 2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-(5-甲氧基-异喹啉-1-氧基)-吡咯烷-1-甲酸叔丁酯,得到63mg产物(收率44%)。
产物:
化合物253
数据:
1H NMR(400MHz,CD3OD)δppm 1.00(m,12H),1.21(m,10H),1.38(m,1H),1.82(dd,J=8.1,5.6Hz,1H),2.20(m,2H),2.56(dd,J=13.6,6.7Hz,1H),2.88(m,1H),3.08(m,2H),3.93(s,3H),4.01(dd,J=11.9,3.3Hz,1H),4.20(d,J=9.1Hz,1H),4.39(d,J=12.2Hz,1H),4.47(dd,J=9.7,7.0Hz,1H),5.06(d,J=10.0Hz,1H),5.23(d,J=16.9Hz,1H),5.70(m,1H),5.79(s,1H),6.55(d,J=9.5Hz,1H),7.08(d,J=7.6Hz,1H),7.37(t,J=8.0Hz,1H),7.54(d,J=5.9Hz,1H),7.68(d,J=8.3Hz,1H),7.89(d,J=5.9Hz,1H);MS:(M+H)+714.
实施例254:化合物254的制备
化合物254
按照实施例250、方案1的方法制备化合物254,不同之处在于在步骤1中使用2-氯肉桂酸代替3-苯基-丁-2-烯酸。
步骤1:
修改:使用25g 2-氯肉桂酸,得到14.6g产物(收率59%)。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 7.22(d,J=7.3Hz,1H),7.42(t,J=7.8Hz,1H),7.73(d,J=7.8Hz,1H),8.34(d,J=8.1Hz,1H),10.61(s,1H);MS:(M+H)+180.
步骤2:
修改:使用14.2g 5-氯-2H-异喹啉-1-酮,得到8.28g产物(收率53%)。
产物:
数据:
1H NMR(400MHz,CDCl3)δppm 7.60(dd,J=8.6,7.6Hz,1H),7.83(m,1H),8.00(d,J=5.9Hz,1H),8.29(dt,J=8.9,1.0Hz,1H),8.38(d,J=5.9Hz,1H);MS:(M+H)+198.
步骤3:
修改:使用594mg 1,5-二氯-异喹啉,得到1174mg产物(收率100%)。
产物:
数据:MS:(M+H)+393。
步骤4:
修改:使用118mg 4-(5-氯-异喹啉-1-氧基)-吡咯烷-1,2-二甲酸1-叔丁酯,得到154mg产物(收率85%)。
产物:
数据:MS:(M+H)+605。
步骤5:
修改:使用150mg 2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-(5-氯-异喹啉-1-氧基)-吡咯烷-1-甲酸叔丁酯,得到91mg产物(收率51%)。
产物:
化合物254
数据:
1H NMR(400MHz,CD3OD)δppm 0.97(m,12H),1.17(m,10H),1.38(dd,J=9.4,5.3Hz,1H),1.82(dd,J=8.0,5.5Hz,1H),2.21(m,2H),2.58(dd,J=13.8,7.0Hz,1H),2.88(m,1H),4.01(dd,J=11.9,2.8Hz,1H,4.16(d,J=9.3Hz,1H),4.47(m,2H),5.06(d,J=10.3Hz,1H),5.24(d,J=16.9Hz,1H),5.70(m,1H),5.82(s,1H),6.52(d,J=9.3Hz,1H),7.42(t,J=8.0Hz,1H),7.57(d,J=6.1Hz,1H),7.76(d,J=7.6Hz,1H),8.05(d,J=6.1Hz,1H),8.13(d,J=8.3Hz,1H);MS:(M+H)+718.
实施例255:化合物255的制备
化合物255
按照实施例250、方案1的方法制备化合物255,不同之处在于在步骤1中使用2-氟肉桂酸代替3-苯基-丁-2-烯酸。
步骤1:
修改:使用16.6g 2-氟肉桂酸,得到8.55g产物(收率51%)。
产物:
数据:
1H NMR(400MHz,CD3COCD3)δppm 6.62(d,J=7.3Hz,1H),732(d,J=7.3Hz,1H),7.47(m,2H),8.09(m,1H).
步骤2:
修改:使用8.4g 5-氟-2H-异喹啉-1-酮,得到7.5g产物(收率80%)。
产物:
数据:
1H NMR(400MHz,CDCl3)δppm 7.43(ddd,J=9.7,7.8,0.9Hz,1H),7.62(td,J=8.2,5.4Hz,1H),7.84(d,J=5.6Hz,1H),8.14(d,J=8.6Hz,1H),8.33(d,J=5.9Hz,1H);MS:(M+H)+182.
步骤3:
修改:使用203mg 1-氯-5-氟-异喹啉,得到384mg产物(收率90%)。
产物:
数据:
1H NMR(400MHz,CD3SOCD3)δppm 1.34,1.36(2s,9H,外消旋物),2.35(m,1H),2.61(m,1H),3.65(d,J=12.23Hz,1H),3.80(m,1H),4.35(m,1H),5.70(s,1H),7.48(d,J=6.11Hz,1H),7.63(m,2H),7.99(m,1H),8.10(d,J=5.87Hz,1H);MS:(M+Na)+399.
步骤4:
修改:使用76mg 4-(5-氟-异喹啉-1-氧基)-吡咯烷-1,2-二甲酸1-叔丁酯,得到116mg产物(收率99%)。
产物:
步骤5:
修改:使用110mg 2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-(5-氟-异喹啉-1-氧基)-吡咯烷-1-甲酸叔丁酯,得到39mg产物(收率30%)。
产物:
化合物255
数据:
1H NMR(400MHz,CD3OD)δppm 1.05(m,12H),1.25(m,10H),1.44(dd,J=9.5,5.4Hz,1H),1.88(dd,J=8.1,5.4Hz,1H),2.28(m,2H),2.63(dd,J=13.8,7.0Hz,1H),2.94(m,1H),4.07(dd,J=11.9,3.1Hz,1H),4.23(d,J=9.3Hz,1H),4.52(m,2H),5.12(dd,J=10.3,1.5Hz,1H),5.29(d,J=17.4Hz,1H),5.75(m,1H),5.89(s,1H),6.59(d,J=9.1Hz,1H),7.47(m,3H),8.02(d,J=8.1Hz,1H),8.06(d,J=6.1Hz,1H);MS:(M+Na)+724.
实施例256:化合物256的制备
化合物256
按照实施例250、方案1的方法制备化合物256,不同之处在于在步骤1中使用2-二氟甲氧基肉桂酸代替3-苯基-丁-2-烯酸。
步骤1:
修改:使用10.7g 2-二氟甲氧基肉桂酸,得到2g产物(收率18%)。
产物:
数据:
1H NMR(400MHz,CD3SOCD3)δppm 6.06(m,2H),6.42(m,2H),6.71(s,2H),7.35(s,1H);MS:(M+H)+212.
步骤2:
修改:使用300mg 5-二氟甲氧基-2H-异喹啉-1-酮,得到300mg产物(收率92%)。
产物:
数据:
1H NMR(400MHz,CDCl3)δppm 6.70(t,J=72.87Hz,1H),7.48(m,1H),7.64(m,1H),7.92(d,J=5.87Hz,1H),8.21(d,J=8.56Hz,1H),8.35(d,J=5.62Hz,1H).
步骤3:
修改:使用230mg 1-氯-5-二氟甲氧基-异喹啉,得到360mg产物(收率96%)。
产物:
步骤4:
修改:使用37mg 4-(5-羟基-异喹啉-1-氧基)-吡咯烷-1,2-二甲酸1-叔丁酯,得到57mg产物(收率99%)。
产物:
步骤5:
修改:使用57mg 2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-(5-羟基-异喹啉-1-氧基)-吡咯烷-1-甲酸叔丁酯,得到10mg产物(收率15%)。
产物:
化合物256
数据:
1H NMR(400MHz,CD3OD)δppm 0.93(m,4H),1.13(s,9H),1.31(m,1H),1.49(s,9H),1.89(dd,J=7.8,5.4Hz,1H),2.16(q,J=8.8Hz,1H),2.40(m,1H),2.81(m,1H),2.90(m,1H),3.76(m,2H),4.30(m,1H),4.59(dd,J=10.2,7.7Hz,1H),5.07(dd,J=10.3,1.7Hz,1H),5.26(dd,J=17.2,1.3Hz,1H),5.77(dt,J=17.2,9.6Hz,1H),5.93(s,1H),7.24(d,J=8.6Hz,1H),7.51(m,2H),7.63(t,J=8.0Hz,1H),7.98(d,J=6.1Hz,1H),8.24(d,J=8.3Hz,1H);MS:(M+H)+700.
实施例257:化合物257的制备
化合物257
按照实施例250、方案1的方法制备化合物257,不同之处在于在步骤1中使用4-氟肉桂酸代替3-苯基-丁-2-烯酸。
步骤1:
修改:使用16.6g 4-氟肉桂酸,得到8.2g产物(收率49%)。
产物:
数据:
1H NMR(400MHz,CD3COCD3)δppm 6.57(d,J=7.09Hz,1H),7.21(d,J=7.09Hz,1H),7.50(m,1H),7.72(dd,J=8.68,5.26Hz,1H),7.90(dd,J=9.54,2.93Hz,1H).
步骤2:
修改:使用8.15g 7-氟-2H-异喹啉-1-酮,得到7.6g产物(收率84%)。
产物:
数据:
1H NMR(400MHz,CDCl3)δppm 7.52(td,J=8.6,2.6Hz,1H),7.59(d,J=5.6Hz,1H),7.86(dd,J=9.1,5.4Hz,1H),7.95(dd,J =9.5,2.5Hz,1H),8.26(d,J=5.6Hz,1H);MS:(M+H)+182.
步骤3:
修改:使用191mg 1-氯-7-氟-异喹啉,得到350mg产物(收率93%)。
产物:
数据:MS:(M+Na)+399。
步骤4:
修改:使用75mg 4-(7-氟-异喹啉-1-氧基)-吡咯烷-1,2-二甲酸1-叔丁酯,得到100mg产物(收率85%)。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 1.16(m,4H),1.41(m,10H),1.88(dd,J=8.1,5.4Hz,1H),2.28(m,2H),2.56(m,1H,)2.94(m,1H),3.87(m,2H),4.41(dd,J=9.7,7.0Hz,1H),5.12(d,J=10.8Hz,1H),5.31(d,J=17.1Hz,1H),5.78(m,2H),7.36(d,J=5.9Hz,1H),7.54(m,1H),7.78(dd,J=9.3,2.5Hz,1H),7.90(dd,J=9.1,5.1Hz,1H),7.96(d,J=5.9Hz,1H);MS:(M+Na)+611.
步骤5:
修改:使用95mg 2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-(7-氟-异喹啉-1-氧基)-吡咯烷-1-甲酸叔丁酯,得到55mg产物(收率44%)。
产物:
化合物257
数据:
1H NMR(400MHz,CD3OD)δppm 1.05(m,12H),1.22(m,10H),1.44(dd,J=9.3,5.4Hz,1H),1.88(dd,J=8.2,5.5Hz,1H),2.27(m,2H),2.63(dd,J=13.8,7.0Hz,1H),2.94(m,1H),4.07(dd,J=11.5,3.2Hz,1H),4.22(d,J=9.5Hz,1H),4.47(d,J=11.7Hz,1H),4.55(dd,J=10.6,7.5Hz,1H),5.12(d,J=10.3Hz,1H),5.29(d,J=17.1Hz,1H),5.75(m,1H),5.87(s,1H),6.61(d,J=9.5Hz,1H),7.36(d,J=5.9Hz,1H),7.52(td,J=8.9,2.5Hz,1H),7.79(dd,J=9.4,2.6Hz,1H),7.88(dd,J=8.7,5.5Hz,1H),7.96(d,J=5.9Hz,1H);MS:(M+Na)+724.
实施例258:化合物258的制备
化合物258
按照实施例250、方案1的方法制备化合物258,不同之处在于在步骤1中使用4-氯肉桂酸代替3-苯基-丁-2-烯酸。
步骤1:
修改:使用9.13g 4-氯肉桂酸,得到4g产物(收率44%)。
产物:
数据:
1H NMR(400MHz,CD3SOCD3)δppm 6.58(d,J=7.1Hz,1H),7.20(dd,J=7.1,5.9Hz,1H),7.72(m,2H),8.10(m,1H).
步骤2:
修改:使用3.5g 7-氯-2H-异喹啉-1-酮,得到2.8g产物(收率72%)。
产物:
数据:
1H NMR(500MHz,CDCl3)δppm 7.59(d,J =5.5Hz,1H),7.69(dd,J=8.9,2.1Hz,1H),7.80(d,J=8.6Hz,1H),8.29(d,J=5.5Hz,1H),8.34(s,1H);MS:(M+H)+198.
步骤3:
修改:使用208mg 1,7-二氯-异喹啉,得到350mg产物(收率89%)。
产物:
数据:MS:(M+Na)+415。
步骤4:
修改:使用79mg 4-(7-氯-异喹啉-1-氧基)-吡咯烷-1,2-二甲酸1-叔丁酯,得到119mg产物(收率99%)。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 1.17(m,4H),1.43(m,10H),1.88(dd,J=8.31 5.4Hz,1H),2.29(m,2H),2.57(dd,J=13.7,6.9Hz,1H),2.95(m,1H),3.87(m,2H),4.42(dd,J=9.9,6.9Hz,1H),5.13(d,J=10.3Hz,1H),5.31(dd,J=17.1,1.2Hz,1H),5.78(m,2H),7.35(d,J=5.9Hz,1H),7.69(dd,J=8.7,2.1Hz,1H),7.84(d,J=8.8Hz,1H),7.99(d,J=5.9Hz,1H),8.12(d,J=1.7Hz,1H);MS:(M+Na)+627.
步骤5:
修改:使用115mg 2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-(7-氯-异喹啉-1-氧基)-吡咯烷-1-甲酸叔丁酯,得到36mg产物(收率25%)。
产物:
化合物258
数据:MS:(M+Na)+740。
实施例259:化合物259的制备
化合物259
按照实施例250、方案1的方法制备化合物259,不同之处在于在步骤1中使用4-甲基肉桂酸代替3-苯基-丁-2-烯酸。
步骤1:
修改:使用25g 4-甲基肉桂酸,得到15.3g产物(收率62%)。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 2.50(s,3H),6.54(d,J=7.1Hz,1H),7.13(d,J=7.1Hz,1H),7.49(m,2H),8.22(s,1H),11.49(s,1H);MS:(M+H)+160.
步骤2:
修改:使用15.3g 7-甲基-2H-异喹啉-1-酮,得到5.15g产物(收率30%)。
产物:
数据:
1H NMR(400MHz,CDCl3)δppm 2.58(s,3H),7.56(m,2H),7.73(d,J=8.3Hz,1H),8.09(s,1H),8.20(d,J=5.6Hz,1H);MS:(M+H)+178.
步骤3:
修改:使用205mg 1-氯-7-甲基-异喹啉,得到350mg产物(收率89%)。
产物:
数据:MS:(M+H)+373。
步骤4:
修改:使用75mg 4-(7-甲基-异喹啉-1-氧基)-吡咯烷-1,2-二甲酸1-叔丁酯,得到107mg产物(收率95%)。
产物:
数据:MS:(M+Na)+607。
步骤5:
修改:使用107mg 2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-(7-甲基-异喹啉-1-氧基)-吡咯烷-1-甲酸叔丁酯,得到53mg产物(收率41%)。
产物:
化合物259
数据:
1H NMR(400MHz,CD3OD)δppm 1.02(m,12H),1.18(s,9H),1.24(m,1H),1.45(dd,J=9.4,5.5Hz,1H),1.88(dd,J=8.2,5.5Hz,1H),2.28(m,2H),2.50(s,3H),2.61(dd,J=13.8,6.7Hz,1H),3.34(s,1H),4.09(dd,J=11.7,3.2Hz,1H),4.23(s,1H),4.42(d,J=12.0Hz,1H),4.57(dd,J=10.0,7.1Hz,1H),5.12(dd,J=10.3,1.5Hz,1H),5.30(d,J=17.1Hz,1H),5.76(m,1H),5.87(s,1H),7.28(d,J=5.9Hz,1H),7.55(d,J=8.3Hz,1H),7.71(d,J=8.3Hz,1H),7.89(d,J=5.9Hz,1H),7.93(s,1H);MS:(M+H)+698.
实施例260:化合物260的制备
化合物260
按照实施例250、方案1的方法制备化合物260,不同之处在于在步骤1中使用4-甲氧基肉桂酸代替3-苯基-丁-2-烯酸。
步骤1:
修改:使用33g 4-甲氧基肉桂酸,得到7g产物(收率33%)。
产物:
数据:
1H NMR(500MHz,CD3COCD3)δppm 3.90(s,3H),6.49(d,J=7.0Hz,1H),7.10(d,J=7.3Hz,1H),7.28(dd,J=8.6,2.8Hz,1H),7.57(d,J=8.9Hz,1H),7.71(d,J=2.8Hz,1H).
步骤2:
修改:使用4g 7-甲氧基-2H-异喹啉-1-酮,得到3g产物(收率68%)。
产物:
数据:
1H NMR(400MHz,CDCl3)δppm 3.98(s,3H),7.38(dd,J=8.9,2.6Hz,1H),7.52(m,2H),7.73(d,J=8.8Hz,1H),8.16(d,J=5.4Hz,1H).
步骤3:
修改:使用533mg 1-氯-7-甲氧基-异喹啉,得到1115mg产物(收率100%)。
产物:
步骤4:
修改:使用78mg 4-(7-甲氧基-异喹啉-1-氧基)-吡咯烷-1,2-二甲酸1-叔丁酯,得到108mg产物(收率99%)。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 1.17(m,4H),1.40(m,1H),1.43(s,9H),1.85(dd,J=8.1,5.4Hz,1H),2.21(m,2H),2.51(dd,J=13.7,6.6Hz,1H),2.93(s,1H),3.80(m,2H),3.94(s,3H),4.41(dd,J=10.0,6.6Hz,1H),4.57(s,1H),5.11(d,J=11.3Hz,1H),5.29(d,J=17.1Hz,1H),5.77(m,2H),7.01(d,J=7.8Hz,1H),7.22(d,J=5.6Hz,1H),7.32(d,J=8.1Hz,1H),7.58(t,J=8.0Hz,1H),7.87(d,J=5.9Hz,1H);MS:(M+H)+601.
步骤5:
修改:使用100mg 2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-(7-甲氧基-异喹啉-1-氧基)-吡咯烷-1-甲酸叔丁酯,得到30mg产物(收率25%)。
产物:
化合物260
数据:
1H NMR(400MHz,CD3SOCD3)δppm 0.90(m,2H),0.95(s,9H),1.05(m,1H),1.12(s,9H),1.35(m,2H),1.70(m,1H),2.18(m,1H),2.92(m,1H),3.86(s,3H),4.00(m,2H),4.27(d,J=12.0Hz,1H),4.45(t,J=8.6Hz,1H),5.09(d,J=10.8Hz,1H),5.23(d,J=16.9Hz,1H),5.62(m,1H),5.79(s,1H),6.55(d,J=8.1Hz,1H),7.35(d,J=6.6Hz,1H),7.39(d,J=2.5Hz,1H),7.43(dd,J=8.8,2.2Hz,1H),7.84(d,J=8.8Hz,1H),7.88(d,J=5.9Hz,1H);MS:(M+H)+714.
实施例261和262:化合物261和262的制备
化合物261 化合物262
按照实施例250、方案1的方法制备化合物261和262,不同之处在于在步骤1中使用4-氟-3-甲氧基肉桂酸代替3-苯基-丁-2-烯酸。
步骤1:
修改:使用19.6g 4-氟-3-甲氧基肉桂酸,得到9.5g产物(收率48%)。
产物:
数据:
1H NMR(400MHz,CD3COCD3)δppm 4.00(s,1H),6.49(d,J=7.34Hz,1H),7.19(d,J=7.09Hz,1H),7.29(d,J=8.07Hz,1H),7.86(d,J=11.74Hz,1H).
步骤2:
修改:使用9g 7-氟-6-甲氧基-2H-异喹啉-1-酮,得到7g产物(70%收率)。
产物:
数据:
1H NMR(400MHz,CDCl3)δppm 4.04(s,3H),7.17(d,J=8.07Hz,1H),7.48(d,J=5.62Hz,1H),7.94(d,J=11.49Hz,1H),8.20(d,J=5.62Hz,1H).
步骤3:
修改:使用222mg 1-氯-7-氟-6-甲氧基-异喹啉,得到406mg产物。
产物:
步骤4:
修改:使用400mg 4-(7-氟-6-甲氧基-异喹啉-1-氧基)-吡咯烷-1,2-二甲酸1-叔丁酯和4-(1-氯-6-甲氧基-异喹啉-7-氧基)-吡咯烷-1,2-二甲酸1-叔丁酯的混合物,得到700mg产物。
产物:
步骤5:
修改:使用700mg 2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-(7-氟-6-甲氧基-异喹啉-1-氧基)-吡咯烷-1-甲酸叔丁酯和4-(1-氯-6-甲氧基-异喹啉-7-氧基)-2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-吡咯烷-1-甲酸叔丁酯的混合物,得到79mg化合物261和80mg化合物262。
产物:
化合物261 化合物262
化合物261的数据:
1H NMR(400MHz,CD3OD)δppm 1.07(m,12H),1.25(m,10H),1.44(m,1H),1.88(dd,J=8.1,5.6Hz,1H),2.25(m,2H),2.60(dd,J=13.7,6.9Hz,1H),2.94(m,1H),4.02(m,4H),4.22(s,1H),4.43(d,J=12.2Hz,1H),4.53(dd,J=10.3,6.6Hz,1H),5.12(d,J=10.5Hz,1H),5.30(d,J=16.6Hz,1H),5.75(m,1H),5.84(s,1H),7.28(d,J=5.9Hz,1H),7.37(d,J=8.1Hz,1H),7.75(d,J=11.7Hz,1H),7.91(d,J=5.9Hz,1H);MS:(M+Na)+754.
化合物262的数据:
1H NMR(400MHz,CD3OD)δppm 1.07(m,12H),1.25(m,10H),1.44(m,J=9.41,5.26Hz,1H),1.87(dd,J=8.31,5.38Hz,1H),2.24(q,J=8.72Hz,2H),2.57(dd,J=13.82,7.21Hz,1H),2.94(m,1H),3.97(d,J=5.14Hz,3H),4.09(m,J=11.00Hz,1H),4.24(s,1H),4.32(m,1H),4.50(m,J=16.87Hz,1H),5.12(dd,J=10.52,1.71Hz,1H),5.30(dd,J=17.12,1.47Hz,1H),5.38(s,1H),5.76(m,1H),7.39(s,1H),7.63(s,1H),7.66(d,J=5.87Hz,1H),8.07(d,J=5.62Hz,1H);MS:(M+H)+732。
实施例263:化合物263的制备
化合物263
按照实施例250、方案1的方法制备化合物263,步骤1和步骤2除外。
步骤3:
修改:使用176mg 1-氯-8-甲基-异喹啉,得到370mg产物(收率100mg%)。
产物:
步骤4:
修改:使用149mg 8-甲基-异喹啉-1-氧基)-吡咯烷-1,2-二甲酸1-叔丁酯,得到230mg产物(收率99%)。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 1.13(m,4H),1.42(m,10H),1.87(dd,J=8.2,5.3Hz,1H),2.25(m,2H),2.58(dd,J=13.9,6.9Hz,1H),2.83(s,3H),2.96(m,1H),3.85(m,2H),4.38(dd,J=10.2,6.7Hz,1H),5.12(dd,J=10.4,1.6Hz,1H),5.30(dd,J=17.1,1.2Hz,1H),5.76(m,2H),7.28(d,J=5.9Hz,1H),7.36(d,J=6.9Hz,1H),7.53(t,J=7.7Hz,1H),7.62(m,1H),7.88(d,J=5.6Hz,1H);MS:(M+Na)+607.
步骤5:
修改:使用220mg 2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-(8-甲基-异喹啉-1-氧基)-吡咯烷-1-甲酸叔丁酯,得到90mg产物(收率35%)。
产物:
化合物263
数据:1H NMR(400MHz,CD3OD)δppm 1.05(m,12H),1.24(m,10H),1.44(dd,J=9.3,5.4Hz,1H),1.87(dd,J=8.1,5.4Hz,1H),2.25(m,2H),2.60(dd,J=13.9,7.3Hz,1H),2.77(s,3H),2.94(m,1H),4.04(dd,J=11.9,3.1Hz,1H),4.27(d,J=9.5Hz,1H),4.42(d,J=12.0Hz,1H),4.54(dd,J=10.8,7.1Hz,1H),5.12(d,J=10.3Hz,1H),5.28(d,J=17.1Hz,1H),5.75(m,1H),5.95(s,1H),6.63(d,J=9.1Hz,1H),7.28(m,2H),7.50(t,J=7.7Hz,1H),7.60(d,J=7.8Hz,1H),7.89(d,J=5.6Hz,1H);MS:(M+Na)+720。
实施例264:化合物264的制备
化合物264
按照实施例250、方案1的方法制备化合物264,步骤1和步骤2除外。
步骤3:
修改:使用203mg 1-氯-8-甲氧基-异喹啉,得到340mg产物(收率85%)。
产物:
数据:
1H NMR(400MHz,CD3SOCD3)δppm 1.34,1.36(2s,9H,外消旋物),2.26(m,1H),2.49(m,1H),3.67(m,2H),3.86(s,3H),4.31(m,1H),5.67(brs,1H),7.04(d,J=7.8Hz,1H),7.30(d,J=5.9Hz,1H),7.38(d,J=8.1Hz,1H),7.62(t,J=8.0Hz,1H),7.93(d,J=5.6Hz,1H),12.64(s,1H);MS:(M+Na)+411.
步骤4:
修改:使用78mg 8-甲氧基-异喹啉-1-氧基)-吡咯烷-1,2-二甲酸1-叔丁酯,得到115mg产物(收率96%)。
产物:
步骤5:
修改:使用110mg 2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-(8-甲氧基-异喹啉-1-氧基)-吡咯烷-1-甲酸叔丁酯,得到45mg产物(收率34%)。
产物:
化合物264
数据:MS:(M+H)+714。
实施例265和266:化合物265和266的制备
化合物265 化合物266
按照以下实施例250、方案1的方法制备化合物265和化合物266,不同之处在于在步骤1中使用3-(2,3-二氢-苯并呋喃-7-基)-丙烯酸代替3-苯基-丁-2-烯酸。
步骤1:
修改:使用3.8g 3-(2,3-二氢-苯并呋喃-7-基)-丙烯酸,得到2g产物(收率53%)。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 3.37(t,J=9.05Hz,1H),4.73(t,J=9.05Hz,2H),6.67(d,J=7.09Hz,1H),7.10(d,J=7.09Hz,1H),7.37(d,J=8.07Hz,1H),7.81(d,J=8.07Hz,1H);MS:(M+H)+188.
步骤2:
修改:使用1.87g 2,3-二氢-7H-呋喃并[2,3-f]异喹啉-6-酮,得到1.84g产物(收率90%)。
产物:
数据:
1H NMR(400Hz,CDCl3)δppm 3.43(t,J=9.05Hz,2H),4.82(t,J=9.05Hz,2H),7.52(d,J=8.56Hz,1H),7.66(d,J=5.62Hz,1H),7.84(d,J=8.31Hz,1H),8.19(d,J=5.62Hz,1H);MS(M+H)+206.
步骤3:
修改:使用206mg 6-氯-2,3-二氢-呋喃并[2,3-f]异喹啉,得到300mg混合产物。
产物:
步骤4:
修改:使用240mg步骤3的混合产物,得到350mg混合产物。
产物:
步骤5:
修改:使用331mg步骤4的混合产物,得到240mg化合物265和24mg化合物266。
产物:
化合物265 化合物266
化合物265的数据:
1H NMR(400Hz,CD3OD)δppm 0.99(m,12H),1.16(m,10H),1.36(m,1H),1.81(dd,J=8.07,5.62Hz,1H),2.18(m,2H),2.54(dd,J=13.69,6.85Hz,1H),2.87(m,1H),3.31(t,J=9.05Hz,2H),4.01(m,1H),4.18(s,1H),4.36(d,J=11.74Hz,1H),4.46(dd,J=10.15,7.21Hz,1H),4.70(m,2H),5.05(d,J=10.27Hz,1H),5.23(d,J=16.87Hz,1H),5.70(m,2H),7.23(d,J=5.87Hz,1H),7.31(d,J=8.31Hz,1H),7.63(d,J=8.31Hz,1H),7.82(d,J=5.87Hz,1H);MS(M+H)+726.
化合物266的数据:
1H NMR(400MHz,CD3OD)δppm 1.06(m,12H),1.24(m,10H),1.44(dd,J=10.03,5.14Hz,1H,1.88(dd,J=7.83,5.38Hz,1H,2.27(m,2H),2.65(dd,J=12.96,6.36Hz,1H),2.94(m,1H),4.08(dd,J=12.35,3.30Hz,1H),4.25(s,1H),4.54(m,2H),5.12(d,J=10.27Hz,1H),5.29(d,J=17.12Hz,1H),5.75(m,1H),5.91(s,1H),7.05(d,J=1.96Hz,1H),7.72(m,2H),8.02(m,2H),8.11(d,J=5.87Hz,1H),9.19(s,1H);MS:(M+H)+724.
实施例267和268:化合物267和268的制备
化合物267 化合物268
按照实施例250、方案1的方法制备化合物267和化合物268,不同之处在于在步骤1中使用3-(2,3-二氢-苯并呋喃-4-基)-丙烯酸代替3-苯基-丁-2-烯酸。
步骤1:
修改:使用1.14g 3-(2,3-二氢-苯并呋喃-4-基)-丙烯酸,得到600mg产物(收率52%)。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 3.35(t,J=8.93Hz,2H),4.74(t,J=8.93Hz,2H),6.49(d,J=7.09Hz,1H),6.95(d,J=8.56Hz,1H),7.25(d,J=7.09Hz,,1H),8.13(d,J=8.80Hz,1H);MS(M+H)+188.
步骤2:
修改:使用560mg 1,7-二氢-2H-呋喃并[3,2-f]异喹啉-6-酮,得到380mg产物(收率48%)。
产物:
数据:
1H NMR(400Hz,CDCl3)δppm 3.47(t,J=9.05Hz,2H),4.84(t,J=9.05Hz,2H),7.24(d,J=8.56Hz,1H),7.33(d,J=5.87Hz,1H),8.20(m,2H);MS(M+H)+206.
步骤3:
修改:使用105mg 6-氯-1,2-二氢-呋喃并[3,2-f]异喹啉,得到390mg混合产物。
产物:
步骤4:
修改:使用216mg步骤3的混合产物,得到330mg混合产物。
产物:
步骤5:
修改:使用330mg步骤4的混合产物,得到140mg化合物267和25mg化合物268。
产物:
化合物267 化合物268
化合物267的数据:
1H NMR(400Hz,CD3OD)δppm 1.07(m,12H),1.24(m,10H),1.43(m,1H),1.88(dd,J=8.07,5.38Hz,1H),2.26(m,2H),2.61(dd,J=13.69,7.09Hz,1H),2.94(m,1H),3.42(t,J=9.05Hz,2H),4.05(dd,J=11.86,3.55Hz,1H),4.24(s,1H),4.50(m,2H),4.77(t,J=8.93Hz,2H),5.12(m,1H),5.29(d,J=17.12Hz,1H),5.76(m,2H),7.03(d,J=8.80Hz,1H),7.12(d,J=6.11Hz,1H),7.91(d,J=5.87Hz,1H),8.06(d,J=8.80Hz,1H);MS:(M+H)+726.
化合物268的数据:
1H NMR(400Hz,CD3OD)δppm 1.06(m,12H),1.19(s,9H)1.26(m,1H),1.44(m,1H),1.88(dd,J=8.07,5.62Hz,1H),2.24(d,J=8.56Hz,2H),2.64(m,1H)2.95(m,1H),4.07(m,J=3.42Hz,1H),4.24(s,1H),4.54(m,2H),5.12(d,J=10.52Hz,1H),5.30(d,J=17.12Hz,1H),5.76(m,1H),5.91(s,1H),7.39(d,J=1.47Hz,1H),7.68(m,2H),7.96(d,J=1.96Hz,1H),8.12(m,2H);MS:(M+H)+724.
实施例269:化合物269的制备
化合物269
方案2
步骤1:
将2-三氟甲氧基肉桂酸(11.6g)、二苯基磷酰叠氮(13.75g)和三乙胺(7.07g)在苯(50mL)中的溶液搅拌1h。经硅胶柱过滤(用苯洗涤)并浓缩,剩余物溶于二苯基甲烷(80mL)中并回流3h。冷却至室温后,经柱收集固体物,用苯洗涤并干燥,得到5.1g(44%)所需的产物,为固体物。1H NMR(400MHz,CD3OD)δppm 6.79(d,J=7.3Hz,1H),7.29(d,J=7.3Hz,1H),7.57(t,J=8.1Hz,1H),7.70(d,J=7.8Hz,1H),8.30(d,J=8.1Hz;1H);MS:(M+H)+230。
步骤2:
将5-三氟甲氧基-2H-异喹啉-1-酮(4.58g)的磷酰氯(50mL)溶液回流3h。冷却并浓缩后,将剩余物用5N NaOH碱化并用二氯甲烷萃取。有机层用盐水洗涤并经硫酸镁干燥。浓缩后,经Biotage快速层析纯化,用5%乙酸乙酯/己烷洗脱得到4.347g(88%)所需的产物,为固体物。1H NMR(400MHz,CDCl3)δppm 7.66(m,2H),7.87(d,J=5.9Hz,1H),8.31(m,1H),8.37(d,J=5.9Hz,1H);MS:(M+H)+248。
步骤3:
在-78℃下,向1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯(56mg)、1-氯-5-三氟甲氧基-异喹啉(25mg)和三氯化镧(25mg)在DMF(1mL)中的悬浮液内加入叔丁醇钾(0.5mL,1M的THF溶液),升温至室温。搅拌30min后,反应物用饱和氯化铵溶液猝灭并用乙酸乙酯萃取。浓缩后,经制备型HPLC纯化,得到35mg(46%)所需的化合物269,为固体物。
1H NMR(400MHz,CD3OD)δppm 1.03(m,12H),1.24(m,10H),1.44(dd,J=9.7,5.3Hz,1H),1.88(dd,J=8.1,5.6Hz,1H,)2.28(m,2H),2.64(dd,J=13.7,7.1Hz,1H),2.94(m,1H),4.09(m,1H),4.21(d,J=9.3Hz,1H),4.53(m,2H),5.12(d,J=11.5Hz,1H),5.30(d,J=17.1Hz,1H),5.75(m,1H),5.92(m,1H),6.60(d,J=9.5Hz,1H),7.49(d,J=6.1Hz,1H),7.60(m,1H),7.69(d,J=7.3Hz,1H),8.11(d,J=6.1Hz,1H),8.22(d,J=8.3Hz,1H);MS:(M+Na)+790.
实施例270:化合物270的制备
化合物270
按照实施例269、方案2的方法制备化合物270,不同之处在于在步骤1中使用2-三氟甲基肉桂酸代替2-三氟甲氧基肉桂酸。
步骤1:
修改:使用10g 2-三氟甲基肉桂酸,得到5g产物(50%收率)。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 6.83(m,1H),7.33(d,J=7.58Hz,1H),7.63(t,J=7.83Hz,1H),8.09(d,J=7.58Hz,1H),8.57(d,J=8.07Hz,1H).
步骤2:
修改:使用4.4g 5-三氟甲基-2H-异喹啉-1-酮,得到3.5g产物(收率73%)。
产物:
数据:
1H NMR(400MHz,CDCl3)δppm 7.75(t,J=7.95Hz,1H),7.90(m,1H),8.12(d,J=7.34Hz,1H),8.41(d,J=6.11Hz,1H),8.60(d,J=8.56Hz,1H).
步骤3:
修改:使用46mg 1-氯-5-三氟甲基-异喹啉和111mg{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基1-2,2-二甲基-丙基}-氨基甲酸叔丁酯,得到70mg产物(收率47%)。
产物:
化合物270
数据:
1H NMR(400MHz,CD3OD)δppm 1.06(m,12H),1.23(m,10H),1.44(dd,J=9.54,5.38Hz,1H),1.88(dd,J=8.07,5.38Hz,1H),2.28(m,2H),2.65(dd,J=13.82,6.97Hz,1H),2.94(m,1H),4.07(m,1H),4.20(m,1H),4.56(m,2H),5.12(m,1H),5.30(d,J=17.12Hz,1H),5.75(m,1H),5.90(s,1H),6.59(d,J=9.05Hz,1H),7.53(d,J=4.40Hz,1H),7.65(t,J=7.83Hz,1H),8.12(d,J=7.09Hz,1H),8.15(d,J=6.36Hz,1H),8.50(d,J=8.31Hz,1H);MS:(M+Na)+774.
实施例271:化合物271的制备
化合物271
按照实施例269、方案2的方法制备化合物271,不同之处在于在步骤1中使用2-氯肉桂酸代替2-三氟甲氧基肉桂酸。
步骤1:
修改:使用7g 2-氯肉桂酸,得到5g产物(71%收率)。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 3.02(m,4H),3.91(m,4H),6.97(d,J=7.34Hz,1H),7.18(d,J=7.34Hz,1H),7.44(m,2H),8.02(d,J=7.83Hz,1H);MS(M+H)+231.
步骤2:
修改:使用2.2g 5-吗啉-4-基-2H-异喹啉-1-酮,得到2.1g产物(收率87%)。
产物:
数据:
1H NMR(400MHz,CCl3D)δppm 3.09(m,4H),3.97(m,4H),7.32(d,J=7.58Hz,1H),7.60(m,1H),7.91(d,J=5.87Hz,1H),8.06(d,J=8.56Hz,1H),8.26(d,J=5.87Hz,1H).
步骤3:
修改:使用50mg 1-氯-5-吗啉-4-基-异喹啉和111mg{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯,得到40mg产物(收率26%)。
产物:
化合物271
数据:
1H NMR(500MHz,CD3OD)δppm 1.07(m,12H),1.26(m,10H),1.44(d,J=7.93Hz,1H),1.88(dd,J=7.93,5.19Hz,1H),2.25(m,2H),2.62(dd,J=13.73,7.02Hz,1H),2.94(m,1H),3.06(d,J=3.97Hz,4H),3.94(m,4H),4.07(d,J=14.04Hz,1H),4.25(s,1H),4.45(d,J=12.21Hz,1H),4.52(m,1H),5.12(d,J=9.46Hz,1H),5.29(d,J=16.79Hz,1H),5.75(m,1H),5.85(s,1H),7.34(d,J=7.32Hz,1H),7.45(t,J=7.78Hz,1H),7.59(d,J=6.10Hz,1H),7.91(d,J=7.63Hz,1H),7.97(d,J=5.80Hz,1H).
实施例272:化合物272的制备
化合物272
按照实施例269、方案2的方法制备化合物272,不同之处在于在步骤1中使用2,3-二甲氧基肉桂酸代替2-三氟甲氧基肉桂酸。
步骤1:
修改:使用10.4g 2,3-二甲氧基肉桂酸,得到4.1g产物(收率40%)。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 3.86(s,3H),3.96(s,3H),6.82(d,J=7.2Hz,1H),7.10(d,J=7.2Hz,1H),7.28(d,J=8.8Hz,1H),8.07(d,J=8.8Hz,1H);MS:(M+H)+206.
步骤2:
修改:使用4.1g 5,6-二甲氧基-2H-异喹啉-1-酮,得到4.03g产物(收率90%)。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 3.97(s,3H),4.05(s,3H),7.65(d,J=9.29Hz,1H),7.90(dd,J=5.87,0.98Hz,1H),8.12(m,2H).
步骤3:
修改:使用22mg 1-氯-5,6-二甲氧基-异喹啉和56mg{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯,得到31mg产物(收率42%)。
产物:
化合物272
数据:
1H NMR(500MHz,CD3OD)δppm 1.06(m,12H),1.26(m,10H),1.44(s,1H),1.88(d,J=7.32Hz,1H),2.24(s,2H),2.60(m,1H),2.94(m,1H),3.92(s,3H),3.99(s,3H),4.06(d,J=11.90Hz,1H),4.23(s,1H),4.43(d,J=10.68Hz,1H).4.53(m,1H),5.12(d,J=10.38Hz,1H),5.30(d,J=17.40Hz,1H),5.77(m,2H),7.35(d,J=9.16Hz,1H),7.46(d,J=5.80Hz,1H),7.89(d,J=5.80Hz,1H),7.97(d,J=8.85Hz,1H).
实施例273:化合物273的制备
化合物273
按照实施例269、方案2的方法制备化合物273,不同之处在于在步骤1中使用4-氯-3-甲氧基肉桂酸代替2-三氟甲氧基肉桂酸。
步骤1:
修改:使用2.5g 4-氯-3-甲氧基肉桂酸,得到1.2g产物(收率48%)。
产物:
数据:
1H NMR(400MHz,CD3OD)δ4.00(s,3H),6.64(d,J=7.09Hz,1H),7.15(d,J=7.34Hz,1H),7.21(s,1H),8.22(s,1H).
步骤2:
修改:使用1.05g 7-氯-6-甲氧基-2H-异喹啉-1-酮,得到0.8g产物(收率70%)。
产物:
数据:
1H NMR(400Hz,CDCl3)δppm 4.05(s,3H),7.13(s,1H),7.48(d,J=5.38Hz,1H),8.21(d,J=5.62Hz,1H),8.34(s,1H);MS:(M+H)+229.
步骤3:
修改:使用44mg 1,7-二氯-6-甲氧基-异喹啉和113mg{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯,得到25mg产物(收率17%)。
产物:
化合物273
数据:
1H NMR(400Hz,CD3OD)δppm 1.07(m,12H),1.24(m,10H),1.44(dd,J=9.54,5.38Hz,1H),1.88(dd,J=8.07,5.38Hz,1H),2.26(m,1H,)2.60(m,J=13.69,6.85Hz,1H),2.94(m,2H),3.98(s,3H),4.06(m,1H),4.20(m,1H),4.42(d,J=12.23Hz,1H),4.57(m,1H),5.12(d,J=11.74Hz,1H),5.30(d,J=17.36Hz,1H),5.76(m,1H),5.86(s,1H),7.28(d,J=5.62Hz,1H),7.33(s,1H),7.92(d,J=5.87Hz,1H),8.09(s,1H);MS:(M+H)+749.
实施例274:化合物274的制备
化合物274
按照实施例269、方案2的方法制备化合物274,不同之处在于在步骤1中使用2-氟-3-肉桂酸代替2-三氟甲氧基肉桂酸。
步骤1:
修改:使用3.92g 2-氟-3-肉桂酸,得到2.4g产物(收率61%)。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 4.00(s,3H),6.72(m,1H),7.16(d,J=7.34Hz,1H),7.35(t,J=8.44Hz,1H),8.09(d,J=8.80Hz,1H).
步骤2:
修改:使用1.93g 5-氟-6-甲氧基-2H-异喹啉-1-酮,得到1.688g产物(收率80%)。
产物:
数据:
1H NMR(CDCl3)δppm 4.08(s,3H),7.44(dd,J=9.29,7.83Hz,1H),7.75(d,J=5.87Hz,1H),8.12(d,J=9.29Hz,1H),8.22(d,J=5.87Hz,1H);MS:(M+H)+212.
步骤3:
修改:使用41mg 1-氯-5-氟-6-甲氧基-异喹啉和133mg{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯,得到70mg产物(收率48%)。
产物:
化合物274
数据:
1H NMR(CD3OD)δppm 1.06(m,13H),1.21(s,9H),1.44(dd,J=9.78,5.38Hz,1H),1.88(dd,J=8.19,5.50Hz,1H),2.24(d,J=9.29Hz,2H),2.62(d,J=13.94Hz,1H),2.94(m,1H),4.05(m,4H),4.22(d,J=9.29Hz,1H),4.45(m,1H),4.54(dd,J=9.66,7.21Hz,1H),5.12(d,J=10.52Hz,1H),5.30(d,J=16.87Hz,1H),5.76(m,1H),5.86(s,1H),7.39(m,2H),7.95(d,J=6.11Hz,1H),8.00(d,J=9.29Hz,1H).
实施例275:化合物275的制备
化合物275
按照实施例269、方案2的方法制备化合物2,不同之处在于在步骤1中使用2-氯-3-甲氧基肉桂酸代替2-三氟甲氧基肉桂酸。
步骤1:
修改:使用658mg 2-氯-3-甲氧基肉桂酸,得到360mg产物(收率54%)。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 4.02(s,3H),6.91(d,J=7.34Hz,1H),7.23(d,J=7.58Hz,1H),7.35(d,J=9.05Hz,1H),8.27(d,J=9.05Hz,1H).
步骤2:
修改:使用350mg 5-氯-6-甲氧基-2H-异喹啉-1-酮,得到300mg产物(收率80%)。
产物:
数据:
1H NMR(400Hz,CDCl3)δppm 4.09(s,3H),7.43(d,J=9.29Hz,1H),7.93(d,J=6.11Hz,1H),8.30(m,2H);MS(M+H)+229.
步骤3:
修改:使用68mg 1,5-二氯-6-甲氧基-异喹啉和167mg{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯,得到130mg产物(收率60%)。
产物:
化合物275
数据:
1H NMR(400MHz,CD3OD)δppm 1.06(m,12H),1.25(m,10H),1.46(d,J=5.62Hz,1H),1.88(dd,J=8.07,5.62Hz,1H),2.27(m,2H),2.62(m,1H),2.94(m,1H),4.05(m,4H),4.22(d,J=9.05Hz,1H),4.46(d,J=11.49Hz,1H),4.54(dd,J=9.78,6.36Hz,1H),5.13(d,J=10.52Hz,1H),5.30(d,J=15.89Hz,1H),5.76(m,1H),5.86(s,1H),7.40(d,J=9.29Hz,1H),7.55(d,J=6.36Hz,1H),8.01(d,J=6.36Hz,1H),8.20(d,J=9.29Hz,1H);MS:(M+H)+749.
实施例276:化合物276的制备
化合物276
按照实施例269、方案2的方法制备化合物276,不同之处在于在步骤1中使用3-氯-2-甲氧基肉桂酸代替2-三氟甲氧基肉桂酸。
步骤1:
修改:使用4.24g 3-氯-2-甲氧基肉桂酸,得到2.4g产物(收率57%)。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 3.93(s,1H),6.85(d,J=7.34Hz,1H),7.24(d,J=7.34Hz,1H),7.52(d,J=8.80Hz,1H),8.03(d,J=8.80Hz,1H);MS:(M+H)+210.
步骤2:
修改:使用2.09g 6-氯-5-甲氧基-2H-异喹啉-1-酮,得到1.9g产物(收率83%)。
产物:
数据:
1H NMR(400Hz,CDCl3)δppm 4.03(s,2H),7.63(d,J=9.05Hz,1H),7.86(d,J=5.14Hz,1H),8.06(d,J=9.05Hz,1H),8.32(d,J=5.62Hz,1H);MS:(M+H)+229.
步骤3:
修改:使用91mg 1,6-二氯-5-甲氧基-异喹啉和226mg{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯,得到114mg产物(收率38%)。
产物:
化合物276
数据:
1H NMR(400Hz,CD3OD)δppm 1.06(m,12H),1.23(m,10H),1.44(t,J=6.72Hz,1H),1.88(dd,J=7.95,5.26Hz,1H),2.25(m,2H),2.62(dd,J=13.33,6.48Hz,1H),2.94(m,1H),3.98(s,3H),4.03(m,1H),4.20(m,1H),4.51(m,2H),5.12(d,J=10.52Hz,1H),5.32(s,1H),5.75(m,1H),5.87(s,1H),7.50(m,2H),7.95(d,J=8.80Hz,1H),8.06(d,J=5.87Hz,1H);MS(MH+)749.
实施例277:化合物277的制备
化合物277
按照实施例269、方案2的方法制备化合物277,不同之处在于在步骤1中使用3-(4-氯-苯基)-3-甲氧基-丙烯酸代替2-三氟甲氧基肉桂酸。
步骤1:
修改:使用4.24g 3-(4-氯-苯基)-3-甲氧基-丙烯酸,得到130mg产物(收率3%)。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 3.96(s,3H),7.19(dd,J=8.80,2.45Hz,1H),7.28(d,J=2.45Hz,1H),7.34(s,1H),8.25(d,J=9.05Hz,1H);MS:(M+H)+210.
步骤2:
修改:使用105mg 7-氯-4-甲氧基-2H-异喹啉-1-酮,得到60mg产物(收率71%)。
产物:
数据:
1H NMR(400Hz,CDCl3)δppm 4.05(s,3H),7.67(dd,J=8.80,1.96Hz,1H),7.80(s,1H),8.16(d,J=9.05Hz,1H),8.24(d,J=1.96Hz,1H);MS:(M+H)+229.
步骤3:
修改:使用46mg 1,7-二氯-4-甲氧基-异喹啉和113mg{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯,得到50mg产物(收率31%)。
产物:
化合物277
数据:
1H NMR(400Hz,CD3OD)δppm 1.06(m,11H),1.16(s,9H),1.24(m,2H),1.44(dd,J=9.54,5.38Hz,1H),1.88(dd,J=8.07,5.62Hz,1H),2.28(m,2H),2.59(dd,J=13.69,6.85Hz,1H),2.94(m,1H),4.00(s,3H),4.05(d,J=11.74Hz,1H),4.19(s,1H),4.43(d,J=11.49Hz,1H),4.56(dd,J=10.03,6.85Hz,1H),5.12(d,J=11.49Hz,1H),5.30(d,J=17.12Hz,1H),5.76(m,2H),7.57(s,1H),7.67(d,J=8.56Hz,1H),8.04(s,1H),8.08(d,J=8.80Hz,1H);MS:(M+H)+749.
实施例278:化合物278的制备
化合物278
按照实施例269、方案2的方法制备化合物278,步骤1除外。
步骤1:
修改:将6-甲氧基-2H-异喹啉-1-酮(700mg)和NCS(532mg)在MeCN(10mL)中的混合物回流3h。过滤得到600mg(72%)所需的产物,为固体物。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 3.96(s,1H),7.19(dd,J=8.80,2.45Hz,1H),7.28(d,J=2.45Hz,1H),7.34(s,1H),8.25(d,J=9.05Hz,1H);MS:(M+H)+210.
步骤2:
修改:使用500mg 4-氯-6-甲氧基-2H-异喹啉-1-酮,得到400mg产物。
产物:
数据:
1H NMR(400Hz,CDCl3)δppm 4.01(s,3H),7.35(d,J=2.45Hz,1H),7.41(d,J=2.45Hz,1H),8.24(d,J=9.29Hz,1H),8.27(s,1H);MS:(M+H)+229.
步骤3:
修改:使用42mg 1,4-二氯-6-甲氧基-异喹啉和117mg{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯,得到70mg产物(收率47%)。
产物:
化合物278
数据:
1H NMR(400Hz,CD3OD)δppm 1.05(m,12H),1.25(m,10H),1.44(m,1H),1.88(dd,J=8.07,5.62Hz,1H),2.24(m,2H),2.61(dd,J=13.82,6.72Hz,1H),2.94(m,1H),3.97(s,3H),4.04(dd,J=11.74,2.69Hz,1H),4.21(s,1H),4.49(m2H),5.12(d,J=10.52Hz,1H),5.29(d,J=17.12Hz,1H),5.75(m,2H),7.19(d,J=8.80Hz,1H),7.37(s,1H),8.00(s,1H),8.13(d,J=9.05Hz,1H);MS:(M+H)+749.
实施例279:化合物279的制备
化合物279
按照实施例269、方案2的方法制备化合物279,不同之处在于在步骤1中使用3-甲氧基-3-(3-甲氧基-苯基)-丙烯酸代替2-三氟甲氧基肉桂酸。
步骤1:
修改:使用4.24g3-甲氧基-3-(3-甲氧基-苯基)-丙烯酸,得到400mg产物(收率10%)。
产物:
步骤2:
修改:使用400mg 4,6-二甲氧基-2H-异喹啉-1-酮,得到300mg产物(收率69%)。
产物:
数据:
1H NMR(400Hz,CDCl3)δppm 3.97(s,3H),4.05(s,3H),7.31(dd,J=9.17,2.57Hz,1H),7.45(d,J=2.69Hz,1H),7.75(s,1H),8.16(d,J=9.29Hz,1H);MS:(M+H)+224.
步骤3:
修改:使用89mg1-氯-4,6-二甲氧基-异喹啉和223mg{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯,得到160mg产物(收率54%)。
产物:
化合物279
数据:
1H NMR(400Hz,CD3OD)δppm 1.07(m,12H),1.21(m,10H),1.43(m,1H),1.87(dd,J=8.07,5.62Hz,1H),2.24(m,2H),2.58(dd,J=13.57,6.97Hz,1H),2.94(m,1H),3.92(s,3H),3.99(s,3H),4.04(dd,J=11.74,2.93Hz,1H),4.24(s,1H),4.39(d,J=11.98Hz,1H),4.50(m,1H),5.12(d,J=10.52Hz,1H),5.29(d,J=16.87Hz,1H),5.75(m,2H),7.12(d,J=9.05Hz,1H),7.40(d,J=2.20Hz,1H),7.48(s,1H),8.04(d,J=9.05Hz,1H);MS:(M+H)+744.
实施例280:化合物280的制备
化合物280
按照实施例269、方案2的方法制备化合物280,不同之处在于在步骤1中使用3-(3-二氟甲氧基-苯基)-丙烯酸代替2-三氟甲氧基肉桂酸。
步骤1:
修改:使用4.28g 3-(3-二氟甲氧基-苯基)-丙烯酸,得到3.1g产物(收率72%)。
产物:
数据:MS:(M+H)+212。
步骤2:
修改:使用2g 6-二氟甲氧基-2H-异喹啉-1-酮,得到1.5g产物(收率61%)。
产物:
数据:
1H NMR(400Hz,CDCl3)δppm 6.69(t,J=72.75Hz,1H),7.49(m,2H),8.28(d,J=5.62Hz,1H),8.36(d,J=9.05Hz,1H);MS:(M+H)+230.
步骤3:
修改:使用46mg 1-氯-6-二氟甲氧基-异喹啉和113mg{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯,得到8mg产物(收率5%)。
产物:
化合物280
数据:
1H NMR(400Hz,CD3OD)δppm 1.05(m,12H),1.23(m,10H),1.44(m,2H),1.88(dd,J=8.19,5.50Hz,1H),2.30(m,2H),2.67(d,J=13.94Hz,1H),2.93(m,1H),4.07(d,J=10.27Hz,1H),4.21(s,1H),4.53(d,J=6.85Hz,2H),5.13(m,1H),5.31(s,1H),5.76(d,J=47.93Hz,2H),7.11(m,2H),7.26(d,J=6.11Hz,1H),7.81(d,J=6.11Hz,1H),8.16(m,1H);MS:(M+H)+700.
实施例281:化合物281的制备
化合物281
按照实施例269、方案2的方法制备化合物281,不同之处在于在步骤1中使用3-氯-3-苯基-丙烯酸代替2-三氟甲氧基肉桂酸。
步骤1:
修改:使用11g 3-氯-3-苯基-丙烯酸,得到3.1g产物(收率29%)。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 7.34(s,1H),7.52(t,J=7.58Hz,1H),7.77(t,J=7.46Hz,1H),7.90(d,J=8.07Hz,1H),8.39(d,J=8.07Hz,1H),11.37(s,1H):MS:MS:(M+H)+180.
步骤2:
修改:使用3.1g 4-氯-2H-异喹啉-1-酮,得到2.3g产物(收率66%)。
产物:
数据:
1H NMR(400MHz,CDCl3)δppm 7.77(ddd,J=8.31,7.09,1.22Hz,1H),7.88(ddd,J=8.31,7.09,1.22Hz,1H),8.23(d,J=8.31Hz,1H),8.34(s,1H),8.36(d,J=8.56Hz,1H);MS:(M+H)+198.
步骤3:
修改:使用20mg 1,4-二氯-异喹啉和56mg{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯,得到33mg产物(收率30%)。
产物:
化合物281
数据:
1H NMR(400MHz,CD3OD)δppm 1.06(m,12H),1.24(m,10H),1.44(dd,J=9.41,5.26Hz,1H),1.88(dd,J=7.83,5.62Hz,1H),2.27(m,2H),2.63(dd,J=13.82,6.97Hz,1H),2.94(m,1H),4.06(dd,J=11.49,2.45Hz,1H),4.22(d,J=9.29Hz,1H),4.53(m,2H),5.12(d,J=10.76Hz,1H),5.29(d,J=17.12Hz,1H),5.75(m,1H),5.85(s,1H),6.60(d,J=8.80Hz,1H),7.63(t,J=7.58Hz,1H),7.86(t,J=7.70Hz,1H),8.06(s,1H),8.11(d,J=8.56Hz,1H),8.25(d,J=8.31Hz,1H);MS:(M+H)+718.
实施例282:化合物282的制备
化合物282
按照实施例269、方案2的方法制备化合物2,不同之处在于在步骤1中使用3-氯-3-苯基-丙烯酸代替2-三氟甲氧基肉桂酸。
步骤1:
修改:使用20g 3-氯-3-苯基-丙烯酸,得到2g产物(收率8%)。
产物:
数据:MS:(M+H)+230。
步骤2:
修改:使用2g 6-三氟甲氧基-2H-异喹啉-1-酮,得到0.7产物(收率33%)。
产物:
数据:
1H NMR(400MHz,CDCl3)δppm 7.51(d,J=9.29Hz,1H),7.59(d,J=5.62Hz,1H),7.64(s,1H),8.31(d,J=5.62Hz,1H),8.40(d,J=9.05Hz,1H);MS:(M+H)+248.
步骤3:
修改:使用50mg 1-氯-6-三氟甲氧基-异喹啉和113mg{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯,得到42mg产物(收率27%)。
产物:
化合物282
数据:
1H NMR(400MHz,CD3OD)δppm 1.05(m,12H),1.24(m,10H),1.44(dd,J=9.17,5.50Hz,1H),1.88(dd,J=8.07,5.62Hz,1H),2.28(m,2H),2.63(dd,J=13.45,7.09Hz,1H),2.94(m,1H),4.06(dd,J=11.25,2.45Hz,1H),4.21(s,1H),4.53(m,2H),5.13(d,J=10.52Hz,1H),5.30(d,J=17.12Hz,1H),5.75(m,1H),5.89(s,1H),7.39(m,2H),7.72(s,1H),8.05(d,J=5.87Hz,1H),8.31(d,J=9.05Hz,1H),9.18(s,1H);MS:(M+H)+768.
实施例283:化合物283的制备
化合物283
按照实施例269、方案2的方法制备化合物283,不同之处在于在步骤1中使用3-(4-氟-苯基)-3-甲氧基-丙烯酸代替2-三氟甲氧基肉桂酸。
步骤1:
修改:使用3.82g 3-(4-氟-苯基)-3-甲氧基-丙烯酸,得到198mg产物(收率5%)。
产物:
数据:MS:(M+H)+194。
步骤2:
修改:使用193mg 7-氟-4-甲氧基-2H-异喹啉-1-酮,得到199mg产物(收率94%)。
产物:
数据:
1H NMR(400MHz,CDCl3)δppm 4.05(s,3H),7.49(m,1H),7.78(s,1H),7.86(dd,J=9.66,2.57Hz,1H),8.23(dd,J=9.29,5.38Hz,1H);MS:(M+H)+212.
步骤3:
修改:使用42mg 1-氯-7-氟-4-甲氧基-异喹啉和112mg{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯,得到40mg产物(收率14%)。
产物:
化合物283
数据:
1H NMR(400MHz,CD3OD)δppm 1.06(m,12H),1.24(m,10H),1.42(m,1H),1.87(dd,J=7.95,5.50Hz,1H),2.23(m,2H),2.55(dd,J=13.08,6.48Hz,1H),2.93(m,1H),4.06(s,3H),4.09(m,1H),4.23(s,1H),4.30(d,J=11.49Hz,1H),4.46(m,1H),5.12(d,J=10.27Hz,1H),5.29(d,J=17.36Hz,1H),5.40(s,1H),5.76(m,1H),7.46(d,J=9.05Hz,1H),7.56(d,J=2.20Hz,1H),7.75(s,1H),8.18(d,J=9.05Hz,1H);MS:(M+H)+749.
实施例284:化合物284的制备
化合物284
按照实施例269、方案2的方法制备化合物284,步骤1除外。
步骤1:
修改:将7-甲氧基-2H-异喹啉-1-酮(876mg)和NCS(665mg)在MeCN(10mL)中的混合物回流3h。过滤得到500mg(47%)所需的产物,为固体物。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 4.00(s,3H),7.58(m,2H),8.14(d,J=10.03Hz,1H),8.17(s,1H).
步骤2:
修改:使用418mg 4-氯-7-甲氧基-2H-异喹啉-1-酮,得到410mg产物(收率90%)。
产物:
数据:
1H NMR(400Hz,CDCl3)δppm 4.00(s,3H),7.49(dd,J=9.16,2.44Hz,1H),7.55(d,J=2.44Hz,1H),8.12(d,J=9.16Hz,1H),8.21(s,1H);MS:(M+H)+229.
步骤3:
修改:使用42mg 1,4-二氯-7-甲氧基-异喹啉和117mg{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯,得到50mg产物(收率33%)。
产物:
化合物284
数据:
1H NMR(400Hz,CD3OD)δppm 1.05(m,20H),1.24(m,2H),1.44(m,1H),1.89(dd,J=8.19,5.50Hz,1H),2.28(m,2H),2.62(dd,J=13.69,6.85Hz,1H),2.94(m,1H),3.92(s,3H),4.07(dd,J=11.98,3.42Hz,1H),4.19(m,1H),4.44(d,J=11.74Hz,1H),4.58(dd,J=10.27,7.09Hz,1H),5.12(m,1H),5.31(d,J=17.12Hz,1H),5.78(m,2H),7.49(m,2H),7.91(s,1H),8.02(m,1H);MS:(M+H)+749.
实施例285:化合物285的制备
化合物285
按照实施例269、方案2的方法制备化合物285,不同之处在于在步骤1中使用2-二氟甲氧基肉桂酸。
步骤1和步骤2:
参见化合物256
步骤3:
修改:使用46mg 1-氯-5-二氟甲氧基-异喹啉和111mg{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯,得到40mg产物(收率27%)。
产物:
化合物285
数据:
1H NMR(400MHz,CD3OD)δppm 1.06(m,12H),1.25(m,10H),1.44(m,1H.1.89(m,1H).2.22(m,2H).2.62(m,1H).2.94(m,1H),4.08(m,1H),4.23(d,J=9.54Hz,1H),4.52(m,2H),5.12(d,J=10.76Hz,1H),5.29(d,J=17.61Hz,1H),5.75(m,J=10.03Hz,1H),5.88(s,1H),6.60(s,1H),7.02(t,J=73.48Hz,1H),7.52(m,3H),8.07(m,J=5.75,5.75Hz,2H);MS:(M+Na)+772.
实施例286:化合物286的制备
化合物286
按照实施例269、方案2的方法制备化合物286,不同之处在于在步骤1中使用3-(2,3-二氢-苯并[1,4]二氧杂环己烯-5-基)-丙烯酸代替2-三氟甲氧基肉桂酸。
步骤1:
修改:使用4.12g 3-(2,3-二氢-苯并[1,4]二氧杂环己烯-5-基)-丙烯酸,得到2.2g产物(收率53%)。
产物:
数据:
1H NMR(400MHz,CD3OD)δppm 4.37(m,4H),6.83(d,J=7.09Hz,1H),7.02(d,J=8.80Hz,1H),7.12(d,J=7.34Hz,1H),7.79(d,J=8.80Hz,1H);MS:(M+H)+204.
步骤2:
修改:使用2.05g 2,3-二氢-7H-1,4-二氧杂-7-氮杂-菲-8-酮,得到1.5g产物(收率68%)。
产物:
数据:
1H NMR(400Hz,CDCl3)δppm 4.42(m,4H),7.24(d,J=9.05Hz,1H),7.77(d,J=5.87Hz,1H),7.84(d,J=9.05Hz,1H),8.18(d,J=5.87Hz,1H);MS:(M+H)+222.
步骤3:
修改:使用88mg 8-氯-2,3-二氢-1,4-二氧杂-7-氮杂-菲和223mg{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯,得到140mg产物(收率47%)。
产物:
化合物286
数据:
1H NMR(400Hz,CD3OD)δppm 1.06(m,12H),1.24(m,10H),1.43(dd,J=9.05,5.14Hz,1H),1.87(m,1H),2.22(d,J=9.29Hz,2H),2.60(dd,J=13.45,7.09Hz,1H),2.94(m,1H),4.05(dd,J=11.62,2.81Hz,1H),4.24(s,1H),4.44(m,6H),5.13(d,J=17.36Hz,1H),5.29(d,J=17.36Hz,1H),5.75(m,2H),7.04(d,J=9.29Hz,1H),7.44(d,J=5.87Hz,1H),7.69(d,J=9.05Hz,1H),7.88(d,J=6.11Hz,1H);MS:(M+H)+742.
实施例287:化合物287的制备
化合物287
按照实施例269、方案2的方法制备化合物287,不同之处在于在步骤1中使用3-(2,2-二氟-苯并[1,3]二氧杂环戊烯-4-基)-丙烯酸代替2-三氟甲氧基肉桂酸。
步骤1:
修改:使用4.56g 3-(2,2-二氟-苯并[1,3]二氧杂环戊烯-4-基)-丙烯酸,得到2.2g产物(收率55%)。
产物:
数据:
1H NMR(400MHz,CD3DOD)δppm 6.63(d,J=7.09Hz,1H),7.29(d,J=7.34Hz,1H),7.40(d,J=8.80Hz,1H),8.19(d,J=8.80Hz,1H);MS:(M+H)+226.
步骤2:
修改:使用2.2g 2,2-二氟-7H-1,3-二氧杂-7-氮杂-环戊烯并[a]萘-6-酮,得到2.1g产物(收率87%)。
产物:
数据:
1H NMR(500Hz,CDCl3)ppm 7.51(d,J=9.29Hz,1H),7.65(d,J=5.87Hz,1H),8.22(d,J=9.05Hz,1H),8.32(d,J=5.87Hz,1H);MS:(M+H)+244.
步骤3:
修改:使用48mg 6-氯-2,2-二氟-1,3-二氧杂-7-氮杂-环戊烯并[a]萘和113mg{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯,得到40mg产物(收率27%)。
产物:
化合物287
数据:
1H NMR(400Hz,CD3OD)δppm 1.02(s,12H),1.24(m,10H),1.43(m,1H),1.88(dd,J=8.07,5.38Hz,1H),2.32(d,J=3.67Hz,2H),2.64(d,J=13.45Hz,1H),2.95(m,1H),4.05(d,J=11.49Hz,1H),4.19(d,J=9.29Hz,1H),4.53(m,2H),5.12(d,J=9.78Hz,1H),5.32(s,1H),5.77(m,2H),7.34(d,J=5.87Hz,1H),7.46(d.J=9.05Hz,1H),8.11(m,2H);MS:(M+H)+764.
实施例288:化合物288的制备
化合物288
按照实施例269、方案2的方法制备化合物288,不同之处在于在步骤1中使用3-(2,2-二氟-苯并[1,3]二氧杂环戊烯-5-基)-丙烯酸代替2-三氟甲氧基肉桂酸。
步骤1:
修改:使用1g 3-(2,2-二氟-苯并[1,3]二氧杂环戊烯-5-基)-丙烯酸,得到0.55g产物。
产物:
数据:
1H NMR(400MHz,CD3DOD)δppm 6.69(d,J=7.09Hz,1H),7.19(d,J=7.09Hz,1H),7.47(s,1H)7.98(s,1H);MS:(M+H)+226.
步骤2:
修改:使用0.5g 2,2-二氟-6H-[1,3]二氧杂环戊烯并[4,5-g]异喹啉-5-酮,得到0.4g产物。
产物:
数据:
1H NMR(400Hz,CDCl3)δ7.41(s,1H),7.57(d,J=5.49Hz,1H),7.94(s,1H),8.27(d,J=5.80Hz,1H);MS(M+H)+244.
步骤3:
修改:使用48mg 5-氯-2,2-二氟-[1,3]二氧杂环戊烯并[4,5-g]异喹啉和112mg{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯,得到30mg产物。
产物:
化合物288
数据:
1H NMR(400Hz,CD3OD)δppm 1.06(m,12H),1.25(m,10H),1.42(m,1H),1.87(dd,J=8.07,5.62Hz,1H),2.26(m,2H),2.61(dd,J=13.57,6.97Hz,1H),2.93(m,1H),4.07(dd,J=11.86,2.81Hz,1H),4.22(m,1H),4.40(d,J=11.98Hz,1H),4.52(m,1H),5.11(d,J=10.52Hz,1H),5.29(d,J=17.12Hz,1H),5.37(s,1H),5.74(m,1H),7.39(s,1H),7.56(s,1H),7.63(d,J=5.62Hz,1H),7.99(d,J=5.62Hz,1H).
实施例289:化合物289的制备
化合物289
方案3:
将化合物287(15mg)和Pt(S)/C(5%,5mg)在乙酸乙酯(5mL)中的悬浮液氢化(压力10psi)30min。过滤后,浓缩,定量得到15mg化合物289,为固体物。
1H NMR(400MH-Hz,CD3OD)δppm 1.09(m,26H),1.57(m,4H),2.30(m,1H),2.61(m,J=13.82,7.21Hz,1H),2.96(m,1H),4.05(m,J=13.94Hz,1H),4.19(d,J=9.54Hz,1H),4.53(m,2H),5.89(s,1H),7.34(d,J=5.87Hz,1H),7.46(d,J=9.05Hz,1H),8.09(d,J=5.87Hz,1H),8.12(d,J=8.80Hz,1H);MS:(M+H)+766.
实施例290:化合物290的制备
化合物290
按照实施例289、方案3的方法,使用15mg化合物286制备化合物290(15mg,100%)。数据:
1H NMR(400Hz,CD3OD)δppm1.02(m,14H),1.23(m,12H),1.58(m,4H),2.25(m,1H),2.58(dd,J=13.82,7.21Hz,1H),2.96(m,1H),4.05(m,J=11.25,2.93Hz,1H),4.25(d,J=9.54Hz,1H),4.39(m,5H),4.52(m,J=10.03,7.34Hz,1H),5.81(s,1H),7.03(d,J=9.05Hz,1H),7.43(d,J=6.11Hz,1H),7.69(d,J=9.05Hz,1H),7.88(d,J=6.11Hz,1H);MS:(M+H)+744.
实施例291:化合物291的制备
化合物291
按照实施例289、方案3的方法,使用28mg化合物251制备化合物291(28mg,100%)。数据:
1H NMR(400MHz,CD3OD)δppm1.01(m,15H),1.26(m,11H),1.37(m,1H),1.58(m,3H),2.25(m,1H),2.58(dd,J=13.6,7.0Hz,1H),2.96(m,1H),3.99(s,3H),4.06(m,1H),4.25(m,1H),4.44(m,1H),4.53(dd,J=10.3,7.6Hz,1H),5.78(s,1H),6.64(d,J=9.8Hz,1H),7.55(m,2H),7.71(t,J=7.3Hz,1H),8.09(d,J=8.6Hz,1H),8.14(d,J=8.1Hz,1H);MS:(M+Na)+738.
实施例292:化合物292的制备
化合物292
按照实施例289、方案3的方法,使用19mg化合物253制备化合物292(16mg,84%)。
1H NMR(400MHz,CD3OD)δppm 0.90(m,15H),1.15(m,12H),1.48(m,3H),2.18(m,1H),2.51(dd,J=13.7,6.9Hz,1H),2.88(m,1H),3.90(s,3H),3.98(dd,J=11.6,3.1Hz,1H),4.18(d,J=9.5Hz,1H),4.36(d,J=11.0Hz,1H),4.45(dd,J=10.2,7.2Hz,1H),5.76(s,1H),6.56(d,J=9.3Hz,1H),7.05(d,J=7.6Hz,1H),7.34(t,J=8.1Hz,1H),7.51(d,J=5.9Hz,1H),7.65(d,J=8.3Hz,1H),7.86(d,J=6.1Hz,1H);MS:(M+Na)+738.
实施例293:化合物293的制备
化合物293
按照实施例289、方案3的方法,使用20mg化合物252制备化合物293(7mg,35%)。
1H NMR(400MHz,CD3OD)δppm 1.04(m,15H),1.27(m,12H),1.58(m,3H),2.27(m,1H),2.60(m,4H),2.96(m,1H),4.07(dd,J=11.7,2.9Hz,1H),4.25(s,1H),4.46(d,J=12.0Hz,1H),4.54(dd,J=10.0,7.6Hz,1H),5.85(s,1H),7.39(t,J=7.7Hz,1H),7.44(d,J=5.9Hz,1H),7.53(d,J=6.9Hz,1H),8.00(d,J=6.1Hz,1H),8.06(d,J=8.6Hz,1H);MS:(M+H)+700.
实施例294:化合物294的制备
化合物294
按照实施例289、方案3的方法,使用18mg化合物254制备化合物294(14mg,78%)。
1H NMR(400MHz,CD3OD)δppm 0.94(m,15H),1.13(m,10H),1.20(m,2H),1.50(m,3H),2.21(m,1H),2.53(dd,J=13.8,7.0Hz,1H),2.88(m,1H),3.99(dd,J=11.4,2.6Hz,1H),4.14(d,J=9.3Hz,1H),4.45(m,2H),5.79(s,1H),6.53(d,J=9.1Hz,1H),7.40(t,J=8.0Hz,1H),7.54(d,J=5.9Hz,1H),7.73(d,J=7.3Hz,1H),8.02(d,J=6.1Hz,1H),8.10(d,J=8.6Hz,1H);MS:(M+Na)+742.
实施例295:化合物295的制备
化合物295
按照实施例289、方案3的方法,使用30mg化合物270制备化合物295(30mg,100%)。MS:(M+Na)+776。
实施例296:化合物296的制备
化合物296
按照实施例289、方案3的方法,使用20mg化合物259制备化合物296(6.3mg,33%)。
1H NMR(400MHz,CD3OD)δppm 1.04(m,15H),1.24(m,12H),1.59(m,3H),2.29(m,1H),2.50(s,3H),2.60(dd,J=13.69,6.85Hz,1H),2.97(m,1H),4.09(dd,J=11.74,2.93Hz,1H)4.22(s,1H)4.43(d,J=11.74Hz,1H),4.59(dd,J=10.27,6.85Hz,1H),5.87(s,1H),7.30(d,J=5.87Hz,1H),7.57(dd,J=8.31,1.47Hz,1H),7.72(d,J= 8.31Hz,1H),7.89(d,J=5.87Hz,1H),7.94(s,1H);MS:(M+H)+700.
实施例297:化合物297的制备
化合物297
按照实施例289、方案3的方法,使用40mg化合物263制备化合物297(40mg,100%)。
1H NMR(400MHz,CD3OD)δppm 0.98(m,13H)1.07(m,2H)1.27(m,12H)1.57(m,3H)2.27(m,1H)2.58(dd,J=14.7,7.1Hz,1H)2.77(s,3H)2.96(m,1H)4.04(m,1H)4.27(m,1H)4.42(d,J=11.5Hz,1H)4.55(dd,J=10.6,7.0Hz,1H)5.94(s,1H)6.65(d,J=9.5Hz,1H)7.28(m,2H)7.50(t,J= 7.6Hz,1H)7.60(d,J=7.6Hz,1H)7.89(d,J=5.6Hz,1H);MS:(M+Na)+722.
实施例298:化合物298的制备
化合物298
按照实施例289、方案3的方法,使用29mg化合物261制备化合物298(29mg,100%)。1H NMR(400MHz,CD3OD)δppm 0.99(m,15H),1.25(m,12H),1.60(m,3H),2.28(m,1H),2.58(m,1H),2.96(m,1H),4.02(m,4H),4.22(dd,J=8.8,4.4Hz,1H),4.48(m,2H),5.83(s,1H),7.27(d,J=5.6Hz,1H),7.36(d,J=8.8Hz,1H),7.75(d,J=11.3Hz,1H),7.90(d,J=5.9Hz,1H);MS:(M+Na)+756。
实施例299:化合物299的制备
化合物299
按照实施例289、方案3的方法,使用35mg化合物274制备化合物299(34mg,97%)。
1H NMR(400MHz,CD3OD)δppm1.01(m,16H),1.28(m,12H),1.58(m,2H),2.27(s,1H),2.59(dd,J=13.82,6.97Hz,1H),2.96(m,1H),4.02(s,3H),4.07(m,1H),4.21(m,1H),4.44(m,J=11.98Hz,1H),4.55(d,J=10.27Hz,1H),5.85(s,1H),7.39(m,2H),7.95(d,J=6.11Hz,1H),8.00(d,J=9.29Hz,1H);MS:(M+Na)+756.
实施例300:化合物300的制备
化合物300
按照实施例289、方案3的方法,使用30mg化合物262制备化合物300(30mg,100%)。
1H NMR(400MHz,CD3OD)δppm 1.27(m,30H),2.25(s,1H),2.54(s,1H),2.96(m,1H),3.97(s,3H),4.20(m,3H),4.51(m,J=10.52,6.85Hz,1H),5.37(s,1H),7.38(s,1H),7.62(s,1H),7.65(d,J=5.38Hz,1H)8.06(d,J=5.62Hz,1H);MS:(M+Na)+773.
部分G:
用于部分G的LC/MS方法如下:
4.6×50mm Xterra,@梯度3min,流速4mL/min
方案1:(通用方案)
方案2:(通用方案)
方案4:(通用方案)
实施例320:化合物320的制备
化合物320
按照上述方案1和3的方法制备化合物320。
步骤1(方案1):
-78℃下,向N,N-二乙基-4-甲氧基-2-甲基-苯甲酰胺(332mg,1.5mmol)的THF(15mL)溶液中加入t-BuLi(1.7M的戊烷溶液,1.3mL,2.25mmol)。-78℃下将所得的红色溶液搅拌10min,随后加入2-氰基吡啶(156mg,1.5mmol)。接着将反应混合物升温至室温并搅拌过夜。反应物用饱和氯化铵溶液猝灭并用乙酸乙酯萃取两次。将合并的有机层干燥(硫酸镁)并浓缩。粗产物经制备型HPLC纯化,得到淡黄色的TFA盐固体(85mg,收率15%)。
1H NMR(400MHz,CD3OD)δ3.91(m,3H),7.09(dd,J=9.05,2.45Hz,1H),7.17(d,J=2.45Hz,1H),7.37(s,1H),7.42(m,1H),7.92(m,1H),8.08(d,J=8.07Hz,1H),8.18(d,J=9.05Hz,1H),8.65(d,J=4.89Hz,1H).
LC-MS(保留时间:2.14min),MS m/z 253(MH+)。
步骤2(方案3,步骤1):
将6-甲氧基-3-吡啶-2-基-2H-异喹啉-1-酮TFA盐(85mg,0.232mmol)与磷酰氯(3.0mL)一起加热回流2天。随后蒸馏出磷酰氯并将剩余物用冰猝灭。接着用10N NaOH溶液中和,收集棕色固体物,为纯产物(62mg,收率99%)。
LC-MS(保留时间:2.063min),MS m/z 271(MH+)。
步骤3(方案3,步骤2):
化合物320
在-78℃下,向{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯(82mg,0.148mmol)和三氯化镧(36mg,0.148mmol)的DMF(1.5mL)溶液中加入叔丁醇钾(1.0M THF溶液,0.74mL,0.74mmol)。将反应混合物搅拌1hr,随后加入1-氯-6-甲氧基-3-吡啶-2-基-异喹啉(40mg,0.148mmol)。升温至室温并搅拌过夜。接着将所得混合物用水猝灭并过滤。浓缩滤液并将剩余物经制备型HPLC纯化,得到类白色固体产物(化合物320)(23mg,收率20%)。
1H NMR(400MHz,CD3OD)δ0.87-1.08(m,11H),1.20-1.30(m,11H),1.43(m,1H),1.87(m,1H),2.22(m,1H),2.35(m,1H),2.69(m,1H),2.93(m,1H),3.94(s,3H),4.16(m,1H),4.27(m,1H),4.45(m,1H),4.56(m,1H),5.10(d,J=11.3Hz,1H),5.27(d,J=15.9Hz,1H),5.74(m,1H),6.07(s,1H),7.12(d,J=7.33Hz,1H),7.31(d,J=1.96Hz,1H),7.40(m,1H),7.94(dd,J=7.8Hz,1.5Hz,1H),8.11(d,J=9.29Hz,1H),8.22(s,1H),8.45(d,J=8.07Hz,1H),8.62(m,1H).
LC-MS(保留时间:2.393min),MS m/z 791(MH+)。
实施例321:化合物321的制备
方案5
使溴代丙酮酸乙酯与乙基硫脲在回流的二_烷中缩合,定量得到单烷基氨基噻唑HBr盐。用EtI在DMF中将2-乙氨基-噻唑-4-甲酸乙酯烷基化,得到2-二乙氨基-噻唑-4-甲酸乙酯。
LC/MSm/z 229(MH)+。
按照上述方案2和3的方法,在方案2的步骤1中使用2-二乙氨基-噻唑-4-甲酸乙酯制备化合物321。
LC/MS(保留时间2.76min):m/z 868(MH+)。
实施例322:化合物322的制备
化合物322
按照实施例321的方法制备化合物322,不同之处在于在方案2的步骤1中使用2-二甲氨基-噻唑-4-甲酸乙酯(按照方案5制备,不同之处在于使用甲基硫脲和甲基碘代替乙基硫脲和乙基碘)代替2-二甲氨基-噻唑-4-甲酸乙酯。
LC/MS(保留时间2.56min):m/z 840(MH+)。
实施例323:化合物323的制备
化合物323
按照实施例324、步骤3的方法制备化合物323,不同之处在于使用3-氯-6-甲氧基-苯并[d]异_唑代替1-氯-6-甲氧基-3-吡啶-2-基-异喹啉。MS m/z 702(M-H)-。
实施例324:化合物324的制备
化合物324
按照实施例324、步骤3的方法制备化合物324,不同之处在于使用3-氯-苯并[d]异噻唑代替1-氯-6-甲氧基-3-吡啶-2-基-异喹啉。
LC/MS(保留时间1.83min):m/z 688(M-H)-。
实施例325:化合物325的制备
化合物325
按照上述方案1和3的方法制备化合物325。
步骤1(方案1):
-78℃下,向N,N-二乙基-4-甲氧基-2-甲基-苯甲酰胺(332mg,1.5mmol)的THF(15mL)溶液中加入t-BuLi(1.7M戊烷溶液,1.3mL,2.25mmol)。将所得的红色溶液在-78℃下搅拌10min,随后加入4-氰基吡啶(164mg,1.575mmol)。接着将反应混合物升温至室温并搅拌过夜。反应物用饱和氯化铵溶液猝灭,收集黄色沉淀物,为纯产物(145mg,收率38%)。
1H NMR(CD3OD,400MHz)δ3.91(s,3H),7.18(dd,J=8.8Hz,2.8Hz,1H),7.26(m,2H),8.06(d,J=6.0Hz,2H),8.16(d,J=8.8Hz,1H),8.84(d,J=6.0Hz,2H).
LC-MS(保留时间:1.300min),MS m/z 253(MH+)。
步骤2(方案3,步骤1):
将6-甲氧基-3-吡啶-4-基-2H-异喹啉-1-酮(134mg,0.531mmol)与磷酰氯(6.0mL)一起加热回流5天。随后蒸馏出磷酰氯并将剩余物用冰骤冷。接着用饱和碳酸氢钠溶液中和,收集棕色固体物,为纯产物(125mg,收率87%)。
1H NMR(DMSO-d6,400MHz)δ3.99(s,3H),7.53(dd,J=9.04Hz,2.44Hz,1H),7.59(d,J=2.69Hz,1H),8.26(d,J=9.05Hz,1H),8.30(d,J=5.38Hz,2H),8.73(s,1H),8-85(d,J=6.36Hz,2H).
LC-MS(保留时间:2.027min),MS m/z 271(MH+)。
步骤3(方案3,步骤2):
化合物325
在-78℃下,向{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯(83.5mg,0.15mmol)和三氯化镧(36.8mg,0.15mmol)的DME(1.5mL)溶液中加入叔丁醇钾(1.0M THF溶液,0.75mL,0.75mmol)。将反应混合物搅拌1hr,随后加入1-氯-6-甲氧基-3-吡啶-4-基-异喹啉(40.6mg,0.15mmol)。升温至室温并搅拌过夜。接着将所得混合物用水猝灭并过滤。浓缩滤液并将剩余物经制备型HPLC纯化,得到灰白色固体产物(化合物325)(1.6mg,收率1.3%)。
1H NMR(400MHz,CD3OD)δ0.90(m,2H),1.02(s,9H),1.17-1.31(m,11H),1.42(m,1H),1.87(m,1H),2.23(m,1H),2.35(m,1H),2.68(m,1H),2.93(m,1H),3.95(s,3H),4.15(m,1H),4.25(m,1H),4.45(m,1H),4.56(m,1H),5.10(d,J=10.76Hz,1H),5.27(d,J=17.61Hz,1H),5.74(m,1H),6.06(s,1H),7.14(d,J=8.07Hz,1H),7.34(s,1H),8.01(s,1H),8.12(d,J=8.81Hz,1H),8.19(d,J=6.12Hz,2H),8.61(d,J=5.63Hz,2H).
LC-MS(保留时间:2.523min),MS m/z 791(MH+)。
实施例326:化合物326的制备
化合物326
按照上述方案1和4的方法制备化合物326。
步骤1(方案1):
-78℃下,向N,N-二乙基-4-甲氧基-2-甲基-苯甲酰胺(332mg,1.5mmol)的THF(15mL)溶液中加入t-BuLi(1.7M戊烷溶液,1.3mL,2.25mmol)。将所得的红色溶液在-78℃下搅拌10min,随后加入4-二甲氨基苄腈(219mg,1.5mmol)。接着将反应混合物升温至室温并搅拌过夜。反应物用饱和氯化铵溶液猝灭,收集黄色沉淀物并在乙醚中研磨,得到灰白色固体,为纯产物(247mg,收率56%)。
1H NMR(DMSO-d6,400MHz)δ2.97(s,6H),3.87(s,3H),6.72(s,1H),6.78(d,J=8.80Hz,2H),6.97(dd,J=8.80,2.45Hz,1H),7.10(d,J=2.45Hz,1H),7.65(d,J=8.80Hz,2H),8.05(d,J=8.80Hz,1H),11.11(s,1H).
LC-MS(保留时间:2.023min),MS m/z 295(MH+)。
步骤2(方案4,步骤1):
将3-(4-二甲氨基-苯基)-6-甲氧基-2H-异喹啉-1-酮(245mg,0.83mmol)与磷酰氯(10.0mL)一起加热回流2天。随后蒸馏出磷酰氯并将剩余物用冰骤冷。接着用10N NaOH溶液中和,并用乙酸乙酯萃取两次。合并各有机层并干燥(硫酸镁)。蒸发溶剂,得到橙色固体产物(215mg,收率83%)。
1H NMR(400MHz,CD3OD)δ3.01(s,6H),3.96(s,3H),6.88(d,J=9.05Hz,2H),7.20(dd,J=9.17,2.57Hz,1H),7.28(d,J=2.45Hz,1H),7.94(s,1H),7.96(d,J=9.05Hz,2H),8.13(d,J=9.29Hz,1H).
LC-MS(保留时间:2.543min),MS m/z 313(MH+)。
步骤3(方案4,步骤2):
将[4-(1-氯-6-甲氧基-异喹啉-3-基)-苯基]-二甲基-胺(110mg,0.35mmol)和氟化四丁基_·氢氟酸盐(tetrabutyl phosphonium hydrogendifluoride)(0.5g)的混合物在Smith微波反应器中在140℃下加热20min。随后加入水并用乙酸乙酯萃取。分离出有机层,用水洗涤并干燥(硫酸镁)。蒸发溶剂,得到棕色固体产物(85mg,收率82%。
LC-MS(保留时间:2.320min),MS m/z 297(MH+)。
步骤4(方案4,步骤3):
化合物326
在-78℃下,向{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基-氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯(111mg,0.2mmol)和三氯化镧(49mg,0.2mmol)在DMF(2.0mL)的溶液内加入叔丁醇钾(1.0M THF溶液,1.0mL,1.0mmol)。将反应混合物搅拌1hr,随后加入[4-(1-氟-6-甲氧基-异喹啉-3-基)-苯基]-二甲胺(59mg,0.2mmol)。升温至室温并搅拌过夜。接着将所得混合物用水猝灭并过滤。浓缩滤液并将剩余物经制备型HPLC纯化,得到淡黄色固体产物(化合物326)(17.5mg,收率11%)。
1H NMR(400MHz,CD3OD)δ0.97-1.08(m,11H),1.23(m,2H),1.31(s,9H),1.44(m,1H),1.87(m,1H),2.22(m,1H),2.34(m,1H),2.68(m,1H),2.93(m,1H),2.99(m,6H),3.91(s,3H),4.17(m,1H),4.29(m,1H),4.39(m,1H),4.52(m,1H),5.10(d,J=10.76Hz,1H),5.27(d,J=17.11Hz,1H),5.74(m,1H),6.03(s,1H),6.83(m,2H),6.95(m,1H),7.16(s,1H),7.59(s,1H),8.01(m,3H).
LC-MS(保留时间:2.850min),MS m/z 834(MH+)。
实施例327:化合物327的制备
化合物327
按照上述方案1和4的方法制备化合物327。
步骤1(方案1):
在-78℃下,向N,N-二乙基-4-甲氧基-2-甲基-苯甲酰胺(332mg,1.5mmol)的THF(15mL)溶液中加入t-BuLi(1.7M戊烷溶液,1.3mL,2.25mmol)。将所得的红色溶液在-78℃下搅拌10min,随后加入4-二乙氨基苄腈(261mg,1.5mmol)。随后将反应混合物升温至室温并搅拌过夜。反应物用饱和氯化铵溶液猝灭,收集黄色沉淀物,为纯产物(215mg,收率44%)。
1H NMR(400MHz,DMSO-d6)δ1.12(m,6H),3.39(m,4H),3.87(s,3H),6.69(s,1H),6.72(d,J=9.05Hz,2H),6.96(dd,J=8.80,2.45Hz,1H),7.09(d,J=2.45Hz,1H),7.61(d,J=9.05Hz,2H),8.04(d,J=8.80Hz,1H),11.06(s,1H).
LC-MS(保留时间:1.883min),MS m/z 323(MH+)。
步骤2(方案4,步骤1):
将3-(4-二乙氨基-苯基)-6-甲氧基-2H-异喹啉-1-酮(207mg,0.642mmo1)与磷酰氯(8.0mL)一起加热回流1天。随后蒸馏出磷酰氯并将剩余物用冰骤冷。接着用饱和碳酸氢钠溶液中和,并用乙酸乙酯萃取两次。合并各有机层并干燥(硫酸镁)。蒸发溶剂,得到褐色固体产物(180mg,收率82%)。
LC-MS(保留时间:2.397min),MS m/z 341(MH+)。
步骤3(方案4,步骤2):
将[4-(1-氯-6-甲氧基-异喹啉-3-基)-苯基]-二乙胺(90mg,0.264mmol)和氟化四丁基鏻·氢氟酸盐(0.5g)的混合物在Smith微波反应器中在140℃下加热20min。随后加入水并用乙酸乙酯萃取。分离出有机层,用水洗涤并干燥(硫酸镁)。蒸发溶剂,得到淡黄色油状产物(70mg,收率82%)。
LC-MS(保留时间:2.253min),MS m/z 325(MH+)。
步骤4(方案4,步骤3):
化合物327
在-78℃下,向{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基-氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯(100mg,0.18mmol)和三氯化镧(66mg,0.27mmol)的DMF(2.0mL)溶液中加入叔丁醇钾(1.0MTHF溶液,0.9mL,0.9mmol),将反应混合物搅拌1hr,随后加入[4-(1-氟-6-甲氧基-异喹啉-3-基)-苯基]-二乙胺(70mg,0.216mmol)。升温至室温并搅拌过夜。接着将所得混合物用水猝灭并过滤。浓缩滤液并将剩余物经制备型HPLC纯化,得到白色固体产物(化合物327)(18mg,收率12%)。
1H NMR(400MHz,CD3OD)δ0.95-1.07(m,11H),1.18(m,6H),1.25-1.38(m,11H),1.58(m,1H),1.85(m,1H),2.19(m,1H),2.34(m,1H),2.68(m,1H),2.92(m,1H),3.42(m,4H),3.90(s,3H),4.16(m,1H),4.28(m,1H),4.37(m,1H),4.53(m,1H),5.07(d,J=11.0Hz,1H),5.25(d,J=17.36Hz,1H),5.74(m,1H),5.99(s,1H),6.77(d,J=8.8Hz,2H),6.94(d,J=9.05Hz,1H),7.14(s,1H),7.56(s,1H),7.95-8.02(m,3H).
LC-MS(保留时间:2.690min),MS m/z 862(MH+)。
实施例328:化合物328的制备
化合物328
按照上述方案2和3的方法制备化合物328。
步骤1(方案2,步骤1):
-78℃下,向N,N-二乙基-4-甲氧基-2-甲基-苯甲酰胺(332mg,1.5mmol)的THF(15mL)溶液中加入t-BuLi(1.7M戊烷溶液,2.12mL,3.6mmol)。将所得的红色溶液在-78℃下搅拌10min,随后加入烟酸甲酯(206mg,1.5mmol)。将反应混合物在-78℃下搅拌2h。随后将反应物用饱和氯化铵溶液猝灭并用乙酸乙酯萃取两次。将合并的有机层干燥(硫酸镁)并浓缩。粗产物经制备型HPLC纯化,得到淡黄色粘稠油状物,为TFA盐(124mg,收率19%)。
LC-MS(保留时间:1.740min),MS m/z 349(M+Na+)。
步骤2(方案2,步骤2):
将N,N-二乙基-4-甲氧基-2-(2-氧代-2-吡啶-3-基-乙基)-苯甲酰胺(120mg,0.272mmol)与乙酸铵(1g)一起加热3h。随后将其冷却并加入水。用乙酸乙酯萃取并分离出有机层。接着干燥(硫酸镁)并浓缩,得到褐色固体产物(65mg,收率95%)。
1H NMR(400MHz,DMSO-d6)δ3.89(s,3H),6.93(s,1H),7.10(dd,J=8.80,2.45Hz,1H),7.19(d,J=2.45Hz,1H),7.52(dd,J=7.46,4.77Hz,1H),8.15(m,2H),8.64(dd,J=4.89,1.47Hz,1H),8.96(d,J=1.71Hz,1H),11.51(s,1H).
LC-MS(保留时间:1.377min),MS m/z 253(MH+)。
步骤3(方案3,步骤1):
将6-甲氧基-3-吡啶-3-基-2H-异喹啉-1-酮(65mg,0.258mmol)与磷酰氯(2.5mL)一起加热回流7天。随后蒸馏出磷酰氯并将剩余物用冰骤冷。接着用10N NaOH溶液中和,并用乙酸乙酯萃取两次。将合并的有机层干燥(硫酸镁)并浓缩,得到黄色固体物产物(27mg,收率39%)。
LC-MS(保留时间:2.090min),MS m/z 271(MH+)。
步骤4(方案3,步骤2):
化合物328
在-78℃下,向{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯(56mg,0.10mmol)和三氯化镧(25mg,0.10mmol)的DMF(1.5mL)溶液中加入叔丁醇钾(1.0M THF溶液,0.5mL,0.5mmol)。将反应混合物搅拌1hr,随后加入1-氯-6-甲氧基-3-吡啶-3-基-异喹啉(27mg,0.10mmol)。升温至室温并搅拌过夜。接着将所得混合物用水猝灭并过滤。浓缩滤液并将剩余物经制备型HPLC纯化,得到白色固体产物(化合物328)(17mg,收率21%)。
1H NMR(400MHz,CD3OD)δ0.95(m,2H),1.02(s,9H),1.20-1.30(m,11H),1.41(m,1H),1.86(m,1H),2.21(m,1H),2.35(m,1H),2.67(m,1H),2.93(m,1H),3.93(s,3H),4.14(m,1H),4.26(m,1H),4.47(d,J=11.99Hz,1H),4.55(m,1H),5.09(d,J=10.02Hz,1H),5.26(d,J=17.85Hz,1H),5.74(m,1H),6.07(s,1H),7.09(m,1H),7.29(d,J=1.96Hz,1H),7.53(m,1H),7.86(s,1H),8.09(d,J=9.05Hz,1H),8.50-8.58(m,2H),9.28(s,1H).
LC-MS(保留时间:2.453min),MS m/z 791(MH+)。
实施例329:化合物329的制备
化合物329
按照上述方案2和4的方法制备化合物329。
步骤1(方案2,步骤1):
在-78℃下,向N,N-二乙基-4-甲氧基-2-甲基-苯甲酰胺(332mg,1.5mmol)的THF(15mL)溶液中加入t-BuLi(1.7M戊烷溶液,2.2mL,3.75mmol)。将所得的红色溶液在-78℃下搅拌10min,随后加入N,N-二甲基邻氨基苯甲酸甲酯(269mg,1.5mmol)。将反应混合物在-78℃下搅拌2h。随后将反应物用饱和氯化铵溶液猝灭并用乙酸乙酯萃取两次。将合并的有机层干燥(硫酸镁)并浓缩。粗产物经制备型HPLC纯化,得到淡黄色粘稠油状产物(256mg,收率46%)。
1H NMR(400MHz,CD3OD)δ0.99-1.13(m,6H),3.23-3.31(m,8H),3.39(m,2H),3.82(s,3H),4.35(s.2H),6.91.(dd,J=8.44,2.57Hz,1H),6.99(d,J=2.45Hz,1H),7.22(d,J=8.56Hz,1H),7.69(t,J=7.70 Hz,1H),7.84(m,1H),7.96(d,J=8.31Hz,1H),8.18(d,J=7.83Hz,1H).
LC-MS(保留时间:1.557min),MS m/z 369(MH+)。
步骤2(方案2,步骤2):
将2-[2-(2-二甲氨基-苯基)-2-氧代-乙基]-N,N-二乙基-4-甲氧基-苯甲酰胺(250mg,0.678mmol)与乙酸铵(1.5g)一起加热2h。随后将其冷却并加入水。用乙酸乙酯萃取并分离出有机层。接着干燥(硫酸镁)并浓缩,得到淡黄色固体产物(125mg,收率63%)。
1H NMR(400MHz,CD3OD)δ2.95(s,6H),3.92(s,3H),6.92(s,1H),7.12(dd,J=8.80,2.45Hz,1H),7.16(d,J=2.45Hz,1H),7.35(m,1H),7.55(m,2H),7.63(d,J=7.83Hz,1H),8.20(d,J=9.05Hz,1H).
LC-MS(保留时间:2.097min,MS m/z 295(MH+)。
步骤3(方案4,步骤1):
将3-(2-二甲氨基-苯基)-6-甲氧基-2H-异喹啉-1-酮(125mg,0.425mmol)与磷酰氯(4.0mL)一起加热回流1天。随后蒸馏出磷酰氯并将剩余物用冰骤冷。接着用10N NaOH溶液中和,并用乙酸乙酯萃取两次。合并各有机层并干燥(硫酸镁)。蒸发溶剂,得到褐色固体产物(82mg,收率62%)。
LC-MS(保留时间:2.040min),MS m/z 313(MH+)。
步骤4(方案4,步骤2):
将[2-(1-氯-6-甲氧基-异喹啉-3-基)-苯基]-二甲基-胺(82mg,0.262mmol)和氟化四丁基_·氢氟酸盐(1.0g)的混合物在Smith微波反应器中在140℃下加热20min。随后加入水并用乙酸乙酯萃取。分离出有机层,用水洗涤并干燥(硫酸镁)。蒸发溶剂,得到粗产物,经制备型HPLC纯化,得到淡黄色油状产物(85mg)。
1H NMR(400MHz,CD3OD)δ3.41(s,6H),4.00(s,3H),7.42(dd,J=9.05,2.45Hz,1H),7.53(s,1H),7.71(m,2H),7.99(m,1H),8.16(m,2H),8.31(s,1H).
LC-MS(保留时间:1.873min),MS m/z 297(MH+)。
步骤5(方案4,步骤3):
化合物329
在-78℃下,向{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基-氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯(56mg,0.1mmol)和三氯化镧(25mg,0.1mmol)的DMF(1.0mL)溶液中加入叔丁醇钾(1.0M THF溶液,0.5mL,0.5mmol),将反应混合物搅拌1hr,随后加入[2-(1-氟-6-甲氧基-异喹啉-3-基)-苯基]-二甲基-胺(30mg,0.1mmol)。升温至室温并搅拌过夜。接着将所得混合物用水猝灭并过滤。浓缩滤液并将剩余物经制型HPLC纯化,得到白色固体产物(化合物329)(4.0mg,收率5%)。
1H NMR(400MHz,CD3OD)δ0.98-1.08(m,11H),1.16-1.32(m,11H),1.40(m,1H),1.85(m,1H),2.16-2.32(m,2H),2.60-2.71(m,7H),2.92(m,1H),3.91(s,3H),4.08(m,1H),4.26(m,1H),4.45(m,1H),4.55(m,1H),5.10(d,J=10.27Hz,1H),5.28(d,J=18.09Hz,1H),5.74(m,1H),5.89(s,1H),7.05(d,J=6.85Hz,1H),7.10-7.20(m,2H),7.29(m,1H),7.63(d,J=7.58Hz,1H),7.78(s,1H),8.07(d,J=8.56Hz,1H).
LC-MS(保留时间:2.550min),MS m/z 834(MH+)。
实施例330:化合物330的制备
化合物330
按照上述方案2和4的方法制备化合物330。
步骤1(方案2,步骤1):
在-78℃下,向N,N-二乙基-4-甲氧基-2-甲基-苯甲酰胺(332mg,1.5mmol)的THF(15mL)溶液中加入t-BuLi(1.7M戊烷溶液,2.2mL,3.75mmol)。将所得的红色溶液在-78℃下搅拌10min,随后加入(3-二甲氨基)苯甲酸甲酯(269mg,1.5mmol)。将反应混合物在-78℃下搅拌2h。随后将反应物用饱和氯化铵溶液猝灭并用乙酸乙酯萃取两次。将合并的有机层干燥(硫酸镁)并浓缩。粗产物经制备型HPLC纯化,得到淡黄色粘稠油状物,为TFA盐(245mg,收率33%)。1H NMR(400MHz,CD3OD)δ1.01(t,J=6.85Hz,3H),1.09(m,3H),3.11(s,6H),3.21(m,2H),3.40(m,2H),3.79(s,3H),4.39(s,2H),6.84-6.91(m,2H),7.19(d,J=8.32Hz,1H),7.35(m,1H),7.49(t,J=8.07Hz,1H),7.66-7.71(m,2H).
LC-MS(保留时间:1.930min),MS m/z 369(MH+)。
步骤2(方案2,步骤2):
将2-[2-(3-二甲氨基-苯基)-2-氧代-乙基]-N,N-二乙基-4-甲氧基-苯甲酰胺(240mg,0.497mmol)与乙酸铵(2.0g)一起加热2.5小时。随后将其冷却并加入水。收集褐色固体,为纯产物(95mg,收率65%)。
1H NMR(400MHz,CD3OD)δ2.98(s,6H),3.88(s,3H),6.74-6.87(m,2H),7.01-7.07(m,3H),7.18(d,J=2.44Hz,1H),7.28(t,J=7.82Hz,1H),8.10(d,J=8.80Hz,1H).
LC-MS(保留时间:1.773min),MS m/z 295(MH+)。
步骤3(方案4,步骤1):
将3-(3-二甲氨基-苯基)-6-甲氧基-2H-异喹啉-1-酮(92mg,0.312mmol)与磷酰氯(3.0mL)一起加热回流2天。随后蒸馏出磷酰氯并将剩余物用冰骤冷。接着用饱和碳酸氢钠溶液中和,并用乙酸乙酯萃取两次。合并各有机层并干燥(硫酸镁)。蒸发溶剂,得到褐色粘稠油状产物(72mg,收率74%)。
LC-MS(保留时间:2.297min),MS m/z 313(MH+)。
步骤4(方案4,步骤2):
将[3-(1-氯-6-甲氧基-异喹啉-3-基)-苯基]-二甲胺(72mg,0.23mmol)和氟化四丁基_·氢氟酸盐(0.5g)的混合物在Smith微波反应器中在140℃下加热20min。随后加入水并用乙酸乙酯萃取。分离出有机层,用水洗涤并干燥(硫酸镁)。蒸发溶剂,得到褐色油状产物(58mg,收率85%)。
LC-MS(保留时间:2.193min),MS m/z 297(MH+)。
步骤5(方案4,步骤3):
化合物330
在-78℃下,向{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基-氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸叔丁酯(86mg,0.155mmol)和三氯化镧(57mg,0.233mmol)的DMF(1.5mL)溶液中加入叔丁醇钾(1.0M THF溶液,0.5mL,0.5mmol)。将反应混合物搅拌lhr,随后加入[3-(1-氟-6-甲氧基-异喹啉-3-基)-苯基]-二甲胺(55mg,0.185mmol)。升温至室温并搅拌过夜。接着将所得混合物用水猝灭并过滤。浓缩滤液并将剩余物经制备型HPLC纯化,得到灰白色固体产物(化合物330)(8.0mg,收率6%)。
1H NMR(400MHz,CD3OD)δ0.99-1.09(m,11H),1.23(m,2H),1.29(s,9H),1.42(m,1H),1.86(m,1H),2.21(m,1H),2.33(m,1H),2.70(m,1H),2.93(m,1H),3.00(s,6H),3.92(s,3H),4.14(m,1H),4.29(m,1H),4.44-4.57(m,2H),5.10(d,J=11.00Hz,1H),5.27(d,J=16.87Hz,1H),5.74(m,1H),6.01(s,1H),6.63(d,J=8.80Hz,1H),7.03(d,J=6.85Hz,1H),7.24(s,1H),7.28(t,J=8.07Hz,1H),7.45(d,J=7.82Hz,1H),7.59(s,1H),7.72(s,1H),8.05(d,J=8.80Hz,1H).
LC-MS(保留时间:2.707min),MS m/z 834(MH+)。
实施例331:化合物331的制备
化合物331
按照本文描述的方法制备化合物331。
实施例334:化合物334的制备
化合物334
按照以下方法制备化合物334:
步骤1:
0℃下,向Boc-顺-HYP-OMe(122.6mg,0.5mmol)的THF(15mL)溶液中加入三苯基膦(196.7mg,0.75mmol)和苯并[d]异_唑-3-醇(81mg,0.6mmol)。接着加入DEAD(0.118mL,0.75mmol)。将反应混合物升温至室温并搅拌3hr。随后蒸发溶剂并将剩余物经制备型HPLC纯化,得到无色粘稠油状物(117mg,收率54%)。
1H NMR(400MHz,CD3OD)δ1.41(m,9H),2.38(m,1H),2.75(m,1H),3.75(m,3H),3.81(m,1H),3.90(m,1H),4.47(m,1H),5.44(m,1H),7.31(t,J=7.46Hz,1H),7.47(d,J=8.56Hz,1H),7.59(t,J=7.83Hz,1H),7.66(d,J=8.07Hz,1H).
LC-MS(保留时间:2.65min),MS m/z 363(MH+)。
接着将一些偶合产物(85mg,0.235mmol)溶于4NHCl的二_烷(1.5mL)溶液中并搅拌3hr。蒸发溶剂,得到黄色油状物,为盐酸盐(85mg,收率>100%)。
LC-MS(保留时间:1.327min),MS m/z 263(MH+)。
步骤2:
向4-(苯并[d]异_唑-3-氧基)-吡咯烷-2-甲酸甲酯·盐酸盐(85mg,0.285mmol)的乙腈(10mL)溶液中加入N-boc-L-t-亮氨酸(99mg,0.427mmol)、DIEA(0.25mL,1.425mmol)和偶合剂HOBt(65mg,0.427mmol)和HBTU(162mg,0.427mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩。接着经制备型HPLC柱纯化,得到无色粘稠油状产物(63mg,收率46%)。
1H NMR(400MHz,CD3OD)δ1.01(s,9H),1.17(s,9H),2.34(m,1H),2.78(dd,J=14.13,7.83Hz,1H),3.72(s,3H),4.00(dd,J=12.22,3.42Hz,1H),4.19(s,1H),4.57(d,J=12.23Hz,1H),4.68(m,1H),5.51(m,1H),7.27(m,1H),7.47(d,J=8.56Hz,1H),7.57(m,1H),7.63(d,J=8.07Hz,1H).
LC-MS(保留时间:2.737min),MS m/z 498(M+Na+)。
步骤3:
化合物334
向4-(苯并[d]异_唑-3-氧基)-1-(2-叔丁氧基羰基氨基-3,3-二甲基-丁酰基)-吡咯烷-2-甲酸甲酯(63mg,0.132mmol)在THF(3.5mL)、甲醇(2.0mL)和水(0.5mL)的混合物中的溶液内加入一水合氢氧化锂(83mg,1.89mmol)。将反应混合物在室温下搅拌过夜。随后用1N盐酸将其酸化至pH=3至5并浓缩。用乙酸乙酯萃取(2×30mL),合并各有机层并干燥(硫酸镁)。蒸发溶剂,得到淡黄色油状物(61mg,收率100%)。产物直接使用。
向上述化合物(61mg,0.132mmol)的乙腈(8mL)溶液中加入(1R,2S)(1-环丙烷磺酰基-氨基羰基-2-乙烯基-环-丙基)-氨基甲酸盐酸盐(42mg,0.158mmol)、DIEA(0.115mL,0.66mmol)和偶合剂HOBt(30mg,0.198mmol)和HBTU(75mg,0.198mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩。接着经制备型HPLC柱纯化,得到黄色薄膜状终产物(化合物334)(24mg,收率27%)。
1H NMR(400MHz,CD3OD)δ1.01(s,9H),1.05(m,2H),1.12-1.26(m,11H),1.43(m,1H),1.86(dd,J=8.07,5.38Hz,1H),2.17-2.33(m,2H),2.67(dd,J=12.96,5.87Hz,1H),2.93(m,1H),4.05(m,1H),4.22(m,1H),4.49(m,2H),5.11(d,J=10.21Hz,1H),5.29(d,J=17.12Hz,1H),5.55(s,1H),5.74(m,1H),7.29(m,1H),7.48(d,J=8.32Hz,1H),7.54-7.64(m,2H).
LC-MS(保留时间:2.767min),MS m/z 696(M+Na+)。
实施例335:化合物335的制备
方案2:
步骤1:(方案1,步骤1)
向(2S,4R)4-羟基-吡咯烷-1,2-二甲酸1-叔丁酯(0.25g,1.08mmol)的乙腈(10mL)溶液中加入(1R,2S)(1-环丙烷磺酰基-氨基羰基-2-乙烯基-环-丙基)-氨基甲酸盐酸盐(0.346g,1.30mmol)、DIEA(0.94mL,5.41mmol)和偶合剂HOBt(0.248g,1.62mmol)和HBTU(0.615mg,1.62mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩,得到黄色油状物。将其经制备型HPLC柱纯化,得到无色粘稠油状物,随后将其溶于4N HCl的二_烷(5mL)溶液中。将反应混合物在室温下搅拌过夜。蒸发溶剂,得到白色固体产物(200mg,收率49%)。产物直接使用。
LC-MS(保留时间:0.647min),MS m/z 344(MH+)。
步骤2:(方案1,步骤2)
向上述化合物(200mg,0.527mmol)的乙腈(10mL)溶液中加入2-甲氧基羰基氨基-3,3-二甲基-丁酸(0.15g,0.79mmol)、DIEA(0.46mL,2.63mmol)和偶合剂HOBt(0.121g,0.79mmol)和HBTU(0.30g,0.79mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩,得到淡黄色油状物。将其经制备型HPLC柱纯化,得到白色固体终产物(中间体2)(145mg,收率54%)。
1H NMR(CD3OD,500MHz)δ0.99-1.10(m,11H),1.24(m,2H),1.41(dd,J=9.5,5.5Hz,1H),1.87(dd,J=7.9,5.5Hz,1H),1.97(m,1H),2.13(m,1H),2.24(m,1H),2.93(m,1H),3.65(s,3H),3.77-3.88(m,2H),4.33-4.39(m,2H),4.49(m,br,1H),5.13(d,J=10.4Hz,1H),5.31(d,J=17.1Hz,1H),5.76(m,1H).
LC-MS(保留时间:1.590min),MS m/z 515(MH+)。
步骤3:(方案2)
向2,4-二氯嘧啶(149mg,1mmol)的THF(5mL)溶液中加入四(三苯基膦)合钯(23mg,2mol%)和溴化苯基锌的THF溶液(0.5M,2.1mL,1.05mmol)。将反应混合物在50℃下搅拌过夜。随后加入饱和氯化铵溶液并用EtOAc萃取两次。合并各有机层,用水洗涤并干燥(硫酸镁)。蒸发溶剂,得到黄色剩余物,该剩余物经制备型HPLC纯化,得到淡黄色油状2-氯-4-苯基-嘧啶,该产物直接使用。
在0℃下,向中间体2(20mg,0.039mmol)的DMF(3mL)溶液中加入NaH(3.9mg,60%分散于矿物油中,0.0975mmol)。随后将反应混合物升温至室温并搅拌1hr。接着加入上述制备的2-氯-4-苯基-嘧啶(18mg,粗产物)。将反应混合物在室温下搅拌过夜。接着用水猝灭并用EtOAc萃取。分离出有机层,用盐水洗涤并干燥(硫酸镁)。蒸发溶剂,得到淡黄色油状物,接着将该油状物经制备型HPLC纯化,得到粘稠无色油状终产物(化合物335),为TFA盐(5.5mg,收率18%)。
1H NMR(CD3OD,300MHz)δ0.92-1.12(m,11H).1.25(m,2H),1.44(dd,J=9.2,5.5Hz,1H),1.89(dd,J=8.1,5.5Hz,1H),2.17-2.37(m,2H),2.57(m,1H),2.95(m,1H),3.52(s,3H),4.14(m,1H),4.24-4.38(m,2H),4.51(m,1H),5.13(d,J=10.2Hz,1H),5.31(d,J=17.2Hz,1H),5.77(m,1H),5.86(s,1H),7.48-7.60(m,3H),7.66(d,J=5.3Hz,1H),8.18(m,2H),8.60(d,J=5.1Hz,1H).
LC-MS(保留时间:1.947min),MS m/z 669(MH+)。
实施例336:化合物336的制备
步骤1:
向2,4-二氯嘧啶(149mg,1mmol)的THF(5mL)溶液中加入四(三苯基膦)合钯(58mg,5mol%)和溴化2-苯基锌(0.5M,2.4mL,1.2mmol)的THF溶液。将反应混合物在50℃下搅拌过夜。随后加入饱和氯化铵溶液并用EtOAc萃取两次。合并各有机层,用水洗涤并干燥(硫酸镁)。蒸发溶剂,得到黄色剩余物,该剩余物经制备型HPLC纯化,得到淡黄色油状产物(11mg,收率3.6%)。
1H NMR(500MHz,CD3OD)δ7.61(m,1H),8.07(m,1H),8.36(d,J=5.19Hz,1H),8.50(d,J=7.94Hz,1H),8.75(d,J=3.97Hz,1H),8.82(d,J=5.19Hz,1H).
LC-MS(保留时间:1.440min),MS m/z 192(MH+)。
步骤2:
在0℃下,向实施例335的中间体2(15mg,0.029mmol)的DMF(3mL)溶液中加入NaH(1.75mg,60%分散于矿物油,0.0728mmol)。随后将反应混合物升温至室温并搅拌1hr。接着加入2-氯-4-吡啶-2-基-嘧啶(9.5mg,0.0311mmol)。将反应混合物在室温下搅拌过夜。接着用水猝灭并用EtOAc萃取。分离出有机层,用盐水洗涤并干燥(硫酸镁)。蒸发溶剂,得到淡黄色油状物,接着将该油状物经制备型HPLC纯化,得到淡黄色膜状终产物(化合物336),为TFA盐(3.5mg,收率15%)。
1H NMR(CD3OD,500MHz)δ1.03(s,9H),1.08(m,2H),1.24(m,2H),1.43(dd,J=9.77,5.50Hz,1H),1.89(m,1H),2.24(m,1H),2.31(m,1H),2.57(m,1H),2.95(m,1H),3.50(s,3H),4.13(m,1H),4.29(s,1H),4.36(d,J=11.91Hz,1H),4.52(m,1H),5.13(d,J=10.08Hz,1H),5.31(d,J=16.79Hz,1H),5.76(m,1H),5.88(m,1H),7.64(m,1H),8.06-8.13(m,2H),8.54(d,J=7.93Hz,1H),8.73-8.76(m,2H).
LC-MS(保留时间:1.787min),MS m/z 670(MH+)。
实施例337:化合物337的制备
步骤1:
向2,4-二氯嘧啶(149mg,1mmol)的DMF(5mL)溶液中加入二氯·双(三苯基膦)合钯(II)(35mg,5mol%)和2-(三丁基甲锡烷基)噻吩(0.38mL,1.2mmol)。将反应混合物在70℃下加热3h。随后加入饱和KF的甲醇溶液(20mL),在室温下搅拌4hr。将反应混合物与少量硅胶一起浓缩,将剩余物经滤纸过滤并用EtOAc洗涤。随后将滤液浓缩,并将剩余物经制备型HPLC纯化,得到类白色固体产物(110mg,收率35%)。
1H NMR(400MHz,CD3OD)δ7.20(dd,J=5.01,3.79Hz,1H),7.74(dd,J=5.01,1.10Hz,1H),7.77(d,J=5.38Hz,1H),7.98(dd,J=3.79,1.10Hz,1H),8.55(d.J=5.38Hz,1H).
LC-MS(保留时间:1.453min),MS m/z 197(MH+)。
步骤2:
在0℃下,向实施例335的中间体2(20mg,0.039mmol)的DMF(3mL)溶液中加入NaH(7.8mg,60%分散于矿物油中,0.195mmol)。随后将反应混合物升温至室温并搅拌1hr。接着加入2-氯-4-噻吩-2-基-嘧啶(16.9mg,0.0544mmol)。将反应混合物在室温下搅拌过夜。接着用水猝灭并用EtOAc萃取。分离出有机层,用盐水洗涤并干燥(硫酸镁)。蒸发溶剂,得到淡黄色油状物,接着将该油状物经制备型HPLC纯化,得到两种产物(化合物337和中间体3)。
化合物337:(淡黄色膜状物,3.0mg,收率11%)。
1H NMR(CD3OD,500MHz)δ0.98-1.07(m,11H),1.22(m,2H),1.41(d,J=9.54,5.62Hz,1H),1.86(dd,J=8.32,5.63Hz,1H),2.19-2.31(m,2H),2.52(m,1H),2.92(m,1H),3.50(s,3H),4.09(m,1H),4.25-4.32(m,2H),4.47(dd,J=10.03,7.34Hz,1H),5.11(dd,J=10.27,1.71Hz,1H),5.28(dd,J=17.11,1.46Hz,1H),5.69-5.79(m,2H),7.20(dd,J=4.89,3.66Hz,1H),7.51(d,J=5.38Hz,1H),7.70(d,J=4.89Hz,1H),7.95(d,J=3.67Hz,1H),8.54(d,J=5.14Hz,1H).
LC-MS(保留时间:1.787min),MS m/z 696(M+Na+)。
中间体3:(10mg,收率35%)。
LC-MS(保留时间:1.477min),MS m/z 617(MH+)。
实施例338:化合物338的制备
中间体3 化合物338
向1-(2-氨基-3,3-二甲基-丁酰基)-4-(4-噻吩-2-基-嘧啶-2-氧基)-吡咯烷-2-甲酸(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基)-酰胺(10mg,0.0137mmol)的乙腈(5mL)溶液中加入环丙基乙酸(2.1mg,0.0205mmol)、DIEA(0.012mL,0.742mmol)和偶合剂HOBt(3.1g,0.0205mmol)和HBTU(7.8mg,0.0205mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩,得到黄色油状物。将其经制备型HPLC柱纯化,得到淡黄色膜状物,为TFA盐(化合物338)(4.6mg,收率41%)。
1H NMR(CD3OD,400MHz)δ0.12(m,2H),0.48(m,2H),0.90(m,1H),1.01-1.09(m,11H),1.23(m,2H),1.43(dd,J=9.29,5.38Hz,1H),1.87(dd,J=8.31,5.62Hz,1H),2.06(m,2H),2.19-2.31(m,2H),2.52(dd,J=13.45,6.85Hz,1H),2.93(m,1H),4.12(dd,J=11,98,3,91Hz,1H),4.27(d,J=11.74Hz,1H),4.47(dd,J=10.27,6.85Hz,1H),4.63(s,1H),5.11(dd,J=10.27,1.47Hz,1H),5.28(dd,J=17.12,1.47Hz,1H),5.71-5.80(m,2H),7.20(dd,J=4.89,3.67Hz,1H),7.51(d,J=5.38Hz,1H),7.70(d,J=5.20Hz,1H),7.95(d,J=3.67Hz,1H),8.48(d,J=5.13Hz,1H).
LC-MS(保留时间:1.833min),MS m/z 699(MH+)。
实施例339:化合物339的制备
化合物339
按照实施例337和实施例338的方案制备化合物342,不同之处
在于使用2-(三丁基甲锡烷基)呋喃代替在实施例337的步骤1中的2-(三丁基甲锡烷基)噻吩。
步骤1:
向2,4-二氯嘧啶(149mg,1mmol)的DMF(5mL)溶液中加入二氯·双(三苯基膦)合钯(II)(35mg,5mol%)和2-(三丁基甲锡烷基)呋喃(0.35mL,1.1mmol)。将反应混合物在70℃下加热3h。随后加入饱和KF的甲醇溶液(20mL),在室温下搅拌4hr。将反应混合物与少量硅胶一起浓缩,将剩余物经滤纸过滤并用EtOAc洗涤。随后将滤液浓缩,并将剩余物经制备型HPLC纯化,得到褐色固体产物(80mg,收率27%)。
1H NMR(400MHz,CD3OD)δ6.68(dd,J=3.67,1.71Hz,1H),7.42(d,J=3.67Hz,1H),7.67(d,J=5.13Hz,1H),7.30(d,J=1.71Hz,1H),8.62(d,J=5.14Hz,1H).
LC-MS(保留时间:1.233min),MS m/z 181(MH+)。
步骤2:
在0℃下,向{1-[2-(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基-氨基甲酰基)-4-羟基-吡咯烷-1-羰基]-2,2-二甲基-丙基}-氨基甲酸甲酯(20mg,0.039mmol)的DMF(3mL)溶液中加入NaH(7.8mg,60%分散于矿物油中,0.195mmol)。随后将反应混合物升温至室温并搅拌1h。接着加入2-氯-4-噻吩-2-基-嘧啶(16.0mg,0.0544mmol)。将反应混合物在室温下搅拌过夜。接着用水猝灭并用EtOAc萃取。分离出有机层,用盐水洗涤并干燥(硫酸镁)。蒸发溶剂,得到淡黄色油状物,接着将该油状物经制备型HPLC纯化,得到脱boc的偶合产物(3mg,收率11%)。
LC-MS(保留时间:1.420min),MS m/z 601(MH+)。
步骤3:
化合物339
向1-(2-氨基-3,3-二甲基-丁酰基)-4-(4-呋喃-2-基-嘧啶-2-氧基)-吡咯烷-2-甲酸(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基)-酰胺(3mg,0.0042mmol)的乙腈(5mL)溶液中加入环丙基乙酸(0.6mg,0.0063mmol)、DIEA(0.004mL,0.021mmol)和偶合剂HOBt(1.0g,0.0063mmol)和HBTU(24mg,0.0063mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩,得到黄色油状物。将其经制备型HPLC柱纯化,得到淡黄色膜状物,为TFA盐(化合物339)(1.0mg,收率30%)。
1H NMR(CD3OD,400MHz)δ0.12(m,2H),0.48(m,2H),0.90(m,1H),0.99-1.09(m,11H),1.23(m,2H),1.43(dd,J=9.05,5.31Hz,1H),1.87(m,1H),2.05(m,2H),2.19-2.29(m,2H),2.50(m,1H),2.93(m,1H),4.10(dd,J=12.23,3.91Hz,1H),4.25(d,J=11.99Hz,1H),4.47(dd,J=10.52,7.09Hz,1H),4.63(,1H),5.11(dd,J=10.52,1.71Hz,1H),5.29(dd,J=17.12,1.47Hz,1H),5.71-5.79(m,2H),6.65(dd,J=3.67,1.96Hz,1H),7.38(d,J=3.67Hz,1H),7.40(d,J=5.38Hz,1H),7.76(m,1H),8.54(d,J=5.38Hz,1H).
LC-MS(保留时间:1.790min),MS m/z 683(MH+)。
实施例340:化合物340的制备
步骤1:
向2,4-二氯嘧啶(149mg,1mmol)的DMF(5mL)溶液中加入二氯·双(三苯基膦)合钯(II)(35mg,5mol%)和2-(三丁基甲锡烷基)噻唑(412mg,1.1mmol)。将反应混合物在80℃下加热3h。随后加入饱和KF的甲醇溶液(20mL),在室温下搅拌4hr。将反应混合物与少量硅胶一起浓缩,将剩余物经滤纸过滤并用EtOAc洗涤。随后将滤液浓缩,并将剩余物经制备型HPLC纯化,得到褐色固体产物(9mg,3%收率)。
LC-MS(保留时间:1.320min),MS m/z 198(MH+)。
步骤2:
在0℃下,向1-[2-(2-环丙基-乙酰氨基)-3,3-二甲基-丁酰基]-4-羟基-吡咯烷-2-甲酸(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基)-酰胺(12.5mg,0.0232mmol)的DMF(3mL)溶液中加入NaH(3.7mg 60%分散于矿物油中,0.0.0928mmol)。随后将反应混合物升温至室温并搅拌1h。接着加入2-氯-4-噻唑-2-基-嘧啶(9.0mg,0.0289mmol)。将反应混合物在室温下搅拌过夜。接着用水猝灭并用EtOAc萃取。分离出有机层,用盐水洗涤并干燥(硫酸镁)。蒸发溶剂,得到粗产物,该产物随后经制备型HPLC纯化,得到白色固体终产物(化合物340)(2.8mg,收率17%)。
1H NMR(CD3OD,400MHz)δ0.12(m,2H),0.47(m,2H),0.89(m,1H),1.00-1.09(m,11H),1.22(m,2H),1.44(dd,J=9.54,5.38Hz,1H),1.87(dd,J=8.07,5.38Hz,1H),2.06(m,2H),2.20-2.32(m,2H),2.52(dd,J=13.70,6.85Hz,1H),2.93(m,1H),4.13(dd,J=11.98,3.91Hz,1H),4.30(d,J=11.98Hz,1H),4.48(dd,J=10.51,7.09Hz,1H).4.63(d.J=9.54Hz,1H),5.11(d,J=10.51Hz,1H),5.29(d,J=17.12Hz,1H),5.73-5.80(m,2H),7.81(d,J=5.14Hz,1H),7.84(d,J=3.18Hz,1H),8.03(d,J=2.93Hz,1H),8.68(d,J=5.13Hz,1H).
LC-MS(保留时间:1.710min),MS m/z 700(MH+)。
实施例341:化合物341的制备
方案1:
方案2:
步骤1(方案1,步骤1):
在0℃下,向Boc-HYP-OH(1.0g,4.324mmol)的DMF(20mL)溶液中加入NaH(0.38g,60%分散于矿物油中,9.513mmol)。将反应混合物搅拌1h。接着加入2,4-二氯嘧啶(0.709g,0.0289mmol)。反应混合物升温至室温并搅拌过夜。随后用1N HCl溶液猝灭并用EtOAc萃取。分离出有机层,用盐水洗涤并干燥(硫酸镁)。蒸发溶剂,得到粗产物,该产物随后经制备型HPLC纯化,得到无色油状产物(0.4g,收率27%)。
1H NMR(CD3OD,300MHz)δ1.13(m,9H),2.37(m,1H),2.62(m,1H),3.70-3.84(m,2H),4.38(m,1H),5.65(m,1H),6.88(d,J=5.86Hz,1H),8.37(d,J=5.86Hz,1H).
LC-MS(保留时间:1.370min),MS m/z 344(MH+)。
步骤2:(方案1,步骤2)
向(2S,4R)4-(2-氯-嘧啶-4-氧基)-吡咯烷-1,2-二甲酸1-叔丁酯(0.34g,0.99mmol)的乙腈(20mL)溶液中加入(1R,2S)/(1S,2R)(1-环丙烷磺酰基-氨基羰基-2-乙烯基-环丙基)-氨基甲酸(0.511g,1.48mmol)、DIEA(0.86mL,4.95mmol)和偶合剂HOBt(0.226g,1.48mmol)和HBTU(0.561g,1.48mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩。接着经制备型HPLC柱纯化,得到黄色固体物(中间体4)(0.33g,收率41%)。
1H NMR(CD3OD,300MHz)δ非对映异构体混合物。
LC-MS(保留时间:2.907min),MS m/z 655(MH+)。
步骤3:(方案2,步骤1)
向中间体4(50mg,0.061mmol)的二氯甲烷(2.5mL)溶液中加入1,2,3,4-四氢异喹啉(0.011mL,0.0915mmol)和三乙胺(0.021mL,0.153mmol)。反应混合物在室温下搅拌过夜和在40℃下搅拌1天。汽提出溶剂并将剩余物经制备型HPLC纯化,得到无色油状物,随后将其溶于4N HCl的二_烷(1mL)溶液中,搅拌过夜。蒸发溶剂,得到无色油状物,为盐酸盐(20mg,收率52%)。
LC-MS(保留时间:1.160min),MS m/z 553(MH+)。
步骤4:(方案2,步骤2)
向4-[2-(3,4-二氢-1H-异喹啉-2-基)-嘧啶-4-氧基]-吡咯烷-2-甲酸(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基)-酰胺盐酸盐(20mg,0.032mmol)的乙腈(5mL)溶液中加入2-甲氧基羰基氨基-3,3-二甲基-丁酸(9.1mg,0.048mmol)、DIEA(0.028mL,0.16mmol)和偶合剂HOBt(7.3mg,0.048mmol)和HBTU(18.2mg,0.048mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩,得到淡黄色油状物。将其经制备型HPLC柱纯化,得到无色油状物,为TFA盐(化合物341)(16mg,收率60%)。
1H NMR(CD3OD,500MHz)δ0.98-1.06(m,13H),1.13(m,1H),1.22-1.32(m,1H),1.35-1.44(m,1H),1.82(dd,J=8.24,5.19Hz,0.5H),1.90(dd,J=8.24,5.49Hz,0.5H),2.26(m,1H),2.32-2.43(m,1H),2.56(m,1H),2.96(m,1H),3.11(m,br,2H),3.56(s,3H),4.14(m,1H),4.21(m,1H),4.38(m,1H),4.47(m,1H),5.15(m,1H),5.31(m,1H),5.75(m,1H),5.94(s,1H),6.47(d,J=7.02Hz,1H),7.29(s,4H),7.49(m,1H),7.56(m,1H),7.74(d,J=8.24Hz,1H),7.88(d,J=8.24Hz,1H),8.11(d,J=7.02Hz,1H).
LC-MS(保留时间:1.517min),MS m/z 724(MH+)。
实施例342:化合物342的制备
化合物342
按照实施例341、方案2的方法制备化合物342,不同之处在于使用异吲哚啉代替在方案2的步骤1中的1,2,3,4-四氢异喹啉。
步骤1:
向实施例341的中间体4(50mg,0.061mmol)的二氯甲烷(2.5mL)溶液中加入异吲哚啉(0.013mL,0.115mmol)和三乙胺(0.026mL,0.19mmol)。将反应混合物在室温下搅拌2天。汽提出溶剂并将剩余物经制备型HPLC纯化,得到无色油状物。随后将其溶于4N HCl的二_烷(1mL)溶液中并搅拌过夜。蒸发溶剂,得到粗产物,该产物再次经制备型HPLC纯化,得到淡黄色的TFA盐固体(8.5mg,收率14%)。
LC-MS(保留时间:1.860min),MS m/z 539(MH+)。
步骤2:
化合物342
向4-[2-(1,3-二氢-异吲哚-2-基)-嘧啶-4-氧基]-吡咯烷-2-甲酸(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基)-酰胺盐酸盐(8.5mg,0.0104mmol)的乙腈(5mL)溶液中加入2-甲氧羰基氨基-3,3-二甲基-丁酸(3.0mg,0.0156mmol)、DIEA(0.009mL,0.052mmol)和偶合剂HOBt(2.4mg,0.0156mmol)和HBTU(5.9mg,0.0156mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩,得到淡黄色油状物。将其经制备型HPLC柱纯化,得到无色油状物,为TFA盐(化合物342)(3mg,收率35%)。
1H NMR(CD3OD,300MHz)δ非对映异构体混合物。
LC-MS(保留时间:2.547min),MS m/z 710(MH+)。
实施例343:化合物343的制备
化合物343
按照实施例341、方案2的方法制备化合物342,不同之处在于使用吗啉代替在方案2的步骤1中的1,2,3,4-四氢异喹啉。
步骤1:
向实施例341的中间体4(50mg,0.061mmol)的二氯甲烷(2.5mL)溶液中加入吗啉(0.008mL,0.0915mmol)和三乙胺(0.021mL,0.153mmol)。将反应混合物在室温下搅拌过夜和在40℃下搅拌1天。汽提出溶剂并将剩余物经制备型HPLC纯化,得到无色油状物。随后将其溶于4N HCl的二_烷(1mL)溶液中,搅拌过夜。蒸发溶剂,得到无色油状物,为盐酸盐(12.6mg,收率36%)。
LC-MS(保留时间:0.810min),MS m/z 507(MH+)。
步骤2:
化合物343
向4-(2-吗啉-4-基-嘧啶-4-氧基)-吡咯烷-2-甲酸(1-环丙烷磺酰氨基羰基-2-乙烯基-环丙基)-酰胺盐酸盐(126mg,0.0217mmol)的乙腈(5mL)溶液中加入2-甲氧羰基氨基-3,3-二甲基-丁酸(6.2mg,0.0326mmol)、DIEA(0.019mL,0.1085mmol)和偶合剂HOBt(5.0mg,0.0326mmol)和HBTU(12.4mg,0.0326mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩,得到淡黄色油状物。将其经制备型HPLC柱纯化,得到无色油状物,为TFA盐(化合物343)(7mg,收率41%)。
1H NMR(CD3OD,500MHz)δ非对映异构体混合物。
LC-MS(保留时间:1.280min),MS m/z 678(MH+)。
实施例344:化合物344的制备
方案1:
方案2:
步骤1:(方案1)
向4-对甲苯硫基羰基-吡咯烷-1,2-二甲酸1-叔丁酯2-甲酯(3.0g,7.91mmol)在乙醇(15mL)和THF(30mL)的混合物中的溶液内加入硼氢化钠(0.6g,15.8mmol)。将反应混合物在室温下搅拌过夜。随后将其浓缩,用1N HCl溶液洗涤并用EtOAc萃取三次。合并各有机层,用饱和碳酸氢钠溶液洗涤并干燥(硫酸镁)。蒸发溶剂,得到淡黄色油状物,该产物经快速柱层析纯化(硅胶,3∶1 EtOAc∶己烷),得到无色油状产物(中间体5)(1.77g,收率86%)。
1H NMR(CD3OD,500MHz)δ1.43(m,9H),2.00-2.13(m,2H),2.46(m,1H),3.19(m,1H),3.47-3.53(m,2H),3.61(m,1H),3.73(m,3H),4.31(m,1H).
LC-MS(保留时间:1.240min),MS m/z 282(M+Na+)。
步骤2:(方案2,步骤1)
0℃下,向中间体5(80mg,0.309mmol)的THF(10mL)溶液中加入三苯基膦(121.4mg,0.463mmol)和4-羟基喹啉(67.2mg,0.463mmol)。接着加入DEAD(80.6mg,0.463mmol)。将反应混合物升温至室温并搅拌2天。随后蒸发溶剂并将剩余物经制备型HPLC纯化,得到无色油状物,随后将其溶于4N HCl的二_烷(3mL)溶液并搅拌2h。蒸发溶剂,得到粘稠无色油状物,为双盐酸盐(110mg,收率99%)。
1H NMR(500MHz, CD3OD)δ2.52(m,1H).2.60(m,1H),3.19(m,1H),3.45(m,1H),3.66(s,3H),3.86(m,1H),4.61-4.75(m,3H),7.56(d,J=6.7Hz,1H),7.94(t,J=7.3Hz,1H),8.10-8.20(m,2H),8.55(d,J=8.2Hz,1H),9.07(d,J=6.7Hz,1H).
LC-MS(保留时间:0.570min),MS m/z 287(MH+)。
步骤3:(方案2,步骤2)
向4-(喹啉-4-氧基甲基)-吡咯烷-2-甲酸甲酯双盐酸盐(110mg,0.306mmol)的乙腈(10mL)溶液中加入2-甲氧羰基氨基-3,3-二甲基-丁酸(87mg,0.46mmol)、DIEA(0.27mL,1.53mmol)和偶合剂HOBt(70mg,0.46mmol)和HBTU(174mg,0.46mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩,得到淡黄色油状物。将其经制备型HPLC柱纯化,得到无色油状物,为TFA盐(105mg,收率60%)。
1H NMR(CD3OD,500MHz)δ1.07(s,9H).2.34(m,1H),2.45(m,1H),3.14(m,1H),3.27(s,3H),3.75(s,3H),4.05(m,1H),4.20(m,1H),4.31(s,1H),4.57-4.63(m,2H),4.73(m,1H),7.53(d,J=6.7Hz,1H),7.91(t,J=7.6Hz,1H),8.06-8.16(m,2H),8.43(d,J=8.6Hz,1H),9.02(d,J=6.4Hz,1H).
LC-MS(保留时间:1.250min),MS m/z 458(MH+)。
步骤4:(方案2,步骤3)
向1-(2-甲氧羰基氨基-3,3-二甲基-丁酰基)-4-(喹啉-4-氧基甲基)-吡咯烷-2-甲酸甲酯(100mg,0.175mmol)在THF(6mL)、甲醇(3.25mL)和水(1.0mL)的混合物中的溶液内中加入一水合氢氧化锂(110mg,2.62mmol)。将反应混合物在室温下搅拌过夜。随后用1N盐酸将其酸化至pH=3-5并浓缩。用乙酸乙酯萃取(3×40mL),合并各有机层并干燥(硫酸镁)。蒸发溶剂,得到粘稠无色油状物(25mg,32%收率)。产物直接使用。
向上述化合物(25mg,0.056mmol)的乙腈(5mL)溶液中加入(1R,2S)(1-环丙烷磺酰基-氨基羰基-2-乙烯基-环-丙基)-氨基甲酸盐酸盐(22.5mg,0.085mmol)、DIEA(0.05mL,0.28mmol)和偶合剂HOBt(12.9mg,0.085mmol)和HBTU(32mg,0.085mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。将合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩,得到黄色油状物。将其经制备型HPLC柱纯化,得到无色粘稠油状物,为TFA盐(化合物344)(20mg,收率46%)。
1H NMR(CD3OD,500MHz)δ1.02-1.10(m,11H).1.24(m,2H),1.40(dd,J=9.2,5.5Hz,1H),1.90(dd,J=7.9,5.5Hz,1H),2.19-2.38(m,3H),2.95(m,1H),3.19(m,1H),3.28(s,3H),4.10(m,1H),4.15(m,1H),4.34(s,1H),4.55(m,1H),4.62(d,J=4.6Hz,2H),5.15(d,J=10.7Hz,1H),5.30(d,J=17.1Hz,1H),5.72(m,1H),7.54(d,J=6.7Hz,1H),7.93(m,1H),8.07-8.18(m,2H),8.41(d,J=8.6Hz,1H),9.03(d,J=6.7Hz,1H),9.09(s,1H).
LC-MS(保留时间:1.617min),MS m/z 656(MH+)。
实施例345:化合物345的制备
化合物345
按照实施例344、方案2的方法制备化合物345,不同之处在于使用3-溴苯酚代替在方案2的步骤1中的4-羟基喹啉。
步骤1:
0℃下,向实施例344的中间体5(150mg,0.578mmol)的THF(15mL)溶液中加入三苯基膦(228mg,0.868mmol)和3-溴苯酚(150mg,0.868mmol)。随后加入DEAD(0.14mL,0.868mmol)。将反应混合物升温至室温并搅拌2天。随后蒸发溶剂并将剩余物经制备型HPLC纯化,得到无色油状产物(105mg,收率44%)。
LC-MS(保留时间:2.023min),MS m/z 436(M+Na+)。
步骤2:
将4-(3-溴-苯氧基甲基)-吡咯烷-1,2-二甲酸1-叔丁酯2-甲酯(35mg,0.085mmol)溶于4N HCl的二_烷(1.5mL)溶液中并搅拌2h。蒸发溶剂,得到粘稠无色油状物。向该油状物在乙腈(10mL)中的溶液内加入2-甲氧羰基氨基-3,3-二甲基-丁酸(21.9mg,0.1155mmol)、DIEA(0.067mL,0.385mmol)和偶合剂HOBt(17.7mg,0.1155mmol)和HBTU(43.8mg,0.1155mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩。接着经制备型HPLC柱纯化,得到无色油状产物(20mg,收率49%)。
1H NMR(CD3OD,400MHz)δ1.03(s,9H),2.15(m,1H),2.24(m,1H),2.83(m,1H),3.54(s,3H),3.70(s,3H),3.87(m,1H),3.91-3.98(m,3H),4.31(s,1H),4.59(dd,J=8.80,5.38Hz,1H),6.89(d,J=8.32Hz,1H),7.03-7.10(m,2H),7.15(t,J=8.07Hz,1H).
LC-MS(保留时间:1.943min),MS m/z 485(MH+)。
步骤3:
化合物345
向4-(3-溴-苯氧基甲基)-1-(2-甲氧羰基氨基-3,3-二甲基-丁酰基)-吡咯烷-2-甲酸甲酯(17mg,0.035mmol)在THF(1.5mL)、甲醇(0.8mL)和水(0.25mL)的混合物中的溶液内加入一水合氢氧化锂(22mg,0.525mmol)。将反应混合物在室温下搅拌3天。随后用1N盐酸将其酸化至pH=3-5并浓缩。用乙酸乙酯萃取(2×20mL),合并各有机层并干燥(硫酸镁)。蒸发溶剂,得到粘稠无色油状物(15mg,91%收率)。产物直接使用。
向上述酸(15mg,0.0318mmol)的乙腈(5mL)溶液中加入(1R,2S)(1-环丙烷磺酰基-氨基羰基-2-乙烯基-环-丙基)-氨基甲酸盐酸盐(12.7mg,0.0477mmol)、DIEA(0.028mL,0.159mmol)和偶合剂HOBt(7.3mg,0.0477mmol)和HBTU(18.1mg,0.0477mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩。接着经制备型HPLC柱纯化,得到无色粘稠油状终产物(化合物345)(14mg,收率64%)。
1H NMR(CD3OD,400MHz)δ1.00-1.06(m,11H),1.21(m,2H),1.37(dd,J=9.53Hz,5.62Hz,1H),1.86(dd,J=8.07Hz,5.62Hz,1H),2.06(m,1H),2.14-2.24(m,2H),2.81-2.94(m,2H),3.53(s,3H),3.91-3.97(m,4H),4.33(s,1H),4.38(m,1H),5.11(dd,J=10.27,1.47Hz,1H),5.28(dd,J=17.12,1.22Hz,1H),5.70(m,1H),6.89(m,1H),7.05-7.11(m,2H),7.16(t,J=8.07Hz,1H).
LC-MS(保留时间:3.500min),MSm/z683(MH+)。
实施例346:化合物346的制备
方案1:
方案2:
步骤1:(方案1,步骤1)
0℃下,向4-羟基甲基-吡咯烷-1,2-二甲酸1-叔丁酯2-甲酯(300mg,1.157mmol)的THF(15mL)溶液中加入三苯基膦(455mg,1.735mmol)和5-溴-吡啶-3-醇(按照F.E.ziegler等,J.Am.Chem.Soc.,(1973),95,7458的描述制备)(302mg,1.735mmol)。接着加入DEAD(0.273mL,1.735mmol)。将反应混合物升温至室温并搅拌2天。随后蒸发溶剂并将剩余物经制备型HPLC纯化,得到淡黄色油状物。接着将其溶于4N HCl的二_烷(3.0mL)溶液中并搅拌4hr。蒸发溶剂,得到粗产物,该产物进一步经制备型HPLC纯化,得到淡黄色油状物,为TFA盐(70mg,收率11%)。
LC-MS(保留时间:0.890min),MS m/z 315(MH+)。
步骤2:(方案1,步骤2)
向4-(5-溴-吡啶-3-氧基甲基)-吡咯烷-2-甲酸甲酯(70mg,0.129mmol)的乙腈(10mL)溶液中加入2-甲氧羰基氨基-3,3-二甲基-丁酸(36.5mg,0.193mmol)、DIEA(0.135mL,0.744mmol)和偶合剂HOBt(30mg,0.193mmol)和HBTU(73mg,0.193mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩。接着经制备型HPLC柱纯化,得到无色油状产物(80mg,收率100%)。
1H NMR(CD3OD,400MHz)δ1.04(s,9H),2.17(m,1H),2.26(m,1H),2.87(m,1H),3.50(s,3H),3.70(s,3H),3.88-3.98(m,2H),4.04-4.12(m,2H),4.28(s,1H),4.60(dd,J=9.05,5.87Hz,1H),7.86(m,1H)8.31-8.35(m,2H).
LC-MS(保留时间:1.697min),MS m/z 486(MH+)。
步骤3:(方案1,步骤3)
向4-(5-溴-吡啶-3-氧基甲基)-1-(2-甲氧羰基氨基-3,3-二甲基-丁酰基)-吡咯烷-2-甲酸甲酯(80mg,0.133mmol)在THF(5.6mL)、甲醇(3mL)和水(1mL)混合物中的溶液内加入一水合氢氧化锂(84mg,2.0mmol)。将反应混合物在室温下搅拌3天。随后用1N盐酸将其酸化至pH=3-5。用乙酸乙酯萃取(2×20mL),合并各有机层并干燥(硫酸镁)。蒸发溶剂,得到粘稠无色油状产物(中间体6)(50mg,收率80%)。
1H NMR(CD3OD,400MHz)δ1.04(s,9H),2.16-2.30(m,2H),2.88(m,1H),3.51(s,3H),3.92(m,2H),4.07(m,2H),4.29(s,1H),4.57(dd,J=8.56,5.87Hz,1H),7.79(m,1H)8.29(m,2H).
LC-MS(保留时间:1.590min),MS m/z 472(MH+)。
步骤4:(方案2)
向4-(5-溴-吡啶-3-氧基甲基)-1-(2-甲氧羰基氨基-3,3-二甲基-丁酰基)-吡咯烷-2-甲酸(5mg,0.0106mmol)的乙腈(5mL)溶液中加入(1R,2S)(1-环丙烷磺酰基-氨基羰基-2-乙烯基-环-丙基)-氨基甲酸盐酸盐(4.2mg,0.0159mmol)、DIEA(0.009mL,0.053mmol)和偶合剂HOBt(2.4mg,0.0159mmol)和HBTU(6.0mg,0.0159mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩。接着经制备型HPLC柱纯化,得到无色粘稠油状终产物(化合物346)(2mg,收率24%)。
1H NMR(CD3OD,400MHz)δ1.00-1.07(m,11H),1.20(m,2H),1.37(dd,J=9.29Hz,5.14Hz,1H),1.86(dd,J=8.07Hz,5.38Hz,1H),2.08(m,1H),2.15-2.25(m,2H),2.87-2.94(m,2H),3.51(s,3H),3.92-3.97(m,2H),4.02-4.07(m,2H),4.31(s,1H),4.39(m,1H),5.10(dd,J=10.27,1.47Hz,1H),5.28(dd,J=17.12,1.46Hz,1H),5.70(m,1H),7.68(m,1H).8.24(m,2H).
LC-MS(保留时间:1.727min),MS m/z 684(MH+)。
实施例347:化合物347的制备
步骤1:
向实施例346的中间体6(16mg,0.0339mmol)的DMF(1mL)溶液中加入3-噻吩硼酸(5.6mg,0.044mmol)、四(三苯基膦)合钯(2.0mg,0.0017mmol)和2M碳酸钠溶液(0.051mL,0.1017mmol)。将反应混合物在110℃下加热4hr。随后过滤并用甲醇洗涤。将滤液浓缩并经制备型HPLC纯化,得到褐色油状产物(6mg,收率37%)。
1H NMR(CD3OD,400MHz)δ1.05(s,9H),2.21-2.30(m,2H),2.95(m,1H),3.42(s,3H),3.93(m,1H),4.01(m,1H),4.20-4.30(m,3H),4.60(dd,J=8.56,5.87 Hz,1H),7.64(m,2H),8.12(m,1H)8.37(m,1H),8.45(m,1H),8.75(s,1H).
LC-MS(保留时间:1.353min),MS m/z 476(MH+)。
步骤2:
向1-(2-甲氧羰基氨基-3,3-二甲基-丁酰基)-4-(5-噻吩-3-基-吡啶-3-氧基甲基)-吡咯烷-2-甲酸(6mg,0.0126mmol)的乙腈(5mL)溶液中加入(1R,2S)(1-环丙烷磺酰基-氨基羰基-2-乙烯基-环-丙基)-氨基甲酸盐酸盐(5.0mg,0.0189mmol)、DIEA(0.011mL,0.063mmol)和偶合剂HOBt(2.9mg,0.0189mmol)和HBTU(7.2mg,0.0189mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩。接着经制备型HPLC柱纯化,得到淡黄色膜状物,为TFA盐(化合物347)(2.2mg,收率22%)。
1H NMR(CD3OD,400MHz)δ1.01-1.07(m,11H),1.19(m,2H),1.36(m,1H),1.88(dd,J=8.07Hz,5.62Hz,1H),2.09-2.24(m,3H),2.91(m,1H),3.00(m,1H),3.45(s,3H),3.98(d,J=5.86Hz,2H),4.20-4.31(m,3H),4.43(m,1H),5.12(dd,J=10.27,1.71Hz,1H),5.29(dd,J=17.12,1.22Hz,1H),5.69(m,1H),7.64(m,2H),8.11(m,1H),8.31(m,1H),8.43(m,1H),8.75(s,1H).
LC-MS(保留时间:1.540min),MS m/z 688(MH+)。
实施例348:化合物348的制备
方案2:
方案3:
步骤1:(方案1)
向实施例344的中间体5(700mg,2.7mmol)在THF(90mL)、甲醇(50mL)和水(12mL)混合物中的溶液内加入一水合氢氧化锂(1700mg,2.0mmol)。将反应混合物在室温下搅拌过夜。随后用1N盐酸将其酸化至pH=3-5。用乙酸乙酯萃取(2×20mL),合并各有机层并干燥(硫酸镁)。蒸发溶剂,得到粘稠无色油状产物(中间体7)(0.58,收率88%)。
1H NMR(CD3OD,400MHz)δ1.42(m,9H),2.00-2.09(m,2H),2.45(m,1H),3.17(m,1H),3.49(m,2H),3.59(m,1H),4.24(m,1H).
LC-MS(保留时间:1.08min),MS m/z 268(M+Na+)。
步骤2:(方案2,步骤1)
向中间体7(270mg,1.1mmol)的DMSO(10mL)溶液中加入叔丁醇钾(309mg,2.75mmol)。将反应混合物在室温下搅拌1h。接着加入2-溴-4-氯-吡啶(254mg,1.32mmol)。将反应混合物在室温下搅拌过夜。接着将所得混合物用水猝灭并用乙酸乙酯洗涤。分离出水层并用1N盐酸溶液酸化至pH=3。用乙酸乙酯萃取两次,合并各有机层并干燥(硫酸镁)。蒸发溶剂,得到橙色油状物。接着在-78℃下将其溶于甲醇中并通入HCl(气体)鼓泡2分钟,随后将反应混合物升温至室温并搅拌过夜。蒸发溶剂,得到橙色油状物,为粗产物,该产物直接使用。
LC-MS(保留时间:0.65min),MS m/z 315(MH+)。
步骤3:(方案2,步骤2)
向4-(2-溴-吡啶-4-氧基甲基)-吡咯烷-2-甲酸甲酯粗品的乙腈(20mL)溶液中加入2-甲氧羰基氨基-3,3-二甲基-丁酸(312mg,1.65mmol)、DIEA(1.15mL,6.6mmol)和偶合剂HOBt(252mg,1.65mmol)和HBTU(626mg,1.65mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩。接着经制备型HPLC柱纯化,得到无色油状产物(270mg,两步总收率41%)。
1H NMR(CD3OD,400MHz)δ1.03(s,9H),2.13-2.19(m,2H),2.87(m,1H),3.51(s,3H),3.70(s,3H),3.93(d,J=6.36Hz,2H),4.11(m,2H),4.27(s,1H),4.60(dd,J=8.80,5.87Hz,1H),7.06(d,J=5.87,2.20Hz,1H)7.32(d,J=2.20Hz,1H),8.18(d,J=6.11Hz,1H).
LC-MS(保留时间:1.657min),MS m/z 486(MH+)。
步骤4:(方案2,步骤3)
向4-(2-溴-吡啶-4-氧基甲基)-1-(2-甲氧羰基氨基-3,3-二甲基-丁酰基)-吡咯烷-2-甲酸甲酯(270mg,0.45mmol)在THF(18mL)、甲醇(10mL)和水(3.3mL)混合物中的溶液内加入一水合氢氧化锂(283mg,6.75mmol)。将反应混合物在室温下搅拌过夜。接着将其浓缩并用1H HCl溶液酸化至pH=3-5。收集类白色固体产物(中间体8)(180mg,收率85%)。
1H NMR(CD3OD,500MHz)δ1.06(s,9H),2.20-2.29(m,2H),2.89(m,1H),3.54(s,3H),3.92(d,J=6.4Hz,2H),4.06-4.13(m,2H),4.31(d,J=8.85Hz,1H),4.59(dd,J=8.85,5.50Hz,1H),7.00(dd,J=6.10,2.24Hz,1H),7.22(d,J=1.83Hz,1H),8.12(d,J=5.80Hz,1H).
LC-MS(保留时间:2.113min),MS m/z 472(MH+)。
步骤5:(方案3)
向中间体8(10mg,0.0212mmol)的乙腈(5mL)溶液中加入(1R,2S)(1-环丙烷磺酰基-氨基羰基-2-乙烯基-环-丙基)-氨基甲酸盐酸盐(8.5mg,0.00318mmol)、DIEA(0.018mL,0.106mmol)和偶合剂HOBt(4.9mg,0.0318mmol)和HBTU(12.1mg,0.0318mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩。接着经制备型HPLC柱纯化,得到无色粘稠油状终产物(化合物348)(9mg,收率53%)。
1H NMR(CD3OD,400MHz)δ1.00-1.06(m,11H),1.20(m,2H),1.36(dd,J=9.54Hz,5.38Hz,1H),1.87(dd,J=8.07Hz,5.38Hz,1H),2.04-2.24(m,3H),2.88-2.94(m,2H),3.52(s,3H),3.93(d,J=5.87Hz,2H),4.09(m,2H),4.30(s,1H),4.38(t,J=7.58Hz,1H),5.11(dd,J=10.27,1.47Hz,1H),5.28(dd,J=17.12,1.47Hz,1H),5.70(m,1H),7.00(dd,J=5.87,2.20Hz,1H),7.24(d,J=2.20Hz,1H),8.14(d,J=5.87Hz,1H).
LC-MS(保留时间:1.670min),MS m/z 684(MH+)。
实施例349:化合物349的制备
化合物349
按照实施例347的方案制备化合物349,不同之处在于在步骤1中使用实施例348的中间体8代替实施例346的中间体6。
步骤1:
向中间体8(20mg,0.0423mmol)的DMF(1mL)溶液中加入3-噻吩硼酸(7.0mg,0.055mmol)、四(三苯基膦)合钯(2.4mg,0.00212mmol)和2M碳酸钠溶液(0.063mL,0.127mmol)。将反应混合物在110℃下加热30h。随后过滤并用甲醇洗涤。将滤液浓缩并经制备型HPLC纯化,得到褐色油状产物(10.5mg,收率42%)(50177-165)。
LC-MS(保留时间:1.690min),MS m/z 476(MH+)。
步骤2:
化合物349
向1-(2-甲氧羰基氨基-3,3-二甲基-丁酰基)-4-(2-噻吩-3-基-吡啶-4-氧基甲基)-吡咯烷-2-甲酸(10mg,0.017mmol)的乙腈(5mL)溶液中加入(1R,2S)(1-环丙烷磺酰基-氨基羰基-2-乙烯基-环-丙基)-氨基甲酸盐酸盐(6.8mg,0.0254mmol)、DIEA(0.015mL,0.085mmol)和偶合剂HOBt(3.9mg,0.0254mmol)和HBTU(9.6mg,0.0254mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩。接着经制备型HPLC柱纯化,得到褐色膜状物,为TFA盐(化合物349)(2.2mg,收率16%)(50177-172)。
1H NMR(CD3OD,400MHz)δ1.00-1.07(m,11H),1.20(m,2H),1.37(dd J=9.04,5.38Hz,1H),1.88(dd,J=8.07Hz,5.38Hz,1H),2.10-2.25(m,3H),2.90(m,1H),3.04(m,1H),3.47(s,3H),3.93-4.02(m,2H),4.29(s,1H),4.354.45(m,3H),5.12(d,J=10.51Hz,1H),5.28(d,J=17.61Hz,1H),5.69(m,1H),7.40(dd,J=6.84,2.44Hz,1H),7.71-7.80(m,3H),8.38(m,1H),8.51(d,J=7.09Hz,1H).
LC-MS(保留时间:1.443min),MS m/z 688(MH+)。
实施例350:化合物350的制备
步骤1:
向实施例348的中间体8(20mg,0.0423mmol)的DMF(2mL)溶液中加入2-噻吩硼酸(7.0mg,0.055mmol)、四(三苯基膦)合钯(2.4mg,0.00212mmol)和氢氧化钡(40mg,0.127mmol)。将反应混合物在Smith微波反应器中在150℃下加热110min。接着过滤并用甲醇洗涤。将滤液浓缩并经制备型HPLC纯化,得到淡黄色油状产物(5.0mg,收率20%)。
LC-MS(保留时间:2.137min),MS m/z 476(MH+)。
步骤2:
向上述羧酸(5.0mg,0.0085mmol)的乙腈(5mL)溶液中加入(1R,2S)(1-环丙烷磺酰基-氨基羰基-2-乙烯基-环-丙基)-氨基甲酸盐酸盐(3.4mg,0.0127mmol)、DIEA(0.007mL,0.0424mmol)和偶合剂HOBt(1.9mg,0.0127mmol)和HBTU(4.8mg,0.0127mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩。接着经制备型HPLC柱纯化,得到淡黄色油状物,为TFA盐(化合物350)(2.6mg,收率38%)。
1H NMR(CD3OD,400MHz)δ0.99-1.07(m,11H),1.19(m,2H),1.37(dd,J=9.54,5.63Hz,1H),1.87(dd,J=8.07Hz,5.38Hz,1H),2.10-2.25(m,3H),2.91(m,1H),3.03(m,1H),3.48(s,3H),3.92-4.02(m,2H),4.30(s,1H),4.32-4.45(m,3 H),5.11(dd,J=10.27,1.22Hz,1H),5.28(d,J=17.11Hz,1H),5.69(m,1H),7.30-7.38(m,2H),7.66(d,J=2.45Hz,1H),7.92(m,1H),7.95(m,1H),8.48(d,J=6.85Hz,1H).
LC-MS(保留时间:2.067min),MS m/z 688(MH+)。
实施例351:化合物351的制备
化合物351
按照实施例350的方案制备化合物351,不同之处在于在步骤1中使用3-呋喃硼酸代替2-噻吩硼酸。
步骤1:
向实施例348的中间体8(20mg,0.0423mmol)的DMF(2mL)溶液中加入3-呋喃硼酸(6.2mg,0.055mmol)、四(三苯基膦)合钯(2.4mg,0.00212mmol)和氢氧化钡(40mg,0.127mmol)。将反应混合物在Smith微波反应器中在150℃下加热30min。接着过滤并用甲醇洗涤。将滤液浓缩并经制备型HPLC纯化,得到淡黄色油状产物(12mg,收率49%)。
LC-MS(保留时间:1.937min),MS m/z 460(MH+)。
步骤2:
化合物351
向上述羧酸(5.0mg,0.0209mmol)的乙腈(5mL)溶液中加入(1R,2S)(1-环丙烷磺酰基-氨基羰基-2-乙烯基-环-丙基)-氨基甲酸盐酸盐(8.4mg,0.0314mmol)、DHEA(0.018mL,0.1046mmol)和偶合剂HOBt(4.8mg,0.0314mmol)和HBTU(11.9mg,0.0314mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩。接着经制备型HPLC柱纯化,得到淡黄色油状物,为TFA盐(化合物351)(4.0mg,收率24%)。
1H NMR(CD3OD,400MHz)δ1.00-1.08(m,11H),1.21(m,2H),1.37(dd,J=8.80,5.62Hz,1H),1.87(dd,J=8.32Hz,5.38Hz,1H),2.11-2.24(m,3H),2.91(m,1H),3.03(m,1H),3.49(s,3H),3.91-4.03(m,2H),4.29(s,1H),4.35-4.46 (m,3H,5.12(dd,J=10.27,1.47Hz,1H),5.28(d,J=17.12Hz,1H),5.69(m,1H),7.11(m,1H),7.38(dd,J=7.10,2.69Hz,1H)7.71(d,J=2.69Hz,1H),7.81(m,1H),8.48(s,1H),8.50(d,J=7.09Hz,1H).
LC-MS(保留时间:1.410min),MS m/z 672(MH+)。
实施例352:化合物352的制备
化合物352
按照实施例348方案2和方案3的方法制备化合物352,不同之处在于使用2,6-二溴吡啶代替在方案2的步骤1中的2-溴-4-氯-吡啶。
步骤1:(方案2,步骤1)
向实施例348的中间体7(270mg,1.1mmol)的DMSO(10mL)溶液中加入叔丁醇钾(309mg,2.75mmol)。将反应混合物在室温下搅拌1h。接着加入2,6-二溴吡啶(313mg,1.32mmol)。将反应混合物在室温下搅拌过夜。接着将所得混合物用水猝灭并用乙酸乙酯洗涤。分离出水层并用1N盐酸溶液酸化至pH=3。用乙酸乙酯萃取两次,合并各有机层并干燥(硫酸镁)。蒸发溶剂,得到橙色油状物。接着在-78℃下将其溶于甲醇中并通入HCl(气体)鼓泡2分钟,随后将反应混合物升温至室温并搅拌过夜。蒸发溶剂,得到橙色油状物,为粗产物,该产物直接使用。
LC-MS(保留时间:1.480min),MS m/z 315(MH+)。
步骤2:(方案2,步骤2)
向粗4-(6-溴-吡啶-2-氧基甲基)-吡咯烷-2-甲酸甲酯的乙腈(20mL)溶液中加入2-甲氧羰基氨基-3,3-二甲基-丁酸(312mg,1.65mmol)、DIEA(1.15mL,6.6mmol)和偶合剂HOBt(252mg,1.65mmol)和HBTU(626mg,1.65mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩。接着经快速柱层析纯化(硅胶,1∶1己烷∶乙酸乙酯),得到无色油状产物(340mg,两步总收率63%)。
1H NMR(CD3OD,400MHz)δ1.03(s,9H),2.10-2.24(m,2H),2.84(m,1H),3.55(s,3H),3.70(s,3H),3.83(m,1H),3.94(m,1H),4.20-4.29(m,2H),4.31(s,1H),4.59(dd,J=8.80,5.13Hz,1H),6.76(d,J=8.07Hz,1H)7.11(d,J=7.58Hz,1H),7.53(t,J=7.83Hz,1H).
LC-MS(保留时间:1.820min),MS m/z 486(MH+)。
步骤3:(方案2,步骤3)
中间体9
向4-(6-溴-吡啶-2-氧基甲基)-1-(2-甲氧羰基氨基-3,3-二甲基-丁酰基)-吡咯烷-2-甲酸甲酯(330mg,0.679mmol)在THF(28mL)、甲醇(15mL)和水(5mL)混合物中的溶液内加入一水合氢氧化锂(427mg,10.18mmol)。将反应混合物在室温下搅拌2天。接着将其浓缩并用1N HCl溶液酸化至pH=3-5。收集白色固体产物(中间体9)(310mg,收率97%)。
1H NMR(CD3OD,500MHz)δ1.06(s,9H),2.18-2.25(m,2H),2.88(m,1H),3.57(s,3H),3.84(m,1H),3.96(m,1H),4.25(m,1H),4.28-4.35(m,2H),4.58(m,1H),6.79(d,J=7.94Hz,1H),7.13(d,J=7.32Hz,1H),7.55(m,1H).
LC-MS(保留时间:3.030min),MS m/z 472(MH+)。
步骤4:(方案3)
化合物352
向中间体9(10mg,0.0212mmol)的乙腈(5mL)溶液中加入(1R,2S)(1-环丙烷磺酰基-氨基羰基-2-乙烯基-环-丙基)-氨基甲酸盐酸盐(8.5mg,0.00318mmol)、DlEA(0.018mL,0.106mmol)和偶合剂HOBt(4.9mg,0.0318mmol)和HBTU(12.1mg,0.0318mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩。接着经制备型HPLC柱纯化,得到淡黄色膜状物,为TFA盐(化合物352)(10.2mg,收率60%)。
1H NMR(CD3OD,400MHz)δ1.00-1.06(m,11H),1.20(m,2H),1.37(dd,J=9.54Hz,5.63Hz,1H),1.86(dd,J=8.07Hz,5.38Hz,1H),2.05(m.1H),2.12-2.25(m,2H),2.86-2.94(m,2H),3.54(s,3H),3.87(m,1H),3.94(m,1H),4.18-4.27(m,2H),4.33(s,1H),4.37(m,1H),5.11(dd,J=10.27,1.72Hz,1H),5.28(dd,J=17.12,1.47Hz,1H),5.70(m,1H),6.76(d,J=8.32Hz,1H),7.11(d,J=7.33Hz,1H),7.53(t,J=7.82Hz,1H).
LC-MS(保留时间:1.837min),MS m/z 684(MH+)。
实施例353:化合物353的制备
化合物353
按照实施例347的方案制备化合物353,不同之处在于在步骤1中使用实施例352的中间体9代替实施例346的中间体6。
步骤1:
向中间体9(25mg,0.053mmol)的DMF(1mL)溶液中加入3-噻吩硼酸(8.8mg,0.0688mmol)、四(三苯基膦)合钯(3.1mg,0.00265mmol)和2M碳酸钠溶液(0.080mL,0.159mmol)。将反应混合物在110℃下加热过夜。随后过滤并用甲醇洗涤。将滤液浓缩并经制备型HPLC纯化,得到褐色油状产物(15mg,收率48%)。
1H NMR(CD3OD,500MHz)δ1.06(s,9H),2.20-2.31(m,2H),2.94(m,1H),3.55(s,3H),3.91(m,1H),3.98(m,1H),4.34(s,1H),4.37-4.46(m,2H),4.61(dd,J=8.85,5.19Hz,1H),6.77(d,J=8.24Hz,1H),7.39(d,J=7.32Hz,1H),7.48(dd,J=5.19,3.05Hz,1H),7.68(dd,J=4.88,1.22Hz,1H),7.77(t,J=7.93 Hz,1H),8.04(m,1H).
LC-MS(保留时间:1.857min),MS m/z 476(MH+)。
步骤2:
化合物353
向1-(2-甲氧羰基氨基-3,3-二甲基-丁酰基)-4-(6-噻吩-3-基-吡啶-2-氧基甲基)-吡咯烷-2-甲酸(15mg,0.0254mmol)的乙腈(5mL)溶液中加入(1R,2S)(1-环丙烷磺酰基-氨基羰基-2-乙烯基-环-丙基)-氨基甲酸盐酸盐(10.2mg,0.0382mmol)、DIEA(0.022mL,0.127mmol)和偶合剂HOBt(5.8mg,0.0382mmol)和HBTU(14.5mg,0.0382mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩。接着经制备型HPLC柱纯化,得到淡黄色膜状物,为TFA盐(化合物353)(6mg,收率29%)。
1H NMR(CD3OD,400MHz)δ1.00-1.06(m,11H),1.20(m,2H),1.36(dd,J=9.29,5.38Hz,1H),1.86(dd,J=8.07Hz,5.38Hz,1H),2.07(m,1H),2.16-2.25(m,2H),2.87-2.99(m,2H),3.54(s,3H),3.87-3.99(m,2H),4.31-4.44(m,4H),5.11(dd,J=10.27,1.46Hz,1H),5.28(d,J=17.12Hz,1H),5.70(m,1H),6.67(d,J=8.31Hz,1H),7.33(d,J=7.34Hz,1H),7.44(dd,J=4.89,2.93Hz,1H),7.63-7.70(m,2H),7.99(m,1H).
LC-MS(保留时间:2.770min),MS m/z 688(MH+)。
实施例354:化合物354的制备
化合物354
按照实施例347的方案制备化合物354,不同之处在于在步骤1中使用实施例352的中间体9代替实施例346的中间体6和使用苯基硼酸代替3-噻吩硼酸。
步骤1:
向中间体9(20mg,0.0423mmol)的DMF(1mL)溶液中加入苯基硼酸(6.7mg,0.0688mmol)、四(三苯基膦)合钯(2.4mg,0.00212mmol)和碳酸铯(41mg,0.127mmol)。将反应混合物在110℃下加热过夜。随后过滤并用甲醇洗涤。将滤液浓缩并经制备型HPLC纯化,得到淡黄色油状产物(12mg,收率49%)。
LC-MS(保留时间:2.733min),MS m/z 470(MH+)。
步骤2:
化合物354
向1-(2-甲氧羰基氨基-3,3-二甲基-丁酰基)-4-(6-苯基-吡啶-2-氧基甲基)-吡咯烷-2-甲酸(12mg,0.0206mmol)的乙腈(5mL)溶液中加入(1R,2S)(1-环丙烷磺酰基-氨基羰基-2-乙烯基-环-丙基)-氨基甲酸盐酸盐(8.2mg,0.0308mmol)、DIEA(0.018mL,0.1028mmol)和偶合剂HOBt(4.7mg,0.0308mmol)和HBTU(11.7mg,0.0308mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩。接着经制备型HPLC柱纯化,得到白色固体物,TFA盐(化合物354)(1.5mg,收率9%)。
1H NMR(CD3OD,400MHz)δ1.00-1.07(m,11H),1.20(m,2H),1.36(m,1H),1.85(m,1H),2.09(m,1H),2.17-2.25(m,2H),2.87-3.00(m,2H),3.52(s,3H),3.84-4.00(m,2H),4.33-4.44(m,4H),5.11(dd,J=10.27,1.71Hz,1H),5.28(d,J=17.12,1.22Hz,1H),5.70(m,1H),6.71(d,J=8.31Hz,1H),6.78(m,1H),7.34-7.44(m,3H),7.74(m,1H),7.95(m,1H),8.01(d,J=8.31Hz,1H).
LC-MS(保留时间:3.553min),MS m/z 682(MH+)。
实施例355:化合物355的制备
化合物355
按照实施例347的方案制备化合物354,不同之处在于在步骤1中使用实施例352的中间体9代替实施例346的中间体6和使用3-呋喃硼酸代替3-噻吩硼酸。
步骤1:
向中间体9(20mg,0.0423mmol)的DMF(1mL)溶液中加入3-呋喃硼酸(6.2mg,0.055mmol)、四(三苯基膦)合钯(2.4mg,0.002115mmol)和2M碳酸钠溶液(0.064mL,0.127mmol)。将反应混合物在110℃下加热2天。接着过滤并用甲醇洗涤。将滤液浓缩并经制备型HPLC纯化,得到淡黄色油状产物(7.0mg,收率29%)。
步骤2:
化合物355
向上述羧酸(6.0mg,0.0109mmol)的乙腈(5mL)溶液中加入(1R,2S)(1-环丙烷磺酰基-氨基羰基-2-乙烯基-环-丙基)-氨基甲酸盐酸盐(4.4mg,0.0163mmol)、DIEA(0.0095mL,0.0544mmol)和偶合剂HOBt(2.5mg,0.0163mmol)和HBTU(6.2mg,0.0163mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩。接着经制备型HPLC柱纯化,得到淡黄色膜状物,为TFA盐(化合物355)(1.5mg,收率18%)。
1H NMR(CD3OD,400MHz)δ0.98-1.07(m,11H),1.20(m,2H),1.35(dd,J=9.54,5.87Hz,1H),1.86(dd,J=8.07Hz,5.62Hz,1H),2.06(m,1H),2.15-2.25(m,2H),2.85-2.98(m,2H),3.55(s,3H),3.89(m,1H),3.95(m,1H),4.28-4.42(m,4H),5.11(dd,J=10.27,1.71Hz,1H),5.28(dd,J=17.12,1.22Hz,1H),5.69(m,1H),6.63(d,J=8.07Hz,1H),6.90(m,1H),7.16(d,J=7.33H.1H).7.53(m,1H),7.63(m,1H),8.08(s,1H).
LC-MS(保留时间:3.340min),MS m/z 672(MH+)。
实施例356:化合物356的制备
化合物356
按照实施例350的方案制备化合物356,不同之处在于在步骤1中使用实施例352的中间体9代替实施例348的中间体8。
步骤1:
向中间体9(20mg,0.0423mmol)的DMF(2mL)溶液中加入2-噻吩硼酸(7.0mg,0.055mmol)、四(三苯基膦)合钯(2.4mg,0.00212mmol)和氢氧化钡(40mg,0.127mmol)。将反应混合物在Smith微波反应器中在150℃下加热30min。接着过滤并用甲醇洗涤。将滤液浓缩并经制备型HPLC纯化,得到褐色油状产物(13.0mg,收率52%)。
1H NMR(CD3OD,400MHz)δ1.03(s,9H),2.18-2.25(m,2H),2.93(m,1H),3.55(s,3H),3.83(m,1H),3.98(m,1H),4.34(s,1H),4.38(m,2H),4.58(dd,J=8.05,5.14Hz,1H),6.63(d,J=8.07Hz,1H),7.07(dd,J=4.89,3.67Hz,1H),7.33(d,J=7.34Hz,1H),7.42(d,J=5.14Hz,1H),7.60-7.66(m,2H).
LC-MS(保留时间:3.393min),MS m/z 476(MH+)。
步骤2:
化合物356
向1-(2-甲氧羰基氨基-3,3-二甲基-丁酰基)-4-(6-噻吩-2-基-吡啶-2-氧基甲基)-吡咯烷-2-甲酸(11.5mg,0.0195mmol)的乙腈(5mL)溶液中加入(1R,2S)(1-环丙烷磺酰基-氨基羰基-2-乙烯基-环-丙基)-氨基甲酸盐酸盐(7.8mg,0.0293mmol)、DIEA(0.017mL,0.0975mmol)和偶合剂HOBt(4.5mg,0.0293mmol)和HBTU(11.1mg,0.0293mmol)。
将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩。接着经制备型HPLC柱纯化,得到灰白色固体,为TFA盐(化合物356)(8.5mg,收率54%)。
1H NMR(CD3OD,400MHz)δ0.99-1.06(m,11H),1.21(m,2H),1.36(dd,J=9.54,5.38Hz,1H),1.86(dd,J=8.07Hz,5.62Hz,1H),2.06(m,1H),2.15-2.25(m,2H),2.87-2.99(m,2H),3.54(s,3H),3.89(m,1H),3.96(m,1H),4.30-4.44(m,4H),5.11(dd,J=10.51,1.71Hz,1H),5.28(dd,J=17.11,1.22Hz,1H),5.70(m,1H),6.62(d,J=8.07Hz,1H),7.07(dd,J=4.89,3.66Hz,1H),7.33(d,J=7.59Hz,1H),7.42(d,J=4.89Hz,1H),7.60-7.66(m,2H).
LC-MS(保留时间:1.967min),MS m/z 688(MH+)。
实施例357:化合物357的制备
步骤1:
向(2S,4R)Fmoc-4-氨基-1-boc-吡咯烷-2-甲酸(400mg,0.884mmol)的乙腈(15mL)溶液中加入五滴吡咯烷。将反应混合物在室温下搅拌3hr。随后将其浓缩并置于高真空中,得到粗4-氨基-1-boc-吡咯烷-2-甲酸。在室温、氮气气氛中,将另一个圆底烧瓶中的Pd2dba3(40mg,5%mol)和外消旋BINAP(56mg,10%mol)在脱气甲苯(8mL)中的溶液搅拌1h。接着加入1-氯异喹啉(216mg,1.326mmol)和叔丁醇钠(340mg,3.536mmol)并将反应混合物搅拌30min。接着加入4-氨基-1-boc-吡咯烷-2-甲酸并将反应混合物加热回流1h。加入水猝灭反应物,分离出水层,经滤纸过滤。接着浓缩并经制备型HPLC纯化,得到偶合产物,为TFA盐(165mg,收率40%)。
1H NMR(CD3OD,400MHz)δ1.44(m,9H),2.51-2.74(m,2H),3.64(m,1H),4.01(m,1H),4.49(m,1H),4.64(m,1H),7.30(d,J=6.85Hz,1H),7.58(d,J=6.85Hz,1H),7.79(m,1H),7.91-7.99(m,2H),8.56(d,J=8.56Hz,1H).
LC-MS(保留时间:1.707min),MS m/z 358(MH+)。
步骤2:
向4-(异喹啉-1-基氨基)-吡咯烷-1,2-二甲酸1-叔丁酯(115mg,0.244mmol)的乙腈(10mL)溶液中加入(1R,2S)(1-环丙烷磺酰基-氨基羰基-2-乙烯基-环-丙基)-氨基甲酸盐酸盐(97mg,0.366mmol)、DIEA(0.255mL,1.464mmol)和偶合剂HOBt(56mg,0.366mmol)和HBTU(139mg,0.366mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩,得到黄色油状物。所述油状物经制备型HPLC柱纯化,得到淡黄色固体,为TFA盐(112mg,收率67%)。
1H NMR(CD3OD,400 MHz)δ1.05(m,2H),1.20(m,2H),1.40-1.48(m,10H),1.87(dd,J=8.19,5.50Hz,1H),2.23(m,1H),2.39(m,1H),2.50(m,1H),2.93(m,1H),3.65(m,1H),4.08(m,1H),4.33(t,J=7.09Hz,1H),4.69(m,1H),5.12(d,J=10.27Hz,1H),5.29(d,J=17.12Hz,1H),5.74(m,1H),7.31(d,J=6.85Hz,1H),7.60(d,J=7.09Hz,1H),7.80(m,1H),7.93-8.00(m,2H),8.56(d,J=8.19Hz,1H).
LC-MS(保留时间:2.023min),MS m/z 570(MH+)。
步骤3::
化合物357
将2-(1-环丙烷磺酰氨基羰基-2-乙烯基环丙基-氨基甲酰基)-4-(异喹啉-1-基氨基)-吡咯烷-1-甲酸叔丁酯(31mg,0.0453mmol)溶于4NHCl的二_烷(1.5mL)溶液中并在室温下搅拌2h。蒸发溶剂,得到淡黄色油状物,为双盐酸盐。向该双盐酸盐的乙腈溶液(5mL)中加入N-boc-L-t-亮氨酸(11.5mg,0.0498mmol)、DIEA(0.047mL,0.272mmol)和偶合剂HOBt(10.4mg,0.068mmol)和HBTU(25.8mg,0.068mmol)。将溶液在室温下搅拌过夜。随后将其浓缩,用水洗涤并用乙酸乙酯萃取两次。合并的有机层用盐水洗涤,经硫酸镁干燥并浓缩,得到淡黄色油状物。将其经制备型HPLC柱纯化,得到类白色固体终产物(化合物357)(9mg,收率29%)。
1H NMR(CD3OD,400MHz)δ0.98(m,2H),1.05(s,9H),1.20(m,2H),1.36-1.43(m,10H),1.84(m,1H),2.10-2.30(m,2H),2.52(m,1H),2.90(m,1H),4.07(m,1H),4.17-4.27(m,2H),4.47(m,1H),4.79(m,1H),5.07(d,J=9.29Hz,1H),5.24(d,J=16.87Hz,1H),5.72(m,1H),6.62(m,1H),6.98(d,J=6.11Hz,1H),7.47(m,1H),7.62(m,1H),7.69(d,J=8.07Hz,1H),7.84(d,J=5.87Hz,1H),8.20(d,J=8.56Hz,1H).
LC-MS(保留时间:2.043min),MS m/z 683(MH+)。
部分H:
用于部分H的LC-MS条件
柱:(方法A)-YMC ODS S7 C18 3.0×50mm
(方法B)-YMC ODS-A S7 C18 3.0×50mm
(方法C)-YMC S7 C18 3.0×50 mm
(方法D)-YMC Xterra ODS S7 3.0×50mm
(方法E)-YMC Xterra ODS S7 3.0×50mm
(方法F)-YMC ODS-A S7 C18 3.0×50mm
(方法H)-Xterra S7 3.0×50mm
(方法I)-Xterra S7 C18 3.0×50mm
(方法G)-YMC C18 S5 4.6×50mm
(方法J)-Xterra ODS S7 3.0×50mm
(方法K)-YMC ODS-A S7 C18 3.0×50mm
梯度:100%溶剂A/0%溶剂B至0%溶剂A/100%溶剂B
梯度时间:2min.(A,B,D,F,G,H,I);8min.(C,E);4min(J);3min(K)
保留时间:1min.(A,B,D,F,G,H,I,J,K);2min.(C,E)
流速:5mL/min(A,B,C,D,E,F,G)
流速:4mL/min(J,K)
检测器波长:220nm
溶剂A:10%甲醇/90%H2O/0.1%TFA
溶剂B:10%H2O/90%甲醇/0.1%TFA。
实施例370:化合物370的制备
化合物370
方案1
步骤1:
将(1R,2S)/(1S,2R)-1-氨基-2-乙烯基环丙烷甲酸乙酯盐酸盐(2.54g,12mmol)的乙腈(70mL)溶液用二异丙基乙胺(9.5mL,67mmol)、[(4R)-(2-甲氧羰基-7-甲氧基喹啉-4-氧代)-S-脯氨酸](5.9g,13.2mmol)和TBTU(3.89g,12.21mmol)在乙腈(50mL)中的溶液处理。将反应混合物搅拌14h并浓缩。 将溶于乙酸乙酯中的剩余物用碳酸氢钠水溶液和盐水反复洗涤,干燥(硫酸镁)并浓缩。将剩余物经Biotage65M柱纯化(EtOAc/己烷:45-100%),得到高Rf立体异构体(Boc-P2[(4R)-(2-甲氧羰基-7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)甲酸乙酯2.0g(52%),为白色固体物:
1H NMR(甲醇-d4)δppm 1.24(t,J=7.02Hz,3H),1.38(m,11H),1.76(m,1H),2.21(m,1H),2.45(m,1H),2.71(m,1H),3.92(m,2H),3.96(s,3H),4.03(s,3H),4.16(q,J=7.22Hz,2H),4.42(m,1H),5.10(m,1H),5.30(m,1H),5.44(s,1H),5.77(m,1H),7.27(d,J=9.16Hz,1H),7.48(s,1H),7.52(s,1H),8.05(s,1H).
方案2
步骤2:
0℃下,将实施例370的步骤1的高Rf产物{Boc-P2[(4R)-(2-甲氧羰基-7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)甲酸乙酯}(3.16g,5.40mmol)溶于甲醇/THF(1/1,13.2mL)中,用1.0N NaOH水溶液(5.5mL,5.5mmol)处理,搅拌1h,加入AcOH中和。真空除去溶剂。再次将剩余物溶于THF/二氯甲烷(1/1,150mL)中,干燥(硫酸镁)并真空浓缩得到产物,该产物直接用于下一步骤:
LC-MS(保留时间:1.53方法D),MS m/z 570(M++1)。
步骤3:
0℃下,向实施例370的步骤2的产物(估计5.4mmol)的THF(35mL)溶液中加入新鲜制备的CH2N2(30mmol)的乙醚(80mL)溶液。将反应混合物在该温度下搅拌0.5h,接着在室温下搅拌18.5h。向反应混合物中通入氮气鼓泡1小时后,真空除去溶剂。将剩余物再次溶于EtOAc(1L)中,用饱和碳酸氢钠水溶液(2×200mL)和盐水(100mL)洗涤,干燥(硫酸镁)。真空除去溶剂,得到产物3.10g(两步总收率97%):LC-MS(保留时间:3.06,方法J),MS m/z 594(M++1)。
步骤4:
0℃下,向实施例370的步骤3的产物{Boc-P2[(4R)-(2-重氮乙酰基-7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)甲酸乙酯}(3.03g,5.10mmol)的THF(110mL)溶液中加入2ml 48%HBr。将混合物搅拌1h,在EtOAc(500mL)和饱和碳酸氢钠水溶液(100mL)间分配。分离出乙酸乙酯层,干燥(硫酸镁)。除去溶剂,得到产物(3.12g,95%):LC-MS(保留时间:1.56方法D)。MS m/z 648(M++1),MSm/z646(M-1)。
步骤5:
将实施例370的步骤4的产物{Boc-P2[(4R)-(2-溴乙酰基-7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)甲酸乙酯}(1.0g,1.54mmol)用异丙基硫脲(0.365g,3.09mmol)的异丙醇(57mL)溶液处理2h,随后除去溶剂。将剩余物溶于1.0N盐酸(30mL)和EtOAC(200mL)中,加入1.0N氢氧化钠水溶液调节pH至7。水层用EtOAc萃取(2×100mL),将合并的萃取液干燥(硫酸镁),浓缩。剩余物经Biotage40+M柱纯化(EtOAc-己烷:30-100%),得到产物870mg(84%),直接用于下一步骤。
步骤6:
将实施例370的步骤5的产物{Boc-P2{(4R)-[2-(2-异丙氨基噻唑-4-基)-7-甲氧基喹啉-4-氧代]-S-脯氨酸}-P1(1R,2S乙烯基Acca)甲酸乙酯}(0.250g,0.375mmol)用4N HCl/二_烷(2.5mL,10mmol)处理2.5h并真空浓缩。向剩余物中加入N-甲基吗啉0.206mL,1.875mmol)DMF(3mL)溶液、N-Boc-L-叔-亮氨酸(0.117g,0.506mmol)和HATU(0.192g,0.506mmol)。将混合物搅拌过夜并在乙酸乙酯和pH4.0的缓冲液间分配。乙酸乙酯层用水和碳酸氢钠水溶液洗涤,干燥(硫酸镁),浓缩。将剩余物经Biotage40M柱纯化(甲醇-二氯甲烷:0-8%),得到产物0.289g(99%):
LC-MS(保留时间:2.53,方法K),MS m/z 779(M++1)。
步骤7:
向实施例370的步骤6的产物{BOCNH-P3(L-t-BuGly)-{[2-(2-异丙氨基噻唑-4-基)-7-甲氧基喹啉-4-氧代]-S-脯氨酸}-P1(1R,2S乙烯基Acca)-甲酸乙酯}(274mg,0.352mmol)在THF(10.6mL)、甲醇(2.6mL)和H2O(5.3mL)中的悬浮液内加入LiOH(0.068g,2.86mmol)。将反应混合物搅拌24,调节至pH6,真空除去有机溶剂。将含水剩余物酸化至pH4,并用二氯甲烷重复萃取。将合并的有机溶剂干燥(硫酸镁)并真空浓缩,得到所需的产物255mg(95%):
LC-MS(保留时间:2.58,方法K),MS m/z 751(M++1)。
方案3
步骤8:
将CDI(0.024g,0.15mmol)和实施例370的步骤7的产物{BOCNH-P3(L-t-BuGly)-{[2-(2-异丙氨基噻唑-4-基)-7-甲氧基喹啉-4-氧代]-S-脯氨酸}-P1(1R,2S乙烯基Acca)-甲酸}(0.0683g,0.09mmol)的THF(2mL)溶液回流60min,随后冷却至室温。依次加入环丙烷磺酰胺(0.022g.0.18mmol)和纯DBU(0.027mL,0.18mmol)。将反应物搅拌过夜,进行如下后处理:用乙酸乙酯稀释、用pH4.0缓冲液洗涤,干燥(硫酸镁)并浓缩。将剩余物经制备型HPLC反复纯化(0-100%溶剂B),并经1000μM的Analtech制备TLC板(20×40cM)纯化,得到0.0032g(4%)所需的产物(化合物370),为浅黄色泡沫体:
LC-MS(保留时间:1.71,方法I)MS m/z 854(M++1)。
实施例371:化合物371的制备
化合物371
方案1
步骤1:
向冷却至0℃下的N-Boc-顺-L-4-羟基脯氨酸甲酯(10g,40.7mmol)和7-氯喹啉-4-醇(8.73g,49.0mmol)的THF(200mL)悬浮液中加入三苯基膦(12.82g,48.9mmol)和DIAD(8.80g,42.13mmol)。将混合物缓慢升温至室温下过夜,总共搅拌30h。将混合物溶于EtOAc(800mL)中,依次用1N盐酸、5%碳酸钾水溶液(3×100mL)、盐水(2×100mL)洗涤并干燥(硫酸镁)及浓缩。将剩余物经Biotage65M(甲醇-二氯甲烷:0-10%)纯化数次,累计得到10.57g(68%)所需的产物,为玻璃状物:
1H NMR(CDCl3)δ1.40(s,9H),2.33-2.42(m,1H),2.61-2.72(m,1H),3.75(s,3H),3.91(m,2H),4.45-4.59(m,1H),5.13(m,1H),6.61-6.64(m,1H),7.41(dd,J=9,2Hz,1H),7.98(d,J=2Hz,1H),8.03(d,J=9Hz,1H),8.67-8.71(m,1H).
LC-MS(保留时间:1.39,方法D),MS m/e 407(M++1)。
步骤2:
向冷却至0℃的实施例371、步骤1的产物{BOC-N-P2[(4R)-(7-氯喹啉-4-氧代)脯氨酸甲酯}(10.57g,26.0mmol)溶于甲醇(800mL)中的溶液内加入1N NaOH水溶液(44.5mL,44.5mmol)。将混合物升温至室温并保持在该温度下6h后,搅拌过夜并使用1.0N盐酸将pH调节至pH7。将溶液浓缩直至只剩下水层,使用6N盐酸将pH调节至4,将混合物反复用EtOAc分配(3×500mL)。将合并的有机层干燥(硫酸镁)并浓缩,得到10.16g(100%),为白色固体物。
1HNMR(DMSO-d6)δ1.32,1.34(两种s(旋转异构体)9H),2.31-2.40(m,1H),2.58-2.69(m,1H),3.65-3.81(m,2H),4.33-4.40(m,1H),5.34(m,1H)7.10-7.11(m,1H),7.57(d,J=9Hz,1H),7.98(s,1H),8.09-8.14(m,1H),8.75(d,J=5Hz,1H),12.88(brs,1H).13C NMR(DMSO-d6)δ27.82,,35.84,51.52,57.75,76.03,79.33,102.95,119.54,123.86,126.34,127.24,134.49,149.32,152.88,153.25,159.08,173.74.
LC-MS(保留时间:1.48,方法D),MS m/e 393(M++1)。
步骤3:
向实施例371、步骤2的产物{Boc-4(R)-(7-氯喹啉-4-氧代)脯氨酸}(5.11g,13mmol)、乙烯基Acca的盐酸盐(3.48g,18.2mmol)(为1∶1的非对映异构体混合物(1R,2S/1S,2R,其中环丙基羧乙基与乙烯基部分呈顺式关系)和NMM(7.1mL 65mmol)在DMF(30mL)中的溶液内加入HATU(6.92g,18.2mmol)。将混合物搅拌3天。反应混合物用EtOAc(180mL)稀释并用pH4.0缓冲液(3×100mL)分配。有机层依次用饱和碳酸氢钠水溶液(2×50mL)、水(2×50mL)和盐水(2×50mL)洗涤。将有机溶液干燥(硫酸镁)并浓缩。将剩余物经Biotage40M柱(EtOAc-己烷∶50%-100%)纯化,得到2.88g产物,为非对映异构体混合物。使用Biotage65M柱(甲醇-EtOAc:0%-9%)将该混合物部分分离,得到BOC-NH-P2[(4R)-(7-氯喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基accaP1部分)-COOEt,为初始洗脱的高Rf异构体(1.20g,17.4%)。
1H NMR(CDCl3/甲醇-d4)δ1.16(t,J=7Hz,3H),1.35(s,9H),1.37-1.43(m,1H),1.76-1.84(m,1H),2.06-2.11(m,1H),2.35-2.45(m,1H),2.63(m,1H),3.72-3.93(m,2H),4.02-4.15(m,1H),4.33-4.40(m,1H),5.06(d,J=9Hz,1H),5.16(m,1H),5.24(d,J=17Hz,1H),5.63-5.70(m,1H),6.74(m,1H),7.39(dd,J=9,2Hz,1H),7.74-7.78(m,1H),7.89(d,J=2Hz,1H),7.97(d,J=9Hz,1H),8.60(d,J=5Hz,1H).1H NMR(甲醇-d4,60/40异构体)δ1.24(t,J=7Hz,3H),1.39,1.43(2s,9H,比率4∶6),1.71-1.74(m,0.4H),178-1.81(m,0.6H),2.18-2.23(m,1H),2.65-2.69(m,0.4H),2.71-2.76(m,0.6H),3.88-3.96(m,2H),4.11-4.18(m,2H),4.39-4.45(m,1H),5.09-5.13(m,1H),5.28-5.33(m,1H),5.37(m,1H),5.73-5.81(m,1H),7.05(d,J=5Hz,1H),7.53(d,J=8.9Hz,1H),7.92(s,1H),8.12(d,J=8.9Hz,1H),8.70(d,J=5Hz,1H).
LC-MS(保留时间:1.54,方法A)MSm/z530(M++1)。剩余物(~1.66g,24%)为各部分的混合物,富含较低Rf异构体。
方案2
步骤4:
将实施例371、步骤3的产物{BOC-P2[(4R)-(7-氯喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca-CO2Et}(0.65g,1.22mmol)溶于4N HCl/二_烷(4.5ml,18mmol)中并在室温下搅拌1h。将反应混合物浓缩,粗产物直接用于下一步骤中:LC-MS(保留时间:0.94,方法A)LC-MSm/z430(M++1)。
步骤5:
室温下,向实施例371、步骤4的产物1{NH2-P2[(4R)-(7-氯喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S-乙烯基Acca)-甲酸乙酯)的双盐酸盐}(0.22mmol)、N-BOC-L-叔-亮氨酸(BOC L-tBuGly)(0.34g,1.47mmol)、DIPEA(1.0ml,5.74mmol)、HOBT·H2O(0.22g,1.47mmol)在二氯甲烷(15mL)中的悬浮液内加入HBTU(0.56g,1.47mmol)。将反应混合物搅拌过夜,用二氯甲烷(50mL)稀释,依次用pH4.0缓冲液(2×20mL)、饱和碳酸氢钠水溶液(50mL)和盐水(50mL)洗涤,干燥(硫酸镁)并浓缩。将剩余物经Biotage40M柱(EtOAc-己烷:15%-60%)纯化,得到607mg(77%)产物,为泡沫状物。
1H NMR(CDCl3-甲醇-d4)δ1.00(s,9H),1.19(t,J=7Hz,1H),1.30(s,9H),1.38(m,1H),1.78-1.83(m,1H),2.01-2.46(m,2H),2.73-2.82(m1H),3.96-4.03(m,1H),4.04(d,J=10Hz,1H),4.11(q,J=7Hz,2H),4.42(d,J=12Hz,1H),4.68-4.73(m,1H),5.09-5.13(m,1H),5.23-5.31(m,2H),5.67-5.79(m,1H),6.78(d,J=9Hz,1H),7.38(d,J=9Hz,1H),7.70(s,1H),7.96(s,1H),8.08(d,J=9Hz,1H),8.68(d,J=5Hz,1H).
LC-MS(保留时间:1.64,方法A),MSm/z643(M++1)。
步骤6:
向实施例371、步骤5的产物{BOCNH-P3(L-t-BuGly)-P2[(4R)-7-氯喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CO2Et}(207mg,0.32mmol)在THF(14mL)、甲醇(2mL)和H2O(8mL)中的悬浮液内加入LiOH(62mg,2.60mmol)。将反应混合物搅拌1天,调节至中性pH,真空浓缩直至仅剩下水层。加入1.0N盐酸将所得的含水剩余物酸化至pH4.0,随后用固体NaCl饱和。用EtOAc反复萃取该含水混合物(3×60mL),将合并的有机溶剂干燥(硫酸镁)并真空浓缩,得到107mg(54%)产物{BOCNH-P3(L-t-BuGly)-P2[(4R)-(7-氯喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CO2H},为白色固体物。
1H NMR(CDCl3)δ1.06(s,9H),1.23(2s,9H),1.31-1.43(m 1H),1.63-1.70(m,1H),1.85-1.89(m,1H),2.19(m,1H),2.65-2.78(m,1H),4.03-4.10(m,1H),4.18-4.21(m,1H),4.55-4.62(m,1H),5.03-5.12(m,1H),5.23-5.31(m,1H),5.51(m,1H),5.88-5.95(m,1H),7.12(m,1H),7.47-7.50(m,1H),7.96(m,1H),8.26(d,J=9Hz,1H),8.75(d,J=5Hz,1H).
LC-MS(保留时间:1.46,方法A),MS m/z 15(M++1)。
步骤7:
向实施例371、步骤6的三肽酸{BOCNH-P3(L-t-BuGly)-P2[(4R)-(7-氯喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CO2H}(0.0453g,0.074mmol)的THF(4mL)溶液中加入CDI(17mg,0.10mmol),将所得的溶液回流45min,冷却至室温。一次性加入环丙基磺酰胺(0.013g,0.10mmol),随后加入纯DBU(0.015mL,0.10mmol)。搅拌反应物18h,用EtOAc(200mL)稀释并依次用pH4.0缓冲液(3×30mL)、水(2×30mL)和盐水(30mL)洗涤,干燥(硫酸镁)并使用一块20×40cM 1000 Analtech PTLC板(甲醇-二氯甲烷:0-2%)纯化,得到所需的产物(化合物371),为泡沫状物(0.040g,76%):
1H NMRδ0.95-1.23(m,4H),1.03(s,9H),1.19(s,9H),1.40-1.43(m,1H),1.85(dd,J=8,5Hz,1H),2.12-2.20.(m,1H),2.43(m,1H),2.82(m,1H),4.07-4.19(m,2H),4.51-4.57(m,2H),5.07(d,J=10Hz,1H),5.25(d,J=17Hz,1H),5.85(m,1H),5.48(s,1H),7.09(d,J=5Hz,1H),7.45(d,J=9Hz,1H),7.92(m,1H),8.20(d,J=9Hz,1H),8.72(d,J=5Hz,1H);
LC-MS(保留时间:1.52,方法B),MS m/z 718(M++1)。HRMSm/z(M+H)+,C41H51N5SO9计算值:718.2677,实测值:718.2674。
实施例372:化合物372的制备
化合物372
方案1:
步骤1:
向L-叔-亮氨酸(2g,15.25mmol)溶于乙腈(50mL)中的溶液内加入TMSCN(7.06mL,56.41mmol)并搅拌15min。将反应混合物在75℃下加热30min。向反应混合物中加入氯甲酸环戊酯(2.83g,19.06mmol)并将反应混合物在80℃下加热过夜,真空浓缩。将剩余物用甲醇(40mL)处理,搅拌10min并真空浓缩。调节剩余物的pH至8.5,并用乙醚(2×200mL)萃取。水层酸化至pH3并用二氯甲烷(2×200mL)萃取。将合并的萃取液干燥(硫酸镁)并真空浓缩。剩余物在尽可能少量的乙醚/己烷中重结晶,得到产物3.48g(94%):
1H NMR(500MHz,甲醇-d4)δppm 1.00(s,9H),1.59(m,2H),1.73(m,4H),1.84(dd,J=5.95,3.20Hz,2H),3.98(s,1H),5.02(m,1H)。
步骤2:
向实施例371的步骤4的产物{P2[(4R)-7-氯喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)甲酸乙酯的盐酸盐}(530.1mg,1.04mmol)、实施例372的步骤1的产物{(L)-2-环戊氧基羰基氨基-3,3-二甲基-丁酸}(328mg,1.35mmol)、HOBT(146mg,1.08mmol)和二异丙基乙胺(0.755mL,4.32mmol)在二氯甲烷(7mL)中的溶液内加入HBTU(512mg,1.35mmol)。将反应混合物搅拌过夜并在二氯甲烷和pH4.0缓冲液间分配。将二氯甲烷层依次用水和饱和碳酸氢钠水溶液洗涤,干燥(硫酸镁)并浓缩。将剩余物经Biotage40M柱(EtOAc-己烷:35-100%)纯化,得到产物640mg(92%):
1H NMR(甲醇-d4)δppm 1.02(s,9H),1.26(m,4H),1.56(m,10H),2.19(q,J=8.75Hz,1H),2.41(m,1H),2.70(dd,J=14.19,8.09Hz,1H),4.01(dd,J=11.90,3.05Hz,1H),4.13(m,2H),4.20(s,1H),4.53(m,1H),4.62(m,1H),5.09(d,J=10.38Hz,1H),5.26(d,J=17.09Hz,1H),5.47(m,1H),5.77(m,1H),7.07(d,J=5.49Hz,1H),7.47(m,1H),7.94(m,1H),8.20(d,J=8.85Hz,1H),8.72(d,J=5.49Hz,1H).
LC-MS(保留时间:1.71,方法B),MS m/z 655(M++1)。
步骤3:
按照实施例370、方案2的步骤7的方法制备三肽酸,不同之处在于使用环戊氧基羰基-NH-P3(L-叔-BuGly)-P2[(4R)-(7-氯喹淋-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-甲酸乙酯代替实施例370、步骤6的产物。
修改:使用实施例372、步骤2的产物0.636g(0.97mmol),得到0.424g产物(收率69%)。
产物:
数据:
1H NMR(甲醇-d4)δppm 1.02(s,9H),1.57(m,11H),2.14(q,J=9.03Hz,1H),2.46(m,1H),2.68(m,1H),4.02(dd,J=11.89,3.11Hz,1H),4.19(m,1H),4.50(d,J=26.35Hz,1H),4.64(t,J=8.42Hz,1H),5.04(m,1H),5.24(d,J=17.20Hz,1H),5.44(s,1H),5.87(m,1H),7.05(d,J=5.12Hz,1H),7.48(m,1H),7.92(m,1H),8.18(d,J=8.78Hz,1H),8.71(d,J=5.49Hz,1H).
LC-MS(保留时间:2.32,方法A),MS m/z 627(M++1)。
方案2
步骤4:
将CDI(0.021g,0.13mmol)和实施例372、步骤3的产物{BOCNH-P3(L-t-BuGly)-P2[(4R)-7-氯喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CO2H}(0.058g,0.09mmol)在THF(2mL)中的溶液回流40min,冷却至室温。总共加入0.016g(0.13mmol)环丙烷磺酰胺,接着加入纯DBU溶液(0.019mL,0.13mmol)。将反应物搅拌过夜,随后用EtOAc(100mL)稀释并用pH4.0缓冲液(2×)洗涤,干燥(硫酸镁),浓缩并经1000μM制备型TLC板(Analtech,20×40cM,依次用50%-0%-2%甲醇/二氯甲烷洗脱)纯化三次,得到产物(化合物372)27.3mg(40%):
1H NMR(甲醇-d4)δppm 0.94(m,2H),1.02(s,9H),1.14(m,1H),1.49(m,11H),1.86(m,1H),2.14(m,1H),2.49(m,1H),2.68(dd,J=13.89,7.48Hz,1H),2.78(m,1H),4.08(m,1H),4.22(s,1H),4.55(m,2H),5.05(d,J=10.07Hz,1H),5.22(d,J=17.09Hz,1H),5.46(m,1H),5.86(m,1H),7.07(d,J=5.19Hz,1H),7.46(d,J=8.55Hz,1H),7.91(s,1H),8.18(d,J=8.85Hz,1H),8.72(d,J=5.19Hz,1H)。LC-MS(保留时间:1.52方法I),MS m/z 730(M++1)。
实施例373:化合物373的制备
化合物373
方案1
步骤1
0℃下将2-氨基-4-甲氧基苯乙酮(4.45g,26.94mmol)溶于二氯甲烷(100mL)中,随后用环丙烷甲酰氯(3.1mL,33.68mmol)、二异丙基乙胺(19mL,107.8mmol)和DMAP(0.780g,6.4mmol)处理。将反应混合物在室温下搅拌过夜,接着真空浓缩。将剩余物溶于二氯甲烷溶液(500mL)中,依次用1N盐酸、水和碳酸氢钠水溶液洗涤,并干燥(硫酸镁)。真空除去溶剂,将固体剩余物用EtOAc/己烷(1/1)处理,得到产物(5.35g,85%):
1H NMR(甲醇-d4)δppm 0.94(m,4H),1.69(m,J=3.97Hz,1H),2.60(s,3H),3.84(s,3H),6.69(d,J=7.93Hz,1H),7.98(d,J=8.85Hz,1H),8.23(s,1H)。
步骤2:
将实施例373、步骤1的产物{环丙烷甲酸(2-乙酰基-5-甲氧基-苯基)-酰胺}(5.35g,22.72mmol)和叔丁醇钾(5.45g,48.6mmol)的叔丁醇(130g)溶液回流6h。将反应混合物冷却,倒入冰冷却的缓冲液并调节至pH7,过滤。收集固体物,在甲醇/乙醚中重结晶,得到产物(1g,20%):1H NMR(甲醇-d4)δppm 0.96(m,2H),1.15(m,2H),1.94(m,1H),3.87(s,3H),5.86(m,1H),6.93(m,2H),8.04(d,J=8.85Hz,1H)。
步骤3:
在30min内,向0℃下的N-Boc-L-3-羟基脯氨酸(1.06g,4.32mmol)和三苯基膦(2.27g,8.64mmol)溶于THF(25mL)中的溶液内加入实施例373、步骤2的产物{2-环丙基-7-甲氧基-喹啉-4-醇}(0.93g,4.32mmol)和DEAD(1.50g,8.64mmol)的THF(25mL)溶液。将反应混合物搅拌过夜并浓缩。将剩余物经Biotage 40+M柱(EtOAc-己烷:20-65%)纯化两次,得到产物1.74g(90%):LC-MS(保留时间:2.56,方法J),MS m/z 443(M++1)。
步骤4:
向实施例373、步骤3的产物{Boc-(4R)-(2-环丙基-7-甲氧基-喹啉-4-氧代)-S-脯氨酸甲酯}(1.70g,3.86mmol)在THF(91mL)、甲醇(18.2mL)和H2O(27mL)中的悬浮液内加入LiOH(0.73g,30mmol)。将反应混合物搅拌16h,调节至pH6,真空除去有机溶剂。将剩余物酸化至pH4,并用EtOAc萃取(4×100mL)。将合并的有机萃取液干燥(硫酸镁)并真空浓缩,得到浆状产物1.64g(100%):
1H NMR(甲醇-d4)δppm 1.32(m,13H),2.37(m,2H),2.71(m,1H),3.86(m,1H),3.95(s,3H),4.14(m,1H),4.43(m,1H),5.41(s,1H),6.65(s,1H),7.19(m,1H),7.30(m,1H),8.02(dd,J=12.63,9.33Hz,1H)。
步骤5:
将实施例373、步骤4的产物{Boc-P2{(4R)-[2-环丙基-7-甲氧基喹啉-4-氧代]-S-脯氨酸}-P1(1R,2S乙烯基Acca)甲酸乙酯}(1.61g,2.79mmol)溶于HCl/二_烷(15mL;60mmol)中并在室温下搅拌3h。将反应混合物浓缩并与无水THF共沸蒸馏,得到产物(1.58g,100%):
LC-MS(保留时间:2.12,方法K),MS m/z 566(M++1)。
方案2
步骤6:
向实施例373的步骤5的产物{P2{(4R)-[2-环丙基-7-甲氧基喹啉-4-氧代]-S-脯氨酸}-P1(1R,2S乙烯基Acca)甲酸乙酯的双盐酸盐}(1.58g,2.79mmol)、二异丙基乙胺(1.65mL,9.25mmol)、N-Boc-L-叔-亮氨酸(0.775g,3.35mmol)、HOBT·H2O(0.515g,3.36mmol)在二氯甲烷(13mL)中的悬浮液内加入HBTU(1.28g,3.36mmol)。将混合物搅拌14h并在乙酸乙酯和pH4.0缓冲液间分配。将EtOAc层干燥(硫酸镁)并浓缩。剩余物经Biotage 40+M柱(EtOAc-己烷:20-100%,接着用甲醇洗脱)纯化,并进一步经20×40cM 1000 Analtech PTLC板(甲醇-二氯甲烷2%)纯化,得到产物1.4g(63%):
1H NMR(甲醇-d4)δppm 1.04(s,9H),1.20(m,5H),1.28(s,9H),1.39(m,2H),1.69(m,1H),2.19(m,2H),2.36(m,1H),2.63(dd,J=13.54,7.68Hz,1H),3.90(s,3H),4.08(m,4H),4.19(d,J=11.34Hz,1H),4.47(d,J=11.71Hz,1H),4.56(t,J=8.60Hz,1H),5.08(m,1H),5.24(m,1H),5.39(s,1H),5.78(m,1H),6.56(s,1H),6.96(dd,J=9.15,2.20Hz,1H),7.21(d,J=2.56Hz,1H),7.97(d,J=9.15Hz,1H).
LC-MS(保留时间:2.34,方法K),MS m/z 679(M++1)。
步骤7:
向实施例373、步骤6的产物Boc-NH-P3(L-叔-BuGly)-P2[(4R)-(2-环丙基-7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-甲酸乙酯(1.28g,1.89mmol)在THF(93mL)、甲醇(23mL)和H2O(45mL)中的悬浮液内加入LiOH(0.491g,20.4mmol)。将反应混合物搅拌18.5h,调节至pH4,真空除去有机溶剂。剩余物用EtOAc萃取(5×100mL)。将合并的有机溶剂干燥(硫酸镁)并真空浓缩,得到所需的产物1.17g(97%):
1H NMR(甲醇-d4)δppm 1.04(s,9H),1.24(s,9H),1.27(m,3H),1.42(m,2H),1.68(dd,J=8.05,5.12Hz,1H),2.17(m,1H),2.33(m,1H),2.47(m,1H),2.66(m,1H),3.95(s,3H),4.09(m,2H),4.51(d,J=11.71Hz,1H),4.59(t,J=8.60Hz,1H),5.07(m,1H),5.26(m,1H),5.52(s,1H),5.85(m,1H),6.69(s,1H),7.10(dd,J=9.15,2.20Hz,1H),7.27(d,J=2.20Hz,1H),8.10(d,J=9.15Hz,1H).
LC-MS(保留时间:2.21,方法K),MS m/z 651(M++1)。
方案3
步骤8:
将CDI(0.058g,0.344mmol)和实施例373、步骤7的产物{Boc-NH-P3(L-叔-BuGly)-P2[(4R)-(2-环丙基-7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-COOH}(0.160g,0.246mmol)在THF(2mL)中的溶液回流60min,冷却至室温。依次加入环丙烷磺酰胺(0.041g,0.344mmol)和纯DBU(0.051mL,0.344mmol)。搅拌反应物24h并进行如下后处理:将反应混合物在pH4.0缓冲液和EtOAc间分配。将有机层干燥(硫酸镁),浓缩并经制备型HPLC(0-100%溶剂B)纯化,得到产物(化合物373)0.086g(46%):
1H NMR(三氟乙酸-D)δppm 1.04(s,9H),1.21(m,16H),1.41(m,1H),1.87(dd,J=8.05,5.49Hz,1H),2.26(m,3H),2.61(dd,J=12.99,6.77Hz,1H),2.93(m,1H),3.92(s,3H),4.09(m,1H),4.21(m,1H),4.49(m,2H),5.11(d,J=11.71Hz,1H),5.27(d,J=17.20Hz,1H),5.46(s,1H),5.76(m,1H),6.62(m,2H),7.01(dd,J=8.97,2.01Hz,1H),7.23(d,J=2.56Hz,1H),8.00(d,J=8.78Hz,1H)。
实施例374:化合物374的制备
化合物374
方案1
步骤1:
向间甲氧基苯胺(58g,471mmol)在800ml乙腈中的溶液内加入Meldrum酸(75g,518mmol)和原甲酸三甲酯(60g,565mmol)。将多相混合物回流2h。真空除去溶剂,加入甲醇(30mL),将所得的沉淀物过滤并用10-15ml甲醇洗涤。对浓缩母液重复加入甲醇/过滤操作。将所得的合并固体物干燥(~20托,45℃,过夜),得到117.6g(90%)中间体5-[(3-甲氧基苯基-氨基)亚甲基]-2,2-二甲基-[1,3]二_烷-4,6-二酮。
步骤2:
在30分钟内向加热至250℃的PH2O溶液(500g)中分次加入108.7g(392mmol)5-[(3-甲氧基苯基-氨基)亚甲基]-2,2-二甲基-[1,3]二_烷-4,6-二酮。再将混合物加热15min,冷却至室温,用己烷(800mL)稀释并将所得的浆状物搅拌过夜。倾倒出己烷,将固体剩余物溶于600ml回流的甲醇中,冷却至室温,将所得的固体过滤并用少量二氯甲烷洗涤。进行类似的重结晶操作,总共得到20.73g(30%)7-甲氧基喹啉-4-醇,为浅棕色固体物。1H NMR(甲醇-d4)δ3.87(s,3H),6.23d,J=7.3Hz,1H),6.68(d,J=2.4Hz,1H),6.96(dd,J=9.0,2.4Hz,1H),8.11(d,J=9Hz,1H);LC-MS(保留时间:0.77,方法D),MS m/z 176(M++1)。
步骤3:
在45分钟内,向冷却至0℃下的N-Boc-顺-L-4-羟基脯氨酸甲酯(12.24g,49.8mmol)和三苯基膦(26.14g,99.7mmol)在THF(200mL)中的溶液内加入DEAD(17.36g,99.7mmol)和7-甲氧基喹啉-4-醇(8.73g,49.8mmol)的THF(700mL)溶液。将混合物缓慢升温至室温下过夜,真空浓缩。将剩余物经Biotage65M柱(甲醇-EtOAc:0-10%)纯化,得到12.78g(64%)产物,为无色玻璃状物:1H NMR(CDCl3)δ1.36(s,9H),2.26-2.35(m,1H),2.57-2.68(m,1H),3.71(s,3H),3.75-3.92(m,2H),3.86、3.87(两个s(旋转异构体),3H),4.41-4.53(m,1H),5.09(m,1H),6.52(d,J=5.5Hz,1H),7.06-7.09(m,1H),7.24-7.26(m,1H),7.94(d,J=9.1Hz,1H),8.50-8.56(m,1H);LC-MS(保留时间:1.34,方法D),MS m/e 403(M++1)。
步骤4:
向实施例374、步骤3的产物{BOC-N-P2[(4R)-(7-甲氧基喹啉-4-氧代)脯氨酸甲酯}(8.54g,21.2mmol)在600ml THF/甲醇(5∶1)中的溶液内加入LiOH(4.0g,167mmol)的水(150ml)溶液。将混合物搅拌过夜,使用6N盐酸将pH调节至pH7,浓缩溶液直至仅剩下水层。使用1N盐酸调节剩余物至pH4,加入NaCl以饱和混合物,由于产物为水溶性,因此依次用乙酸乙酯和THF反复分配。将合并的有机层干燥(硫酸镁)并浓缩,得到产物8.18g(99%),为白色固体物。
1H NMR(CDCl3-甲醇-d4)δ1.42(s,9H),2.40-2.49(m,1H),2.68-2.77(m,1H),3.88(m,2H),3.94(s,3H),4.41-4.53(m,1H),5.32(m,1H),6.86-6.92(m,1H),7.21(dd,J=9,2Hz,1H),7.30(d,J=2Hz,1H),8.05-8.10(m,1H),8.62(d,J=6Hz,1H);
LC-MS(保留时间1.20,方法A),MS m/z 389(M++1)。
步骤5:
向实施例374、步骤4的产物{Boc-4(R)-(7-甲氧基喹啉-4-氧代)脯氨酸}(4.50g,11.60mmol)、2.66g(13.9mmol)乙烯基Acca的盐酸盐(为1∶1非对映异构体的混合物(1R,2S/1S,2R,其中环丙基羧乙基与乙烯基部分呈顺式构型)、10mL(57.4mmol)DIPEA和2.13g(13.9mmol)HOBT·H2O在150ml二氯甲烷中的溶液内加入5.27g(13.9mmol)HBTU,将所得混合物搅拌过夜。将溶液用200ml二氯甲烷稀释并用pH4.0缓冲液(2×50mL)分配。有机层依次用饱和碳酸氢钠水溶液(2×50mL)、水(2×50mL)和盐水(2×50mL)洗涤。将有机溶液干燥(硫酸镁),浓缩并使用Biotage65M柱(洗脱液:0-9%甲醇/EtOAc)纯化,得到初始洗脱的异构体BOC-NH-P2[(4R)-(7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基accaP1部分)-COOEt(2.21g,总共36%),接着为1.13g(19%)纯低Rf异构体BOC-NH-P2[(4R)-(7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1S,2R乙烯基AccaP1部分)-CO2Et。还得到混合组分。BOCN-P2[(4R)-(7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S)-(乙烯基Acca)-甲酸乙酯的数据:
1H NMR(CDCl3)δ1.16(t,J=7Hz,3H),1.35(s,9H),1.37-1.47(m,1H),1.74-1.88(m,1H),2.04-2.13(m,1H),2.32-2.46(m,1H),2.58-2.69(m,1H),3.76(m,1H),3.87(s,3H),4.02-4.13(m,2H),4.30-4.44(m,1H),5.05-5.19(m,2H),5.24(d,J=17Hz,1H),5.63-5.71(m,1H),6.61(m,1H),7.07(dd,J=9,2Hz,1H),7.22(d,J=2Hz,1H),7.76-7.83(m,1H),7.92(d,J=9Hz,1H),8.50(d,J=5Hz,1H).
LC-MS(保留时间:1.38,方法A),MS m/z 526(M++1)。
方案2
步骤6:
将实施例374、步骤5的全部产物{BOC-P2[(4R)-(7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-甲酸乙酯}(1.35g,2.90mmol)溶于4N HCl/二_烷(15ml,60mmol)中并在室温下搅拌2.5h。将反应混合物真空浓缩,得到1.3g(100%)产物,为黄褐色固体,该产物直接用于下一步骤。
1H NMR(甲醇-d4)δ1.25(t,J=7Hz,1H),1.47-1.52(m,1H),1.78(dd,J=8,5Hz,1H),2.21-2.32(m,1H),2.55-2.64(m,1H),2.99(dd,J=15,7Hz,1H),3.96(s,2H),4.06(s,3H),4.14(q,J=7Hz,2H),4.69-4.75(m,1H),5.13(d,J=10Hz,1H),5.33(d,J=17Hz,1H),5.71-5.83(m,1H),5.89(m,1H),7.44(m,1H),7.49-7.52(m,1H),8.51-8.55(m,1H),8.94-8.96(m,1H);13CNMR(甲醇-d4)δ14.62,23.08,30.89,34.73,36.97,41.03,52.42,57.11,60.17,62.70,81.13,100.06,103.07,117.02,118.53.122.70,126.86,134.74,143.15,146.75,166.62,167.71,169.37,171.18.
LC-MS(保留时间:0.94,方法D),MS m/z 426(M++1)。
步骤7:
室温下,向实施例374的步骤6的产物{NH2-P2[(4R)-(7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S-乙烯基Acca)-甲酸乙酯,双盐酸盐}(1.3g,2.61mmol)、N-BOC-L-叔-亮氨酸(BOCL-tBuGly)(0.94g,4.25mmol)、NMM(1.7ml,15.5mmol)在DMF(20mL)中的悬浮液内加入HATU(1.55g,3.40mmol)。将反应混合物搅拌过夜,用75%EtOAc-THF(300mL)稀释,依次用pH4.0缓冲液(2×50mL)、饱和碳酸氢钠水溶液(50mL)和盐水(50mL)洗涤,干燥(硫酸镁),经Biotage40M柱(洗脱液:15%-100%EtOAc/己烷)纯化,得到产物0.702g(42%){BOCNH-P3(L-t-BuGly)-P2[(4R)-(7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1-CO2Et,为泡沫状物。
1HNMR(甲醇-d4)δ1.06(s,9H),1.22-1.32(m,3H),1.28(s,9H),1.42-1.46(m,1H),1.73(dd,J=8,5Hz,1H),2.19-2.25(m,1H),2.67-2.72(m,1H),3.95(s,3H),4.03-4.07(m,1H),4.10-4.18(m,2H),4.20-4.24(m,1H),4.54(d,J=12Hz,1H),4.60-4.63(m,1H),5.11(dd,J=10,2Hz,1H),5.28-5.30(m,1H),5.43(m,1H),5.76-5.83(m,1H),6.50(d,J=9Hz,NH),6.93(d,J=5Hz,1H),7.10(dd,J=9,2Hz,1H),7.28(m,1H),7.99(m,1H),8.11(d,J=9Hz,1H),8.62(d,J=5H);
LC-MS m/z 639(保留时间:1.53方法D)。
步骤8:
向实施例374、步骤7的产物{BOCNH-P3(L-t-BuGly)-P2[(4R)-7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-甲酸乙酯}(702mg,1.1mmol)在THF(50mL)、甲醇(7mL)和H2O(22mL)中的悬浮液内加入LiOH(211mg,8.80mmol)。将反应混合物搅拌1天,酸化至中性pH,真空浓缩直至仅剩下水层。加入1.0N盐酸将所得的含水剩余物酸化至pH4.0,随后用固体NaCl饱和。将该含水混合物用乙酸乙酯和THF反复萃取,将合并的有机溶剂用盐水(50mL)洗涤,干燥(硫酸镁),过滤并真空浓缩,得到631mg(92%)产物BOCNH-P3(L-t-BuGly)-P2[(4R)-(7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CO2H,为固体物。
1H NMR(甲醇-d4)δ1.04(s,9H),1.22(s,9H),1.34-1.39(m,1H),1.67(dd,J=8,5Hz,1H),2.03-2.13(m,1H),2.43-2.49(m,1H),2.67-2.73(m,1H),3.96(8,3H),4.00-4.05(m,1H),4.15-4.21(m,1H),4.56-4.62(m,2H),5.02(d,J=10Hz,1H),5.20(d,J=17Hz,1H),5.52(m,1H),5.87-5.99(m,1H),6.47(d,J=8Hz,1H),6.91(s,1H),7.12(d,J=5Hz,1H),7.19(dd,J=9,2Hz,1H),7.31(d,J=2Hz,1H),8.22(d,J=9Hz,1H),8.72(d,J=5Hz,1H).
LC-MS(保留时间:1.44,方法D),MS m/z 611(M++1)。
步骤9:
向实施例374、步骤8的三肽酸(0.120g,0.195mmol)的THF(2mL)溶液中加入CDI(44.3mg,0.27mmol),将所得的溶液回流60min,冷却至室温。一次性加入环丁基磺酰胺(0.037g,0.273mmol),随后加入纯DBU(0.041mL,0.273mmol)。搅拌反应物24h,加入另一当量的CDI和环丁基磺酰胺,再将混合物搅拌48h。混合物用50%THF/EtOAc(200mL)稀释,用盐水饱和的pH4.0缓冲液(30mL)洗涤,干燥(硫酸镁)并真空浓缩。将剩余物溶于2ml50%THF-二氯甲烷中,加入75mg(0.39mmol)EDAC、48mg(0.39mmol)4-DMAP、58□L(0.39mmol)DBU和53mg(0.39mmol)环丁基磺酰胺,将混合物搅拌4天。使混合物经1000 Analtech PTLC板(20×40cM,洗脱液:2%甲醇/二氯甲烷)纯化一次,得到所需的产物:化合物374,BOCNH-P3(L-t-BuGly)-P2[(4R)-(7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丁烷,为泡沫状物2mg(2%):
1H NMR(甲醇-d4)δ1.07、1.08(两个s(旋转异构体),9H),1.20、1.21(两个s(旋转异构体),9H),1.41-1.48(m,1H),1.64-1.70(m,1H),1.72-1.91(m,2H),1.95-2.11(m,2H),2.23-2.37(m,2H),2.40-2.58(m,2H),2.72-2.75(m,1H),4.06(s,3H),4.12-4.17(m,2H),4.35-4.38(m,1H),4.58-4.62(m,1H),4.65-4.70(m,1H),5.16-5.18(m,1H),5.24-5.37(m,1H),5.69-5.76(m,2H),7.40-7.46(m,3H),8.35-8.40(m,1H),8.92(d,J=7Hz,1H)。
LC-MS(保留时间:1.58方法B),MS m/z 728(M++1)。
实施例375:化合物375的制备
化合物375
方案1
步骤1:
向实施例374、步骤5的产物{N-BOC-P2[(4R)-(7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CO2Et}(794mg,1.51mmol)在68ml 12%甲醇/THF中的溶液内加入218mg(9.08mmol)氢氧化锂在30ml水中的溶液,并将混合物搅拌16h。加入6N盐酸将pH调节至中性,浓缩直至仅剩下水,使用1N盐酸调节溶液pH至4,随后用50%THF-EtOAc(5×200-mL/份)萃取。将合并的有机层干燥(硫酸镁)并浓缩,得到产物752mg(100%){N-BOC-P2[(4R)-(7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CO2H}:
1H NMR(甲醇-d4)δ13.7-1.43(m,1H),1.39(s,9H),1.69-1.78(m,1H),2.16-2.24(m,1H),2.44-2.54(m,1H),2.64-2.74(m,1H),3.89-3.94(m,2H),3.96(s,3H),4.40-4.43(m,1H),5.11(d,J=10Hz,1H),5.31(d,J=17Hz,1H),5.40(m,1H),5.79-5.87(m,1H),6.91(s,1H),7.04(d,J=6Hz,1H),7.25(dd,J=9.1,2Hz,1H),7.29(m,1H),8.09(d,J=9.1Hz,1H),8.66(d,J=6Hz,1H).
LC-MS(保留时间:1.05,方法H)。MS m/z 498(M++1)。
步骤2:
将实施例375、步骤1的产物{N-BOC-P2[(4R)-(7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CO2H}(399.5mg,0.668mmol)的THF(4mL)溶液和CDI(434mg,2.68mmol)回流60min,冷却至室温。一次性加入环丙基磺酰胺(406mg,3.35mmol),随后加入纯DBU(0.50mL,3.35mmol)。搅拌反应物16h,用50%THF-EtOAc(200mL)稀释并用盐水饱和的pH4.0缓冲液(2×40mL)洗涤。将有机层干燥(硫酸镁),浓缩并经Biotage25M柱(甲醇/二氯甲烷,0%-15%)纯化,得到217mg(54%)所需的产物{N-BOC-P2[(4R)-(7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷}:
1H NMR(甲醇-d4)δ1.01-1.10(m,2H),1.11-1.18(m,1H),1.20-1.27(m,1H),1.39-1.48(m,1H),1.44(s,9H),1.87(dd,J=8,5Hz,1H),2.(01-2.38(m,2H),2.57(dd,J=14,7Hz,1H),2.91-2.96(m,1H),3.83-3.92(m,2H),3.94(s,3H),4.36-4.39(m,1H),5.11(d,J=10Hz,1H),5.29(d,J=17Hz,1H),5.38(m,1H),5.74-5.81(m,1H),6.91(d,J=5.5Hz,1H),7.20(dd,J=9.2,2.4Hz,1H),729(m,1H),8.07(d,J=9.2Hz,1H),8.60(d,J=5.5Hz,1H).
LC-MS(保留时间:1.28,方法I)。MS m/z 601(M++1)。
步骤3:
将全部实施例375、步骤2的产物{BOC-P2[(4R)-(7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷}(198mg,0.33mmol)溶于4N HCl/二_烷(4ml,16mmol)中并在室温下搅拌2h。浓缩反应混合物,得到粗产物,为黄褐色固体,该产物立即用于下一反应步骤中。
步骤4:
将实施例375、步骤3的粗产物{HN-P2[(4R)-(7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷,双盐酸盐}悬浮在10mL二氯甲烷中。室温下,向该混合物中加入N-BOC-L-叔-亮氨酸(BOCL-tBuGly)[120mg,0.52mmol]、HOAT(30mg,0.20mmol)、DIPEA(0.29ml,1.65mmol)和HATU(160mg,0.43mmol)。将反应混合物搅拌16h,用50%EtOAc-THF(300mL)稀释,用盐水饱和的pH4.0缓冲液(3×50mL)洗涤,干燥(硫酸镁),浓缩。剩余物经Isco35g柱(洗脱液:0%-15%甲醇/二氯甲烷)纯化,得到产物(130.1mg,47%),为Hunning碱盐(化合物375):
1H NMR(甲醇-d4)δppm 1.00-1.48(m,29H),1.47(s,9H),1.89(m,1H),2.26(m,1H),2.36(m,1H),2.69(m,1H),2.97(m,1H),3.25(q,J=7.43Hz,2H),3.74(m,2H),3.97(s,3H),4.10(m,1H),4.23(dd,J=19.68,9.92Hz,1H),4.57(m,2H),5.15(m,1H),5.31(m,1H),5.50(s,1H),5.77(m,1H),7.01(t,J=5.34Hz,1H),7.16(d,J=9.16Hz,1H),7.31(d,J=1.83Hz,1H),8.14(m,1H),8.67(d,J=5.49Hz,1H).
LC-MS(保留时间:1.49方法d),MS m/z 714(M++1)。
实施例376:化合物376的制备
化合物376
方案1
步骤1:
将化合物375(130mg)溶于EtOAc中,用pH4缓冲液和盐水洗涤数次,随后干燥(硫酸镁)。将粗品混合物经1000PTLC板(Analtech,20×40cm,洗脱液:3%甲醇/二氯甲烷)纯化两次,得到产物(化合物376)54mg(收率23%,以三肽酸计算):
1HNMR(甲醇-d4)δ0.88-1.00(m,2H),1.01-1.14(m,2H),1.03(s,9H),1.25(s,9H),1.34(dd,J=9,5Hz,1H),1.81-1.89(m,1H),2.06-2.13(m,1H),2.45-2.50(m,1H),2.65-2.75(m,1H),3.91(s,3H),3.98-4.11(m,1H),4.21-4.22(m,1H),4.46-4.50(m,1H),4.54-4.57(m,1H),4.97-5.02(m,1H),5.14-5.22(m,1H),5.33-5.41(m,1H),5.81-5.99(m,1H),6.87-6.95(m,1H),7.06-7.09(m,1H),7.25(m,1H),8.07-8.10(m,1H),8.59(d,J=5.2Hz,1H).
HRMS m/z(M-H):C35H46N5O9S的计算值:712.3016,实测值:712.3024;LC-MS m/e 714(保留时间:1.42,方法I)。
实施例377:化合物377的制备
化合物377
方案1
步骤1:
室温下,向实施例375、步骤2的全部1.0mmol产物{HN-P2[(4R)-(7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷,双盐酸盐}在20mL二氯甲烷中的悬浮液内加入352mg(1.30mmol)2-(S)-叔丁氧基羰基氨基-8-壬烯酸(购自RSP AminoAcids)、HOAT(82mg,0.60mmol)、DIPEA(0.74ml,5.0mmol)和HATU(494mg,1.30mmol)。将反应混合物搅拌16h,真空除去大部分的二氯甲烷。混合物用饱和的pH4.0缓冲液(150mL)稀释,并萃取入EtOAc(4×200mL)中。将合并的有机层干燥(硫酸镁),浓缩。将剩余物经Biotage 40M柱(洗脱液:0%-15%甲醇/二氯甲烷)纯化,得到产物(化合物377)574mg(76%):LC-MS m/z 754(保留时间:1.64,方法I)。
实施例378:化合物378的制备
化合物378
方案1
步骤1:
将实施例375、步骤2的全部0.34mmol产物{HN-P2[(4R)-(7-甲氧基喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丙烷,双盐酸盐}悬浮在3mL二氯甲烷中。室温下,向该混合物中加入N-BOC-L-缬氨酸(L-Val)(120mg,0.55mmol)、HOAT(30mg,0.20mmol)、DIPEA(0.29ml,1.65mmol)和HATU(160mg,0.43mmol)。将反应混合物搅拌16h,用饱和的pH4.0缓冲液(150mL)稀释,并萃取入EtOAc(3×200mL)中。将合并的有机层用盐水洗涤,干燥(硫酸镁)并浓缩。将剩余物经Isco35g柱(甲醇/二氯甲烷:0%-15%)纯化。该物质进一步经两δPTLC板(Analtech,20×40cm,洗脱液:3%甲醇/二氯甲烷)纯化,得到产物104.1mg(44%),化合物378:HRMSm/z(M-H)-:C34H44N5O9S的计算值:698.2860,实测值:698.2865。LC-MSm/e 700(保留时间:1.60,方法D)。
实施例379:化合物379的制备
化合物379
方案1
步骤1:
-30℃下,在5min内向2-吡啶甲酸(3.73g,30.3mmol)和2-氨基-4-甲氧基二苯甲酮(5.0g,30.3mmol)溶于吡啶(150mL)中的悬浮液内加入磷酰氯(3.7mL,45.4mmol)。在该温度下将反应混合物搅拌3hr并在室温下搅拌过夜。将反应混合物倒入冷水中并用EtOAc萃取(3×)。将合并的萃取液干燥,得到产物(7.67g,93%):1H NMR(甲醇-d4)δppm 2.65(s,3H),3.92(s,3H),6.78(m,1H),7.60(m,1H),8.00(m,lH),8.06(m,1H),8.21(d,J=7.63Hz,1H),8.59(t,J=2.29Hz,1H),8.76(d,J=3.97Hz,1H)。LC-MS(保留时间:1.56,方法D),MS m/z271(M++1)。
步骤2:
向N-(2-乙酰基-5-甲氧基-苯基)-吡啶-2-甲酰胺(2.90g,10.7mmol)的THF(50mL)悬浮液中加入t-BuOK/THF(1M,24mL,24mmol)。将反应混合物在70℃下加热3h并搅拌过夜。真空除去溶剂。向剩余物中加入冷水并用1.0N盐酸调节pH至4.6,过滤。固体剩余物经Biotage65M柱(甲醇/二氯甲烷:0-15%)纯化,得到产物(2.26g,84%):
LC-MS(保留时间:1.19,方法D),MS m/z 253(M++1)。
步骤3:
将7-甲氧基-2-吡啶-2-基-喹啉-4-醇(2.2g,8.71mmol)在磷酰氯(92mL)中的混合物回流3h,随后真空除去溶剂。向剩余物中加入冰水,用1.0N NaOH调节至pH>10,用EtOAc萃取(2×)。将合并的萃取液用水和盐水洗涤,干燥(硫酸镁),除去溶剂,得到产物,为黄色固体物(89%,2.1g):
DMSO-D6)δppm3.97(s,3H),7.40(dd,J=9.16,2.44Hz,1H),7.53(m,1H),8.01(m,1H),8.09(d,J=9.16Hz,1H),8.46(s,1H),8.56(d,J=7.93Hz,1H),8.74(d,J=3.97Hz,1H).
LC-MS(保留时间:1.50,方法D),MS m/z 271(M++1)。
步骤4:
向N-Boc-4-羟基脯氨酸(1.6g,6.7mmol)的DMSO(20mL)溶液中加入t-BuOK(1.9g,16.8mmol)。将得到的混合物搅拌1.5h,加入4-氯-7-甲氧基-2-吡啶-2-基-喹啉(2.0g,7.4mmol)和DMSO(10mL)。将反应混合物搅拌38h,用冷水稀释并用EtOAc/乙醚(1/4,2×)萃取。将水层酸化至pH4并用EtOAc/THF(5×)萃取。将合并的萃取液干燥(硫酸钠/硫酸镁),真空除去溶剂,剩余物经制备型HPLC(0-80%溶剂B)纯化,得到产物(1.6g,50%):LC-MS(保留时间:1.23,方法I),MS m/z 466(M++1)。
步骤5:
将实施例379、步骤4的产物{N-boc-(1R,2S)-1-氨基-2-乙烯基环丙烷甲酸乙酯}(0.21g,0.65mmol)在HCl/二_烷(4M,5mL,20mmol)中的溶液搅拌3h,真空除去溶剂。向剩余物中加入二氯甲烷(10mL)、二异丙基乙胺(0.4mL,3.23mmol)、HOBT(0.20g,1.35mmol)、Boc-(4R)-(2-环丙基-7-甲氧基-喹啉-4-氧代)-S-脯氨酸(0.20g,0.5mmol)和HATU(0.415g,1.07mmol)。将反应混合物搅拌过夜并用pH4.0缓冲液稀释,用EtOAc萃取。将萃取液干燥(硫酸镁)并经Biotage 40M柱(使用甲醇/二氯甲烷(0-15%)为洗脱液)纯化,得到产物(204.7mg,70%):
1H NMR(甲醇-d4)δppm 0.64(m,1H),0.96(m,2H),1.33(m,8H),1.39(m,9H),1.90(m,2H),2.18(m,1H),2.54(m,1H),2.81(m,1H),4.01(m,5H),4.44(d,J=28.99Hz,1H),5.08(m,1H),5.31(m,1H),5.57(s,1H),6.03(m,1H),6.94(s,1H),7.27(d,J=8.24Hz,1H),7.64(m,1H),7.92(m,1H),8.14(m,2H),8.66(s,1H),8.74(s,1H).
步骤6:
将P2 Boc-(4R)-(7-甲氧基-2-吡啶-2-基-喹啉-4-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2(1-环丙基甲基环丙烷-1-基)(实施例379、步骤5)(203mg,0.3mmol)的4M HCl/二_烷(3.5mL,14mmol)浆状物搅拌2h,真空除去溶剂。向剩余物中加入二氯甲烷(2mL)、二异丙基乙胺(0.63mL,3.6mmol)、Boc-L-叔-亮氨酸(83mg,0.36mmol)、HOAt(41mg,0.3mmol)和HATU(148mg,0.39mmol)。反应混合物在室温下搅拌7h并真空除去溶剂。剩余物经制备型HPLC(35-85%溶剂B)纯化,得到25.1mg(11%)所需的产物(化合物379):
1H NMR(甲醇-d4)δppm-0.05(m,1H),0.30(m,1H),0.66(m,1H),0.91(m,2H),1.05(s,9H),1.28(s,9H),1.67(m,8H),2.15(m,1H),2.58(m,1H),2.77(m,1H),3.96(s,3H),4.19(d,J=40.25Hz,2H),4.51(d,J=16.47Hz,2H),4.95(m,1H),5.15(m,1H),5.53(s,1H),5.89(dd,J=16.65,9.33Hz,1H),7.09(d,J=8.42Hz,1H),7.43(d,J=1.83Hz.1H),7.50(m,1H),7.82(s,1H),7.99(m,1H),8.10(d,J=9.15Hz,1H),8.48(d,J=7.68Hz,1H),8.72(s,1H).
LC-MS(保留时间:1.59,方法I),MS m/z 791(M++1)。
实施例380:化合物380的制备
化合物380
方案1
步骤1:
该实施例方案1的原料通过偶合部分B、实施例11、步骤3的产物和P1(1R,2S乙烯基Acca)-甲酸乙酯的氨基末端制备。将所述偶合产物P2 Boc-(4R)-(6-甲氧基-异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-甲酸乙酯(7.88g,14.99mmol)在4M HCl/二_烷(120mL,480mmol)中的浆状物搅拌2h,真空并在无水二_烷中共沸蒸馏除去溶剂。向剩余物中加入DMF(75mL)、N-甲基吗啉(6.27mL,57.07mmol)、Boc-L-叔-亮氨酸(5.20g,22.49mmol)和HATU(8.53g,22.49mmol)。将反应混合物在室温下搅拌过夜并进行如下后处理:将反应混合物倒入冰水中并用1.0N盐酸调节至pH为5,用EtOAc萃取。萃取液用碳酸氢钠水溶液和盐水洗涤,干燥(硫酸镁)并浓缩。将剩余物经Biotage 65M柱(EtOAc-己烷:5-100%)纯化,得到产物(8.07g,84%):保留时间:1.88方法I)MS m/z 639(M++1)。
步骤2:
向实施例384、步骤1的产物{Boc-NH-P3(L-叔-BuGly)-P2[(4R)-(6-甲氧基-异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-甲酸乙酯}(4.0g,6.26mmol)在THF(250mL)、甲醇(31mL)和H2O(125mL)中的悬浮液内加入LiOH(2.4g,100.2mmol)。将反应混合物搅拌过夜,随后用1.0N盐酸调节至pH7。真空除去有机溶剂。将含水剩余物酸化至pH4并用EtOAc萃取(2×)。将合并的有机溶剂干燥(硫酸钠/硫酸镁)并真空浓缩,得到产物(3.79g,99%):
1H NMR(甲醇-d4)□ppm 1.05(s,9H),1.25(m,1H),1.29(s,9H),1.46(m,1H),1.72(dd,J=8.24,5.19Hz,1H),2.23(q,J=8.55Hz,1H),2.68(dd,J=13.89,7.78Hz,1H),3.94(s,3H),4.05(dd,J=11.60,3.05Hz,1H),4.23(d,J=8.85Hz,1H),4.46(d,J=11.60Hz,1H),4.63(t,J=8.39Hz,1H),5.10(d,J=10.38Hz,1H),5.29(d,J=17.40Hz,1H),5.85(m,2H),7.10(d,J=9.16Hz,1H),7.19(s,1H),7.26(d,J=5.49Hz,1H),7.91(d,J=5.80Hz,1H),8.12(d,J=9.16Hz,1H).
保留时间:1.81方法I)MS m/z 611(M++1)。
步骤3:
将CDI(0.052g,0.32mmol)和实施例384、步骤2的产物{BOCNH-P3(L-t-BuGly)-P2[(4R)-6-甲氧基-异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CO2H}(0.130g,0.21mmol)在THF(2mL)中的溶液回流60min,冷却至室温。依次加入环丁烷磺酰胺(0.043g,0.32mmol)和纯DBU溶液(0.048mL,0.32mmol)。将反应物搅拌过夜,随后经注射器过滤器过滤并经制备型HPLC纯化(30%-100%溶剂B),得到所需的产物0.1422mg(92%):
1H NMR(甲醇-d4)δppm 1.04(s,9H),1.26(d,J=13.43Hz,9H),1.39(m.1H),1.85(dd,J=7.63.5.19Hz,1H).1.98(m,2H),2.26(m,4H),2.50(m,2H),2.61(m,1H),3.92(s,3H),4.05(m,1H),4.24(m,1H),4.33(m,1H),4.43(d,J=11.60Hz,1H),4.52(m,1H),5.13(m,1H),5.30(m,1H),5.71(m,1H),5.82(s,1H),7.08(d,J=8.85Hz,1H),7.18(s,1H),7.24(d,J=5.80Hz,1H),7.88(m,1H),8.08(d,J=9.16Hz,1H).
保留时间:1.89方法I)MS m/z 728(M++1)。
方案2
步骤4:
将实施例380、步骤3的产物{(BOCNH-P3(L-t-BuGly)-P2-(4R)-(6-甲氧基-异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丁基}(0.196mg,0.27mmol)溶于HCl/二_烷(5mL;20mmol)中并在室温下搅拌2h。真空除去溶剂,得到0.1887g(100%)题述产物,该产物直接用于下一步骤中。
步骤5:
向实施例380、步骤4的产物{NH2-P3(L-t-BuGly)-P2-(4R)-(6-甲氧基-异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丁基的盐酸盐}(0.037g,0.053mmol)和二异丙基乙胺(0.046mL,0.26mmol)在二氯甲烷(2mL)中的混合物内加入氯甲酸环戊酯(0.7M,0.151mL,0.069mmol)。将反应混合物搅拌过夜并经制备型HPLC(30%-100%溶剂B)纯化,得到所需的产物(化合物380)(0.0303g,77%):
1H NMR(甲醇-d4)δppm 1.03(s,9H),1.48(m,9H),1.86(dd,J=8.24,5.49Hz,1H),1.99(m,2H),2.27(m,4H),2.51(m,2H),2.60(dd,J=13.89,6.87Hz,1H),3.92(s,3H),4.05(dd,J=12.21,3.97Hz,1H),4.32(m,2H),4.41(d,J=11.90Hz,1H),4.53(m,1H),4.69(m,1H),5.12(d,J=10.38Hz,1H),5.29(d,J=17.09Hz,1H),5.71(m,1H),5.83(s,1H),7.11(d,J=9.46Hz,1H),7.19(s,1H),7.25(d,J=5.80Hz,1H),7.88(d,J=5.80Hz,1H),8.08(d,J=9.16Hz,1H).
保留时间:1.85方法H),MS m/z 740(M++1)。
实施例381:化合物381的制备
化合物381
步骤1
步骤1:
向实施例380、步骤4的产物{NH2-P3(L-t-BuGly)-P2-(4R)-(6-甲氧基-异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丁基的盐酸盐}(0.037g,0.053mmol)和异丙基乙胺(0.046mL,0.26mmol)在二氯甲烷(2mL)中的混合物内加入氯甲酸新戊酯(0.012mL,0.069mmol)。将反应混合物搅拌过夜并直接经制备型HPLC纯化(30%-100%溶剂B),得到所需的产物(化合物381)(0.0252g,64%):
1H NMR(甲醇-d4)δppm 0.84(s,9H),1.05(s,9H),1.40(m,1H),1.86(m,1H),2.00(m,2H),2.28(m,4H),2.51(m,2H),2.57(m,1H),3.39(d,J=10.07Hz,1H),3.55(d,J=10.38Hz,1H),3.92(s,3H),4.05(m,1H),4.33(m,2H),4.41(d,J=11.29Hz,1H),4.53(m,1H),5.12(d,J=10.07Hz,1H),5.29(d,J=17.09Hz,1H),5.71(m,1H),5.82(s,1H),7.10(d,J=9.16Hz,1H),7.19(s,1H),7.25(d,J=5.80Hz,1H),7.88(d,J=5.80Hz,1H),7.97(s,1H),8.07(d,J=8.85Hz,1H).
保留时间:1.89方法H),MS m/z 742(M++1)。
实施例382:化合物382的制备
化合物382
方案1
步骤1:
向实施例380、步骤4的产物{NH2-P3(L-t-BuGly)-P2-(4R)-(6-甲氧基-异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丁基的盐酸盐}(0.037g,0.053mmol)和二异丙基乙胺(0.046mL),0.26mmol)在二氯甲烷(2mL)中的混合物内加入二碳酸二-叔戊基酯(0.0169g,0.069mmol)。将反应混合物搅拌过夜并直接经HPLC(30%-100%溶剂B)纯化,得到所需的产物(化合物382)(0.0175g,44%):
1H NMR(甲醇-d4)δppm 0.79(t,J=6.87Hz,3H),1.04(s,8H),1.21(s,3H),1.23(s,3H),1.41(m,2H),1.64(m,2H),1.83(m,1H),2.00(m,2H),2.26(m,4H),2.51(m,2H),2.60(m,1H),3.92(s,3H),4.07(m,1H),4.24(m,1H),4.33(m,1H),4.43(d,J=11.60Hz,1H),4.52(m,1H),5.13(m,1H),5.29(m,1H),5.71(m,1H),5.82(s,1H),7.09(d,J=8.85Hz,1H),7.18(s,1H),7.25(d,J=5.49Hz,1H),7.88(d,J=5.80Hz,1H),8.08(d,J=8.85Hz,1H).
保留时间:1.90,方法H),MS m/z 742(M++1)。
实施例383:化合物383的制备
化合物383
方案1
步骤1
向实施例380、步骤4的产物{NH2-P3(L-t-BuGly)-P2-(4R)-(6-甲氧基-异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丁基的盐酸盐}(0.037g,0.053mmol)和异丙基乙胺(0.046mL、0.26mmol)的二氯甲烷(2mL)混合物中加入叔丁基-异氰酸酯(isocianate)(0.008mL,0.069mmol)。将反应混合物搅拌过夜并直接经制备型HPLC(30%-100%溶剂B)纯化,得到所需的产物(化合物383)(0.024g,62%):
1H NMR(甲醇-d4)δppm 1.05(s,9H),1.19(s,9H),1.37(m,1H),1.85(dd,J=8.09,5.34Hz,1H),2.00(m,2H),2.26(m,4H),2.50(m,2H),2.58(m,1H),3.92(s,3H),4.06(m,1H),4.32(m,2H),4.49(m,2H),5.11(d,J=10.38Hz,1H),5.27(d,J=17.40Hz,1H),5.69(m,1H),5.83(s,1H),7.08(dd,J=9.16,2.44Hz,1H),7.17(d,J=2.44Hz,1H),7.24(d,J=5.80Hz,1H),7.87(d,J=6.10Hz,1H),8.12(d,J=8.85Hz,1H).
保留时间:1.77,方法H),MS m/z 727(M++1)。
实施例384:化合物384的制备
化合物384
方案1
步骤1:
将二异丙基乙胺(0.031mL,0.018mmol)、碳酸N,N’-二琥珀酰亚胺酯(0.0274g,0.107mmol)和实施例380、步骤4的产物{NH2-P3(L-t-BuGly)-P2-(4R)-(6-甲氧基-异喹啉-1-氧代)-S-脯氨酸]-P1(1R,2S乙烯基Acca)-CONHSO2环丁基的盐酸盐}(0.050g,0.0714mmol)在THF(2mL)中的悬浮液在80℃下超声15min。加入KH(0.046g,1.14mmol)和1-甲基环戊醇(0.079mL,0.714mmol)。将反应混合物搅拌20min并进行如下后处理:用冷水稀释,调节pH至4,用EtOAc萃取,将萃取液干燥(硫酸镁)并将剩余物经制备型HPLC(30%-100%溶剂B)纯化,得到所需的产物(化合物384)(0.018g,33%):(甲醇-d4)δppm
δppm 1.04(s,9H),1.29-1.79(m,10H),1.84(m,2H),1.99(m,3H),2.26(m,4H),2.49(m,2H),2.60(dd,J=13.73,7.02Hz,1H),3.92(s,3H),4.05(dd,J=11.29,2.44Hz,1H),4.26(s,1H),4.32(m,1H),4.44(d,J=11.90Hz,1H),4.52(m,1H),5.12(d,J=10.07Hz,1H),5.28(d,J=16.79Hz,1H),5.71(m,1H),5.82(s,1H),7.10(d,J=8.85Hz,1H),7.19(s,1H),7.26(d,J=5.80Hz,1H),7.88(d,J=5.80Hz,1H),8.08(d,J=9.16Hz,1H).
LC-MS保留时间:1.91方法H),MS m/z 754(M++1)。
实施例385:化合物385的制备
化合物385
方案1
步骤1:
将2-氰基甲基-4-甲氧基-苯甲酸甲酯(1.9g)和TsOH.H2O(0.15g,mmol)在吗啉(5mL)中的悬浮液回流4h,真空除去溶剂。将剩余物在EtOAc/己烷及数滴甲醇中重结晶,得到产物(0.43g,17%):LC-MS保留时间:1.07方法H),MS m/z 266(M++1)。
步骤2:
将6-甲氧基-3-吗啉-4-基-异喹啉-1-醇(0.298g,1.15mmol)在磷酰氯(20mL)中的混合物回流2h,真空除去溶剂,加入冷水。加入1.0N NaOH调节pH>11。水层用EtOAc萃取。将萃取液干燥(硫酸镁),真空除去溶剂得到产物(0.299g,94%):LC-MS保留时间:1.68方法H),MS m/z 279(M++1)。
步骤3:
将1-氯-6-甲氧基-3-吗啉-4-基-异喹啉(0.050g,0.18mmol)和氟化四丁基_·氟化氢(0.8g,2.8mmol)[Synlett 1992,(4),345-6]的混合物在140℃下,微波加热10min。将反应混合物用乙酸乙酯稀释并经ISCO 25g预柱(顶部有一层硅胶)过滤,除去溶剂得到产物(0.037mg,77%):1H NMR(氯仿-D)δppm 3.48(m,4H),3.84(m,4H),3.89(s,3H),6.46(d,J=1.22Hz,1H),6.85(s,1H),6.90(dd,J=9.16,2.44Hz,1H),7.82(d,J=8.85Hz,1H)。LC-MS保留时间:1.56方法H),MSm/z 263(M++1)。
步骤4:
将1-氟-6-甲氧基-3-吗啉-4-基-异喹啉(0.037g,0.14mmol)、三氯化镧(0.020g,0.8mmol)、t-BuOK(1M/THF,0.32mL,0.3mmol)和Boc-NH-P3(L-叔-BuGly)-P2[(4R)-4-羟基-S-脯氨酸]-P1(R,2S乙烯基Acca)-CONHSO2环丙烷(0.045g,0.08mmol)在THF(3mL)中的混合物搅拌3天。反应混合物用甲醇稀释,经注射器过滤器过滤并经制备型HPLC纯化得到产物,为浅黄色泡沫状物(0.0158g,24%):
1H NMR(甲醇-d4)δppm 1.03(s,9H),1.24(m,4H),1.31(s,9H),1.43(m,2H),1.88(m,1H),2.24(m,2H),2.59(dd,J=13.43,6.71Hz,1H),2.94(m,1H),3.47(m,4H),3.83(m,4H),3.86(s,3H),4.08(m,1H),4.28(s,1H),4.48(m,1H),5.12(d,J=10.38Hz,1H),5.29(d,J=16.48Hz,1H),5.76(m,2H),6.74(d,J=9.16Hz,1H),6.94(s,1H),7.85(d,J=8.85Hz,1H),9.19(s,1H).
保留时间:1.86方法H),MS m/z 799(M++1)。
实施例386:化合物386的制备
化合物386
使用本文中描述的方法制备化合物386。
部分I:
在部分I中的所有化合物均采用LC/MS方法分析测试,使用以下条件。
方法A:Xtela C18 S7 3.0×50mm
梯度:100%溶剂A/0%溶剂B至0%溶剂A/100%溶剂B
梯度时间:3min
保持时间:1min
流速:4mL/min
检测器波长:220nm
溶剂A:10%甲醇/90%H2O/0.1%TFA
溶剂B:10%H2O/90%甲醇/0.1%TFA
实施例410:化合物410的制备
化合物410
方案1
步骤1:
在氮气气氛中,向Boc-L-羟基脯氨酸(0.723g,3.13mmol)的DMSO溶液中加入叔丁醇钾(0.808g,7.2mmol)。在室温下搅拌悬浮液1.5小时,分两批加入4-氯-6-甲基-2-(三氟甲基)喹啉(0.916g,3.75mmol)。混合物在室温下搅拌3小时,使用1.3当量HCl(1N)中和反应液。加入pH4.0的缓冲溶液并将pH调节至pH4-5。水层用乙酸乙酯萃取(3×25mL),将合并的有机层用盐水(20mL)洗涤并经硫酸镁干燥,得到题述化合物,为白色固体物(粗产物收率未作计算)。将粗产物用于下一步骤中。
LC/MS Rt-min(MH+):2.48(441.5)(方法A)。
步骤2:
将步骤1的粗产物4-(6-甲基-2-三氟甲基-喹啉-4-氧基)-吡咯烷-1,2-二甲酸1-叔丁酯(估计3.13mmol)在THF(10mL)和甲醇(10mL)中的溶液冷却至0℃。在氮气气氛中,将TMSCN2的己烷溶液(2M,~1.3eq)缓慢加入所述搅拌着的溶液中,直至溶液中不再有气体释放出。随后将完全反应的溶液真空浓缩,经Biotage40M柱(用10%-30%乙酸乙酯/己烷洗脱)纯化,得到纯题述化合物,为白色泡沫状物(976mg,步骤1和2的总收率69%)。
LC/MS Rt-min(MH+):2.60(477)(方法A)。
步骤3:
将步骤2的产物(0.976g,2.15mmol)在DCM(7mL)和TFA(6.62mL)中的溶液在室温下搅拌1小时。真空除去溶剂并将剩余物悬浮在1N HCl/乙醚(8mL)中,温和搅拌并真空浓缩。重复该步骤并将所得的产物用油泵抽真空过夜,定量得到白色固体物。
1H NMR:(DMSO-d6)δ2.50(s,3H),2.57-2.6(m,1H),2.66-2.71(m,1H),3.62-3.65(br d,J=15Hz,1H),3.80-3.81(m,4H),4.8(br s,1H),5.7(s,1H),7.46(s,1H),7.72-7.75(d,J=7.5Hz,1H),7.98-7.8(d,J=8.5Hz,1H),8.24(s,1H),9.54(br s,1H);
LC/MS Rt-min(MH+):1.61(355)(方法A)。
步骤4:
在氮气气氛中,将步骤3的产物(估计定量收率,2.746mmol)加入BOC-叔丁基-L-甘氨酸(0.635g,2.746mmol)的DCM(20mL)溶液中。该步骤完成后,加入HOBt(0.408g,3.02mmol)、DIPEA(3.35mL,19.2mmol)和HBTU(1.56g,4.12mmol)。立即得到桃红色溶液,将反应物在室温下搅拌过夜。将10mL DCM加入到所述完全反应的反应物中以增加体积,将反应物用pH4.00的缓冲溶液(25mL)猝灭。使用1N HCl将混合物酸化至pH4.5,水相用DCM(3×20mL)萃取。将有机相用pH4.00的缓冲溶液(20mL)、饱和NaOH(25mL)和盐水(20mL)洗涤两次,随后用硫酸镁干燥。将所得的溶液真空浓缩并经Biotage40M柱(用10%-40%乙酸乙酯/己烷洗脱)纯化,得到纯题述化合物,为白色固体物(1.11g,89%)。
1H NMR:(DMSO-d6)δ0.96-1.02(旋转异构体,3∶2,s,18H),2.27-2.33(m,1H),2.50(s,3H),2.68-2.72(m,1H),3.67(s,3H),4.02-4.04(m,1H),4.43-4.45(br d,J=15Hz,2H),4.58-4.61(t,1H),5.60(br s,1H),6.72-6.74 (br d,J=15Hz,1H),7.38(s,1H),7.68-7.73(m,1H),7.95-7.97(m,2H);
LC/MS Rt-min(MH+):2.61(590)(方法A)。
步骤5:
通过加热和超声将LiOH(0.138g,5.78mmol)溶于水(10mL)中。
将该LiOH溶液和甲醇(10mL)加入步骤4的纯物质(1.09g,1.93mmol)的THF(10mL)溶液中。混合物立即变成鲜蓝色。将反应物在室温下搅拌3小时,随后用1N HCl(5.78mL,5.78mmol)酸化。反应物用pH4.00缓冲溶液猝灭并使用1N氢氧化钠水溶液将pH调节至pH4.5。水层用EtOAc萃取(3×25mL),用盐水(20mL)洗涤并经硫酸镁干燥。过滤溶液,将滤液真空浓缩并抽真空过夜。将粗产物(957mg,90%收率)直接用于下一步骤中。
LC/MS Rt-min(MH+):2.51(577)(方法A)。
步骤6:
将步骤5的粗产物(60mg,0.11mmol)溶于DCM(5mL)中,加入N-(1R)-氨基-2(S)-乙烯基-环丙烷羰基)-环丙烷磺酰胺·盐酸盐(实施例1,步骤8)(0.029g,0.11mmol)。接着在氮气气氛中依次加入DIPEA(0.094mL,0.541mmol)和HATU(0.0575,0.151mmol)。将反应物在室温下搅拌大约8h。向溶液中加入10mL DCM以增加体积,将反应物用pH4.00缓冲溶液(10mL)猝灭。使用1N HCl将混合物酸化至pH为4-5,水相用DCM(3×20mL)萃取。有机相用pH4.00缓冲溶液(10mL)和盐水(10mL)洗涤两次,随后经硫酸镁干燥。将所得的溶液真空浓缩并经Biotage 12M柱(用10%-40%丙酮/己烷洗脱)纯化。该纯化操作得到化合物410,为白色粉末(29mg,35%)。
1H NMR:(DMSO-d6)δ0.976-1.12(m,24H),1.36-1.39(m,1H),1.70-1.72(m,1H),2.15-2.25(m,2H),2.50-2.52(m,4H),2.91-2.96(m,1H),3.97-4.01(m,2H),4.40-4.47(m,2H),5.09-5.11(d,J=10Hz,1H),5.21-5.24(d,J=15Hz,1H),5.59-5.66(m,2H),6.65-6.67(d,NH),7.43(s,1H),7.72-7.74(d,J=10Hz,1H),7.90(s,1H),7.98-8.0(d,J=10Hz,1H),8.87(s,NH),10.35(s,NH);LC/MS Rt-min(MH+):2.65(789.61)(方法A)。
实施例411:化合物411的制备
化合物411
步骤1:
在氮气气氛中,向Boc-L-羟基脯氨酸(2.00g,8.65mmol)的THF(25mL)溶液中加入NaH(0.795g,19.87mmol)。将悬浮液在室温下搅拌15分钟,加入4,6-二氯嘧啶(2.58g,17.30mmol,分两批加入)。将混合物在室温下搅拌1小时,使用1.3当量HCl(1N)中和反应物。加入pH4.0缓冲溶液将pH调节至pH5。使用乙酸乙酯萃取水相(3×25mL),有机层用盐水(20mL)洗涤并经硫酸镁干燥,得到题述化合物,为白色固体物(粗产物收率未作计算)。该粗产物直接用于下一步骤中。
LC/MS Rt-min(MH+):1.91(366.2)(方法A)。
步骤2:
将步骤1的粗产物(大约8.65mmol)在THF(40mL)和2,2-二甲砜基丙烷(methonal)(40mL)中的溶液冷却至0℃。在氮气气氛中,TMSCN2的己烷溶液(2M,~1.3eq)缓慢加至该搅拌着的溶液中,直至溶液中不再有气体释放出。随后将该完全反应的溶液真空浓缩,经Biotage40M柱(用20%-40%乙酸乙酯/己烷洗脱)纯化,得到纯题述化合物,为白色泡沫状物(497mg,步骤2a-2b总收率16%)。
1H NMR:(DMSO-d6)δ1.34-1.38(旋转异构体,2∶1,9H),2.25-2.29(m,1H),2.53-2.56(m,1H),3.58-3.75(m,2H),3.69(s,3H),4.28-4.33(m,1H),5.59(s,1H),7.24(s,1H),8.69(s,1H);
LC/MS Rt-min(MH+):2.08(380.14)(方法A)。
步骤3:
在室温下,将步骤2的纯产物(472mg,1.83mmol)在DCM(3mL)和TFA(5.65mL)中的溶液搅拌1小时。真空除去溶剂,将剩余物悬浮在1N HCl/乙醚(7.33mL)中,温和搅拌,真空浓缩。重复该操作,将所得的产物在油泵抽真空下过夜,定量得到白色固体物。
LC/MS Rt-min(MH+):0.55(258.35)(方法A)。
步骤4:
在氮气气氛中,将步骤3的产物(估计为定量收率,1.83mmol)加至BOC-叔丁基-L-甘氨酸(0.424g,1.83mmol)的DCM(11mL)溶液中。随后加入HOBt(0.272g,2.02mmol)、DIPEA(2.23mL,12.82mmol)和HBTU(1.04g,2.75mmol)。将反应物在室温下搅拌15h。向溶液中加入15mL DCM以增加体积,将反应物用pH4.00缓冲溶液(15mL)猝灭。使用1N HCl将混合物酸化至pH为4.5,水相用DCM(3×20mL)萃取。有机相依次用pH4.00缓冲溶液(20mL)、饱和的NaOH(25mL)和盐水(20mL)洗涤两次,经硫酸镁干燥。将所得的溶液真空浓缩并经Biotage 40S柱(用20%-50%乙酸乙酯/己烷洗脱)纯化。该纯化步骤得到纯的题述化合物,为白色固体物(454mg,53%)。
1H NMR:(DMSO-d6)δ0.94(s,9H),1.25(s,9H),2.21-2.27(m,1H),2.48-2.55(m,1H),3.64(s,3H),3.86-4.02(m,2H),4.29-4.31(d,J=10Hz,1H),4.46-4.49(t,1H),5.75(brs,1H),6.72-6.74(d,NH),7.12(s,1H),8.71(s,1H);
LC/MS Rt-min(MH+):2.27(493.5)(方法A)。
步骤5:
通过加热和超声将LiOH(0.0141g,0.589mmol)溶于水(7.5mL)中。向该LiOH溶液中加入步骤4的纯物质(252mg,0.535mmol)的THF(7.5mL)溶液,在室温下搅拌。3小时后反应完全。用pH4.0缓冲液猝灭并用1N HCl酸化至pH为约4.5。水相用EtOAc(3×25mL)萃取,有机相用盐水(20mL)洗涤并经硫酸镁干燥。将滤液真空浓缩并保持在真空下过夜。粗产物(231mg,收率95%)直接用于下一步骤中。
1H NMR(DMSO-d6)δ0.94(s,9H),1.25(s,9H),2.14-2.22(m,1H),2.50-2.54(m,1H),3.84-3.876(d,J=150Hz,1H),3.97-3.99(d,J=10Hz,1H),4.27-4.30(d,J=15Hz,1H),4.37-4.40(t,1H),5.63(br s,1H),6.69-6.71(d,NH),7.12(s,1H),8.71(s,1H),12-56(br s,OH);
LC/MS Rt-min(MH+):2.24(479.5)(方法A)。
步骤6:
将步骤5的纯物质(80mg,0.146mmol)和苯基硼酸(0.0178g,0.146mmol)在DMF(2mL)中溶剂化。将溶液置于氮气气氛中并加入2M碳酸钠水溶液(0.146mL,0.292mmol)。加入5%摩尔的四(三苯基膦)合钯(0)(8.44mg,0.0073mmol),并通过微波(使用PersonalChemisty Emrys Optimizer的设备)将混合物在140℃下加热50分钟。反应完全后将沉积出的钯黑除去。用1当量的1N HCl将混合物酸化,经注射器过滤,使用甲醇萃取产物。产物经制备型HPLC(柱-4 Xterra S530×75mm,溶剂-70% A/30% B-30% A/70% B(其中溶剂A为10%甲醇、90%H2O、0.1%TFA,溶剂B为90%甲醇、10%H2O、0.1%TFA),梯度时间-15min,保持时间-1min.,流速-40mL/min,纯产物保留时间-10.45-11.37)纯化。用1N NaOH中和含所需产物的洗脱液,并置于离心真空(speed vacuum)下约4h。合并各洗脱液,加入pH4.0的缓冲液(15mL)。使用1N HCl将pH调节至pH4-5,水层用乙酸乙酯(3×20mL)萃取。有机层用盐水(15mL)洗涤,经硫酸镁干燥并真空浓缩。产物在油泵抽真空下干燥过夜,得到粘稠油状物(37mg,50%)。
LC/MS Rt-min(MH+):2.37(499.3)(方法A)。
步骤7:
将实施例411,步骤6的产物(36.7mg,0.061mmol)溶于DCM(4mL)中,加入N-(1(R)-氨基-2(S)-乙烯基-环丙烷羰基)-环丙烷磺酰胺·盐酸盐(实施例1,步骤8)(0.0164g,0.161mmol)。接着在氮气气氛中依次加入DIPEA(0.0534mL,0.307mmol)和HATU(0.0326,0.0858mmol)。将反应物在室温下搅拌3h。向溶液中加入10mL DCM以增加体积,用pH4.00缓冲溶液(10mL)猝灭反应物。使用1N HCl将混合物酸化至pH为4-5,水相用DCM(3×15mL)萃取。有机相用pH4.00缓冲溶液(10mL)和盐水(10mL)洗涤两次,随后经硫酸镁干燥。将所得的溶液真空浓缩并经Biotage 12M柱(用20%-40%丙酮/己烷洗脱)纯化。该纯化操作得到纯化合物411,为白色粉末(7mg,15%)。
1H NMR:δ1.07-1.46(m,24H),1.87-1.90(m,1H),2.22-2.32(m,2H),2.51-2.55(m,1H),2.92-2.97(m,1H),4.04-4.07(d,J=15Hz,1H),4.20-4.22(d,J=10Hz,1H),4.36-4.38(d,J=10Hz,1H),4.47-4.50(t,1H),5.12-5.14(d,J=10Hz,1H),5.29-5.33(d,J=20Hz,1H),5.73-5.82(m,2H),6.59-6.60(d,NH),7.29(s,1H),7.50-7.5 1(m,3H),8.03-8.05(m,2H),8.81(s,1H);
LC/MS Rt-min(MH+):2.50(711.4)(方法A)。
实施例412:化合物412的制备
化合物412
步骤1:
将实施例411、步骤5的产物(80mg,0.146mmol)在DMF(2mL)中溶剂化,并向该溶液中加入2-噻吩硼酸(0.028g,0.219mmol)。将反应物置于氮气气氛中,加入2M碳酸钠水溶液(0.146mL,0.292mmol)和5%摩尔的四(三苯基膦)合钯(0)(8.44mg,0.0073mmol)。使用微波(使用Personal Chemistry Emrys Optimizer设备)将反应物在150℃下加热30分钟。反应完成后将沉积出的钯黑除去。用1当量的1N HCl将混合物酸化,经注射器过滤,使用甲醇萃取产物。产物经制备型HPLC(柱-4 Xterra S5 30×75mm,溶剂-70% A/30% B-30% A/70% B(其中溶剂A为10%甲醇、90%H2O、0.1%TFA,溶剂B为90%甲醇、10%H2O、0.1%TFA),梯度时间-15min.,保持时间-1min,流速-40mL/min,纯产物保留时间-10.45-11.37)纯化。用1N NaOH中和含所需产物的洗脱液,并置于离心真空(speed vacuum)下约4h。合并各洗脱液,加入pH4.0的缓冲液(15mL)。使用1N HCl将pH调节至pH为4-5,水层用乙酸乙酯(3×20mL)萃取。有机层用盐水(15mL)洗涤,经硫酸镁干燥并真空浓缩。产物在油泵抽真空下干燥过夜,得到油状液体(37mg,50%)。
LC/MS Rt-min(MH+):2.37(499.3)(方法A)。
步骤2:
将实施例412、步骤1的产物(39mg,0.0773mmol)溶于DCM(4mL)中,加入N-(1(R)-氨基-2(S)-乙烯基-环丙烷羰基)-环丙烷磺酰胺·盐酸盐(实施例1,步骤8)(0.0206g,0.0773mmol)。接着在氮气气氛中依次加入DIPEA(0.015mL,0.387mmol)和HATU(0.0411g,0.108mmol)。将反应物在室温下搅拌15h。向溶液中加入10mL DCM以增加体积,将反应物用pH4.00缓冲溶液(10mL)猝灭。使用lN HCl将混合物酸化至pH4-5,水相用DCM(3×15mL)萃取。有机相用pH4.00缓冲溶液(10mL)和盐水(10mL)洗涤两次,随后经硫酸镁干燥。将所得的溶液真空浓缩并经Biotage 12M柱(用20%-50%丙酮/己烷洗脱)纯化。该纯化操作得到纯化合物412,为白色粉末(4mg,7%)。
1H NMR:δ1.04-1.29(m,24H),1.45-1.47(m,1H),1.89-1.91(m,1H),2.26-2.31(m,2H),2.51-2.53(m,1H),2.92-2.95(m,1H),4.05-4.07(d,J=10Hz,1H),4.22-4.24(d,J=10Hz,1H),4.38-4.40(d,J=10Hz,1H),4.48-4.52(m,1H),5.15-5.17(d,J=10Hz,1H),5.32-5.36(d,J=20Hz,1H),5.76-5.83(m,2H),6.65-6.67(d,NH),7.19-7.21(m,2H),7.66-7.67(d,J=5Hz,1H),7.86-7.87(d,J=5Hz,1H),8.69(s,1H);
LC/MS Rt-min(MH+):2.45(739.4)(方法A)。
实施例413:化合物413的制备
化合物413
步骤1:
将2-溴-6-氯吡啶(3.0g,15.55mmol)和苯基硼酸(1.896g,15.55mmol)在EtOH、甲苯和水(2∶1∶1;120mL)的混合物中溶剂化。在氮气气氛中加入1M碳酸钠水溶液(15.55mL,31.10mmol)和5%摩尔的四(三苯基膦)合钯(0)(0.896g,0.775mmol)。反应物在90℃下回流1小时。加入水(20mL)猝灭反应物,水层用乙醚(4×25mL)萃取。随后将有机层用盐水洗涤,经硫酸镁干燥并真空浓缩。粗混合物经Biotage40S柱(用2%-10%乙酸乙酯/己烷洗脱)纯化,得到纯的题述化合物(1.45g,74%)。
1H NMR:δ7.24-7.26(m,1H),7.42-7.48(m,3H),7.63-7.70(m,2H),7.98-8.00(m,2H);
LC/MS Rt-min(MH+):2.04(190.18)(方法A)。
步骤2:
向在步骤1中得到的纯固体(3.27g,17.24mmol)中加入TFA(20mL)。在氮气气氛中,将30%的H2O2溶液(5.55mL,48.9mmol)缓慢滴入搅拌着的溶液中。将反应物在100℃下加热回流3小时,向溶液中再加入0.5当量的H2O2(2.27mL,25mmol)。反应物继续在100℃下搅拌2h。将烧瓶冷却至室温,接着将溶液真空浓缩至原始体积的约一半。加入水(40mL)以猝灭反应物,水层用乙酸乙酯(5×30mL)萃取。有机层用盐水(20mL)洗涤一次,经硫酸镁干燥并真空浓缩。粗产物经Biotate 40M柱(用10%-75%乙酸乙酯/己烷洗脱)纯化,得到浅黄色液体(0.81g,23%)。还回收到纯原料以重复使用(1.895g,58%)。
1H NMR:δ7.40-7.42(t,1H),7.49-7.50(m,3H),7.59-7.61(d,J=10Hz,1H),7.77-7.82(m,3H);
LC/MS Rt-min(MH+):1.12(206.37)(方法A)。
步骤3:
将步骤2的纯化产物(0.81g,3.94mmol)加入SOCl2(25mL)的溶液中并在60℃下搅拌2h。随后将温度升高至80℃使反应完全,接着再加热30min。将溶液真空浓缩并用冰小心猝灭。使用10N NaOH将pH调节至pH为4-5,将烧瓶保持在冰浴中。水相用乙醚(4×25mL)萃取,有机层用盐水(20mL)洗涤,经硫酸镁干燥并真空浓缩。粗黄色液体经Biotage 40S柱(用2%-10%乙酸乙酯/己烷洗脱)纯化。得到浅黄色的粘稠液体(538mg,61%)。
1H NMR:δ7.52-7.55(m,3H),7.73(s,1H),8.10-8.11(m,2H),8.17(s,1H);
LC/MS Rt-min(MH+):2.62(225.33)(方法A)。
步骤4:
在氮气气氛中,向Boc-L-羟基脯氨酸(555mg,2.40mmol)的DMSO(4mL)溶液中加入叔丁醇钾(0.619g,5.52mmol)。在室温下搅拌悬浮液45min,分两批加入2,4-二氯-6-苯基-吡啶(在步骤3中纯化)(538mg,2.40mmol)。在室温下搅拌混合物2小时,使用1.3当量HCl(1N)中和反应物。加入pH4.0的缓冲溶液,并将pH调节至pH4-5。使用乙酸乙酯(3×25mL)萃取水相,有机相用盐水(20mL)洗涤并经硫酸镁干燥,得到题述化合物,为白色固体物(粗产物=962mg)。粗产物直接用于下一步骤中。
LC/MS Rt-min(MH+):2.55(419.27)(方法A)。
步骤5:
将步骤4的粗产物(估计2.4mmol)在THF(10mL)和2,2-二甲砜基丙烷(methonal)(10mL)中的溶液冷却至0℃。在氮气气氛中,将TMSCN2的己烷溶液(2M,~1.3eq)缓慢加至搅拌着的溶液中,直至溶液中不再有气体释放出。随后将所述完全反应的溶液真空浓缩,经Biotage 25S柱(用10%-50%乙酸乙酯/己烷洗脱)纯化,得到纯题述化合物,为白色泡沫状物(503mg,步骤4d-4e的总收率48%)。
1H NMR:(DMSO-d6)δ1.35-1.38(旋转异构体,3∶2,9H),2.21-2.26(m,1H),2.50-2.58(m,1H),3.67-3.70(m,5H),4.26-4.34(m,1H),5.35(br s,1H),7.14-7.15(m,1H),7.47-7.55(m,4H),8.05-8.09(m,2H);
LC/MS Rt-min(MH+):2.69(455.52)(方法A)。
步骤6:
将步骤5的纯产物(503mg,1.16mmol)在DCM(2.5mL)和TFA(3.58mL)中的溶液在室温下搅拌1小时。真空除去溶剂并将剩余物悬浮在1N HCl/乙醚(6mL)中,温和搅拌并真空浓缩。重复该步骤,将所得的产物采用油泵抽真空,定量得到白色固体物。粗产物直接用于下一步骤中。
步骤7:
在氮气气氛中,将步骤6的产物(估计为定量收率,1.83mmol)加入BOC-叔丁基-L-甘氨酸(0.424g,1.83mmol)的DCM(11mL)溶液中。随后加入HOBt(0.272g,2.02mmol)、DIPEA(2.23mL,12.82mmol)和HBTU(1.04g,2.75mmol)。将反应物在室温下搅拌15h。向溶液中加入15mL DCM以增加体积,反应物用pH4.00缓冲溶液(15mL)猝灭。使用1N HCl将混合物酸化至pH4.5,水相用DCM(3×20mL)萃取。有机相用pH4.00缓冲溶液(20mL)、饱和的NaOH(25mL)和盐水(20mL)洗涤两次,经硫酸镁干燥。将所得的溶液真空浓缩并经Biotage 40S柱(用20%-50%乙酸乙酯/己烷洗脱)纯化。该纯化步骤得到纯的题述化合物,为白色固体物(454mg,53%)。
1H NMR:(DMSO-d6)δ0.96(s,9H),1.19(s,9H),2.17-2.29(m,1H),2.48-2.58(m,1H),3.65(s,3H),3.81-3.89(m,1H),4.05-4.08(m,1H),4.21-4.26(m,1H),4.44-4.50(t,1H),5.45(brs,1H),6.72-6.75(d,J=15Hz,1H),7.09(s,1H),7.46-7.51(m,4H),8.02-8.06(m,2H);
LC/MS Rt-min(MH+):2.27(493.5)(方法A)。
步骤8:
通过加热和超声将LiOH(0.0233g,0.973mmol)溶于水(10mL)中。将该LiOH溶液加入步骤7的纯物质(483mg,0.885mmol)的THF(10mL)溶液中,混合物的颜色立即转变为桃红色。将反应物在室温下搅拌1小时,用1N HCl(0.973mL,0.974mmol)酸化。反应物用pH4.00缓冲溶液猝灭并将pH调节至4至5之间。水层用EtOAc萃取(3×20mL),用盐水(15mL)洗涤并经硫酸镁干燥。将滤液真空浓缩并保持在真空下过夜。粗产物(480mg,收率>95%)直接用于下一步骤中。
1H NMR:(DMSO-d6)δ0.94(s,9H),1.20(s,9H),1.32-1.34(m,1H),2.12-2.4(m,1H),2.51-2.55(m,1H),3.81-3.83(d,J=10Hz,1H),4.21-4.23(d,J=10Hz,1H),4.32-4.35(t,1H),5.4(brs,1H),6.63-6.65(d,NH),7.09(s,1H),7.47-7.49(m,4H),8.03-8.06(m,2H),12.56(s,1H).
步骤9:
将步骤8的二肽(100mg,0.188mmol)和2-噻吩硼酸(0.0481g,0.376mmol)在DMF(2mL)中溶剂化。将溶液置于氮气气氛中,加入2MKF水溶液(0.282mL,0.376mmol)。加入5%摩尔的四(三苯基膦)合钯(0)(0.011mg,0.0094mmol),使用微波(使用Personal ChemistryEmrys Optimizer设备)将混和物在150℃下加热30分钟。反应完成后将沉积出的钯黑除去。用1当量的1N HCl将混合物酸化,经注射器过滤,使用甲醇萃取产物。产物经制备型HPLC(柱-4Xterra S5 5μm30×75mm,溶剂-85%A/15%B-10%A/90%B(其中溶剂A为10%甲醇、90%H2O、0.1%TFA,溶剂B为90%甲醇、10%H2O、0.1%TFA),梯度时间-15min.,保持时间-1min.,流速-40mL/min,纯产物保留时间-16.23)纯化。用1N NaOH中和含所需产物的洗脱液,并置于离心真空(speed vacuum)下约2h。合并各洗脱液,加入pH4.0的缓冲液(15mL)。使用1N HCl将pH调节至pH4-5,水层用乙酸乙酯(3×20mL)萃取。有机层用盐水(15mL)洗涤,经硫酸镁干燥并真空浓缩。产物在油泵抽真空下干燥过夜,得到浅黄色油状物(44mg,40%)。
LC/MS Rt-min(MH+):2.7(580.54)(方法A)。
步骤10:
将实施例413、步骤9的产物(43mg,O.0742mmol)溶于DCM(2mL)中,加入N-(1(R)-氨基-2(S)-乙烯基-环丙烷羰基)-环丙烷磺酰胺·盐酸盐(实施例1,步骤8)(0.0198g,0.0742mmol)。接着在氮气气氛中依次加入DIPEA(0.0646mL,0.371mmol)和HATU(0.039g,0.0104mmol)。将反应物在室温下搅拌4.5h。向溶液中加入10ml DCM以增加体积,反应物用pH4.00缓冲溶液(10mL)猝灭。使用1N HCl将混合物酸化至pH为4-5,水相用DCM(3×15mL)萃取。有机相用pH4.00缓冲溶液(10mL)和盐水(10mL)洗涤两次,随后经硫酸镁干燥。将所得的溶液真空浓缩并经Biotage 12S柱(用10%-50%丙酮/己烷洗脱)纯化。所述化合物经制备型HPLC(柱-YMC ODS-A 20×50mm s5,溶剂-60% A/40% B-10% A/90% B(其中溶剂A为10%甲醇、90%H2O、0.1%TFA,溶剂B为90%甲醇、10%H2O、0.1%TFA),梯度时间-8min.,保持时间-2min.,流速-25mL/min,纯产物保留时间-8.702)分离和预纯化。该纯化操作得到纯化合物413,为浅橙色油状物(22.3mg,38%)。
1H NMR:δ1.02-1.29(m,24H),1.42-1.45(m,1H),1.87-1.89(m,1H),2.24-2.30(m,2H),2.53-2.57(m,1H),2.92-2.97.(m,1H),4.064.08(d,J=10Hz,1H),4.25(s,1H),4.31-4.33(d,J=10Hz,1H),4.45-4.49(t,1H),5.12-5.14(d,J=10Hz,1H),5.29-5.32(d,J=15Hz,1H),5.46(brs,1H),5.73-5.80(m,1H),7.13-7.15(m,1H),7.29-7.30(d,J=5Hz,2H),7.42-7.53(m,4H),7.75-7.56(d,J=5Hz,1H),8.07-8.09(d,J=10Hz,2H).
LC/MS Rt-min(MH+):2.79(792.72)(方法A)。
实施例414:化合物414的制备
化合物414
步骤1:
将实施例413、步骤8的纯物质(100mg,0.188mmol)和4-甲氧基苯基硼酸(0.0429g,0.282mmol)在DMF(2.5mL)中溶剂化。将溶液置于氮气气氛中,加入2M碳酸钠水溶液(0.188mL,0.376mmol)。加入5%摩尔的四(三苯基膦)合钯(0)(0.011mg,0.0094mmol),并使用微波(使用Personal Chemistry Emrys Optimizer设备)将混和物在150℃下加热30分钟。反应完成后将沉积出的钯黑除去。用1当量的1NHCl将混合物酸化,经注射器过滤,使用甲醇萃取产物。产物经制备型HPLC(柱-Xterra MS C18 5μm 30×50mm,溶剂-90% A/10% B-10%A/90% B(其中溶剂A为10%甲醇、90%H2O、0.1%TFA,溶剂B为90%甲醇、10%H2O、0.1%TFA),梯度时间-15min,保持时间-1min.,流速-45mL/min,纯产物保持时间-13.72)纯化。用1N NaOH中和含所需产物的洗脱液,并置于离心真空(speed vacuum)下约2h。合并各洗脱液,加入pH4.0的缓冲液(15mL)。使用lN HCl将pH调节至pH为4-5,水层用乙酸乙酯(3×20mL)萃取。有机层用盐水(15mL)洗涤,经硫酸镁干燥并真空浓缩。产物在油泵抽真空下干燥过夜,得到粘稠油状物(44mg,50%)。
LC/MS Rt-min(MH+):2.23(604.61)(方法A)。
步骤2:
将实施例414、步骤1的产物(43mg,0.0712mmol)溶于DCM(2mL)中,加入N-(1(R)-氨基-2(S)-乙烯基-环丙烷羰基)-环丙烷磺酰胺盐酸盐(实施例1,步骤8)(0.01899g,0.0712mmol)。随后在氮气气氛中加入DIPEA(0.062mL,0.356mmol)和HATU(0.038g,0.0997mmol)。将反应物在室温下搅拌4.5h。向溶液中加入10mL DCM以增加体积,反应物用pH4.00缓冲溶液(10mL)猝灭。使用1N HCl将混合物酸化至pH为4-5,水相用DCM(3×15mL)萃取。有机相用pH4.00缓冲溶液(10mL)和盐水(10mL)洗涤两次,随后经硫酸镁干燥。将所得的溶液真空浓缩并经Biotage 12S柱(用20%-50%丙酮/己烷洗脱)纯化。所述化合物经制备型HPLC(柱-YMC ODS-A20×50mm s5,溶剂-60%A/40%B-10%A/90%B(其中溶剂A为10%甲醇、90%H2O、0.1%TFA和溶剂B为90%甲醇、10%H2O、0.1%TFA),梯度时间-10min,保持时间-2min、流速-25mL/min,纯产物保留时间-7.43-8.24)分离和预纯化。该纯化操作得到纯化合物414,为淡橙色油状物(19.4mg,34%)。
1H NMR:δ1.02-1.23(m,24H),1.29-1.44(m,1H),1.89-1.95(m,1H),2.24-2.30(q,1H),2.32-2.40(m,1H),2.59-2.62(m,1H),3.90(s,3H),4.10-4.12(d,J=10Hz,1H),4.21(s,1H),4.42-4.56(m,2H),5.12-5.14(d,J=10Hz,1H),5.28-5.31(d,J=15Hz,1H),5.61(brs,1H),5.72-5.80(m,1H),7.14-7.16(d,J=10Hz,2H),7.52-7.54(d,J=10Hz,2H),7.61-7.62 (m,2H),7.95-7.98(m,4H);
LC/MS Rt-min(MH+):2.35(816.76)(方法A)。
实施例415:化合物415的制备
化合物415
步骤1:
将实施例413、步骤8的二肽(174mg,0.327mmol)和苯基硼酸(0.06g,0.491mmol)在DMF(4mL)中溶剂化。将溶液置于氮气气氛中,加入2M碳酸钠水溶液(0.33mL,0.654mmol)。加入5%摩尔的四(三苯基膦)合钯(0)(0.019mg,0.0164mmol),使用微波(使用PersonalChemistry Emrys Optimizer设备)将混和物在150℃下加热30分钟。反应完成后将沉积出的钯黑除去。用1当量的1N HCl将混合物酸化,经注射器过滤,使用甲醇萃取产物。产物经制备型HPLC(柱-5 Xterrac-185um 30×100mm,溶剂-80%A/20%B-0%A/100%B(其中溶剂A为10%甲醇、90%H2O、0.1%TFA,溶剂B为90%甲醇、10%H2O、0.1%TFA),梯度时间-20min.,保持时间-1min.,流速-40mL/min,纯产物保留时间-11.28-11.72)纯化。用1N NaOH中和含所需产物的洗脱液,并置于离心真空(speed vacuum)下约2h。合并各洗脱液,加入pH4.0的缓冲液(15mL)。使用1N HCl将pH调节至pH4-5,水层用乙酸乙酯(4×15mL)萃取。有机层用盐水(15mL)洗涤,经硫酸镁干燥并真空浓缩。产物在油泵抽真空下干燥过夜,得到产物(31.5mg,17%)。
LC/MS Rt-min(MH+):2.54(574.37)(方法A)。
步骤2:
将实施例415、步骤1的产物(31.5mg,0.055mmol)溶于DCM(3mL)中,加入N-(1(R)-氨基-2(S)-乙烯基-环丙烷羰基)-环丙烷磺酰胺盐酸盐(实施例1,步骤8)(0.0147g,0.055mmol)。接着在氮气气氛中依次加入DIPEA(0.048mL,0.275mmol)和HATU(0.029g,0.077mmol)。将反应物在室温下搅拌3.5小时,再加入0.3当量的N-(1(R)-氨基-2(S)-乙烯基-环丙烷羰基)-环丙烷磺酰胺盐酸盐。再将反应物搅拌8h。向溶液中加入10ml DCM以增加体积,反应物用pH4.00缓冲溶液(10mL)猝灭。使用1N HCl将混合物酸化至pH为4-5,水相用DCM(4×10mL)萃取。有机相用pH4.00缓冲溶液(10mL)和盐水(10mL)洗涤两次,随后经硫酸镁干燥。将所得的溶液真空浓缩并经制备型HPLC(柱-YMC ODS-A30×50mm,溶剂-80% A/20% B-10% A/90%B(其中溶剂A为10%甲醇、90%H2O、0.1%TFA和溶剂B为90%甲醇、10%H2O、0.1%TFA),梯度时间-20min.,保持时间-3min.,流速-30mL/min,纯产物保留时间-19.6)纯化。该纯化操作并不能得到纯的化合物,因而再经Biotage12S柱(用10%-50%丙酮/己烷洗脱)纯化。该纯化步骤得到纯的题述化合物,为淡橙色油状物(22.3mg,38%)。
1H NMR:δ1.02-1.45(m,24H),1.85-1.86(m,1H),2.03-2.11(m,2H),2.42-2.46(m,1H),2.77-2.85(m,1H),4.10-4.12(m,1H),4.25(s,1H),4.31-4.33(d,J=10Hz,1H),4.49-4.54(m,1H),5.03-5.05(d,J=10Hz,1H),5.20-5.24(d,J=20Hz,1H),5.44(brs,1H),5.82-5.94(m,1H),7.33(s,2H),7.43-7.50(m,6H),8.09-8.10(d,J=5Hz,4H);
LC/MS Rt-min(MH+):2.37(786.37)(方法A)。
部分J:
在部分J中使用以下LC/MS方法:
柱:方法A:YMC ODS-A C18 S7(4.6×33mm)
方法B:YMC Xterra ODS S7(3.0×50mm)
方法C:Xterra ms C18(4.6×33mm)
方法D:YMC ODS-A C18 S3(4.6×33mm)
梯度:100%溶剂A/0%溶剂B-0%溶剂A/100%溶剂B
梯度时间:3min
保持时间:1min
流速:5mL/min
检测器波长:220nm
溶剂:溶剂A:10%甲醇/90%水/0.1%TFA
溶剂B:90%甲醇/10%水/0.1%TFA。
实施例420:化合物420的制备
化合物420
方案1
步骤1:
向三苯基膦(16.8g,63.9mmol)的THF(150mL)溶液中滴加入DEAD(10.1mL,63.9mmol)并将溶液在室温下搅拌30min。缓慢加入Boc-顺-(L)-Hyp-OMe(10.5g,42.6mmol)的THF(50mL)溶液,接着分批加入5-溴-吡啶-3-醇(按照F.E.Ziegler等,J.Am.Chem.Soc.,(1973),95,7458中描述的方法制备)(8.90g,51.1mmol)。所得的溶液在45℃下搅拌18hr。将溶液真空浓缩并将剩余物在乙醚(200mL)和1N NaOH(50mL)间分配。将有机相干燥(硫酸镁),将乙醚溶液过滤,随后浓缩至其一半的体积。过滤除去形成的沉淀物。将滤液真空浓缩并经Biotage 65M柱(洗脱液:己烷-EtOAc 2∶1,3∶2,1∶1)纯化,得到题述化合物(11.9g,69%),为红色油状物。
1H NMR:(CDCl3)δ1.42,1.45(s,9H(旋转异构体)),2.25-2.30(m,1H),2.49-2.58(m,1H),3.75,3.81(s,3H(旋转异构体)),3.66-3.81(m,2H(覆盖)),4.42,4.50(t,J=8Hz,1H(旋转异构体)),4.92(m,1H),7.36(s,1H),8.20(s,1H),8.32(s,1H)。
LC/MS Rt-min(MH+):2.26(401,403)(方法B)。
步骤2:
将步骤1的产物(2.34g,5.83mmol)溶于DCM(20mL)和TFA(18mL,0.23mole)中并在室温下搅拌2hr。真空除去挥发份,将剩余物在乙酸乙酯和水间分配。有机相用1N HCl(25mL)萃取,将合并的含水萃取液用饱和碳酸氢钠溶液(40mL)中和。水相用EtOAc萃取(两次),合并的有机萃取液用盐水洗涤并干燥(硫酸镁),得到题述化合物(1.02g,58%,游离碱),为无色油状物。
1H NMR:(DMSO-d6)δ2.12-2.20(m,2H),2.94(d,J=12Hz,1H),3.19(dd,J=4.5,12Hz,1H),3.64(s,3H),3.90(t,J=8Hz,1H),5.07(m,1H),7.69(s,1H),8.26(s,1H),8.29(s,1H).
LC/MS Rt-min(MH+):0.78(301,302)(方法B)。
步骤3:
向步骤2的产物(1.02g,3.39mmol)、N-BOC-L-叔-亮氨酸(862mg,3.73mmol)和HOBt(458mg,3.39mmol)在DCM(15mL)中的悬浮液内依次加入DIPEA(2.36mL,13.6mmol)和HBTU(1.61g,4.24mmol)。所得的溶液在室温下搅拌15hr并用DCM猝灭,用pH4缓冲液和一些1N HCl调节pH至4-5。有机相用pH4缓冲液、饱和碳酸氢钠溶液(两次)和盐水洗涤并干燥(硫酸镁)。使用Biotage 40M柱(用己烷-EtOAc 3∶2、1∶1洗脱)纯化,得到题述化合物(1.48g,85%),为白色固体物。
1H NMR:(DMSO-d6)δ0.94(s,9H),1.17(s,9H),2.16-2.21(m,1H),2.50(m,(覆盖),1H),3.64(s,3H),3.82(d,J=12Hz,1H),4.06(d,J=9.5 Hz,1H),4.16(d,J=12Hz,1H),4.45(dd,J=8,9.5Hz,1H),5.26(m,1H),6.71(d,J=9Hz,NH),7.75(s,1H),8.28(s,1H),8.32(s,1H).
LC/MS Rt-min(MH+):2.35(514,516)(方法B),
步骤4:
向步骤3的产物(98mg,0.19mmol)、四(三苯基膦)合钯(6.6mg,0.00573mmol)、2M碳酸钠水溶液(0.191mL,0.381mmol)在甲苯(2mL)中的混合物内加入苯基硼酸(29mg,0.24mmol)的甲醇(0.1mL)溶液。将溶液在氮气气氛、85℃下加热6小时。冷却至室温后,将混合物用pH4缓冲液猝灭,用EtOAc(2×10mL)萃取,并干燥(硫酸钠)。使用Biotage 12M柱(用己烷-EtOAc2∶3洗脱)纯化,得到题述化合物(72mg,74%),为无色油状物。
1H NMR:(甲醇-d4)δ1.05(s,9H),1.31(s,9H),2.29-2.35(m,1H),2.68-2.72(m,1H),3.77(s,3H),4.00(d,J=12Hz,1H),4.24(d,J=9.5Hz,1H),4.41(d,J=12Hz,1H),4.67(dd,J=7.5,10Hz,1H),5.36(m,1H),6.5 1(d,J=9.5Hz,NH),7.79-7.85(m,3H),8.39(s,1H),8.59(s,1H),8.67-8.68(m,2H).
LC/MS Rt-min(MH+):2.16(512)(方法B)。
步骤5:
向步骤4的产物(150mg,0.293mmol)在THF(2mL)和甲醇(2mL)中的溶液内加入LiOH(14mg,0.59mmol)的水(2mL)溶液。将混合物在室温下搅拌2h并用1N HCl猝灭,直至pH值呈中性。真空除去有机挥发份,向剩余物中加入pH4缓冲液。产物萃取至EtOAc(3×10mL)中,用盐水/pH4缓冲液洗涤并干燥(硫酸镁),在戊烷中研磨后定量得到题述化合物,为白色固体物。
1H NMR:(甲醇-d4)δ1.05(s,9H),1.32(s,9H),2.32-2.38(m,1H),2.69-2.74(m,1H),3.99(dd,J=3,12Hz,1H),4.25(s,1H),4.40(d,J=12Hz,1H),4.64(dd,J=7.5,9.5Hz,1H),5.36(m,1H),7.85-7.87(m,3H),8.40(s,1H),8.60(s,1H),8.69-8.70(m,2H).
LC/MS Rt-min(MH+):2.03(499)(方法B)。
步骤6:
向步骤5的产物(93mg,0.19mmol)和实施例1、步骤8的产物(50mg,0.19mmol)在DCM(2mL)中的悬浮液内依次加入DIPEA(0.163mL,0.935mmol)和HATU(92mg,0.243mmol)。所得的混合物在室温下搅拌18hr并用DCM、pH4缓冲液和一些lN HCl猝灭以调节pH为4-5。分离各层,水相用DCM(10mL)萃取并干燥(硫酸钠)。使用Biotage 12M柱(用梯度己烷-丙酮20-60%洗脱)纯化,得到题述化合物(65mg,49%),为白色粉末。
1H NMR:(DMSO-d6)δ0.95(s,9H),1.03-1.05(m,2H),1.08-1.10(m,2H),1.26(s,9H),1.36-1.39(m,1H),1.69-1.72(m,1H),2.09-2.21(m,2H),2.41-2.45(m,1H),2.92-2.94(m,1H),3.91(d,J=11.5Hz,1H),4.08(d,J=7.5Hz,1H),4.16(d,J=11.5Hz,1H),4.36(t,J=8.5Hz,1H),5.10(d,J=10Hz,1H),5.23(d,J=17.5Hz,1H),5.38(m,1H),5.59-5.67(m,1H),6.54(s,NH),7.41-7.46(m,1H),7.50-7.53(m,2H),7.65(s,1H),7.74-7.75(m,2H),8.28(s,1H),8.52(s,1H),8.92(s,NH)。
LC/MS Rt-min(MH+):2.08(710)(方法B)。
实施例421:化合物421的制备
化合物421
方案1
步骤1:
向冷(-5℃)的48%HBr水溶液(35mL)中加入4-氯-吡啶-2-胺(4.18g,32.5mmol;按照K.S.Gudmundsson等;Synth.Commun.(1997),27,861的方法制备),接着缓慢加入溴(6.7mL,0.13mole)。20分钟后,在相同的温度下加入亚硝酸钠(8.63g,0.125mole)的水(40mL)溶液,继续搅拌30min。并在冰冷却下,用10N NaOH(约40mL)猝灭反应物至呈碱性pH。产物萃取至EtOAc(2×100mL)中并干燥(硫酸钠)。粗产物使用Biotage40M柱(洗脱液:10%EtOAc/己烷)纯化,得到题述化合物(2.50g,40%),为无色油状物,在静置时固化。
1H NMR:(DMSO-d6)δ 7.64(dd,J=1.5,5.5Hz,1H),7.94(d,J=1.5Hz,1H),8.40(d,J=5.5Hz,1H);
LC/MS Rt-min(MH+):1.65(192,194,196)(方法B)。
步骤2:
室温、氮气气氛中,向N-BOC-反式-L-Hyp-OH(3.22g,13.9mmol)的DMSO(30mL)溶液中分批加入叔丁醇钾(3.90g,34.8mmol)。1.5h后,加入步骤1的产物的DMSO(5mL)溶液。将混合物搅拌过夜并用水(150mL)猝灭。将溶液用EtOAc(100mL)洗涤。用1N HCl将水相酸化至pH4。将所述粗甲酸萃取至EtOAc中(三次)并干燥(硫酸镁)。剩余物为棕色固体物,将其悬浮在THF(20mL)和甲醇(20mL)中并冷却至0℃。滴加入三甲基甲硅烷基重氮甲烷的己烷溶液(2M,12mL),15分钟后,将溶液真空浓缩。使用Biotage40M柱(洗脱液:己烷-EtOAc2∶1,1∶1)纯化,得到题述化合物(3.47g,62%),为灰白色粉末。
1H NMR:(DMSO-d6)δ1.34,1.38(s,9H(旋转异构体)),2.24-2.29(m,1H),2.43-2.50(m,1H),3.57(d,J=12.5Hz,1H),3.64-3.68(m,1H),3.66,3.69(s,3H(旋转异构体)),4.27-4.34(m,1H),5.21(m,1H),7.06(d,J=6Hz,1H),7.28(s,1H),8.20(d,J=6Hz,1H);
LC/MS Rt-min(MH+):2.59(401,403)(方法B)。
步骤3:
将步骤2的产物(2.00g,4.98mmol)悬浮在4N HCl/二_烷(10mL)和1N HCl/乙醚(40mL)中,在室温下搅拌过夜。将所得的悬浮液真空浓缩,并将剩余物在戊烷中研磨,定量得到题述化合物,为白色粉末。
LC/MS Rt-min(MH+):0.24(301,303)(方法B)。
步骤4:
向步骤3的产物(估计4.98mmol)、N-环戊氧基羰基-L-叔-亮氨酸(1.24g,5.10mmol)和HOBt(673mg,4.98mmol)在DCM(25mL)中的悬浮液内依次加入DIPEA(4.34mL,24.9mmol)和HBTU(2.36g,6.23mmol)。所得的溶液在室温下搅拌15hr并用DCM、pH4缓冲液和10mL 1N HCl猝灭以调节pH值为4。水相用DCM萃取(两次)。合并的有机萃取液用饱和碳酸氢钠溶液(两次)和盐水洗涤并干燥(硫酸镁)。使用Biotage 40M柱(用己烷-EtOAc 3∶2 1∶1洗脱)纯化,得到题述化合物(2.09g,80%),为白色泡沫状物。
1H NMR:(DMSO-d6)δ0.94(s,9H),1.43-1.74(m,8H),2.18-2.23(m,1H),2.46-2.49(m,1H),3.64(s,3H),3.87(d,J=12Hz,1H),4.08-4.13(m,1H),4.42(dd,J=7.5,9.5Hz,1H),4.78(m,1H),5.30(m,1H),7.03-7.05(m,1H),7.24(s,1H),8.120(d,J=6Hz,1H).
LC/MS Rt-min(MH+):2.34(526,528)(方法C),
步骤5:
向步骤4的产物(206mg,0.391mmol)在THF(2mL)和甲醇(2mL)中的溶液内加入LiOH(28mg,1.2mmol)/水(2mL)。室温下将混合物搅拌2h并用1N HCl猝灭直至pH值呈中性。真空除去有机挥发份,向剩余物中加入pH4缓冲液。将产物萃取至EtOAc(2×20mL)中并干燥(硫酸镁),得到题述化合物(196mg,100%),为白色固体物。
1H NMR:(甲醇-d4)δ1.05(s,9H),1.60-1.85(m,8H),2.30-2.36(m,1H),2.62-2.67(m,1H),3.97(d,J=12Hz,1H),4.26(s,1H),4.32(d,J=12Hz,1H),4.58(dd,J=7.5,9.5Hz,1H),4.89(m(覆盖),1H),5.29(m,1H),7.03-7.05(m,1H),7.26(s,1H),8.19(d,J=6Hz,1H);
LC/MS Rt-min(MH+):2.17(512,514)(方法B)。
步骤6:
该产物按照实施例420、步骤4的方法制备(反应时间20h),以实施例421、步骤5的产物为原料,收率为20%。
1H NMR:(甲醇-d4)δ1.03(s,9H),1.45-1.74(m,8H),2.31-2.37(m,1H),2.65-2.69(m,1H),3.99(dd,J=12,3.0Hz,1H),4.26(d,J=9.5Hz,1H),4.33(d,J=11.5Hz,1H),4.60(dd,J=8.0,9.5Hz,1H),4.80(m,1H),5.35(m,1H),6.72(d,J=9.0Hz,NH),7.02(dd,J=2.5,6.0Hz,1H),7.39(s,1H),7.50(m,3H(覆盖)),7.91(d,J=6.5Hz,1H),8.46(d,J=6.0Hz,1H).
LC/MS Rt-min(MH+):2.03(510)(方法A)。
步骤7:
化合物421按照实施例420、步骤6的方法制备,以实施例421、步骤6的产物为原料,收率68%。
1H NMR:(甲醇-d4)δ1.03(s,9H),1.06-1.09(m,2H),1.23-1.26(m,2H),1.43(dd,J=5.5,9.5Hz,1H),1.47-1.77(m,8H),1.88(dd,J=5.5,8.5Hz,1H),2.21-2.29(m,2H),2.50-2.54(m,1H),2.91-2.96(m,1H),4.06(dd,J=3.0,11.5Hz,1H),4.28-4.30(m,2H),4.44(dd,J=7.0,10.5Hz,1H),4.82(m,1H(覆盖)),5.12(d,J=10Hz,1H),5.30(d,J=17Hz,1H),5.37(m,1H),5.73-5.80(m,1H),6.92(d,J=9.5Hz,NH),6.98(dd,J=5.5,2.0Hz,1H),7.37(s,1H),7.43-7.50(m,3H),7.90(d,J=7.0Hz,1H),8.45(d,J=5.5Hz,1H).
LC/MS Rt-min(MH+):2.37(723)(方法A)。
实施例422:化合物422的制备
化合物422
步骤1:
向实施例420、步骤3的产物(102mg,0.198mmol)和四(三苯基膦)合钯(23mg,0.0198mmol)在甲苯(2mL)中的混合物内加入4-三丁基锡烷基-吡啶(87mg,0.24mmol)。在氮气气氛中,将溶液在105℃下加热20h。冷却至室温后,将混合物用饱和碳酸氢钠溶液猝灭,用EtOAc萃取(2×10mL)并干燥(硫酸钠)。使用Biotage12M柱(用梯度己烷-丙酮20-60%洗脱)纯化,得到题述化合物(49mg,49%),为白色固体物。
1H NMR:(甲醇-d4)δ1.05(s,9H),1.31(s,9H),2.29-2.35(m,1H),2.68-2.72(m,1H),3.77(s,3H),4.00(d,J=12Hz,1H),4.24(d,J=9.5Hz,1H),4.41(d,J=12Hz,1H),4.67(dd,J=7.5,10Hz,1H),5.36(m,1H),6.51(d,J=9.5Hz,NH),7.79-7.85(m,3H),8.39(s,1H),8.59(s,1H),8.67-8.68(m,2H).
LC/MS Rt-min(MH+):1.77(514)(方法C)。
步骤2:
该产物按照实施例420、步骤5的相同方法以定量收率制备,不同之处在于使用实施例422、步骤1的产物。
1H NMR:(甲醇-d4)δ1.05(s,9H),1.32(s,9H),2.32-2.38(m,1H),2.69-2.74(m,1H),3.99(dd,J=3,12Hz,1H),4.25(s,1H),4.40(d,J=12Hz,1H),4.64(dd,J=7.5,9.5Hz,1H),5.36(m,1H),7.85-7.87(m,3H),8.40(s,1H),8.60(s,1H),8.69-8.70(m,2H).
LC/MS Rt-min(MH+):1.57(500)(方法B)。
步骤3:
按照实施例420、步骤6的相同方法制备化合物422,收率51%,为白色固体物,不同之处在于使用实施例422、步骤2的产物。
1H NMR:(DMSO-d6)δ0.95(s,9H),1.03(m,1H),1.08(m,1H),1.23(s,9H),1.34-1.37(m,1H),1.68-1.70(m,1H),2.12-2.18(m,2H),2.41-2.45(m,1H),2.93(m,1H),3.90(d,J=11Hz,1H),4.06(d,J=9Hz,1H),4.17(d,J=11Hz,1H),4.36(dd,J=7,10Hz,1H), 5.10(d,J=12Hz,1H),5.23(d,J=16.5Hz,1H),5.40(m,1H),5.59-5.66(m,1H),6.60(d,J=9Hz,NH),7.81-7.82(m,3H),8.38(s,1H),8.65(s,1H),8.68-8.70(m,2H),8.93(s,NH),10.4(s,NH).
LC/MS Rt-min(MH+):2.14(711)(方法D)。
实施例423:化合物423的制备
化合物423
步骤1:
向实施例420、步骤3的产物(1.00g,1.94mmol)在THF(5mL)和甲醇(5mL)中的溶液内加入LiOH(140mg,5.83mmol)的水(5mL)溶液。将混合物在室温下搅拌2h并用1N HCl猝灭,直至pH值呈中性。真空除去有机挥发份,加入pH4缓冲液,将产物萃取至EtOAc(3×25mL)中并干燥(硫酸镁),得到题述化合物(1.0g,100%),为白色固体物。
1H NMR:(DMSO-d6)δ0.95(s,9H),1.27(s,9H),2.14-2.19(m,1H),2.47-2.50(m,1H),3.80(d,J=11.5Hz,1H),3.99-4.07(m,2H),4.15(d,J=11.5Hz,1H),4.36(dd,J=8,10Hz,1H),5.25(m,1H),6.66(d,J=9Hz,NH),7.75(s,1H),8.28(s,1H),8.31(s,1H),12.5(s,1H).
LC/MS Rt-min(MH+):2.47(500,502)(方法A)。
步骤2:
按照实施例420、步骤6的方法制备化合物423,以实施例423、步骤1的产物和实施例8、步骤3的产物(外消旋混合物P1,(1R,2S)和(1S,2R))为原料,收率为52%。
1H NMR:(DMSO-d6)δ0.93,0.95(s,9H),1.00-1.09(m,4H),1.28,1.29(s,9H),1.37-1.39(m,1H),1.69-1.72(m,1H),2.09-2.22(m,2H),2.36-2.46(m,1H),2.88-2.94(m,1H),3.82-3.87(m,1H),4.02-4.04(m,1H),4.10-4.15(m,1H),4.31-4.34(m,1H),5.10(d,J=10.5Hz,1H),5.22-5.28(m,1H),5.54-5.66(m,1H),6.56,6.60(d,J=9.5Hz,NH),7.74,7.76(s,1H),8.28-8.29(m,1H),8.32(s,1H),8.79,8.89(s,1H).
LC/MS Rt-min(MH+):2.42(712,714)(方法B)。
实施例424:化合物424的制备
化合物424
步骤1:
向实施例423、步骤1的产物(91mg,0.18mmol)、四(三苯基膦)合钯(10.5mg,0.0091mmol)和3-呋喃基硼酸(25.4mg,0.227mmol)的DMF(2mL)溶液中加入2M碳酸钠水溶液(0.273mL,0.546mmol)。将混合物在氮气气氛、110℃下加热2.5h。冷却至室温后,过滤除去固体,将滤液真空浓缩。剩余物经制备型HPLC纯化(梯度30-80%B),得到题述化合物(64mg,72%),为白色固体物。
1H NMR:(DMSO-d6)δ0.95(s,9H),1.25(s,9H),2.17-2.21(m,1H),2.50(m(覆盖),1H),3.84-3.86(m,1H),4.09-4.15(m,2H),4.37(t,J=9Hz,1H),5.28(m,1H),6.67(d,J=9Hz,NH),7.08(s,1H),7.62(s,1H),7.79(s,1H),8.17(s,1H),8.33(s,1H),8.51(s,1H),12.6(s,1H).
LC/MS Rt-min(MH+):1.60(488)(方法B)。
步骤2:
按照实施例420、步骤6的方法制备化合物424,以实施例424、步骤1的产物为原料,收率为55%。
1H NMR:(甲醇-d4)δ1.02(s,9H),1.05-1.09(m,2H),1.22-1.25(m,2H),1.34(s,9H),1.42-1.45(m,1H),1.86-1.89(m,1H),2.21-2.26(m,1H),2.48-2.52(m,1H),2.91-2.96(m,1H),4.02(d,J=12Hz,1H),4.24-4.29(m,2H),4.43-4.47(dd,J=7.5,10.5Hz,1H),5.12(d,J=10.5Hz,1H),5.28-5.32(m,1H),5.73-5.81(m,1H),6.63(d,J=9Hz,NH),6.90(s,1H),7.60-7.62(m,2H),8.07(s,1H),8.12(s,1H),8.39(s,NH).
LC/MS Rt-min(MH+):2.30(700)(方法E)。
实施例425:化合物425的制备
化合物425
按照实施例421、步骤7的方法制备化合物425,以实施例421、步骤5的产物为原料,收率为45%。
1H NMR:(DMSO-d6)δ0.95 (s,9H),1.03-1.04(m,1H),1.08-1.09(m,1H),1.33-1.36(m,1H),1.46-1.75(m,9H),2.08-2.12(m,1H),2.17,(q,J=9Hz,1H),2.35-2.38(m,1H),2.88-2.93(m,1H),3.91(d,J=9.5Hz,1H),4.05-4.10(m,2H),4.27(dd,J=10,7Hz,1H),4.80(m,1H),5.10(d,J=12Hz,1H),5.23(d,J=16.5Hz,1H),5.32(m,1H),5.58-5.66(m,1H),6.94(d,J=9Hz,NH),7.03-7.05(m,1H),7.26(s,1H),8.21(d,J=6Hz,1H),8.86(s,NH),10.4(s,NH).
LC/MS Rt-min(MH+):2.56(724,726)(方法D)。
实施例426:化合物426的制备
化合物426
步骤1:
按照实施例421、步骤6的方法制备该产物,收率65%,不同之处在于使用3-呋喃硼酸。
1H NMR:(甲醇-d4)δ1.03(s,9H),1.49-1.76(m,8H),2.30-2.35(m,1H),2.63-2.67(m,1H),3.98(d,J=12Hz,1H),4.26(t,J=9Hz,1H),4.31(d,J=12Hz,1H),4.58(t,J=8.0Hz,1H),5.32(m,1H),6.71(d,J=9.5Hz,NH),6.92(dd,J=2.5,6.0Hz,1H),6.98(s,1H),7.25(d,J=2.5Hz,1H),7.60(s,1H),8.15(s,1H),8.35(d,J=5.5Hz,1H).
LC/MS Rt-min(MH+):1.94(500)(方法D)。
步骤2:
按照实施例421、步骤7的方法制备化合物426,以实施例426、步骤1的产物为原料,收率为57%。
1H NMR:(DMSO-d6)δ0.95(s,9H),1.03-1.04(m,2H),1.08-1.09(m,2H),1.34-1.71(m,8H),2.09-2.20(m,2H),2.37-2.41(m,1H),2.93(m,1H),3.96(d,J=9.5 Hz,1H),4.07-4.11(m,2H),4.29(m,1H),4.78(m,1H),5.10(d,J=10.5Hz,1H),5.23(d,J=17Hz,1H),5.34(m,1H),5.59-5.66(m,1H),6.87-6.91(m,2H),7.08(s,1H),7.27(s,1H),7.75(s,1H),8.33(s,1H),8.39(d,J=6Hz,1H),8.90(s,NH),10.4(s,1H).
LC/MS Rt-min(MH+):2.38(712)(方法D)。
实施例427:化合物427的制备
化合物427
步骤1:
按照实施例421、步骤6的方法制备该产物,收率86%,不同之处在于使用4-氟苯基硼酸。
1H NMR:(甲醇-d4)δ1.03(s,9H),1.44-1.75(m,8H),2.30-2.36(m,1H),2.64-2.69(m,1H),3.99 dd,J=12,3.5Hz,1H),4.26(d,J=9.5Hz,1H),4.32(d,J=12Hz,1H),4.59(dd,J=8.0,9.5Hz,1H),4.81(m,1H),5.34(m,1H),6.73(d,J=9.5Hz,NH),6.99(dd,J=2.5,6.0Hz,1H),7.20(t,2H(覆盖)),7.36(s,1H(覆盖)),7.94-7.97(m,2H),8.44(d,J=6.0Hz,1H).
LC/MS Rt-min(MH+):2.22(528)(方法A)。
步骤2:
按照实施例421、步骤7的方法制备化合物427,以实施例427、步骤1的产物为原料,收率为53%。
1H NMR:(甲醇-d4)δ1.03(s,9H),1.06-1.09(m,2H),1.23-1.26(m,2H),1.43(dd,J=5.0,9.5Hz,1H),1.47-1.78(m,8H),1.88(dd,J=5.5,8.0Hz,1H),2.21-2.29(m,2H),2.49-2.53(m,1H),2.91-2.96(m,1H),4.05(dd,J=3.0,11.5Hz,1H),4.27-4.29(m,2H),4.44(dd,J=7.0,10.5Hz,1H),4.83(m,1H(覆盖)),5.12(d,J=10Hz,1H),5.29(d,J=17Hz,1H),5.37(m,1H),5.73-5.80(m,1H),6.93(d,J=9.5Hz,NH),6.98(m,1H),7.21(t,J=9.0Hz,2H),7.36(s,1H),7.94-7.97(m,2H),8.44(d,J=6.0Hz,1H).
LC/MS Rt-min(MH+):2.42(741)(方法A)。
实施例428:化合物428的制备
化合物428
步骤1:
按照实施例421、步骤6的方法制备该产物,收率37%不同之处在于使用4-甲氧基苯基硼酸。
1H NMR:(甲醇-d4)δ1.03(s,9H),1.45-1.74(m,8H),2.31-2.36(m,1H),2.64-2.68(m,1H),3.85(s,3H),3.98(dd,J=12,3.5Hz,1H),4.25(d,J=9.0Hz,1H),4.33(d,J=11.5Hz,1H),4.59(dd,J=8.0,10Hz,1H),4.77(m,1H),5.36(m,1H),6.72(d,J=9.0Hz,NH),7.01(m,1H),7.04(d,J=9.0Hz,2H),7.37(d,J=2.0 Hz),7.86(d,J=9.0Hz,2H),8.42(d,J=6.0Hz,1H).
LC/MS Rt-min(MH+):2.09(540)(方法A)。
步骤2:
按照实施例420、步骤6的方法制备化合物428,以实施例428、
步骤1的产物为原料,收率为72%。
1H NMR:(:甲醇-d4)δ1.03(s,9H),1.06-1.09(m,2H),1.23-1.25(m,2H),1.43(dd,J=5.5,9.5Hz,1H),1.48-1.77(m,8H),1.88(dd,J=5.0,8.0Hz,1H),2.21-2.28(m,2H),2.50-2.54(m,1H),2.91-2.96(m,1H),3.85(s,3H),4.05(d,J=12Hz,1H),4.27-4.29(m,2H),4.44(dd,J=7.0,10.5Hz,1H),4.84(m,1H(覆盖)),5.12(d,J=10Hz,1H),5.30(d,J=17Hz,1H),5.37(m,1H),5.72-5.80(m,1H),6.89(d,J=9.5Hz,NH),6.95(dd,J=2.5,6.0Hz,1H),7.03(d,J=9.0Hz,1H),7.33(d,J=2.5Hz,1H),7.86(d,J=9.0Hz,2H),8.41(d,J=6.0Hz,1H).
LC/MS Rt-min(MH+):2.40(753)(方法A)。
实施例429:化合物429的制备
化合物429
步骤1:
按照实施例421、步骤6的方法制备该产物,收率22%,不同之处在于使用2-噻吩硼酸。
1H NMR:(甲醇-d4)δ1.03(s,9H),1.46-1.74(m,8H),2.29-2.35(m,1H),2.63-2.67(m,1H),3.97(d,J=12Hz,1H),4.26(d,J=8.5Hz,1H),4.3 1(d,J=12Hz,1H),4.60(t,J=8.5Hz,1H),4.81(m,1H(覆盖)),5.31(m,1H),6.87-6.89(m,2H),7.13(d,J=5.0Hz,1H),7.52(s,1H(覆盖)),7.70(d,J=2.5Hz,1H),8.32(d,J=6.0Hz,1H).
LC/MS Rt-min(MH+):1.96(516)(方法A)。
步骤2:
按照实施例420、步骤6的方法制备化合物429,以实施例429、步骤1的产物为原料,收率为57%。
1H NMR:(甲醇-d4)δ1.02(s,9H),1.07(m,2H),1.24(m,2H),1.43(dd,J=5.5,9.5Hz,1H),1.53-1.78(m,8H),1.88(dd,J=5.5,8.0Hz,1H),2.21-2.28(m,2H),2.49-2.53(m,1H),2.92-2.96(m,1H),4.04(d,J=10Hz,1H),4.26-4.29(m,2H),4.43(t,J=9.0Hz,1H),4.82(m,1H(覆盖)),5.12(d,J=10.5Hz,1H),5.29(d,J=17.5Hz,1H),5.35(m,1H),5.73-5.80(m,1H),6.89(d,J=4.5Hz,1H),6.92(d,J=4.5Hz,NH),7.13(s,1H),7.35(s,1H),7.51(d,J=4.5Hz,1H),7.70(s,1H),8.32(d,J=5.5Hz,1H)。
LC/MS Rt-min(MH+):2.37(728)(方法A)。
实施例430:化合物430的制备
化合物430
步骤1:
按照实施例421、步骤6的方法制备该产物,收率17%,不同之处在于使用3-噻吩硼酸。
1H NMR:(甲醇-d4)δ1.03(s,9H),1.43-1.74(m,8H),2.30-2.36(m,1H),2.64-2.68(m,1H),3.98(dd,J=11.5,3.0Hz,1H),4.25(d,J=9.0Hz,1H),4.32(d,J=11.5Hz,1H),4.60(dd,J=8.0,9.5Hz,1H),4.80(m,1H(覆盖)),5.33(m,1H),6.96(dd,J=2.5,6.0Hz,1H),7.36(s,1H(覆盖)),7.51(s,1H(覆盖)),7.67(d,J=5.0Hz,1H),8.03(s,1H),8.39(d,J=6.0Hz,1H).
LC/MS Rt-min(MH+):1.94(516)(方法A)。
步骤2:
按照实施例420、步骤6的方法制备化合物430,以实施例430、步骤1的产物为原料,收率为45%。
1H NMR:(甲醇-d4)δ1.02(s,9H),1.06(m,2H),1.29(m,2H),1.43(dd,J=5.5,9.5Hz,1H),1.43-1.75(m,8H),1.87(dd,J=6.0,7.5Hz,1H),2.20-2.27(m,2H),2.49-2.53(m,1H),2.93-2.95(m,1H),4.05(d,J=9.0Hz,1H),4.27-4.29(m,2H),4.42-4.45(m,1H),4.85(m,1H(覆盖)),5.12(d,J=10.0Hz,1H),5.29(d,J=17.5Hz,1H),5.36(m,1H),5.73-5.80(m,1H),6.92-6.94(m,2H),7.35(s,1H),7.5 1(s,1H),7.66(d,J=4.5Hz,1H),8.00(s,1H),8.38(d,J=5.5Hz,1H).
LC/MS Rt-min(MH+):2.36(728)(方法A)。
实施例431:化合物431的制备
化合物431
步骤1:
向实施例421、步骤5的产物(100mg,0.195mmol)和四(三苯基膦)合钯(23mg,0.0195mmol)在二_烷(3mL)中的混合物内加入2-三丁基甲锡烷基噻唑(95mg,0.254mmol)和三乙胺(82μL,0.585mmol)。将溶液在氮气气氛、95℃下加热5h,随后在105℃下加热15h。冷却至室温后,将混合物过滤并浓缩。剩余物经制备型HPLC(梯度30-80%B)纯化。合并的洗脱液在pH4缓冲液和二氯甲烷间分配。水相用二氯甲烷萃取,合并的有机萃取液用盐水洗涤并干燥(硫酸镁)。得到题述化合物(31mg),为无色油状物,包含大量的三丁基甲锡烷基剩余杂质。
1H NMR:(甲醇-d4)δ1.03(s,9H),1.4(m,8H(覆盖)),2.32-2.36(m,1H),2.64-2.68(m,1H),4.00(d,J=11.5Hz,1H),4.25(s,1H),4.33(d,J=11.5Hz,1H),4.60(t,J=8.5Hz,1H),4.80(m,1H(覆盖)),5.33(m,1H),7.03(brs,1H),7.70(s,1H),7.73(s,1H),7.93(s,1H),8.41(d,J=5.5Hz,1H).
LC/MS Rt-min(MH+):2.25(517)(方法A)。
步骤2:
按照实施例420、步骤6的方法制备化合物431,以实施例431、步骤1的产物为原料,收率为41%。
1H NMR:(甲醇-d4)δ1.02(s,9H),1.06-1.70(m,13H),1.86-1.89(m,1H),2.22-2.31(m,2H),2.51-2.55(m,1H),2.92-2.94(m,1H),4.06(d,J=11.5Hz,1H),4.23-4.32(m,2H),4.44-4.47(m,1H),4.82(m,1H(覆盖)),5.12(d,J=11Hz,1H),5.30(d,J=17Hz,1H),5.36(m,1H),5.73-5.81(m,1H),6.90(d,J=9.0Hz,NH),7.04(m,1H),7.71(m,2H),7.93(s,1H),8.42(d,J=5.5Hz,1H).
LC/MS Rt-min(MH+):2.46(729)(方法A)。
实施例432:化合物432的制备
化合物432
步骤1:
向实施例423、步骤1的产物(102mg,0.204mmol)和四(三苯基膦)合钯(24mg,0.0204mmol)在二_烷(3mL)中的混合物内加入2-三丁基甲锡烷基噻唑(99mg,0.265mmol)和三乙胺(85μL,0.612mmol)。将溶液在氮气气氛、95℃下加热5h,随后在105℃下加热15h。冷却至室温后,将混合物过滤并浓缩。剩余物经制备型HPLC(梯度30-80%B)纯化。合并的洗脱液用浓氨水中和并浓缩。剩余物在pH4缓冲液和二氯甲烷间分配。水相用二氯甲烷萃取,合并的有机萃取液用盐水洗涤并干燥(硫酸镁)。得到题述化合物(33mg),为无色油状物,包含大量的三丁基甲锡烷基剩余杂质。
1H NMR:(甲醇-d4)δ1.03(s,9H),1.30(s,9H),2.28-2.32(m,1H),2.62-2.67(m,1H),3.98(d,J=11.5Hz,1H),4.21(s,1H),4.34(d,J=11.5Hz,1H),4.58(t,J=9.0Hz,1H),5.31(m,1H),7.71(d,J=2.5Hz,1H),7.94-7.96(m,2H),8.33(s,1H),8.73(s,1H).
LC/MS Rt-min(MH+-Boc):2.21(405)(方法A)。
步骤2:
按照实施例420、步骤6的方法制备化合物432,以实施例432、步骤1的产物为原料,收率为42%。
1H NMR:(甲醇-d4)δ1.02(s,9H),1.07-1.08(m,2H),1.24(m,2H),1.32(s,9H),1.44(m,1H(覆盖)),1.86-1.89(m,1H),2.21-2.29(m,2H),2.51-2.55(m,1H),2.93-2.95(m,1H),4.04(d,J=12Hz,1H),4.23(d,J=9.5Hz,1H),4.33(d,J=12Hz,1H),4.47(t,J=9.5Hz,1H),5.12(d,J=10.0Hz,1H),5.30(d,J=18Hz,1H),5.36(m,1H),5.72-5.81(m,1H),6.62(d,J=8.5Hz,NH),7.73(s,1H),7.96(m,1H),8.34(s,1H),8.74(s,1H).
LC/MS Rt-min(MH+):2.42(717)(方法A)。
实施例433:化合物433的制备
化合物433
步骤1:
向实施例420、步骤3的产物(1.00g,1.94mmol)和四(三苯基膦)合钯(112mg,0.097mmol)在二_烷(15mL)中的混合物内加入三丁基(1-乙氧基乙烯基)锡(876mg,2.43mmol)。将溶液在氮气气氛、105℃下加热6h。冷却至室温后,将混合物过滤并浓缩。剩余物在饱和碳酸氢钠溶液和乙酸乙酯间分配。水相用乙酸乙酯萃取,合并的有机萃取液用5%KF水溶液和盐水洗涤并干燥(硫酸镁)。使用Biotage 40M柱(洗脱梯度己烷-EtOAc40-70%)纯化,得到题述化合物(624mg,64%),为黄色油状物。
1H NMR:(DMSO-d6)δ0.95(s,9H),1.27(s,9H),1.35(t,J=7.0Hz,3H),2.16-2.21(m,1H),2.50(m,1H(覆盖)),3.64(s,3H),3.85(d,J=11Hz,1H),3.90(q,J=7.0Hz,2H),4.09(d,J=9.0Hz,1H),4.13(d,J=11Hz,1H),4.40(d,J=2.5Hz,1H),4.45(t,J=8.0Hz,1H),4.91(d,J=2.5Hz,1H),5.27(m,1H),6.69(d,J=9.0Hz,NH),7.48(s,1H),8.25(s,1H),8.46(s,1H).
LC/MS Rt-min(MH+):2.14(507)(方法B)。
步骤2:
向实施例433、步骤1的产物(125mg,0.247)在THF(3mL)和水(111μL,6.18mmol)中的溶液内加入NBS(44mg,0.247mmol)。在室温下搅拌20min后,将混合物浓缩并在乙酸乙酯和盐水间分配。将有机相干燥(硫酸镁),得到中间体溴代甲基酮。将该中间体溶于DMF,用硫代乙酰胺(24mg,0.321mmol)和碳酸氢钠(31mg,0.371mmol)处理。将混合物在室温下搅拌2h,浓缩并悬浮于饱和碳酸氢钠溶液中。产物用乙酸乙酯萃取(2×),用盐水洗涤并干燥(硫酸镁)。使用Biotage12M柱(洗脱梯度己烷-EtOAc 50-70%)纯化,得到题述化合物(50mg,38%),为浅色油状物。
1H NMR:(甲醇-d4)δ1.02(s,9H),1.31(s,9H),2.27-2.31(m,1H),2.63-2.67(m,1H),2.77(s,3H),3.74(s,3H),3.98(d,J=10.0Hz,1H),4.22(d,J=9.5Hz,1H),4.34(d,J=10.5Hz,1H),4.64(t,J=9.0Hz,1H),5.29(m,1H),6.38(s,NH),7.87(s,1H),7.91(s,1H),8.20(s,1H),8.71(s,1H).
LC/MS Rt-min(MH+):1.96(533)(方法B)。
步骤3:
按照实施例420、步骤5的方法制备该产物,不同之处在于使用实施例433、步骤2的产物。
1H NMR:(DMSO-d6)δ0.96(s,9H),1.25(s,9H),2.18-2.23(m,1H),2.50(m,1H(覆盖)),2.73(s,3H),3.86(d,J=11.5Hz,1H),4.10(d,J=9.0Hz,1H),4.14(d,J=11.5Hz,1H),4.39(1,J=8.5Hz,1H),5.30(m,1H),6.60(brs,NH),7.84(s,1H),8.14(s,1H),8.24(s,1H),8.78(s,1H).
LC/MS Rt-min(MH+):1.84(519)(方法B)。
步骤4:
按照实施例420、步骤6的方法制备化合物433,以实施例433、步骤3的产物为原料,收率46%。
1H NMR:(甲醇-d4)δ1.05(s,9H),1.09-1.11(m,2H),1.26-1.29(m,2H),1.36(s,9H),1.45-1.48(m,1H),1.89-1.92(m,1H),2.25-2.30(m,2H),2.53-2.57(m,1H),2.80(s,3H),2.94-3.00(m,1H),4.06(d,J=10.5Hz,1H),4.27(s,1H),4.32(d,J=12Hz,1H),4.48(dd,J=10.5,7.0Hz,1H),5.15(d,J=10.5Hz,1H),5.33(d,J=17Hz,1H),5.35(m,1H),5.76-5.83(m,1H),7.93(s,1H),7.95(m,1H),8.22(s,1H),8.74(s,1H).
LC/MS Rt-min(MH+):2.21(732)(方法B)。
实施例434:化合物434的制备
化合物434
步骤1:
向实施例421、步骤1的产物(300mg,1.56mmol)和四(三苯基膦)合钯(90mg,0.078mmol)在DMF(6mL)中的溶液内加入1-甲基-2-(三丁基甲锡烷基)-1H-吡咯(750mg,2.03mmol)和三乙胺(0.435mL,3.12mmol)。将溶液在微波炉(Emrys,Personal Chemistry)中,在氮气气氛、150℃下加热30min。冷却至室温后,混合物用乙醚和5%KF水溶液稀释并过滤。水相用乙醚(2×)萃取。合并的有机萃取液用5%KF水溶液、水和盐水洗涤并干燥(硫酸镁)。使用Biotage25S柱(洗脱梯度己烷-乙醚0-5%)纯化,得到题述化合物(169mg,56%),为无色油状物。
1H NMR:(DMSO-d6)δ 3.94(s,3H),6.10(s,1H),6.77(d,J=2.0Hz,1H),6.93(s,1H),7.27(d,J=4.0Hz,1H),7.77(s,1H),8.49(d,J=5.0Hz,1H),
LC/MS Rt-min(MH+):1.15(193,195)(方法B)。
步骤2:
按照实施例421、步骤2的方法制备该产物,以实施例434、步骤1的产物为原料,收率为39%。
1H NMR:(甲醇-d4)δ1.42,1.43(s,9H(旋转异构体)),2.27-2.34(m,1H),2.55-2.62(m,1H),3.75(m,2H),3.76、3.74(s,3H(旋转异构体)),3.84(s,3H),4.39-4.45(m,1H),5.19(m,1H),6.10(m,1H),6.49(m,1H),6.78(s,1H),6.80-6.82(m,1H),7.07(s,1H),8.35(d,J=6.0Hz,1H)。
LC/MS Rt-min(MH+,甲酸):1.49(389)(方法B)。
步骤3:
将实施例434、步骤2的产物(90mg,0.22mmol)溶于二氯甲烷(1.5mL)和TFA(1.0mL,9.0mmol)中。将溶液在室温下搅拌45min并浓缩。剩余物用1N HCl/乙醚(5mL)处理并浓缩。定量收率得到题述化合物,为浅色油状物。
LC/MS Rt-min(MH+):0.32(302)(方法B)。
步骤4:
按照实施例420、步骤3的方法制备该产物,以实施例434、步骤3的产物为原料,收率为80%。
1H NMR:(甲醇-d4)δ1.02(s,9H),1.31(s,9H),2.25-2.31(m,1H),2.61-2.64(m,1H),3.73(s,3H),3.84(s,3H),3.97-3.99(m,1H),4.22(d,J=9.5Hz,1H),4.32(d,J=11.5Hz,1H),4.61(t,J=7.5Hz,1H),5.27(m,1H),6.10(m,1H),6.42(br d,J=9.0Hz,NH),6.49(m,1H),6.78(m,1H),6.81(m,1H),7.07(s,1H),8.34(d,J=5.5Hz,1H)。
LC/MS Rt-min(MH+):1.81(516)(方法B)。
步骤5:
向实施例434、步骤4的产物(92mg,0.179mmol)在THF(1mL)和甲醇(1mL)中的溶液内加入LiOH(13mg,0.536mmol)的水(1mL)溶液。将混合物在室温下搅拌1.5h,用1N HCl猝灭,直至呈中性pH值。真空除去有机挥发份,剩余物经制备型HPLC(梯度10-80%B)纯化。合并的洗脱份用浓氨水中和并浓缩。剩余物在pH4缓冲液和乙酸乙酯间分配。水相用乙酸乙酯萃取,合并的有机萃取液用盐水洗涤并干燥(硫酸镁)。得到题述化合物(56mg,62%),为白色固体物。
1H NMR:(甲醇-d4)δ1.03(s,9H),1.32(s,9H),2.31-2.35(m,1H),2.62-2.67(m,1H),3.85(s,3H),3.98(d,J=11.5Hz,1H),4.21-4.23(m,1H),4.33(d,J=11.5Hz,1H),4.58(t,J=8.0Hz,1H),5.31(m,1H),6.12(m,1H),6.40(brd,J=8.0Hz,NH),6.52(m,1H),6.82(m,1H),6.87(m,1H),7.12(s,1H),8.36(d,J=5.5Hz,1H).LC/MS Rt-min(MH+):1.73(502)(方法B)。
步骤6:
按照实施例420、步骤6的方法制备化合物434,以实施例434、步骤5的产物为原料,收率为68%。
1H NMR:(甲醇-d4)δ1.02(s,9H),1.06-1.09(m,2H),1.23-1.26(m,2H),1.33(s,9H),1.42-1.45(m,1H),1.86-1.89(m,1H),2.21-2.27(m,2H),2.47-2.51(m,1H),2.91-2.96(m,1H),3.85(s,3H),4.04(d,J=12Hz,1H),4.24(d,J=10.0Hz,1H),4.28(d,J=12Hz,1H),4.43(dd,J=10.0,7.0Hz,1H),5.12(d,J=10.0Hz,1H),5.30(d,J=17Hz,1H),5.32(m,1H),5.73-5.80(m,1H),6.10(m,1H),6.49(m,1H),6.64(brd,J=9.0Hz,NH),6.79(m,1H),6.82(m,1H),7.08(s,1H),8.25(d,J=5.5Hz,1H).
LC/MS Rt-min(MH+):2.08(714)(方法B)。
实施例435:化合物435的制备
化合物435
步骤1:
按照实施例421、步骤2的方法制备该产物,以2,6-二溴吡啶为原料,收率为74%。
1H NMR:(DMSO-d6)δ1.34、1.38(s,9H(旋转异构体)),2.23-2.31(m,1H),2.43-2.47(m,1H(覆盖)),3.53(d,J=12Hz,1H),3.66、3.69(s,3H(旋转异构体)),3.72-3.75(m,1H),4.29-4.34(m,1H),5.42(m,1H),6.89(d,J=7.5Hz,1H),7.26(d,J=7.5Hz,1H),7.68(t,J=7.5Hz,1H)。
LC/MS Rt-min(MH+):2.62(523,425)(方法A)。
步骤2:
按照实施例420、步骤2的方法制备该产物,以实施例435、步骤1的产物为原料,定量收率。
LC/MS Rt-min(MH+):1.42(301,303)(方法B)。
步骤3:
按照实施例420、步骤3的方法制备该产物,以实施例435、步骤2的产物为原料,收率为96%。
1H NMR:(DMSO-d6)δ0.95(s,9H),1.28(s,9H),2.20-2.26(m,1H),2.45-2.48(m,1H),3.64(s,3H),3.94(d,J=9.5 Hz,1H),4.01-4.08(m,2H),4.45(t,J=8.5Hz,1H),5.53(m,1H),6.66(d,J=7.0Hz,1H),6.82(d,J=7.0Hz,1H),7.25(d,J=7.0Hz,1H),7.64-7.69(m,1H).
LC/MS Rt-min(MH+):2.73(514,516)(方法A)。
步骤3:
按照实施例420、步骤5的方法制备该产物,以实施例435、步骤2的产物为原料,定量收率。
1H NMR:(DMSO-d6)δ0.95(s,9H),1.28(s,9H),2.19-2.25(m,1H),2.43-2.47(m,1H),3.94(m,1H),4.01-4.08(m,2H),4.36(t,J=8.5Hz,1H),5.52(m,1H),6.65(d,J=8.0Hz,1H),6.82(d,J=7.5Hz,1H),7.25(d,J=7.5Hz,1H),7.67(t,J=7.5Hz,1H),12.6(s,1H).
LC/MS Rt-min(MNa+):2.51(522,524)(方法B)。
步骤4:
向实施例435、步骤3的产物(125mg,0.250mmol)、四(三苯基膦)合钯(14.4mg,0.0125mmol)和3-呋喃基硼酸(35mg,0.313mmol)在DMF(2mL)和水(0.025mL)中的溶液内加入碳酸铯(244mg,0.750mmol)。将混合物在氮气气氛、105℃下加热3h。冷却至室温后过滤除去固体物,将滤液真空浓缩。剩余物经制备型HPLC(梯度30-100%B)纯化。合并的洗脱份用浓氨水中和并浓缩。剩余物在pH4缓冲液和二氯甲烷间分配。水相用二氯甲烷萃取,合并的有机萃取液用盐水洗涤并干燥(硫酸镁)。得到题述化合物(90mg,76%),为白色泡沫状物。
1H NMR:(甲醇-d4)δ1.06(s,9H),1.39(s,9H),2.36-2.42(m,1H),2.61-2.65(m,1H),4.12(dd,J=4.0.11Hz,1H),4.19(d,J=11Hz,1H),4.28(s,1H),4.62(t,J=8.5Hz,1H),5.79(m,1H),6.64(d,J=8.0Hz,1H),6.96(s,1H),7.22(d,J=8.0Hz,1H),7.58(m,1H),7.66(t,J=8.0Hz,1H),8.13(s,1H).
LC/MS Rt-min(MNa+):2.53(511)(方法B)。
步骤5:
按照实施例420、步骤6的方法制备化合物435,以实施例435、步骤4的产物为原料,收率为57%。
1H NMR:(DMSO-d6)δ0.96(s,9H),1.02-1.05(m,2H),1.09-1.11(m,2H),1.19(s,9H),1.35-1.38(m,1H),1.71(dd,J=5.5,8.0Hz,1H),2.15-2.22(m,2H),2.37-2.41(m,1H),2.93(br m,1H),4.03-4.08(m,3H),4.35(br t,1H),5.10(d,J=10.5Hz,1H),5.24(d,J=17Hz,1H),5.60-5.67(m,1H),5.73(m,1H),6.47(br s,1H),6.64(d,J=7.5Hz,1H),7.04(s,1H),7.31(d,J=7.5Hz,1H),7.72(t,J=7.5Hz,1H),7.77(s,1H),8.32(s,1H),8.92(s,NH),10.4(s,NH).
LC/MS Rt-min(MNa+):2.64(723)(方法B)。
实施例436:化合物436的制备
化合物436
步骤1:
按照实施例435、步骤4的方法制备该产物,收率73%,不同之处在于使用苯基硼酸。
1H NMR:(甲醇-d4)δ1.06(s,9H),1.39(s,9H),2.39-2.45(m,1H),2.65-2.77(m,1H),4.18-4.30(m,3H),4.64(t,J=8.0Hz,1H),5.72(m,1H),6.74(d,J=8.0Hz,1H),7.44-7.55(m,4H),7.75(t,J=8.0Hz,1H),8.07(d,J=7.5Hz,2H).
LC/MS Rt-min(MNa+):2.72(521)(方法B)。
步骤2:
按照实施例420、步骤6的方法制备化合物436,以实施例436、步骤1的产物为原料,收率为66%。
1H NMR:(DMSO-d6)δ0.96(s,9H),1.04-1.05(m,2H),1.09(m,2H),1.30(s,9H),1.36-1.39(m,1H),1.71(t,J=7.5Hz,1H),2.16-2.24(m,2H),2.41-2.45(m,1H),2.93(m,1H),4.06-4.09(m,3H),4.38(brt,1H),5.10(d,J=10.5Hz,1H),5.30(d,J=17Hz,1H),5.60-5.67(m,1H),5.80(m,1H),6.49(brs,1H),6.75(d,J=7.5Hz,1H),7.43-7.51(m,3H),7.59(d,J=7.5Hz,1H),7.81(t,J=7.5Hz,1H),8.09(d,J=7.0Hz,2H),8.91(s,NH),10.4(s,NH).
LC/MS Rt-min(MH+):2.77(711)(方法B)。
实施例437:化合物437的制备
化合物437
步骤1:
向2-溴-6-甲基-吡啶(7.65g,44.4mmol)的二氯甲烷(50mL)溶液中加入mCPBA(77%,12.9g,57.7mmol)的二氯甲烷(100mL)溶液。将溶液在环境温度下搅拌18h。将混合物用固体碳酸钠中和并加入水。水相用二氯甲烷(2×)萃取。合并的有机萃取液用5%硫代硫酸钠、5%碳酸钠和盐水洗涤并干燥(硫酸镁)。使用Biotage 40M柱(洗脱梯度己烷-乙酸乙酯40-70%)纯化,得到题述化合物(5.0g,60%)为无色油状物,该产物在静置时固化。
1H NMR:(DMSO-d6)δ2.43(s,3H),7.15(t,J=8.0Hz,1H),7.50(s,1H),7.78(s,1H)。
LC/MS Rt-min(MH+):0.32(188,190)(方法B)。
步骤2:
向实施例437、步骤1的产物(4.5g,24mmol)的DMF(20mL)溶液中分批加入POBr3(8.2g,29mmol)。发生剧烈的放热反应并形成沉淀。将混合物在环境温度下静置2h。混合物用水和饱和碳酸氢钠溶液猝灭,直至呈中性pH值。水相用乙醚(2×)萃取。合并的有机萃取液用盐水洗涤并干燥(硫酸镁)。使用Biotage 40M柱(洗脱梯度己烷-乙醚0-10%)纯化,得到题述化合物(1.78g,30%),为无色油状物。
1H NMR:(DMSO-d6)δ2.45(s,3H),7.64(s,1H),7.80(s,1H)。
LC/MS Rt-min(MH+):1.85(250,252,254)(方法B)。
步骤3:
按照实施例421、步骤2的方法制备该产物,以实施例437、步骤1的产物为原料,收率为46%。
1H NMR:(DMSO-d6)δ1.34,1.38(s,9H(旋转异构体)),2.20-2.27(m,1H),2.38(s,3H),2.43-2.50(m,1H),3.55(d,J=12.5Hz,1H),3.64-3.67(m,1H),3.66、3.69(s,3H(旋转异构体)),4.26-4.32(m,1H),5.17(m,1H),6.93(s,1H),7.08(s,1H)。
LC/MS Rt-min(MH+):2.17(415,417)(方法B)。
步骤4:
按照实施例420、步骤3的方法制备该产物,以实施例437、步骤3的产物为原料,定量收率。
步骤4:
按照实施例420、步骤3的方法制备该产物,以实施例437、步骤3的产物为原料,收率为75%。
1H NMR:(DMSO-d6)δ0.94(s,9H),1.27(s,9H),2.16-2.21(m,1H),2.36(s,3H),2.48(m,1H(覆盖)),3.64(s,3H),3.83(d,J=10.5Hz,1H),4.06(d,J=8.5Hz,1H),4.14(d,J=10.5Hz,1H),4.42(t,J=9.0Hz,1H),5.27(m,1H),6.68(brs,NH),6.88(s,1H),7.03(s,1H).
LC/MS Rt-min(MH+):2.19(528,530)(方法B)。
步骤5:
按照实施例420、步骤5的方法制备该产物,以实施例437、步骤4的产物为原料,定量收率。
1H NMR:(DMSO-d6)δ0.95(s,9H),1.27(s,9H),2.15-2.19(m,1H),2.36(s,3H),2.44-2.48(m,1H),3.81(d,J=10.5Hz,1H),4.04-4.06(m,1H),4.13(d,J=10.5Hz,1H),4.33(t,J=8.5Hz,1H),5.26(m,1H),6.66(br d,J=9.0Hz,NH),6.88(s,1H),7.03(s,1H).
LC/MS Rt-min(MNa+):2.14(536,538)(方法B)。
步骤6:
向实施例437、步骤5的产物(101mg,0.196mmol)、四(三苯基膦)合钯(11.3mg,0.0098mmol)和苯基硼酸(34mg,0.275mmol)在DMF(2mL)中的溶液内加入2M碳酸钠水溶液(0.294mL,0.588mmol)。将试管密封并在微波炉(Emrys,Personal Chemistry)中,在氮气气氛、150℃下加热15min。冷却至室温后,混合物用1N HCl(0.5mL)酸化。过滤除去固体,将滤液真空浓缩。剩余物经制备型HPLC(梯度20-80%B)纯化。合并的洗脱份用浓氨水中和并浓缩。剩余物在pH4缓冲液和乙酸乙酯间分配。水相用乙酸乙酯萃取(2×),合并的有机萃取液用盐水洗涤并干燥(硫酸镁)。得到题述化合物(115mg,>100%),为白色固体物。
1H NMR:(甲醇-d4)δ1.06(s,9H),1.30(s,9H),2.36-2.41(m,1H),2.69(s,3H),2.65-2.76(m,1H),4.01(dd,J=3.0,12Hz,1H),4.21(s,1H),4.44(d,J=12Hz,1H),4.64(dd,J=8.0,10Hz,1H),5.46(m,1H),6.91(s,1H),7.29(s,1H),7.39(m,3H),7.57(d,J=7.5Hz,2H).
LC/MS Rt-min(MH+):1.84(513)(方法B)。
步骤7:
按照实施例420、步骤6的方法制备化合物437,以实施例437、步骤6的产物为原料,收率为31%。
1H NMR:(DMSO-d6)δ0.93(s,9H),0.99-1.04(m,4H),1.25(s,9H),1.34-1.37(m,1H),1.69-1.71(m,1H),2.1-2.20(m,2H),2.39-2.43(m,H),2.48(s,3H),2.93(m,H),3.92(d,J=8.5Hz,1H),4.07(d,J=9.0Hz,1H),4.11(d,J=12Hz,1H),4.32(t,J=7.0Hz,1H),5.10(d,J=10.5Hz,1H),5.23(d,J=17.5Hz,1H),5.39(m,1H),5.60-5.67(m,1H),6.58(d,J=8.5Hz,1H),6.83(s,1H),7.27(s,1H),7.40-7.48(m,3H),8.04-8.06(m,2H),8.92(s,NH),10.4(s,NH)。
LC/MS Rt-min(MH+):2.10(725)(方法B)。
实施例438:化合物438的制备
化合物438
步骤1:
按照实施例437、步骤6的方法制备该产物,以2-噻吩硼酸为原料,定量收率。
1H NMR:(甲醇-d4)δ1.05(s,9H),1.32(s,9H),2.32-2.38(m,1H),2.56(s,3H),2.66-2.70(m,1H),3.99(dd,J=3.0,12Hz,1H),4.23(s,1H),4.36(d,J=12Hz,1H),4.61(t,J=8.5Hz,1H),5.35(m,1H),5.86(s,1H),7.16-7.18(m,1H),7.21(s,1H),7.56(m,1H),7.72(d,J=3.0Hz,1H).
LC/MS Rt-min(MH+):1.80(519)(方法B)。
步骤2:
按照实施例420、步骤6的方法制备化合物438,以实施例438、步骤1的产物为原料,收率为41%。
1H NMR:(DMSO-d6)δ0.91(s,9H),0.99-1.04(m,4H),1.20(s,9H),1.35-1.37(m,1H),1.69-1.71(m,1H),2.09-2.20(m,2H),2.33(m,1H),2.42(s,3H),2.93(m,1H),3.92(d,J=8.5Hz,1H),4.07-4.11(m,2H),4.29-4.32(m,1H),5.10(d,J=10.5Hz,1H),5.23(d,J=17.0Hz,1H),5.36(m,1H),5.60-5.67(m,1H),6.58(d,J=8.5Hz,1H),6.75(s,1H),7.14(t,J=4.5Hz,1H),7.28(s,1H),7.59(d,J=5.5Hz,1H),7.80(d,J=3.5Hz,1H),8.92(s,NH),10.4(s,NH).
LC/MS Rt-min(MH+):2.06(731)(方法B)。
部分K:
实施例450:化合物450的制备
化合物450
按照实施例8、步骤5的方法制备化合物450,不同之处在于使用4-氯-6-氟-2-三氟甲基喹啉。
1H NMR(CD3OD)δ0.97-1.04(m,12H),1.17-1.24(m,10H),1.39-1.46(m,1H),1.82-1.87(m,1H),2.20-2.23(m,1H),2.35-2.39 9m,1H),2.55-2.65(m,1H),2.91-2.96(m,1H),4.09-4.11(m,1H),4.18-4.21(m,1H),4.56(b,2H),5.10-5.14(m,1H),5.28-5.31(m,1H),5.60(b,1H),5.70-5.80(m,1H),7.41(s,1H),7.65-7.68(m,1H),7.86(s,1H),8.13-8.15(m,1H)。
实施例451:化合物451的制备
化合物451
方案1
步骤1:
按文献所述方法制备甲苯磺酸盐(Patchett,A.A.;Witkof,B.J.Am.Chem.Soc.1957,185-192)并未经进一步纯化直接使用。
向NaH(76mg,1.90mmol)的DMF(20ml)浆状物中加入1-萘硫酚(0.29mg,1.80mmol)并将混合物搅拌30分钟。加入所述甲苯磺酸盐溶液(0.61g,1.80mmol),将混合物在23℃下搅拌12h。将混合物浓缩,剩余物在EtOAc/H2O间分配。将有机萃取液干燥(硫酸镁)并浓缩。剩余物经柱层析(用5%EtOAc/己烷至30%EtOAc/己烷洗脱)纯化,得到261mg(38%)产物,为黄色油状物。
1H NMR(CDCl3,3∶2旋转异构体的混合物)δ1.41(s,9H),1.44(s,9H),2.25-2.29(m,2H),3.69(s,3H),3.35-3.42(m,1H),3.51-3.53(m,1H),3.80-3.86(m,2H),4.38-4.39(m,1H),4.46-4.48(m,1H),7.41-7.46(m,1H),7.42-7.54(m,1H),7.57-7.59(m,1H),7.58(d,J=4Hz,1H),7.82-7.88(m,2H),8.46(d,J=5Hz,1H);
LC-MS(保留时间:1.93),MS m/z 388(M++1)。
步骤2:
将4-(萘-1-硫基)-吡咯烷-1,2-二甲酸1-叔丁酯2-甲酯(0.38g,0.98mmol)和4N HCl(1.0ml)的混合物在23℃下搅拌2h。除去溶剂并将剩余物溶于乙腈(20ml)中,用酸(0.37g,2.16mmol)、TBTU(0.23g,0.98mmol)和DIPEA(0.37g,2.16mmol)处理并搅拌12h。将混合物浓缩,并将剩余物溶于乙酸乙酯中并用1N HCl、饱和碳酸氢钠洗涤,随后经硫酸镁干燥并浓缩。剩余物未经进一步纯化直接使用。
1H NMR(CDCl3,1∶1旋转异构体的混合物)δ0.99(s,9H),1.02(s,9H),1.44(s,9H),1.46(s,9H)2.2-2.25(m,2H).70(s,3H),3.82-3.86(m,1H),3.89-3.92(m,2H)4.26(s,1H),4.28(s,1H),4.70-4.75(m,1H),7.40-7.48(m,1H),7.54-7.55(m,1H),7.59-7.62(m,1H),7.72-7.74(m,1H),7.86 7.89(m,2H),8.48-8.50(m,1H);
LC-MS(保留时间:1.59),MS m/z 523(M+Na)。
步骤3:
向1-(2-叔丁氧羰基氨基-3,3-二甲基-丁酰基)-4-(萘-1-硫基)-吡咯烷-2-甲酸甲酯(实施例451、步骤2产物)(0.49g,0.98mmol)在THF/H2O(2∶1)中的混合物内加入LiOH水合物(O.20g,4.9mmol),将混合物搅拌12h。将溶液浓缩,用EtOAc洗涤。水层用1N HCl酸化并用EtOAc萃取。在第一次EtOAc萃取液中观测到产物。所述第一次有机萃取液经硫酸镁干燥并浓缩,得到328mg(71%)黄褐色固体。
1H NMR(DMSO-d6,2∶1旋转异构体的混合物)δ0.88(s,9H),0.92(s,9H),1.34(s,9H),1.38(s,9H),2.18-2.25(m,2H),3.66-3.75(m,1H),3.89-4.00(m,2H),4.10-4.13(m,1H),4.25-4.32(m,1H),7.45-7.7.51(m,1H),7.56-7.61(m,2H),7.65-7.7.69(m,1H),7.88-7.91(m,1H),7.97(d,J=4.8Hz,1H),8.27-8.35(m,1H);
LC-MS(保留时间:1.52),MS m/z 486(M+1)。
步骤4:
向所述酸(实施例451、步骤3产物)(0.32g,0.87mmol)在乙腈(10mL)和DMF(2mL)中的溶液内加入1(R)-2(S)和1-(S)-2(R)1-氨基-2-乙烯基-环丙烷甲酸乙酯盐酸盐的非对映异构体混合物(240mg,0.87mmol)、TBTU(201mg,0.87mmol)和DIPEA(0.32mL,0.742mmol),将所得混合物在23℃下搅拌12h。将混合物浓缩,剩余物在乙酸乙酯和水间分配。分离出有机层,经硫酸镁干燥并浓缩。以30%EtOAc/己烷为洗脱液,将剩余物经柱层析纯化,得到240mg(38%),为浅黄色固体物。
1H NMR(DMSO-d6,旋转异构体和非对映异构体的混合物)δ0.87(s,9H),0.88(s,9H),0.97-1.04(m,3H),1.33(s,9H),1.38(s,9H),2.11-2.20(m,2H),3.74-3.85(m,1H),3.89-3.96(m,2H),3.98-4.03(m,4H),4.00-4.09(m,1H),4.40-4.42(m,1H),5.04-5.09(m,1H),5.17-5.29(m,2H),5.50-5.70(m,1H),6.60-6.62(m,1H),6.72-6.75(m,1H),7.50-7.56(m,1H),7.56-7.72(m,2H),7.70-7.80(m,1H),7.92-8.00(m,1H),8.00-8.06(m,1H),8.29-8.40(m,1H),8.65(s,1H),8.79(s,1H);
LC-MS(保留时间:1.59),MS m/z 623(M+1)。
步骤5:
按照实施例451、步骤3的方法,使用LiOH在THF/甲醇/H2O(4/2/1)中的溶液制备所述酸,不同之处在于使用实施例451、步骤4的产物。
1H NMR(DMSO-d6,旋转异构体和非对映异构体的混合物)δ0.90(s,9H),1.17-1.23(m,2H),1.32-1.37(m,9H),2.10-2.12(m,1H),2.20-2.31(m,2H),3.97-4.05(m,2H),4.10-4.12(m,1H),4.32-4.40(m,1H),4.55-4.61(m,1H),4.80-4.98(m,2H),5.03-5.08(m,1H),5.10-5.20(m,1H, 5.75-5.90(m,1H),6.55-6.70(m,1H),7.42-7.57(m,1H),7.60-7.64(m,2H),7.70-7.72(m,1H),7.80-7.97(m,1H),7.96-7.99(m,1H),8.20-8.50(m,2H);
LC-MS(保留时间:1.52),MS m/z 595(M+1)。
步骤6:
将所述酸(实施例451、步骤5的产物)(172mg,0.29mmol)、甲烷磺酰胺(110mg,1.16mmol)、EDAC(110mg,0.58mmol)和DMAP(71mg,0.58mmol)的混合物溶于THF(10ml)并搅拌12h。加入DBU(0.087mL,0.58mmol)并将混合物搅拌48h。除去溶剂并将剩余物溶于乙酸乙酯中,用水和1N HCl洗涤,经硫酸镁干燥并浓缩。剩余物经制备型薄层层析纯化,得到15mg(8%)化合物451,为黄褐色固体物。
1H NMR(DMSO-d6,旋转异构体和非对映异构体的混合物)δ0.98(s,9H),1.27-1.42(m,2H),1.46(s,9H),1.76-1.79(m,1H),1.83-1.86(m,1H),1.92-2.10(m,1H),2.16-2.25(m,2H),3.01-3.10(m,1H),3.80-3.83(m,1H),3.86-3.89(m,1H),3.98-3.99(m,1H),3.99-4.05(m,1H),4.24-4-29(m,1H),4.44-4.53(m,1H),4.86(s,3H),5.08-5.15(m,1H),5.25-5.29(m,1H),5.65-5.85(m,1H),6.5-6.8(m,1H),7.48(t,J=7.7Hz,1H),7.54(t,J=7.1Hz,1H),7.59(t,J=7.1Hz,1H),7.75-7.77(m,1H),7.88-7.91(m,2H),8.46(d,J=8.25Hz,1H);
LC-MS(保留时间:1.52),MS m/z 672(M+1,次峰),m/z693(M+Na主峰)。
实施例452:化合物452的制备
化合物452
步骤1:
向NaH(76mg,1.90mmol)在DMF(20mL)中的浆状物内加入2-萘硫酚(0.29g,1.80mmol),将混合物搅拌30分钟。加入所述甲苯磺酸盐(实施例451、步骤1)(0.61g,1.79mmol)的DMF(2ml)溶液,将所得混合物在23℃下搅拌12h。将混合物浓缩并随后使其在EtOAc/H2O间分配。有机层用饱和碳酸氢钠洗涤,干燥(硫酸镁)并浓缩。将剩余物经层析纯化,依次用5%EtOAc/己烷和30%EtOAc/己烷洗脱,得到261mg(38%)产物,为澄清油状物。
1H NMR(DMSO-d6)δ1.32(s,9H),2.29-2.35(m,2H),3.33-3.47(m,2H),3.66(s,3H),3.71-3.81(m,1H),4.29-4.32(s,1H),7.49-7.55(m,3H),7.70-7.80(m,1H),7.81-7.97(m,3H);
LC-MS(保留时间:1.54),MS m/z 387(M+1)。
步骤2:
在4-(萘-2-硫基)-吡咯烷-1,2-二甲酸1-叔丁酯·2-甲酯(310mg,0.80mmol)和4N HCl在二_烷(1.49ml,2.69mmol)的混合物在23℃下搅拌2h,随后浓缩。将剩余物溶于乙腈(10mL)中,随后加入N-Boc-叔丁基甘氨酸(196mg,0.85mmol)、TBTU(0.27g,0.85mmol)和DIPEA(0.32mL,1.85mmol),并将混合物搅拌过夜。将混合物浓缩并将剩余物溶于EtOAc中,用1N HCl、饱和碳酸氢钠洗涤,干燥并浓缩,得到300mg(90%)产物,为黄色油状物。
1H NMR(甲醇-d4)δ0.99(s,9H),1.44(s,9H),2.20-2.35(m,2H),3.75(s,3H),3.92-4.08(m,2H),4.26(d,J=9.4Hz,1H),4.57(t,J=9.5Hz,1H)6.46(d,J=9.5Hz,1H),7.48-7.60(m,3H).7.83-7.90(m,3H),8.02(s,1H)
LC-MS(保留时间:1.98),MS m/z 523(M+Na)。
步骤3:
将4-(萘-2-硫基)-吡咯烷-1,2-二甲酸1-叔丁酯·2-甲酯(0.48g,0.96mmol)溶于甲醇(20mL)中,并与LiOH(0.2g,4.8mmol)一起搅拌12h。将所得溶液浓缩、酸化并用EtOAc萃取。有机萃取液经硫酸镁干燥并浓缩,得到418mg(91%)黄色固体物。
1H NMR(DMSO-d6,1∶2的旋转异构体混合物)δ0.86、0.93(s,9H)(1∶2的旋转异构体混合物),1.35、1.38(s,9H)(1∶2的旋转异构体混合物),2.01-2.18、2.25-2.35(m,2H),3.25-3.40(m,2H),3.70-3.80(m,1H),4.00-4.20(m,J=9.4Hz,2H),4.30-4.40(s,1H),5.61-5.70、6.42-6.50(m,1H)(1∶2的旋转异构体混合物),7.50-7.54(m,3H),7.87-7.89(m,3H),7.98(s,1H);
LC-MS(保留时间:1.93),MS m/z 487(M+1)。
步骤4:
方案1
将1-叔丁氧羰基氨基-2-乙烯基-环丙烷甲酸乙酯(4.3g,17.8mmol)的甲醇(50mL)溶液用LiOH(0.84g,20.0mmol)和水(5mL)处理,将所得混合物搅拌12h。除去溶剂,将剩余物酸化并用EtOAc萃取。有机层经硫酸镁干燥、过滤并浓缩,得到2.1g(52%)所述酸,为黄色油状物。将所述酸(2.1g,9.25mmol)溶于THF中,用CDI(7.25g,13.8mmol)处理,加热回流3h,随后冷却至23℃。依次加入甲烷磺酰胺(1.76g,18.5mmol)和DBU(2.77ml,18.5mmol)并在23℃下搅拌72h。将混合物浓缩,将剩余物酸化至pH4(1N HCl)并用EtOAc萃取。有机萃取液经硫酸镁干燥并浓缩,得到1.99g(71%)黄色油状物,该产物静置时发生固化。
1H NMR(DMSO-d6)δ1.16-1.23(m,1H),1.43(s,9H),1.65-1.75(m,1H),2.15-2.25(m,2H),3.16(s,3H),5.08(d,J=9.9Hz,1H),5.22(d,J=17.1Hz,1H),5.40-5.52(m,1H);
LC-MS(保留时间:1.14),MS m/z 304(M+1)。
步骤5:
将实施例452、步骤4的产物在二_烷/4N HCl(2ml)中的溶液搅拌2h并随后浓缩。将剩余物溶于乙腈(5mL)中,加入所述酸(实施例452、步骤3产物)(120mg,0.25mmol)、TBTU(58mg,0.25mmol)和DIPEA(0.06ml,0.35mmol)的混合物,将混合物搅拌12h。除去溶剂并将剩余物溶于乙酸乙酯中,用1N HCl和饱和碳酸氢钠洗涤,经硫酸镁干燥并浓缩。剩余物经制备型TLC(Analtech,20×40cM,1000SiO2)纯化,得到128mg(70%)化合物452,为黄褐色固体物。
1H NMR(DMSO-d6,非对映异构体的混合物)δ0.97,0.99(s,9H),1.23-1.43(m,2H),1.45(s,9H),1.82-1.83(m,1H),2.00-2.51(m,2H),2.90-2.99(m,1H),3.33(s,3H),3.90-3.99(m,1H),4.01-4.20(m,2H),4.25-4.30(m,1H),4.45-4.55(m,1H),4.95-5.10(m,1H),5.12-5.25(m,1H),5.71-5.85(m,1H),6.4-6.8(brm,1H),7.46-7.53(m,3H),7.80-7.86(m,3H),7.98-7.99(m,1H);
LC-MS(保留时间:1.95),MS m/z 672(M+1)。
实施例453:化合物453的制备
化合物453
步骤1:
将实施例452、步骤3的产物(110mg,0.23mmol)的DCM(20ml)溶液、3-氯过苯甲酸(121.6mg,0.57mmol,85%过酸)、KHPO4(0.13g,0.94mmol)和K2HPO4(0.18g,1.05mmol)的混合物在23℃下搅拌12h。将所得溶液用DCM稀释,用水和饱和碳酸氢钠溶液洗涤,经硫酸镁干燥并浓缩,得到产物110mg。(92%),为澄清油状物。
1H NMR(DMSO-d6)δ0.91(s,9H),1.48(s,9H),2.23-2.28(m,1H),2.65-2.80(m,1H),3.88-3.90(m,1H),4.12(t,J=8.0Hz,1H),4.20(d,J=9.5Hz,1H),4.27(d,J=9.9Hz,1H),6.76(d,J=9.3Hz,1H),7.70-7.80(m,2H),7.88-7.95(m,1H),8.11(d,J=8.1Hz,1H),8.17(d,J=8.6Hz,1H),8.26(d,J=8.1Hz,1H),8.71(s,1H);
LC-MS(保留时间:1.72),MS m/z 519(M+1)。
步骤2:
将实施例453、步骤1的产物(110mg,0.212mmol)的混合物中依次加入胺(实施例452、步骤5a的产物)(0.65mg,0.212mmol)、TBTU(48.5mg,0.21mmol)和DIPEA(60.8ml,0.35mmol)并在23℃下搅拌12h。除去溶剂并将剩余物溶于乙酸乙酯,用1N HCl和饱和碳酸氢钠溶液洗涤,经硫酸镁干燥并浓缩。剩余物经制备型TLC(洗脱液:10%甲醇/二氯甲烷)纯化,得到25mg(17%)化合物453,为白色固体物。
1H NMR(DMSO-d6,非对映异构体的混合物)δ0.93,0.96(s,9H),1.38-1.45(m,2H),1.53,1.55(m,9H),1.76-1.85(m,1H),2.21-2.40(m,2H),3.13-3.15(m,2H),3.34(s,3H),3.91-3.99(m,1H),4.15(m,1H),4.25(m,1H),4.30(m,1H),5.09-5.12(m,1H),5.26-5.31(m,1H),5.72-5.76(m,1H),6.65-6.68(m,1H),6.71-6.76(m,1H),7.67-7.76(m,2H),7.91-7.95(m,1H),8.03(d,J=7.8Hz,1H),8.13(d,J=8.5Hz,1H),8.20(d,J=7.6Hz,1H),8.7(s,1H);
LC-MS(保留时间:1.76),MS m/z 705(M+1)。
实施例454:化合物454的制备
化合物454
步骤1:
向氢化钠(O.91g,22.7mmol)在THF(50mL)中的浆状物内加入N-BOC-反式-4(R)-羟基-L-脯氨酸(2.5g,10.8mmol),将混合物在23℃下搅拌1h。加入2-氯甲基萘(1.9g,10.8mmol)并在室温下将混合物搅拌12h。除去溶剂并将剩余物倒入水中,用己烷洗涤。将水层酸化(1N HCl)并用EtOAc萃取。分离出EtOAc层,干燥(硫酸镁)并浓缩,得到浅黄色剩余物。将所述油状物经快速层析纯化,用1∶1EtOAc/己烷(含1%乙酸)为洗脱液,得到1.56g(39%)所需的产物,为粘稠油状物。
1H NMR(DMSO-d6,3∶1的旋转异构体混合物)δ1.35、1.37(s,9H,分别为主峰和次峰),1.92-2.02、2.15-2.20(m,2H,分别为主峰和次峰),2.35-2.50(m,2H),3.41-3.49(m,2H),4.12-4.16,4.20-4.21(m,2H),4.65-4.68(m,2H),7.46-7.52(m,3H),7.74-7.91(m,4H),(没有观察到酸羟基);LC-MS(保留时间:1.44,YMC ODS-A C18 S7 3.0×50mm,梯度:10%甲醇/H2O 0.1%TFA至90%甲醇/H2O 0.1%TFA),MSm/z394(M++1+Na)。
步骤2:
向2-(1-乙氧羰基-2-乙烯基-环丙基氨基甲酰基)-4-(萘-2-甲氧基)-吡咯烷-1-甲酸叔丁酯盐酸盐的1∶1的非对映异构体混合物(1R,2S/1S,2R,其中羧基与乙烯基部分呈顺式构型)(0.54g,1.3mmol)[将N-Boc胺与HCl(4N)/二_烷搅拌1h,随后真空除去溶剂制备]在乙腈(50mL)中的溶液内依次加入Boc-4(R)-(2-甲基萘基)脯氨酸(0.5g,1.3mmol)、TBTU(0.45g,1.4mmol)和DIPEA(0.78mL,4.5mmol)。将混合物搅拌12h并浓缩。将剩余物溶于EtOAc/H2O中,用饱和碳酸氢钠溶液和饱和的NaCl溶液洗涤,干燥(硫酸镁)并浓缩,得到粘稠黄色油状产物(0.6g,91%),为非对映异构体的混合物。
1H NMR(DMSO-d6)δ:1.08-1.22(m,7H),1.23-1.39(m,9H),2.02-2.18(m,1H),2.25-2.35(m,1H),3.33-3.53(m,2H);3.90-4.14(m,4H),4.45-4.70(m,2H),5.07-5.11(m,1H),5.24-5.30(m,1H),5.58-5.63(m,1H),7.43-7.51(m,4H),7.84-7.96(m,3H);MS m/z 531(M++1+Na).
步骤3:
将实施例454、步骤2的产物的溶液(600mg,1.18mmol)与HCl(4N,3mL,11.8mmol)/二_烷一起搅拌1h,随后真空除去溶剂。将剩余物溶于乙腈(10mL)中,依次用Boc-L-t-Bu-Gly(0.42g,1.38mmol)、TBTU(0.27g,1.18mmol)和DIPEA(0.71mL,4.1mmol)处理。将混合物搅拌12h并浓缩。将剩余物溶于EtOAc/H2O中,用1N HCl、饱和碳酸氢钠溶液和饱和NaCl溶液洗涤,干燥(硫酸镁)并浓缩,得到粘稠黄色油状物。产物经快速层析纯化,使用梯度洗脱液:5%EtOAc/己烷、10%EtOAc/己烷,最后使用30%EtOAc/己烷,得到粘稠油状产物(0.243g,33%),为非对映异构体和旋转异构体的混合物。
1H NMR(DMSO-d6)δ:0.83-1.00(m,10H),1.34(s,9H),1.58-1.59,1.65-1.67(m,2H),1,95-1.99,2.04-2.06,2.10-2.19,2.24-2.56(m,2H),3.97-4.04(m,3H),4.08-4.17(m,3H),4.29-4.31(m,2H),4.594.72(m,3H),5.06-5.10(m,1H),5.18-5.30(m,1H),5.60-5.63(m,1H),6.59-6.65,6.70-6.74(m,1H),7.43-7.51(m,4H),7.84-7.96(m,3H),8.66,8.76(s,1H);
MS m/z 531(M++1+Na)。
步骤4:
向实施例454、步骤3的产物(240mg,0.39mmol)在THF(15mL)和H2O(2mL)中的悬浮液内加入LiOH(82mg,1.95mmol)。将反应混合物搅拌12h,随后真空浓缩直至仅剩下水层。加入1.0N盐酸将所得的含水剩余物酸化至pH3.0,并用EtOAc萃取(2×80mL)。将合并的有机萃取液干燥(硫酸镁),过滤并真空浓缩,得到产物,为黄褐色固体物(200mg,0.33mmol,85%):
1H NMR(DMSO-d6)δ:0.86、0.94(s,9H分别为次峰和主峰),1.23-1.42(m,2H),1.34(s,9H),1.8-2.1(m,2H),2.18-2.30(m,1H),3.59-3.73(m,3H),4.0-4.09(m,1H),4.18-4.34(m,3H),4.56-4.62(m,1H),4.66-4.67(m,1H),4.82-4.92(m,1H),5.0-5.20(m,1H),5.91-6.08(m,1H),6.5-6.7(m,1H),7.45-7.59(m,3H),7.82-7.97(m,4H),8.2-8.3,8.3-8.4(s,1H);LC-MS(保留时间:1.50),MS m/z 593(M++1)。
步骤5:
向实施例454、步骤4的产物(190mg,0.32mmol)、EDAC(122mg,0.64mmol)和4-DMAP(78mg,0.64mmol)在THF(20mL)中的溶液内加入商品甲烷磺酰胺(122mg,1.28mmol)。将所得的溶液搅拌2天,随后加入DBU(95μL,0.64mmol)。将反应物搅拌24h并接着浓缩。剩余物在EtOAc(80mL)和水间分配,用1N HCl和碳酸氢钠水溶液(2×30mL)洗涤,干燥(硫酸镁)并经制备型HPLC纯化(65-90%甲醇/水/0.1%TFA),得到56mg产物和其中已除去BOC基团的物质的混合物。该物质经制备型TLC(洗脱液:10%甲醇/二氯甲烷,使用20×40cM板,来自Analtech)进一步纯化,得到化合物454,为黄褐色固体物(12mg,6%)。
1H NMR(MeOD-d4 50/50P1的非对映异构体混合物)δ0.88-0.99(m,2H),1.01、1.02(s,9H分别为非对映异构体的次峰和主峰),1.23-1.42(m,2H),1.38(s,9H),1.72-1.79(m,1H),1.86-1.88(m,1H),2.00-2.10(m,2H),2.10-2.23(m,1H),2.3-2.5(m,1H),3.12、3.17(s,3H),3.72-3.79(m,1H),4.26-4.41(m,3H),4.72(d,J=8.2Hz,1H),4.76(d,J=8.2Hz,1H),5.09-5.12(t,J=9.3Hz,1H),5.28(dd,J=3.5,17.6Hz,1H)5.7-5.8(m,1H),6.55-6.80(m,1H),7.45-7.47(m,3H),7.79-7.83(m,4H);
LC-MS(保留时间:1.48),MS m/z 670(M++1)。
实施例470:化合物470的制备
化合物470
方案1
步骤1:
在10分钟内,向商品N-Boc-(4S)-(顺式)-羟基脯氨酸-OMe(200mg,0.82mmol)、三苯基膦(320mg,1.22mmol)和1-萘酚(176mg,1.22mmol)在2.5mL四氢呋喃中的溶液内滴加偶氮二甲酸二乙酯(190μL,1.22mmol)在1.0mL THF中的溶液。搅拌5.5天后,将反应物真空浓缩。将粗品黄色油状物经20×40cM制备型TLC板(AnaltechSiO2)层析纯化,用6-1己烷-乙酸乙酯洗脱,得到所需的产物,为浅黄色油状物(150mg,33%)。
1H NMR(CDCl3,500MHz)δ1.44(s,9H)2.33(1H,m),2.72(1H,m),3.77和3.38(2s,3H,旋转异构体),3.88(dd,1H,J=4.3,12.4Hz),3.97(bd,1H),4.53和4.62(2t,1H,J=7.8Hz,旋转异构体),5.10(bd,1H),6.76(t,1H,J=9.5Hz),7.37(m,1H),7.46(m,3H),7.80(d,1H,J=7.7Hz),8.18(m,1H);
LC-MSA(保留时间:1.86;MS m/z 394(M+Na)+
步骤2:
向搅拌下的Boc-(4R)-萘-l-氧代)-Pro-OEt(150mg,0.40mmol)在1.5mL THF和0.5mL水中的红色溶液内加入氢氧化锂(10mg)。将溶液在室温下搅拌21小时,随后用0.5N碳酸氢钠稀释。将该碱性溶液用乙酸乙酯萃取,随后滴加浓盐酸将水层酸化至pH2。接着将酸化层再次用乙酸乙酯萃取。将这二次的乙酸乙酯层用硫酸镁干燥,过滤,随后真空浓缩,得到Boc-(4R)-萘-1-氧代)-Pro-OH,为浅桃红色结晶(147mg,100%)。
1H NMR(CDCl3,500MHz)δ1.47和1.48(2s,9H,旋转异构体),2.40和2.52(2m,1H),2.68和2.78(2m,1H),3.78-4.07(m,2H),4.57和4.69(2t,1H,J=7.6和8.0Hz,旋转异构体),5.12(bd,1H),6.77(dd,1H,J=7.6,21.2Hz),7.37(m,1H),7.46(m,3H),7.81(t,1H,J=5.8Hz),8.19(m,1H);
LC-MSA(保留时间:1.79;MS m/z 358(M+H)+
步骤3:
向Boc-((4R)-萘-1-氧代)-Pro-OH(147mg,O.41mmol)和外消旋(1R/2S)/(1S/2R)-1-氨基-2-乙烯基环丙烷甲酸乙酯·盐酸盐(79mg,0.41mmol)在2.8mL二氯甲烷中的溶液内加入DIPEA(250μL,1.44mmol)和TBTU(158mg,0.49mmol)。将所得的溶液在氮气气氛中搅拌20小时,随后用40mL二氯甲烷稀释。有机层依次用水、1N碳酸氢钠溶液、1N HCl、水和盐水洗涤。随后将溶液用硫酸钠干燥并真空浓缩。经制备型TLC纯化,得到两种分离的非对映异构体,较高Rf非对映异构体A(P2[Boc(4R)-(萘-1-氧代)脯氨酸]-P1(1R,2S乙烯基Acca)-OEt,78mg,38%)和较低Rf非对映异构体B(P2[Boc(4R)-(萘-1-氧代)脯氨酸]-P1(1S,2R乙烯基Acca)-OEt,91mg,45%),为灰白色固体物:
非对映异构体A:P2[Boc(4R)-(萘-1-氧代)脯氨酸]-P1(1R,2S乙烯基Acca)-OEt:
1H NMR(CDCl3,500MHz)δ1.24(t,3H),1.43(s,9H),1.52(m,1H),1.84(m,1H),2.02(m,1H),2.14(m,1H),2.81(m,1H),3.88(m,2H),4.11(q,1H,J=7.15),4.19(m,1H),4.54(m,1H),5.15(m,1H),5.31(dd,1H,J=17,0.8Hz),5.77(m,1H),6.83(m,1H),7.36(t,1H,J=7.8Hz),7.46(m,3H),7.78(d,1H,J=7.6Hz),8.14(d,1H,J=8.15Hz);
LC-MSB(保留时间:1.85;MS m/z 495(M+H)+
非对映异构体B,实施例10B:P2[Boc(4R)-(萘-1-氧代)脯氨酸]-P1(1S,2R乙烯基Acca)-Oet:
1H NMR(dl-CHCl3,500MHz)δ1.24(t,3H),1.42(s,9H),1.85(m,1H),2.15(q,1H,J=8.9Hz),2.40(m,1H),2.78(m,1H),3.78(m,1H),4.12(m,2H),4.52(m,1H),5.15(m,1H),5.31(m,1H),5.79(m,1H),6.80(m,1H),7.35(t,1H,J=7.6Hz),7.46(m,3H),7.78(d,1H;J=7.6Hz),8.14(d,1H,J=8.10Hz).
LC-MSB(保留时间:1.85;MS m/zm 495(M+H)+
方案2
步骤4:
向P2[Boc(4R)-(萘-1-氧代)脯氨酸]-P1(1R,2S乙烯基Acca)-OEt(A,较高Rf)(78mg,0.16mmol)中加入4N HCl/二_烷(2.0mL),将溶液搅拌30分钟。真空浓缩,得到P2[(4R)-(萘-4-氧代)脯氨酸)]-P1(1R,2S乙烯基Acca)-OEt的盐酸盐,为黄色油状物,该产物未经进一步纯化直接用于下一步反应中。
向BOCL-tBuGly(73mg,0.32mmol)和所述P2[(4R)-(萘-4-氧代)脯氨酸)-P1(1R,2S乙烯基Acca)-OEt盐酸盐(0.16mmol)在11mL乙腈中的溶液内加入DIPEA(140μL,0.79mmol)和HATU(132mg,0.35mmol)。将所得的溶液在氮气气氛中搅拌17小时,随后用100mL乙酸乙酯稀释。有机层依次用水、1N碳酸氢钠溶液、1N HCl、水和盐水洗涤。接着将溶液用硫酸钠干燥并真空浓缩,得到题述化合物,为浅黄色油状薄膜状物(92mg,96%)。
1H NMR(CDCl3,500MHz)δ1.06.(s,9H),1.22(t,3H,J=7.1),1.38(s,9H),1.41(m,1H),1.82(m,1H),2.13(m,1H),2.42(m,1H),2.79(m,1H),3.92-4.2(m,1H),4.12(q,2H,J=6.6Hz),4.38(bt,1H),5.12(d,1H,J=10.3Hz)5.2-5.39(m,3H),5.75(m,1H),6.82(d,1H,J=7.5Hz),7.34-7.46(m,4H),7.59(bs,1H,NH),7.76(d,1H,J=7.9Hz),8.13(d,1H,J=8.3Hz);
LC-MSC(保留时间:2.82;MS m/z 608(M+H)+
步骤5:
向实施例470、步骤4的产物(92mg,0.15mmol)在750mL四氢呋喃和250mL水中的溶液内加入氢氧化锂(4mg)。将所得的溶液搅拌28.5小时,进行常规后处理,随后在相同的条件下处理,不同之处再于加入两倍量的氢氧化锂(8mg)。24小时后,将反应物用乙酸乙酯稀释并用水洗涤。将有机层用硫酸钠干燥并真空浓缩。所得的半固体经快速层析纯化,用3-1己烷-乙酸乙酯洗脱,得到BocNH-P3(t-BuGly)-P2[(Boc(4R)-(萘-1-氧代)脯氨酸)]-P1(1R,2S乙烯基Acca)-OH,为透明半固体物(30mg,34%)。
1H NMR(d4-MeOH,500MHz)δ1.04(s,9H),1.24(t,1H,J=3.9Hz),1.32(s,9H),1.66(m,1H),2.07(m,1H),2.40(m,1H),2.71(m,1H),4.04-4.07(m,1H),4.28(m,1H),4.42(m,1H),4.55(m,1H),5.02(m,1H),5.18-5.29(m,2H),5.90(m,1H),6.54(m,1H),6.92(m,1H),4.26(1m,4H),7.77(m,1H),8.15(m,1H);
LC-MSC(保留时间:2.65;MS m/z 580(M+H)+
步骤6:
向BocNH-P3(t-BuGly)-P2[(Boc(4R)-(萘-1-氧代)脯氨酸)]-P1(1R,2S乙烯基Acca)-OH(实施例470、步骤5产物)(65mg,0.11mmol)在3.7mL四氢呋喃中的溶液内加入1,1′-羰基二咪唑(22mg,0.135mmol)。将所得的混合物回流30分钟,随后冷却至室温。此时,加入甲烷磺酰胺(27mg,0.28mmol)和DBU(34□L,0.224mmol)。搅拌反应物2天,随后再加入DBU(10□L)和甲烷磺酰胺(9mg)。24小时后,反应物用50mL乙酸乙酯稀释并用50mL 0.25N HCl和50mL盐水洗涤。将溶液用硫酸钠干燥并真空浓缩。将粗产物经制备型TLC(3-2乙酸乙酯-己烷)纯化,得到化合物470(21mg,28%),为白色膜状固体物。
1H NMR(d4-MeOH,500MHz)δ1.04(s,9H),1.36(s,9H),1.88(t,1H),2.18(m,1H),2.31(m,1H),2.63(m,1H),3.11(bs,3H),4.076(m,1H),4.30(bd,1H),4.41(bd,1H),4.52(表观t,1H),5.07(m,1H),5.24-5.30(m,2H),5.80(m,1H),6.92(d,1H,J=7.45Hz),7.35-7.46(m,4H),7.76,(d,1H,J=8.1Hz),8.13(d,1H,J=8.3Hz);
LC-MSC(保留时间:2.57;MS m/z 657(M+H)+
实施例471:化合物471的制备
化合物471
方案2
步骤1:
向P2[Boc(4R)-(萘-1-氧代)脯氨酸]-P1(1S,2R乙烯基Acca)-OEt(实施例470、步骤3产物,低Rf)(91mg,0.18mmol)中加入.4N HCl/二_烷(2.0mL)并将溶液搅拌30分钟。真空浓缩,得到P2[(4R)-(萘-1-氧代)脯氨酸)]-P1(1S,2R乙烯基Acca)-OEt盐酸盐,为黄色油状物,该产物未经进一步纯化直接用于下一步反应中。
向N-Boc-L-叔-亮氨酸-OH或BOCL-tBuGly(85mg,0.37mmol)和P2[(4R)-(萘-1-氧代)脯氨酸)]-P1(1S,2R乙烯基Acca)-OEt盐酸盐(得自上述反应的产物)(0.18mmol)在13mL乙腈中的溶液内加入DIPEA(160μL,0.92mmol)和HATU(154mg,0.41mmol)。将所得的溶液在氮气气氛中搅拌17小时,随后用100mL乙酸乙酯稀释。有机层依次用水、1N碳酸氢钠溶液、1N HCl、水和盐水洗涤。随后将溶液用硫酸钠干燥并真空浓缩,得到题述化合物,为透明膜状物(53mg,47%)。
1H NMR(d1-CHCl3,500MHz)δ1.02(s,9H),1.22(t,3H,J=7.0Hz),1.39(s,9H),1.47(m,1H),1.88(dd,1H,J=8.0,5.5Hz),2.07(m,1H),2.42(m,1H),2.80(dt,J=13.8,6.0Hz,1H),3.96(m,1H),4.14(m,2H),4.34(m,2H),4.77(t,1H,J=7.2Hz),5.09-5.33(m,3H),5.72(m,1H),6.82(d,1H,J=7.6Hz),7.34-7.50(m,4H),7.77(d,1H,J=8.0Hz),8.15(d,1H,J=8.25Hz);
LC-MSC(保留时间:2.81;MS m/z 608(M+H)+
步骤2:
按照实施例470、步骤5中描述的方法制备该产物(5mg,10%),不同之处在于使用实施例471、步骤1的产物。
1H NMR(d4-MeOH,500MHz)δ0.99(s,9H),1.28(m,1H),1.37(s,9H),1.60(m,1H),2.06(m,1H),2.28(m,1H),2.66(m,1H),3.91(m,1H),4.33(m,2H),4.61(bt,1H),4.97(d,1H,J=11.0Hz),5.19(m2H),6.09(m,1H),6.88(d,1H,J=7.1Hz),7.35-7.46(m,4H),7.78(d,1H,J=8.2Hz),8.12(d,1H,J=8.3Hz);
LC-MSC(保留时间:2.60;MS m/z 580(M+H)+
步骤3:
向BocN-P3(L-tBuGly)-P2[Boc(4R)-(萘-1-氧代)脯氨酸)]-P1(1S,2R乙烯基Acca)-COOH(38mg,0.066mmol)(实施例471、步骤2产物)在2.2mL四氢呋喃中的溶液内加入1,1′-羰基二咪唑(13mg,0.079mmol)。将所得的混合物回流30分钟,随后冷却至室温。此时,加入甲烷磺酰胺(16mg,0.16mmol)和DBU(20μL,0.13mmol)。搅拌反应物2天,随后再加入DBU(10μL)和甲烷磺酰胺(9mg)。24小时后,将反应物用50mL乙酸乙酯稀释并用50mL 0.25N HCl和50mL盐水洗涤。将溶液用硫酸钠干燥,真空浓缩。粗产物使用一块20×40cM制备型TLC板(Analtech,3-2乙酸乙酯-己烷洗脱)纯化,得到化合物471(25mg,58%),为白色膜状固体物。
1H NMR(d4-MeOH,500MHz)δ1.03(s,9H),1.34(s,9H),1.80(m,1H),2.18(m,1H),2.31(m,1H),2.68(m,1H),3.09(bs,3H),4.04(m,1H),4.20-4.44(m,2H),4.51(表观t,1H),5.08(m,1H),5.25-5.31(m,2H),5.77(m,1H),6.93(d,1H,J=7.6Hz),7.36-7.45(m,4H),7.77(d,1H,J=8.0Hz),8.15m,1H);
LC-MSC(保留时间:2.57;MS m/z 657(M+H)+
部分L:
实施例472:生物学研究
重组HCV NS3/4A蛋白酶复合物FRET肽试验
该体外试验的目的是测定本发明的化合物对来源于BMS、H77C或者J416S菌株的HCV NS3蛋白酶复合物(如以下描述)的抑制作用。该试验提供本发明的化合物如何有效地抑制HCV蛋白水解活性的指征。
自旧金山医院(San Francisco Hospital)的T.Wright博士处得到来源于HCV感染的患者的血清。采用从血清RNA(核糖核酸)的逆转录-PCR(RT-PCR)得到的DNA片段,并使用以其它的基因型1a菌株之间的同源物为基础而选择的引物,构建HCV基因组(BMS菌株)的工程化全长cDNA(互补脱氧核糖核酸)模板。按照Simmonds等(参见PSimmonds,KA Rose,S Graham,SW Chan,F McOmish,BC DoW,EAFollett,PL Yap和H Marsden,J.Clin.Microbiol.,31(6),1493-1503(1993))的分类,自全部基因组序列测定中,将基因型1a指定为HCV分离物。非结构区域NS2-5B的氨基酸序列显示与HCV基因型1a(H77C)有>97%的相同和与基因型1b(J4L6S)有87%的相同。感染克隆物H77C(1a基因型)和J4L6S(1b基因型)来源于R.Purcell(NIH),其序列在Genbank中公开(AAB67036,参见Yanagi,M.,Purcell,R.H,Emerson,S.U.和Bukh,J.Proc.Natl.Acad.Sci.U.S.A.94(16),8738-8743(1997);AF054247,参见Yanagi,M.,St Claire,M.,Shapiro,M.Emerson,S.U.,Purcell,R.H.和Bukh,J,Virology 244(1),161-172(1998))。
BMS、H77C和J4L6S菌株用于制备重组NS3/4A蛋白酶复合物。根据P.Gallinari等(参见Gallinari P,Paolini C,Brennan D,Nardi C,Steinkuhler C,De Francesco R.,Biochemistry.38(17):5620-32(1999))所述,处理编码这些菌株的的重组HCV NS3/4A蛋白酶复合物(氨基酸1027-1711)的DNA。简言之,将三个赖氨酸的增溶尾部加入到NS4A编码区的3’-末端。将NS4A-NS4B裂解部位(氨基酸1711)的P1位点上的半胱氨酸转化为甘氨酸,以避免赖氨酸标记物的蛋白水解裂解。此外,通过PCR,在氨基酸位点1454,引发半胱氨酸至丝氨酸的突变,以阻止NS3解旋酶功能区的自溶裂解。在pET21b细菌表达载体(Novagen)上,克隆变体DNA片段,然后按照P.Gallinari等(参见Gallinari P,Brennan D,Nardi C,Brunetti M,Tomei L,Steinkuhler C,DeFrancesco R.,J Virol.72(8):6758-69(1998))描述的改进方法,在大肠杆菌菌株(Escherichia.Coli strain)BL21(DE3)(Invitrogen)上表达NS3/4A复合物。简言之,于20℃下,用0.5mM异丙基β-D-1-硫代吡喃型半乳牛糖苷IPTG诱发NS3/4A表达22小时。一般的发酵液(10L)得到约80g的湿细胞沉淀。将该细胞重悬浮于溶解缓冲液(10mL/g)中,后者由25mM N-(2-羟乙基)哌嗪-N′-(2-乙烷磺酸)(HEPES),pH7.5、20%甘油、500mM氯化钠(NaCl)、0.5%Triton-X100、1ug/ml溶菌酶、5mM氯化镁(MgCl2)、1ug/ml DnaseI、5mMβ-巯基乙醇(βME)和不含乙二胺四乙酸(EDTA)的蛋白酶抑制剂(Roche)构成,将该悬浮液匀浆化,并于4℃温育20分钟。将匀浆超声处理,并通过在235000g速度下,于4℃超声离心1小时澄清。将咪唑加入到上清液中,使其最终浓度为15mM,然后将pH调至8.0。将粗的蛋白提取物装载到预先用缓冲液B(25mM HEPES,pH8.0,20%甘油,500mMNaCl,0.5% Triton-X100,15mM咪唑,5mM βME)平衡的镍-次氮基三乙酸(Ni-NTA)柱上。在1mL/min的流速下,装载样品。用15倍柱体积的缓冲液C(除0.2%Triton-X100以外,与缓冲液B相同)冲洗该柱。用5倍柱体积的缓冲液D(除采用200mM咪唑外,其余与缓冲液C相同)洗脱蛋白。
收集含NS3/4A蛋白酶复合物的流分,然后装载到预先用缓冲液D(25mM HEPES,pH7.5,20%甘油,300mM NaCl,0.2%Triton-X100,10mMβME)平衡的脱盐柱Superdex-S200上。在1mL/min的流速下,装载样品。收集含NS3/4A蛋白酶复合物的流分,然后浓缩至约0.5mg/ml。经SDS-PAGE和质谱分析判断,来源于BMS、H77C和J4L6S菌株的NS3/4A蛋白酶复合物的纯度在90%以上。
将所述酶在-80℃下贮存,于冰上融化并在使用前在试验缓冲液中稀释。用于NS3/4A蛋白酶试验的底物为RETS1(Resonance EnergyTransfer Depsipeptide Substrate;AhaSpec,Inc.cat#22991)(FRETpeptide),由Taliani等在Anal.Biochem.240(2):60-67(1996)中描述。所述肽序列松散地基于NS4A/NS4B天然裂解部位,但在裂解部位为酯键而非酰胺键。在本发明化合物的不存在或者存在下,将所述肽底物与三种重组NS3/4A复合物中之一共同温育,并采用CytofluorSeries 4000在有效时间内跟踪荧光反应产物的形成。
使用的试剂如下:自GIBCO-BRL处得到HEPES和甘油(Ultrapure)。自Sigma处得到二甲亚砜(DMSO)。自Bio Rad处得到β-巯基乙醇。
试验缓冲液:50mM HEPES,pH7.5;0.15 NaCl;O.1%Triton;15%甘油;10mM βME。底物:2μM最终浓度(得自于-20℃下DMSO中贮存的2mM储备液)。HCV NS3/4A型1a(1b)、2-3nM最终浓度(得自在25mM HEPES,pH7.5、20%甘油、300mM NaCl、0.2%Triton-X100、10mM βME中的5μM储备液)。
该试验在来源于Falcon的96孔聚苯乙烯黑板上进行。每孔含25μl在试验缓冲液中的NS3/4A蛋白酶复合物、50μl在10%DMSO/试验缓冲液中的本发明化合物和25μl在试验缓冲液中的底物。在相同的试验板上也制备对照组(没有化合物)。在启动通过加入底物的酶促反应前,将所述酶复合物与化合物或者对照溶液混合1分钟。采用Cytofluor Series 4000(Perspective Biosystems)迅速读取试验板。于25℃下,设置仪器以读取340nm的发射波长和490nm的激发波。长下的读取值。反应通常进行约15分钟。
用以下方程计算抑制百分率:
100-[(δFinh/δFcon)×100]
其中δF为曲线的线性范围内的荧光变化。非线性曲线拟合应用于抑制率-浓度数据,通过使用ExcelXl-拟合软件,按照方程y=A+((B-A)/(1+((C/x)^D)))计算50%有效浓度(IC50)。
发现所有的受试化合物具有10μM或更小的IC50值。另外,对本发明化合物进行了抗一种以上类型的NS3/4A复合物的试验,虽然一致证实所述化合物对菌株1b比对菌株1a具有更大的效力,但发现本发明的化合物具有类似的抑制性质。
特异性试验
进行特异性试验以证实:与其它的丝氨酸或者半胱氨酸蛋白酶相比较,本发明化合物在抑制HCV NS3/4A蛋白酶上的选择性。
本发明化合物对以下多种丝氨酸蛋白酶的特异性被证实:人白细胞弹性蛋白酶(HLE)、猪胰弹性蛋白酶(PPE)、人-胰凝乳蛋白酶和一种半胱氨酸蛋白酶:人肝组织蛋白酶B。在所有这些试验情况下,按先前描述的(专利WO 00/09543)、其中对丝氨酸蛋白酶试验进行某些改进的方法,采用对每种酶具有特异性的比色剂对硝基苯胺(pNA)底物,以96孔滴定格式板方法进行。
每种试验包括:在RT下将酶抑制剂预温育2小时随后加入底物,然后水解至~30%的转变,该转变通过在Spectramax Pro微量滴定板读数器上测定。依其效力而定,化合物浓度范围为100~0.4μM。
每一个实验的最终条件如下:
50mM三(羟甲基)氨基甲烷盐酸盐(Tris-HCl)pH8,0.5M硫酸钠(Na2SO4),50mM NaCl,0.1mM EDTA,3%DMSO,0.01%Tween-20133μM succ-AAA-pNA和20nM HNE或8nM PPE;133μMsucc-AAV-pNA和15nM HLE;100μM succ-AAPF-pNA和250pM胰凝乳蛋白酶
100mM NaHPO4(磷酸氢钠)pH6,0.1mM EDTA,3%DMSO,1mM TCEP(三(2-羧基乙基)膦盐酸盐),0.01%吐温-20,30μM Z-FR-pNA和5nM组织蛋白酶B(酶原液,使用前在含20mM TCEP的缓冲液中激活)。
用以下方程计算抑制百分率:
[1-((UVinh-UVblank)/(UVctl-UVblank))]×100
将非线性曲线拟合应用于抑制率-浓度数据,通过采用Excel X1-拟合软件,计算50%有效浓度(IC50)。
基于HCV复制子细胞的试验
按Lohmann V,Komer F,Koch J,Herian U,Theilmann L,Bartenschlager R.,Science 285(5424):110-3(1999)中所述,建立HCV复制子全细胞系统。这个系统能够使我们评价我们的HCV蛋白酶化合物对HCV RNA复制的作用。简言之,采用在Lohmann论文(Assession号:AJ238799)中描述的HCV菌株1B序列,生成编码5’内核糖体入口位点(IRES)、新霉素抗性基因、EMCV(脑心肌炎病毒)-IRES和HCV非结构蛋白NS3-NS5B和3’非转录区(NTR)的HCV cDNA。体外转录的cDNA被转染到人肝癌细胞系HuhT中。然后在选择性标记物新霉素(G418)存在下,实现组成性表达HCV复制子细胞的选择。得到的细胞系特征在于正链和负链RNA产生并在规定时间内有蛋白产生。
使Huh7细胞,即组成型表达HCV的复制子,在含10%胎牛血清(FCS)和1mg/ml G418(Gibco-BRL)的Dulbecco修改的Eagle培养基(DMEM)中生长。过夜前将细胞(1.5×104细胞/孔)接种到96孔组织培养灭菌板上。在置于稀释板中的含4%FCS、1∶100青霉素/链霉素、1∶100 L-谷氨酰胺和5%DMSO(在试验时的最终浓度为0.5%DMSO)的DMEM中,制备化合物和不含化合物的对照物。将化合物/DMSO混合物加入到细胞中,并且于37℃温育4天。4天后,将所述板用磷酸盐缓冲盐水(PBS)充分洗涤(3次,150μL)。用25μL含所述FRET肽(RET S1,如体外酶试验中的描述)的溶胞试验试剂将所述细胞溶胞。所述溶胞试验试剂如下制备:将5×细胞虫萤光素酶细胞培养溶胞试剂(Promega #E153A)用蒸馏水稀释至1×,加入NaCl使最终浓度达150mM,将在100%DMSO中的2mM FRET肽原液稀释至最终浓度为10μM。随后将所述板置于Cytofluor4000仪器中,该仪器的设置如下:激发340nm/发射490,21次循环自动模式,在动态模式下读数。EC50的测定同IC50测定。
作为第二种试验,复制子FRET试验中的EC50测定得到定量RNA试验的确证。采用Rneasy试剂盒(Qiagen),将细胞溶胞。采用RiboGreen(Jones LJ,YueST,Cheung CY,Singer VL,Anal.Chem.,265(2):368-74(l998))将纯化的总RNA归一化,并且采用Taqman方法(KolykhalovAA,Mihalik K,Feinstone SM,Rice CM,Journal of Virology 74,2046-2051(2000))和Platinum QuantitatiVe RT-PCR Thermoscript One-Step试剂盒(Invitrogen cat#11731-015)测定HCV RNA表达的相对量。简言之,将制备成5μl(≤1ng)体积的RNA加入到20μl含有以下组分的Ready-MiX中:1.25X Thermoscript反应混合物(含硫酸镁和2-脱氧核苷5′-三磷酸酯(dNTPs)),3mM dNTPs,200nM正向引物(序列:
5′-gggagagccatagtggtctgc-3′),600nM反向引物(5′-cccaaatctccaggcattga-3′),100nM探针(5′-6-FAM-cggaattgccaggacgaccgg-BHQ-1-3′)(FAM-荧光素-氨己基脒(amidite);BHQ:Black Hole Quencher),1μM Rox参比染料(Invitrogencat#12223-012)和Thermoscript Plus Platinum Taq聚合酶混合物。所有引物均用ABI Prism7700软件设计,得自Biosearch Technologies,Novato,CA。含已知浓度的HCV RNA转录物的样品被用作标准物。采用以下循环方法(50℃,30分钟;95℃,5分钟;在95℃下40个周期,15秒,60℃,1分钟),按Perkin Elmer年报中描述的方法,使用ABI Prism 7700序列检测器将HCV RNA表达定量。
生物实施例
在HCV复制子细胞试验中和/或在几个所述特异性试验中,评价本发明的代表性化合物。例如,发现化合物34对酶试验中的NS3/4ABMS菌株具有23nM的IC50。用已公开的H77C(3nM的IC50)和J4L6S(2.9nM的IC50)菌株得到相似的效力值。在复制子试验中的EC50值为166nM。
在特异性试验中,发现相同的化合物具有以下活性:HLE>100μM;PPE>200μM;胰凝乳蛋白酶>200μM;组织蛋白酶B>200μM。这些结果表明本族化合物对NS3蛋白酶具有高特异性,并且这些成员中有许多可抑制HCV复制子的复制。
测试本发明的化合物并发现受试化合物具有如下范围内的活性:
IC50活性范围(NS3/4ABMS菌株):A为10-100μM;B为1-10μM;
C为0.1-1μM;D<0.1μM
EC50活性范围(用于受试化合物):
A为10-100μM;
B为1-10μM;
C为0.1-1μM;
D<0.1μM;
注意:按照表中所示的本发明实施例编号及本发明化合物编号可查找各化合物的结构。
本发明化合物的生物活性(EC50)优选为10μM或以下,更优选1μM或以下,最优选100nM或以下。
表1
生物活性
|
IC50范围 |
EC50范围 |
本发明实施例编号 |
本发明化合物编号 |
|
D |
D |
1 |
1 |
|
D |
C |
2 |
2 |
|
D |
D |
3 |
3 |
|
D |
D |
4 |
4 |
|
D |
D |
5 |
5 |
|
C |
C |
6 |
6 |
|
D |
C |
7 |
7 |
|
C |
B |
8 |
8 |
|
D |
C |
9 |
9 |
|
D |
D |
10 |
10 |
|
D |
D |
11 |
11 |
|
D |
D |
12 |
12 |
|
D |
D |
13 |
13 |
|
D |
D |
14 |
14 |
|
C |
B |
15 |
15 |
| D | C | 16 | 16 |
|
D |
D |
17 |
17 |
|
D |
D |
18 |
18 |
|
D |
D |
19 |
19 |
|
D |
C |
20 |
20 |
|
D |
D |
21 |
21 |
|
D |
C |
22 |
22 |
|
D |
D |
23 |
23 |
|
D |
D |
24 |
24 |
|
D |
D |
25 |
25 |
|
D |
D |
26 |
26 |
|
D |
D |
27 |
27 |
|
D |
D |
28 |
28 |
|
D |
D |
29 |
29 |
|
D |
D |
30 |
30 |
|
D |
D |
31 |
31 |
|
D |
D |
32 |
32 |
|
D |
D |
33 |
33 |
|
D |
C |
34 |
34 |
|
D |
D |
35 |
35 |
|
D |
D |
36 |
36 |
|
D |
D |
37 |
37 |
|
D |
D |
38 |
38 |
|
D |
D |
39 |
39 |
|
D |
C |
40 |
40 |
|
D |
D |
41 |
41 |
|
D |
D |
42 |
42 |
| | | | |
|
C |
B |
45 |
45 |
|
C |
B |
46 |
46 |
|
D |
C |
47 |
47 |
|
B | |
48 |
48 |
|
B | |
49 |
49 |
|
C |
B |
50 |
50 |
|
B | |
52 |
52 |
|
C |
B |
53 |
53 |
|
D |
C |
55 |
55 |
|
D |
D |
56 |
56 |
|
D |
C |
57 |
57 |
|
D |
D |
58 |
58 |
|
D |
C |
59 |
59 |
|
D |
B |
60 |
60 |
|
C |
B |
61 |
61 |
|
D |
D |
62 |
62 |
|
D |
B |
63 |
63 |
|
D |
D |
64 |
64 |
|
D |
C |
65 |
65 |
|
D |
C |
66 |
66 |
|
D |
D |
67 |
67 |
|
D |
D |
68 |
68 |
|
D |
D |
69 |
69 |
|
D |
D |
70 |
70 |
|
D |
D |
71 |
71 |
|
D |
C |
72 |
72 |
|
D |
C |
73 |
73 |
|
D |
D |
74 |
74 |
|
D |
D |
75 |
75 |
|
D |
D |
76 |
76 |
|
D |
C |
77 |
77 |
|
D |
B |
78 |
78 |
| D | C | 79 | 79 |
|
D |
B |
80 |
80 |
|
D |
C |
81 |
81 |
|
D |
C |
82 |
82 |
|
D |
C |
83 |
83 |
|
D |
C |
84 |
84 |
|
C |
B |
85 |
85 |
|
B |
A |
86 |
86 |
|
B |
A |
87 |
87 |
|
B |
A |
88 |
88 |
|
D |
D |
89 |
89 |
|
D |
C |
91 |
91 |
|
D |
D |
92 |
92 |
|
C |
C |
93 |
93 |
|
D |
D |
94 |
94 |
|
D |
C |
95 |
95 |
|
D |
D |
96 |
96 |
|
D |
D |
97 |
97 |
|
B | |
99 |
99 |
|
C |
B |
100 |
100 |
| D | D | 101 | 101 |
|
D |
D |
102 |
102 |
|
D |
D |
103 |
103 |
|
D |
D |
104 |
104 |
|
D |
D |
105 |
105 |
|
D |
D |
106 |
106 |
|
D |
C |
107 |
107 |
|
D |
D |
108 |
108 |
|
D |
D |
109 |
109 |
|
D |
C |
110 |
110 |
|
D |
C |
120 |
120 |
|
D |
C |
121 |
121 |
|
D |
C |
122 |
122 |
|
D |
C |
123 |
123 |
|
C |
B |
124 |
124 |
|
D |
D |
125 |
125 |
|
D |
C |
126 |
126 |
|
D |
C |
127 |
127 |
|
D |
C |
128 |
128 |
|
C |
C |
129 |
129 |
| D | C | 130 | 130 |
|
C |
B |
131 |
131 |
|
D |
B |
132 |
132 |
|
C |
B |
133 |
133 |
|
D |
C |
134 |
134 |
|
D |
C |
135 |
135 |
|
D |
C |
136 |
136 |
|
D |
D |
137 |
137 |
|
D |
D |
138 |
138 |
|
D |
C |
139 |
139 |
|
D |
B |
140 |
140 |
|
D |
D |
141 |
141 |
|
D |
D |
142 |
142 |
|
D |
D |
143 |
143 |
|
D |
C |
144 |
144 |
|
D |
C |
145 |
145 |
|
D |
D |
146 |
146 |
|
D |
B |
147 |
147 |
|
D |
D |
148 |
148 |
|
D |
D |
149 |
149 |
|
D |
D |
150 |
150 |
|
D |
D |
151 |
151 |
|
D |
D |
152 |
152 |
|
D |
D |
153 |
153 |
|
D |
C |
154 |
154 |
|
D |
D |
155 |
155 |
| | | | |
|
D |
C |
180 |
180 |
|
D |
C |
181 |
181 |
|
D |
C |
182 |
182 |
|
D |
D |
183 |
183 |
|
D |
D |
185 |
185 |
|
D |
D |
186 |
186 |
|
D |
C |
187 |
187 |
|
D |
C |
188 |
188 |
|
D |
C |
189 |
189 |
|
D |
C |
190 |
190 |
|
D |
C |
191 |
191 |
|
D |
D |
192 |
192 |
|
D |
C |
193 |
193 |
|
D |
C |
194 |
194 |
|
D |
D |
195 |
195 |
|
D |
B |
196 |
196 |
|
C |
A |
197 |
197 |
|
D |
C |
198 |
198 |
|
D |
C |
199 |
199 |
|
D |
D |
200 |
200 |
|
D |
D |
201 |
201 |
|
D |
B |
202 |
202 |
|
D |
C |
204 |
204 |
|
D |
D |
206 |
206 |
|
D |
D |
207 |
207 |
|
D |
C |
209 |
209 |
|
D |
D |
210 |
210 |
|
D |
D |
211 |
211 |
|
D |
D |
212 |
212 |
|
D |
D |
213 |
213 |
|
D |
D |
215 |
215 |
|
D |
D |
219 |
219 |
|
D |
D |
220 |
220 |
|
D |
D |
223 |
223 |
|
D |
C |
224 |
224 |
|
D |
C |
225 |
225 |
|
D |
D |
227 |
227 |
|
D |
D |
229 |
229 |
|
C |
C |
230 |
230 |
|
B | |
231 |
231 |
|
D |
D |
232 |
232 |
|
C |
C |
233 |
233 |
|
D |
C |
235 |
235 |
|
D |
C |
237 |
237 |
|
D |
C |
238 |
238 |
|
D |
D |
239 |
239 |
|
D |
D |
240 |
240 |
|
D |
D |
241 |
241 |
|
D |
B |
242 |
242 |
|
C |
A |
243 |
243 |
|
D |
A |
244 |
244 |
|
C | |
245 |
245 |
| | | | |
|
D |
D |
250 |
250 |
|
D |
D |
251 |
251 |
|
D |
D |
252 |
252 |
|
D |
D |
253 |
253 |
|
D |
D |
254 |
254 |
|
D |
C |
255 |
255 |
|
A | |
256 |
256 |
|
D |
D |
257 |
257 |
|
D |
D |
258 |
258 |
|
D |
D |
259 |
259 |
|
D |
C |
260 |
260 |
|
D |
D |
261 |
261 |
|
D |
D |
262 |
262 |
|
C |
C |
263 |
263 |
|
C |
B |
264 |
264 |
|
D |
D |
265 |
265 |
|
D |
D |
266 |
266 |
|
D |
D |
267 |
267 |
|
D |
D |
268 |
268 |
|
D |
C |
269 |
269 |
|
D |
C |
270 |
270 |
|
D |
C |
271 |
271 |
|
D |
D |
272 |
272 |
|
D |
D |
273 |
273 |
|
D |
C |
274 |
274 |
|
D |
D |
275 |
275 |
|
D |
D |
276 |
276 |
|
D |
D |
277 |
277 |
|
D |
D |
278 |
278 |
|
D |
D |
279 |
279 |
|
D |
D |
280 |
280 |
|
D |
D |
281 |
281 |
|
D |
C |
282 |
282 |
|
D |
C |
283 |
283 |
|
C |
A |
284 |
284 |
|
D |
D |
285 |
285 |
|
D |
D |
286 |
286 |
|
D |
C |
287 |
287 |
|
D |
B |
288 |
288 |
|
D |
C |
289 |
289 |
|
D |
D |
290 |
290 |
|
D |
D |
291 |
291 |
|
D |
C |
292 |
292 |
|
D |
C |
293 |
293 |
|
D |
C |
294 |
294 |
|
C |
B |
295 |
295 |
|
D |
C |
296 |
296 |
|
C |
B |
297 |
297 |
|
D |
D |
298 |
298 |
|
D |
C |
299 |
299 |
|
D |
C |
300 |
300 |
| | | | |
|
D |
D |
320 |
320 |
|
D |
C |
321 |
321 |
|
D |
D |
322 |
322 |
|
D |
C |
323 |
323 |
|
D |
C |
324 |
324 |
|
D |
D |
325 |
325 |
|
D |
D |
326 |
326 |
|
D |
D |
327 |
327 |
|
D |
D |
328 |
328 |
|
D |
D |
329 |
329 |
|
D |
D |
330 |
330 |
|
D | |
331 |
331 |
|
C |
C |
334 |
334 |
|
D |
B |
335 |
335 |
|
C |
B |
336 |
336 |
|
C |
B |
337 |
337 |
|
D |
B |
338 |
338 |
|
D |
B |
339 |
339 |
|
D |
A |
340 |
340 |
|
B | |
341 |
341 |
|
B | |
342 |
342 |
|
B | |
343 |
343 |
|
C | |
344 |
344 |
|
B |
A |
345 |
345 |
|
B | |
346 |
346 |
|
C |
A |
347 |
347 |
|
A | |
348 |
348 |
|
B | |
349 |
349 |
| C | A | 350 | 350 |
|
B | |
351 |
351 |
|
A | |
352 |
352 |
|
A | |
353 |
353 |
|
B | |
354 |
354 |
|
C |
A |
355 |
355 |
|
C |
B |
356 |
356 |
|
D |
C |
357 |
357 |
| | | | |
|
C |
B |
370 |
370 |
|
D |
D |
371 |
371 |
|
D |
D |
372 |
372 |
|
D |
D |
373 |
373 |
|
D |
C |
374 |
374 |
|
D |
D |
375 |
375 |
|
D |
D |
376 |
376 |
|
D |
C |
377 |
377 |
|
D |
D |
378 |
378 |
|
D |
D |
379 |
379 |
|
D |
C |
380 |
380 |
|
D |
C |
381 |
381 |
|
D |
C |
382 |
382 |
|
D |
D |
383 |
383 |
|
D |
D |
384 |
384 |
|
D |
D |
385 |
385 |
|
C |
B |
386 |
386 |
| | | | |
|
D |
D |
410 |
410 |
|
D |
C |
411 |
411 |
|
D |
D |
412 |
412 |
|
D |
D |
413 |
413 |
|
D |
D |
414 |
414 |
|
D |
D |
415 |
415 |
|
D |
C |
420 |
420 |
|
D |
C |
421 |
421 |
|
D |
B |
422 |
422 |
|
C | |
423 |
423 |
|
D |
C |
424 |
424 |
|
C |
B |
425 |
425 |
|
D |
C |
426 |
426 |
|
D |
C |
427 |
427 |
|
D |
C |
428 |
428 |
|
D |
D |
429 |
429 |
|
D |
C |
430 |
430 |
|
D |
C |
431 |
431 |
|
D |
C |
432 |
432 |
|
C |
B |
433 |
433 |
|
D |
C |
434 |
434 |
|
D |
C |
435 |
435 |
|
D |
C |
436 |
436 |
|
D |
C |
437 |
437 |
|
D |
C |
438 |
438 |
| | | | |
|
D |
C |
450 |
450 |
|
B | |
451 |
451 |
|
B | |
452 |
452 |
| | |
453 |
453 |
|
C |
B |
454 |
454 |
|
C | |
470 |
470 |
|
B | |
471 |
471 |
部分M:
表2
以下化合物可按照本文中描述的方法,尤其是在示例部分A至K,更尤其是在B、E、F和G部分所描述的方法制备。此外,应理解以下所示的各基团B、R3、R2和R1可被部分A至K及本文中其它地方所示例的任何基团或在式1中所标出的任何基团所代替。例如,在表2中所示的R3基因为叔丁基,但本领域技术人员会认识到在以下所有编号化合物中该基团可被异丙基或烷氧基取代的C1-6烷基所替代。或在以下所有编号化合物中的B基团可被叔丁基脲部分所替代。