WO1991018596A1 - Methods for treating inflammation and compounds and compositions suitable for use therein - Google Patents
Methods for treating inflammation and compounds and compositions suitable for use therein Download PDFInfo
- Publication number
- WO1991018596A1 WO1991018596A1 PCT/US1991/003319 US9103319W WO9118596A1 WO 1991018596 A1 WO1991018596 A1 WO 1991018596A1 US 9103319 W US9103319 W US 9103319W WO 9118596 A1 WO9118596 A1 WO 9118596A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hydrogen
- alkyl
- solution
- aryl
- mmol
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 116
- 238000000034 method Methods 0.000 title claims abstract description 44
- 239000000203 mixture Substances 0.000 title abstract description 67
- 206010061218 Inflammation Diseases 0.000 title description 3
- 230000004054 inflammatory process Effects 0.000 title description 3
- 239000001257 hydrogen Substances 0.000 claims abstract description 73
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 73
- 150000002431 hydrogen Chemical class 0.000 claims abstract description 46
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 35
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims abstract description 34
- 239000001301 oxygen Chemical group 0.000 claims abstract description 34
- 229910052760 oxygen Chemical group 0.000 claims abstract description 34
- 125000003118 aryl group Chemical group 0.000 claims abstract description 31
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims abstract description 23
- -1 methylene, ethylene, ethyleneoxy Chemical group 0.000 claims abstract description 23
- 150000003839 salts Chemical class 0.000 claims abstract description 22
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 16
- 150000002367 halogens Chemical class 0.000 claims abstract description 16
- 150000001413 amino acids Chemical class 0.000 claims abstract description 15
- 230000004968 inflammatory condition Effects 0.000 claims abstract description 15
- 125000003545 alkoxy group Chemical group 0.000 claims abstract description 14
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims abstract description 11
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims abstract description 10
- 125000002877 alkyl aryl group Chemical group 0.000 claims abstract description 9
- 125000004183 alkoxy alkyl group Chemical group 0.000 claims abstract description 8
- 125000003710 aryl alkyl group Chemical group 0.000 claims abstract description 8
- 229960003136 leucine Drugs 0.000 claims description 51
- 241001465754 Metazoa Species 0.000 claims description 37
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 26
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 17
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 claims description 16
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 claims description 16
- 229940024606 amino acid Drugs 0.000 claims description 15
- 125000001909 leucine group Chemical group [H]N(*)C(C(*)=O)C([H])([H])C(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 10
- 239000008194 pharmaceutical composition Substances 0.000 claims description 10
- LRQKBLKVPFOOQJ-YFKPBYRVSA-N L-norleucine Chemical group CCCC[C@H]([NH3+])C([O-])=O LRQKBLKVPFOOQJ-YFKPBYRVSA-N 0.000 claims description 7
- 239000004480 active ingredient Substances 0.000 claims description 7
- JTTHKOPSMAVJFE-VIFPVBQESA-N L-homophenylalanine Chemical group OC(=O)[C@@H](N)CCC1=CC=CC=C1 JTTHKOPSMAVJFE-VIFPVBQESA-N 0.000 claims description 6
- 230000003110 anti-inflammatory effect Effects 0.000 claims description 6
- 229960005190 phenylalanine Drugs 0.000 claims description 5
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 4
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical group OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 claims description 4
- 239000003937 drug carrier Substances 0.000 claims description 4
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 claims description 3
- LPBSHGLDBQBSPI-YFKPBYRVSA-N (2s)-2-amino-4,4-dimethylpentanoic acid Chemical group CC(C)(C)C[C@H](N)C(O)=O LPBSHGLDBQBSPI-YFKPBYRVSA-N 0.000 claims description 2
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical group CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 claims description 2
- 239000003085 diluting agent Substances 0.000 claims description 2
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Chemical group CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 claims description 2
- 229960000310 isoleucine Drugs 0.000 claims description 2
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Chemical group OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 claims description 2
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 claims 2
- NPDBDJFLKKQMCM-SCSAIBSYSA-N (2s)-2-amino-3,3-dimethylbutanoic acid Chemical group CC(C)(C)[C@H](N)C(O)=O NPDBDJFLKKQMCM-SCSAIBSYSA-N 0.000 claims 1
- 125000003440 L-leucyl group Chemical group O=C([*])[C@](N([H])[H])([H])C([H])([H])C(C([H])([H])[H])([H])C([H])([H])[H] 0.000 claims 1
- 125000002435 L-phenylalanyl group Chemical group O=C([*])[C@](N([H])[H])([H])C([H])([H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 claims 1
- 241000124008 Mammalia Species 0.000 claims 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 341
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 215
- 239000000243 solution Substances 0.000 description 205
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 185
- 235000019439 ethyl acetate Nutrition 0.000 description 131
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 118
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 114
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 114
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 101
- 239000011541 reaction mixture Substances 0.000 description 84
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 73
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 65
- 239000000377 silicon dioxide Substances 0.000 description 55
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 54
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 47
- 238000006243 chemical reaction Methods 0.000 description 47
- 239000003921 oil Substances 0.000 description 43
- 235000019198 oils Nutrition 0.000 description 43
- 239000007787 solid Substances 0.000 description 42
- 239000000047 product Substances 0.000 description 41
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 38
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 38
- 238000012360 testing method Methods 0.000 description 38
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 36
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 36
- 239000010410 layer Substances 0.000 description 35
- 235000019341 magnesium sulphate Nutrition 0.000 description 35
- 239000002904 solvent Substances 0.000 description 35
- 239000004395 L-leucine Substances 0.000 description 34
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 33
- 239000002085 irritant Substances 0.000 description 33
- 231100000021 irritant Toxicity 0.000 description 33
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 30
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 30
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 28
- 230000005764 inhibitory process Effects 0.000 description 25
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 24
- 238000004587 chromatography analysis Methods 0.000 description 24
- 239000012044 organic layer Substances 0.000 description 23
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 22
- 239000012267 brine Substances 0.000 description 22
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 22
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 22
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 21
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical compound ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 description 21
- 239000002244 precipitate Substances 0.000 description 21
- 235000019454 L-leucine Nutrition 0.000 description 18
- 239000007864 aqueous solution Substances 0.000 description 17
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 16
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 15
- 229910000027 potassium carbonate Inorganic materials 0.000 description 15
- NIHNNTQXNPWCJQ-UHFFFAOYSA-N fluorene Chemical compound C1=CC=C2CC3=CC=CC=C3C2=C1 NIHNNTQXNPWCJQ-UHFFFAOYSA-N 0.000 description 14
- 241000699670 Mus sp. Species 0.000 description 12
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 12
- 238000000746 purification Methods 0.000 description 12
- 238000001953 recrystallisation Methods 0.000 description 12
- 229910000029 sodium carbonate Inorganic materials 0.000 description 12
- 238000003756 stirring Methods 0.000 description 12
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 12
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 11
- 235000001014 amino acid Nutrition 0.000 description 11
- 239000012043 crude product Substances 0.000 description 11
- 230000000694 effects Effects 0.000 description 11
- 229920000609 methyl cellulose Polymers 0.000 description 11
- 239000001923 methylcellulose Substances 0.000 description 11
- CBPJQFCAFFNICX-IBGZPJMESA-N (2s)-2-(9h-fluoren-9-ylmethoxycarbonylamino)-4-methylpentanoic acid Chemical compound C1=CC=C2C(COC(=O)N[C@@H](CC(C)C)C(O)=O)C3=CC=CC=C3C2=C1 CBPJQFCAFFNICX-IBGZPJMESA-N 0.000 description 10
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 10
- 206010030113 Oedema Diseases 0.000 description 10
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 10
- YZXBAPSDXZZRGB-DOFZRALJSA-N arachidonic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O YZXBAPSDXZZRGB-DOFZRALJSA-N 0.000 description 10
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 10
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 9
- 241000700159 Rattus Species 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 230000037396 body weight Effects 0.000 description 9
- 229910002092 carbon dioxide Inorganic materials 0.000 description 9
- 238000001704 evaporation Methods 0.000 description 9
- 230000008020 evaporation Effects 0.000 description 9
- 239000000284 extract Substances 0.000 description 9
- PHEDXBVPIONUQT-RGYGYFBISA-N phorbol 13-acetate 12-myristate Chemical compound C([C@]1(O)C(=O)C(C)=C[C@H]1[C@@]1(O)[C@H](C)[C@H]2OC(=O)CCCCCCCCCCCCC)C(CO)=C[C@H]1[C@H]1[C@]2(OC(C)=O)C1(C)C PHEDXBVPIONUQT-RGYGYFBISA-N 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- 229910052786 argon Inorganic materials 0.000 description 8
- 230000005587 bubbling Effects 0.000 description 8
- 230000003197 catalytic effect Effects 0.000 description 8
- WGLPBDUCMAPZCE-UHFFFAOYSA-N chromium trioxide Inorganic materials O=[Cr](=O)=O WGLPBDUCMAPZCE-UHFFFAOYSA-N 0.000 description 8
- 238000007912 intraperitoneal administration Methods 0.000 description 8
- 238000005406 washing Methods 0.000 description 8
- SJHPCNCNNSSLPL-CSKARUKUSA-N (4e)-4-(ethoxymethylidene)-2-phenyl-1,3-oxazol-5-one Chemical compound O1C(=O)C(=C/OCC)\N=C1C1=CC=CC=C1 SJHPCNCNNSSLPL-CSKARUKUSA-N 0.000 description 7
- 239000000741 silica gel Substances 0.000 description 7
- 229910002027 silica gel Inorganic materials 0.000 description 7
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 6
- 102000008946 Fibrinogen Human genes 0.000 description 6
- 108010049003 Fibrinogen Proteins 0.000 description 6
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 6
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- 239000002671 adjuvant Substances 0.000 description 6
- 239000008346 aqueous phase Substances 0.000 description 6
- YKPUWZUDDOIDPM-SOFGYWHQSA-N capsaicin Chemical compound COC1=CC(CNC(=O)CCCC\C=C\C(C)C)=CC=C1O YKPUWZUDDOIDPM-SOFGYWHQSA-N 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- 229940012952 fibrinogen Drugs 0.000 description 6
- 239000008096 xylene Substances 0.000 description 6
- KXENKHNKGVYMCW-UHFFFAOYSA-N 3-(9h-fluoren-9-yl)propanoyl chloride Chemical compound C1=CC=C2C(CCC(=O)Cl)C3=CC=CC=C3C2=C1 KXENKHNKGVYMCW-UHFFFAOYSA-N 0.000 description 5
- 229940114079 arachidonic acid Drugs 0.000 description 5
- 235000021342 arachidonic acid Nutrition 0.000 description 5
- 239000003054 catalyst Substances 0.000 description 5
- 238000004440 column chromatography Methods 0.000 description 5
- 238000002425 crystallisation Methods 0.000 description 5
- 230000008025 crystallization Effects 0.000 description 5
- 239000003814 drug Substances 0.000 description 5
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 5
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 5
- 229960002317 succinimide Drugs 0.000 description 5
- QAOWNCQODCNURD-UHFFFAOYSA-N sulfuric acid Substances OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 5
- HBEJJYHFTZDAHZ-QMMMGPOBSA-N tert-butyl (2s)-2-amino-4-methylpentanoate Chemical compound CC(C)C[C@H](N)C(=O)OC(C)(C)C HBEJJYHFTZDAHZ-QMMMGPOBSA-N 0.000 description 5
- FCTLDPGHTJJJIS-UHFFFAOYSA-N (1-methyl-9h-fluoren-9-yl)methanol Chemical compound C12=CC=CC=C2C(CO)C2=C1C=CC=C2C FCTLDPGHTJJJIS-UHFFFAOYSA-N 0.000 description 4
- SJTWIOAYIHRXFQ-UHFFFAOYSA-N (1-methyl-9h-fluoren-9-yl)methyl carbonochloridate Chemical compound C12=CC=CC=C2C(COC(Cl)=O)C2=C1C=CC=C2C SJTWIOAYIHRXFQ-UHFFFAOYSA-N 0.000 description 4
- QNCRNFGWICOFHL-UHFFFAOYSA-N (2,7-dimethyl-9h-fluoren-9-yl)methanol Chemical compound CC1=CC=C2C3=CC=C(C)C=C3C(CO)C2=C1 QNCRNFGWICOFHL-UHFFFAOYSA-N 0.000 description 4
- BJZXQAPTSUYLDF-UHFFFAOYSA-N 2,7-dimethyl-9h-fluorene-9-carboxylic acid Chemical compound CC1=CC=C2C3=CC=C(C)C=C3C(C(O)=O)C2=C1 BJZXQAPTSUYLDF-UHFFFAOYSA-N 0.000 description 4
- GGZQLTVZPOGLCC-UHFFFAOYSA-N 2-(2-bromoethyl)-1,3-dioxolane Chemical compound BrCCC1OCCO1 GGZQLTVZPOGLCC-UHFFFAOYSA-N 0.000 description 4
- RKJHJMAZNPASHY-UHFFFAOYSA-N 2-methyl-9h-fluorene Chemical compound C1=CC=C2C3=CC=C(C)C=C3CC2=C1 RKJHJMAZNPASHY-UHFFFAOYSA-N 0.000 description 4
- PCYFMWUUBUXZOJ-UHFFFAOYSA-N 2-methyl-9h-fluorene-9-carboxylic acid Chemical compound C1=CC=C2C3=CC=C(C)C=C3C(C(O)=O)C2=C1 PCYFMWUUBUXZOJ-UHFFFAOYSA-N 0.000 description 4
- LUKYXDUWPLXNQD-UHFFFAOYSA-N 3-(9h-fluoren-9-yl)propanoic acid Chemical compound C1=CC=C2C(CCC(=O)O)C3=CC=CC=C3C2=C1 LUKYXDUWPLXNQD-UHFFFAOYSA-N 0.000 description 4
- XXSCONYSQQLHTH-UHFFFAOYSA-N 9h-fluoren-9-ylmethanol Chemical compound C1=CC=C2C(CO)C3=CC=CC=C3C2=C1 XXSCONYSQQLHTH-UHFFFAOYSA-N 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 4
- 239000003810 Jones reagent Substances 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- 238000000692 Student's t-test Methods 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 239000000427 antigen Substances 0.000 description 4
- 102000036639 antigens Human genes 0.000 description 4
- 108091007433 antigens Proteins 0.000 description 4
- ARUVKPQLZAKDPS-UHFFFAOYSA-L copper(II) sulfate Chemical compound [Cu+2].[O-][S+2]([O-])([O-])[O-] ARUVKPQLZAKDPS-UHFFFAOYSA-L 0.000 description 4
- 229910000366 copper(II) sulfate Inorganic materials 0.000 description 4
- 239000012954 diazonium Substances 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-O diazynium Chemical compound [NH+]#N IJGRMHOSHXDMSA-UHFFFAOYSA-O 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 239000007789 gas Substances 0.000 description 4
- 230000002757 inflammatory effect Effects 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 235000017557 sodium bicarbonate Nutrition 0.000 description 4
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- FKQLIPCECGFKBX-UHFFFAOYSA-N (4-methyl-9h-fluoren-9-yl)methanol Chemical compound OCC1C2=CC=CC=C2C2=C1C=CC=C2C FKQLIPCECGFKBX-UHFFFAOYSA-N 0.000 description 3
- XZKPFRIEWDWYDH-UHFFFAOYSA-N 1-(methoxymethyl)-9h-fluorene Chemical compound C1C2=CC=CC=C2C2=C1C(COC)=CC=C2 XZKPFRIEWDWYDH-UHFFFAOYSA-N 0.000 description 3
- UXJJEBHGAFYLGC-UHFFFAOYSA-N 1-(methoxymethyl)-9h-fluorene-9-carboxylic acid Chemical compound C12=CC=CC=C2C(C(O)=O)C2=C1C=CC=C2COC UXJJEBHGAFYLGC-UHFFFAOYSA-N 0.000 description 3
- JJUQKWSFCOFJQU-UHFFFAOYSA-N 1-methyl-9h-fluorene-9-carboxylic acid Chemical compound C12=CC=CC=C2C(C(O)=O)C2=C1C=CC=C2C JJUQKWSFCOFJQU-UHFFFAOYSA-N 0.000 description 3
- KRPHZIRPXLJJIZ-UHFFFAOYSA-N 2,7-dimethyl-9h-fluorene Chemical compound CC1=CC=C2C3=CC=C(C)C=C3CC2=C1 KRPHZIRPXLJJIZ-UHFFFAOYSA-N 0.000 description 3
- NCXCGGKCYZSYEU-UHFFFAOYSA-N 2,7-dimethylfluoren-1-one Chemical compound CC1=CC=C2C3=CC=C(C)C(=O)C3=CC2=C1 NCXCGGKCYZSYEU-UHFFFAOYSA-N 0.000 description 3
- IJLRCJSWVQQRDY-UHFFFAOYSA-N 2-methoxy-9h-fluoren-9-ol Chemical compound C1=CC=C2C3=CC=C(OC)C=C3C(O)C2=C1 IJLRCJSWVQQRDY-UHFFFAOYSA-N 0.000 description 3
- APWAMQGDVJNMRK-UHFFFAOYSA-N 2-methoxy-9h-fluorene Chemical compound C1=CC=C2C3=CC=C(OC)C=C3CC2=C1 APWAMQGDVJNMRK-UHFFFAOYSA-N 0.000 description 3
- ZPVGUJILHATTOY-UHFFFAOYSA-N 2-methoxy-9h-fluorene-9-carboxylic acid Chemical compound C1=CC=C2C3=CC=C(OC)C=C3C(C(O)=O)C2=C1 ZPVGUJILHATTOY-UHFFFAOYSA-N 0.000 description 3
- DWPLPINHDBWTAE-UHFFFAOYSA-N 3-(1-methyl-9h-fluoren-9-yl)propanoic acid Chemical compound C12=CC=CC=C2C(CCC(O)=O)C2=C1C=CC=C2C DWPLPINHDBWTAE-UHFFFAOYSA-N 0.000 description 3
- ABGIWTVOONWQLB-UHFFFAOYSA-N 3-(2-methoxy-9h-fluoren-9-yl)propanoic acid Chemical compound C1=CC=C2C3=CC=C(OC)C=C3C(CCC(O)=O)C2=C1 ABGIWTVOONWQLB-UHFFFAOYSA-N 0.000 description 3
- BKCZZTBNVHTNMX-UHFFFAOYSA-N 3-(4-methyl-9h-fluoren-9-yl)propanoic acid Chemical compound OC(=O)CCC1C2=CC=CC=C2C2=C1C=CC=C2C BKCZZTBNVHTNMX-UHFFFAOYSA-N 0.000 description 3
- HMXJAHRAHSSFMI-UHFFFAOYSA-N 4,5-dimethyl-9h-fluorene Chemical compound C1C2=CC=CC(C)=C2C2=C1C=CC=C2C HMXJAHRAHSSFMI-UHFFFAOYSA-N 0.000 description 3
- REGZNJBPISAMNA-UHFFFAOYSA-N 4,5-dimethylfluoren-1-one Chemical compound C1=CC(C)=C2C3=C(C)C=CC(=O)C3=CC2=C1 REGZNJBPISAMNA-UHFFFAOYSA-N 0.000 description 3
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 3
- OPFILOLCFWFBNT-UHFFFAOYSA-N 4-methyl-9h-fluorene Chemical compound C1C2=CC=CC=C2C2=C1C=CC=C2C OPFILOLCFWFBNT-UHFFFAOYSA-N 0.000 description 3
- OQKYEMHWZYHWBL-UHFFFAOYSA-N 9h-fluoren-1-ylmethanol Chemical compound C1C2=CC=CC=C2C2=C1C(CO)=CC=C2 OQKYEMHWZYHWBL-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- 206010002091 Anaesthesia Diseases 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- PIWKPBJCKXDKJR-UHFFFAOYSA-N Isoflurane Chemical compound FC(F)OC(Cl)C(F)(F)F PIWKPBJCKXDKJR-UHFFFAOYSA-N 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 239000000443 aerosol Substances 0.000 description 3
- 230000037005 anaesthesia Effects 0.000 description 3
- 229960002504 capsaicin Drugs 0.000 description 3
- 235000017663 capsaicin Nutrition 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- FZFAMSAMCHXGEF-UHFFFAOYSA-N chloro formate Chemical compound ClOC=O FZFAMSAMCHXGEF-UHFFFAOYSA-N 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 229960002725 isoflurane Drugs 0.000 description 3
- 150000002576 ketones Chemical class 0.000 description 3
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 3
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 3
- 239000003960 organic solvent Substances 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 230000000699 topical effect Effects 0.000 description 3
- WMSUFWLPZLCIHP-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 9h-fluoren-9-ylmethyl carbonate Chemical compound C12=CC=CC=C2C2=CC=CC=C2C1COC(=O)ON1C(=O)CCC1=O WMSUFWLPZLCIHP-UHFFFAOYSA-N 0.000 description 2
- UCSMUQIPMXOZBG-UHFFFAOYSA-N (2-methoxy-9h-fluoren-9-yl)methanol Chemical compound C1=CC=C2C3=CC=C(OC)C=C3C(CO)C2=C1 UCSMUQIPMXOZBG-UHFFFAOYSA-N 0.000 description 2
- FHEOTCKQUSUSOM-UHFFFAOYSA-N (2-methyl-9h-fluoren-9-yl)methanol Chemical compound C1=CC=C2C3=CC=C(C)C=C3C(CO)C2=C1 FHEOTCKQUSUSOM-UHFFFAOYSA-N 0.000 description 2
- SJVFAHZPLIXNDH-QFIPXVFZSA-N (2s)-2-(9h-fluoren-9-ylmethoxycarbonylamino)-3-phenylpropanoic acid Chemical compound C([C@@H](C(=O)O)NC(=O)OCC1C2=CC=CC=C2C2=CC=CC=C21)C1=CC=CC=C1 SJVFAHZPLIXNDH-QFIPXVFZSA-N 0.000 description 2
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 2
- GKEUODMJRFDLJY-UHFFFAOYSA-N 1-Methylfluorene Chemical compound C12=CC=CC=C2CC2=C1C=CC=C2C GKEUODMJRFDLJY-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- KPRMWCFKBHNUEP-UHFFFAOYSA-N 2-(2-carboxyphenyl)-5,5-dimethylcyclohexa-1,3-diene-1-carboxylic acid Chemical compound C1=CC(C)(C)CC(C(O)=O)=C1C1=CC=CC=C1C(O)=O KPRMWCFKBHNUEP-UHFFFAOYSA-N 0.000 description 2
- WKBKTKRNNSFTMV-UHFFFAOYSA-N 2-(9h-fluoren-9-yl)ethanol Chemical compound C1=CC=C2C(CCO)C3=CC=CC=C3C2=C1 WKBKTKRNNSFTMV-UHFFFAOYSA-N 0.000 description 2
- LRVQJCQTPOUJNW-UHFFFAOYSA-N 2-(9h-fluoren-9-yl)ethyl carbonochloridate Chemical compound C1=CC=C2C(CCOC(=O)Cl)C3=CC=CC=C3C2=C1 LRVQJCQTPOUJNW-UHFFFAOYSA-N 0.000 description 2
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 2
- SUOYPPZLPLEKAC-UHFFFAOYSA-N 2-[2-(1-methyl-9h-fluoren-9-yl)ethyl]-1,3-dioxolane Chemical compound C1=2C(C)=CC=CC=2C2=CC=CC=C2C1CCC1OCCO1 SUOYPPZLPLEKAC-UHFFFAOYSA-N 0.000 description 2
- HABYRTMJEFDISD-UHFFFAOYSA-N 2-[2-(2-methoxy-9h-fluoren-9-yl)ethyl]-1,3-dioxolane Chemical compound C=1C(OC)=CC=C(C2=CC=CC=C22)C=1C2CCC1OCCO1 HABYRTMJEFDISD-UHFFFAOYSA-N 0.000 description 2
- IZHVBANLECCAGF-UHFFFAOYSA-N 2-hydroxy-3-(octadecanoyloxy)propyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)COC(=O)CCCCCCCCCCCCCCCCC IZHVBANLECCAGF-UHFFFAOYSA-N 0.000 description 2
- QNBZCSMULZKFNV-UHFFFAOYSA-N 2-methoxyfluoren-9-one Chemical compound C1=CC=C2C(=O)C3=CC(OC)=CC=C3C2=C1 QNBZCSMULZKFNV-UHFFFAOYSA-N 0.000 description 2
- AIJPUDMCOIGRBM-UHFFFAOYSA-N 3-(2-methyl-9h-fluoren-9-yl)propanoic acid Chemical compound C1=CC=C2C3=CC=C(C)C=C3C(CCC(O)=O)C2=C1 AIJPUDMCOIGRBM-UHFFFAOYSA-N 0.000 description 2
- VQLOWPVIHVZKSU-UHFFFAOYSA-N 4,5-dimethyl-9h-fluorene-9-carboxylic acid Chemical compound OC(=O)C1C2=CC=CC(C)=C2C2=C1C=CC=C2C VQLOWPVIHVZKSU-UHFFFAOYSA-N 0.000 description 2
- WRHFEDIHGKCTNT-UHFFFAOYSA-N 4-(hydroxymethyl)-9h-fluoren-9-ol Chemical compound OC1C2=CC=CC=C2C2=C1C=CC=C2CO WRHFEDIHGKCTNT-UHFFFAOYSA-N 0.000 description 2
- RYWPRMPRWAQECQ-UHFFFAOYSA-N 4-methyl-9h-fluorene-9-carboxylic acid Chemical compound OC(=O)C1C2=CC=CC=C2C2=C1C=CC=C2C RYWPRMPRWAQECQ-UHFFFAOYSA-N 0.000 description 2
- NFKTVNJUXAVPFX-IBGZPJMESA-N 9H-fluoren-9-ylmethyl N-[(2S)-2-amino-4-methylpentanoyl]carbamate Chemical compound C1=CC=CC=2C3=CC=CC=C3C(C1=2)COC(=O)NC([C@@H](N)CC(C)C)=O NFKTVNJUXAVPFX-IBGZPJMESA-N 0.000 description 2
- WXMGVJAOLIDKGZ-UHFFFAOYSA-N 9h-fluoren-9-ylmethyl n-(1-hydroxy-4-methylpentan-2-yl)carbamate Chemical compound C1=CC=C2C(COC(=O)NC(CO)CC(C)C)C3=CC=CC=C3C2=C1 WXMGVJAOLIDKGZ-UHFFFAOYSA-N 0.000 description 2
- KWOLFJPFCHCOCG-UHFFFAOYSA-N Acetophenone Chemical compound CC(=O)C1=CC=CC=C1 KWOLFJPFCHCOCG-UHFFFAOYSA-N 0.000 description 2
- 102000009027 Albumins Human genes 0.000 description 2
- 108010088751 Albumins Proteins 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- HAPOJKSPCGLOOD-UHFFFAOYSA-N Benzo[b]fluorene Chemical compound C1=CC=C2C=C3CC4=CC=CC=C4C3=CC2=C1 HAPOJKSPCGLOOD-UHFFFAOYSA-N 0.000 description 2
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 2
- 206010012438 Dermatitis atopic Diseases 0.000 description 2
- 206010012442 Dermatitis contact Diseases 0.000 description 2
- 208000009386 Experimental Arthritis Diseases 0.000 description 2
- 102000009123 Fibrin Human genes 0.000 description 2
- 108010073385 Fibrin Proteins 0.000 description 2
- BWGVNKXGVNDBDI-UHFFFAOYSA-N Fibrin monomer Chemical compound CNC(=O)CNC(=O)CN BWGVNKXGVNDBDI-UHFFFAOYSA-N 0.000 description 2
- 206010018364 Glomerulonephritis Diseases 0.000 description 2
- 102000008100 Human Serum Albumin Human genes 0.000 description 2
- 108091006905 Human Serum Albumin Proteins 0.000 description 2
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 2
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- WVMBPWMAQDVZCM-UHFFFAOYSA-N N-methylanthranilic acid Chemical compound CNC1=CC=CC=C1C(O)=O WVMBPWMAQDVZCM-UHFFFAOYSA-N 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 201000004681 Psoriasis Diseases 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- BKYQTMPQPGXCMU-UHFFFAOYSA-N [1-(methoxymethyl)-9h-fluoren-9-yl]methanol Chemical compound C12=CC=CC=C2C(CO)C2=C1C=CC=C2COC BKYQTMPQPGXCMU-UHFFFAOYSA-N 0.000 description 2
- ATNOAWAQFYGAOY-GPTZEZBUSA-J [Na+].[Na+].[Na+].[Na+].Cc1cc(ccc1\N=N\c1ccc2c(cc(c(N)c2c1O)S([O-])(=O)=O)S([O-])(=O)=O)-c1ccc(\N=N\c2ccc3c(cc(c(N)c3c2O)S([O-])(=O)=O)S([O-])(=O)=O)c(C)c1 Chemical compound [Na+].[Na+].[Na+].[Na+].Cc1cc(ccc1\N=N\c1ccc2c(cc(c(N)c2c1O)S([O-])(=O)=O)S([O-])(=O)=O)-c1ccc(\N=N\c2ccc3c(cc(c(N)c3c2O)S([O-])(=O)=O)S([O-])(=O)=O)c(C)c1 ATNOAWAQFYGAOY-GPTZEZBUSA-J 0.000 description 2
- 230000003187 abdominal effect Effects 0.000 description 2
- 238000002835 absorbance Methods 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 239000000908 ammonium hydroxide Substances 0.000 description 2
- 239000002260 anti-inflammatory agent Substances 0.000 description 2
- 229940121363 anti-inflammatory agent Drugs 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 208000006673 asthma Diseases 0.000 description 2
- 201000008937 atopic dermatitis Diseases 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- 239000003637 basic solution Substances 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 239000001045 blue dye Substances 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 229940117975 chromium trioxide Drugs 0.000 description 2
- GAMDZJFZMJECOS-UHFFFAOYSA-N chromium(6+);oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[Cr+6] GAMDZJFZMJECOS-UHFFFAOYSA-N 0.000 description 2
- 239000012230 colorless oil Substances 0.000 description 2
- 208000010247 contact dermatitis Diseases 0.000 description 2
- 239000010779 crude oil Substances 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 229960004397 cyclophosphamide Drugs 0.000 description 2
- 238000006114 decarboxylation reaction Methods 0.000 description 2
- 239000007933 dermal patch Substances 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- ZKQFHRVKCYFVCN-UHFFFAOYSA-N ethoxyethane;hexane Chemical compound CCOCC.CCCCCC ZKQFHRVKCYFVCN-UHFFFAOYSA-N 0.000 description 2
- 229960003699 evans blue Drugs 0.000 description 2
- 229950003499 fibrin Drugs 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 239000003862 glucocorticoid Substances 0.000 description 2
- SHFJWMWCIHQNCP-UHFFFAOYSA-M hydron;tetrabutylazanium;sulfate Chemical compound OS([O-])(=O)=O.CCCC[N+](CCCC)(CCCC)CCCC SHFJWMWCIHQNCP-UHFFFAOYSA-M 0.000 description 2
- WTDHULULXKLSOZ-UHFFFAOYSA-N hydroxylamine hydrochloride Substances Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 2
- WCYJQVALWQMJGE-UHFFFAOYSA-M hydroxylammonium chloride Chemical compound [Cl-].O[NH3+] WCYJQVALWQMJGE-UHFFFAOYSA-M 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 208000002551 irritable bowel syndrome Diseases 0.000 description 2
- 206010025135 lupus erythematosus Diseases 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- GBMDVOWEEQVZKZ-UHFFFAOYSA-N methanol;hydrate Chemical compound O.OC GBMDVOWEEQVZKZ-UHFFFAOYSA-N 0.000 description 2
- 229960000485 methotrexate Drugs 0.000 description 2
- XMJHPCRAQCTCFT-UHFFFAOYSA-N methyl chloroformate Chemical compound COC(Cl)=O XMJHPCRAQCTCFT-UHFFFAOYSA-N 0.000 description 2
- TZIHFWKZFHZASV-UHFFFAOYSA-N methyl formate Chemical compound COC=O TZIHFWKZFHZASV-UHFFFAOYSA-N 0.000 description 2
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 2
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 201000008482 osteoarthritis Diseases 0.000 description 2
- 239000007800 oxidant agent Substances 0.000 description 2
- 230000001590 oxidative effect Effects 0.000 description 2
- 210000005164 penile vein Anatomy 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 206010039073 rheumatoid arthritis Diseases 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 239000012047 saturated solution Substances 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- 235000010288 sodium nitrite Nutrition 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 230000003637 steroidlike Effects 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 229940037128 systemic glucocorticoids Drugs 0.000 description 2
- 238000011200 topical administration Methods 0.000 description 2
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 2
- 238000005303 weighing Methods 0.000 description 2
- VZOHGJIGTNUNNC-GOSISDBHSA-N (2s)-2-(9h-fluoren-9-ylmethoxycarbonylamino)-3,3-dimethylbutanoic acid Chemical compound C1=CC=C2C(COC(=O)N[C@@H](C(C)(C)C)C(O)=O)C3=CC=CC=C3C2=C1 VZOHGJIGTNUNNC-GOSISDBHSA-N 0.000 description 1
- VCFCFPNRQDANPN-IBGZPJMESA-N (2s)-2-(9h-fluoren-9-ylmethoxycarbonylamino)hexanoic acid Chemical compound C1=CC=C2C(COC(=O)N[C@@H](CCCC)C(O)=O)C3=CC=CC=C3C2=C1 VCFCFPNRQDANPN-IBGZPJMESA-N 0.000 description 1
- SEHPSBSPFWAVPK-NRFANRHFSA-N (2s)-2-[(2,7-dimethyl-9h-fluoren-9-yl)methoxycarbonylamino]-4-methylpentanoic acid Chemical compound C1=C(C)C=C2C(COC(=O)N[C@@H](CC(C)C)C(O)=O)C3=CC(C)=CC=C3C2=C1 SEHPSBSPFWAVPK-NRFANRHFSA-N 0.000 description 1
- BUJQSIPFDWLNDC-FQEVSTJZSA-N (2s)-2-[9h-fluoren-9-ylmethoxycarbonyl(methyl)amino]-4-methylpentanoic acid Chemical compound C1=CC=C2C(COC(=O)N(C)[C@@H](CC(C)C)C(O)=O)C3=CC=CC=C3C2=C1 BUJQSIPFDWLNDC-FQEVSTJZSA-N 0.000 description 1
- MUNHVMZDPGYKES-UHFFFAOYSA-N (4,5-dimethyl-9h-fluoren-9-yl)methanol Chemical compound OCC1C2=CC=CC(C)=C2C2=C1C=CC=C2C MUNHVMZDPGYKES-UHFFFAOYSA-N 0.000 description 1
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- WECGLUPZRHILCT-GSNKCQISSA-N 1-linoleoyl-sn-glycerol Chemical compound CCCCC\C=C/C\C=C/CCCCCCCC(=O)OC[C@@H](O)CO WECGLUPZRHILCT-GSNKCQISSA-N 0.000 description 1
- JURAOGMOBVQPBP-UHFFFAOYSA-N 2-(2-carboxy-4-methylphenyl)-5-methylbenzoic acid Chemical compound OC(=O)C1=CC(C)=CC=C1C1=CC=C(C)C=C1C(O)=O JURAOGMOBVQPBP-UHFFFAOYSA-N 0.000 description 1
- WNAJXPYVTFYEST-UHFFFAOYSA-N 2-Amino-3-methylbenzoate Chemical compound CC1=CC=CC(C(O)=O)=C1N WNAJXPYVTFYEST-UHFFFAOYSA-N 0.000 description 1
- HBASPXYOBZJODB-UHFFFAOYSA-N 2-[2-(4-methyl-9h-fluoren-9-yl)ethyl]-1,3-dioxolane Chemical compound C12=CC=CC=C2C=2C(C)=CC=CC=2C1CCC1OCCO1 HBASPXYOBZJODB-UHFFFAOYSA-N 0.000 description 1
- WZFUQSJFWNHZHM-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)CC(=O)N1CC2=C(CC1)NN=N2 WZFUQSJFWNHZHM-UHFFFAOYSA-N 0.000 description 1
- NBUUUJWWOARGNW-UHFFFAOYSA-N 2-amino-5-methylbenzoic acid Chemical compound CC1=CC=C(N)C(C(O)=O)=C1 NBUUUJWWOARGNW-UHFFFAOYSA-N 0.000 description 1
- OFSBZLRBRIAIKL-UHFFFAOYSA-N 2-ethyl-2-(9h-fluoren-9-yl)-1,3-dioxolane Chemical compound C12=CC=CC=C2C2=CC=CC=C2C1C1(CC)OCCO1 OFSBZLRBRIAIKL-UHFFFAOYSA-N 0.000 description 1
- LJDJBBQTXASCSG-UHFFFAOYSA-N 2-hydroxyfluoren-1-one Chemical compound C1=CC=C2C3=CC=C(O)C(=O)C3=CC2=C1 LJDJBBQTXASCSG-UHFFFAOYSA-N 0.000 description 1
- RPEBFZNZXWYPEL-UHFFFAOYSA-N 3-(1-methyl-9h-fluoren-9-yl)propanoyl chloride Chemical compound C12=CC=CC=C2C(CCC(Cl)=O)C2=C1C=CC=C2C RPEBFZNZXWYPEL-UHFFFAOYSA-N 0.000 description 1
- VDONEUQRJMUOJE-UHFFFAOYSA-N 3-(2-methoxy-9h-fluoren-9-yl)propanoyl chloride Chemical compound C1=CC=C2C3=CC=C(OC)C=C3C(CCC(Cl)=O)C2=C1 VDONEUQRJMUOJE-UHFFFAOYSA-N 0.000 description 1
- FBXVEUBPMISTKS-UHFFFAOYSA-N 3-(4-methyl-9h-fluoren-9-yl)propanoyl chloride Chemical compound ClC(=O)CCC1C2=CC=CC=C2C2=C1C=CC=C2C FBXVEUBPMISTKS-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- XKTYXVDYIKIYJP-UHFFFAOYSA-N 3h-dioxole Chemical compound C1OOC=C1 XKTYXVDYIKIYJP-UHFFFAOYSA-N 0.000 description 1
- AFQYQSWTVCNJQT-UHFFFAOYSA-N 9-oxofluorene-4-carboxylic acid Chemical compound O=C1C2=CC=CC=C2C2=C1C=CC=C2C(=O)O AFQYQSWTVCNJQT-UHFFFAOYSA-N 0.000 description 1
- BULODOHSYVQOJP-UHFFFAOYSA-N 9h-fluoren-9-ylmethyl 2,5-dioxopyrrolidine-1-carboxylate Chemical compound C12=CC=CC=C2C2=CC=CC=C2C1COC(=O)N1C(=O)CCC1=O BULODOHSYVQOJP-UHFFFAOYSA-N 0.000 description 1
- JABUROMNIPPAQJ-FQEVSTJZSA-N 9h-fluoren-9-ylmethyl n-[(2s)-4-methyl-1-(methylamino)-1-oxopentan-2-yl]carbamate Chemical compound C1=CC=C2C(COC(=O)N[C@@H](CC(C)C)C(=O)NC)C3=CC=CC=C3C2=C1 JABUROMNIPPAQJ-FQEVSTJZSA-N 0.000 description 1
- HTPXFGUCAUTOEL-UHFFFAOYSA-N 9h-fluorene-1-carboxylic acid Chemical compound C1C2=CC=CC=C2C2=C1C(C(=O)O)=CC=C2 HTPXFGUCAUTOEL-UHFFFAOYSA-N 0.000 description 1
- MNQGEQSXFDKAPY-UHFFFAOYSA-N 9h-fluorene-2-carbaldehyde Chemical compound C1=CC=C2C3=CC=C(C=O)C=C3CC2=C1 MNQGEQSXFDKAPY-UHFFFAOYSA-N 0.000 description 1
- RNDGADNGORZKGH-UHFFFAOYSA-N 9h-fluorene-9-carbaldehyde Chemical compound C1=CC=C2C(C=O)C3=CC=CC=C3C2=C1 RNDGADNGORZKGH-UHFFFAOYSA-N 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- ZCQCZXDCHLPWCN-UHFFFAOYSA-N CC1(C)CC=CC(C(O)=O)=C1C1=CC=CC=C1C(O)=O Chemical compound CC1(C)CC=CC(C(O)=O)=C1C1=CC=CC=C1C(O)=O ZCQCZXDCHLPWCN-UHFFFAOYSA-N 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical compound NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical class NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-N Formic acid Chemical group OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 241000186359 Mycobacterium Species 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- 208000001388 Opportunistic Infections Diseases 0.000 description 1
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 229910006124 SOCl2 Inorganic materials 0.000 description 1
- 206010039710 Scleroderma Diseases 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- NOUDPBCEONUCOV-FJXQXJEOSA-N [(2s)-1-ethoxy-4-methyl-1-oxopentan-2-yl]azanium;chloride Chemical compound Cl.CCOC(=O)[C@@H](N)CC(C)C NOUDPBCEONUCOV-FJXQXJEOSA-N 0.000 description 1
- PQLVXDKIJBQVDF-UHFFFAOYSA-N acetic acid;hydrate Chemical compound O.CC(O)=O PQLVXDKIJBQVDF-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 150000001370 alpha-amino acid derivatives Chemical class 0.000 description 1
- 235000008206 alpha-amino acids Nutrition 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- 125000005605 benzo group Chemical group 0.000 description 1
- QTQGHKVYLQBJLO-YDALLXLXSA-N benzyl (2s)-2-amino-4-methylpentanoate;4-methylbenzenesulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1.CC(C)C[C@H](N)C(=O)OCC1=CC=CC=C1 QTQGHKVYLQBJLO-YDALLXLXSA-N 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 1
- 230000000711 cancerogenic effect Effects 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- IVNKOMARKNDHKI-UHFFFAOYSA-N carbonyl dichloride;dichloromethane Chemical compound ClCCl.ClC(Cl)=O IVNKOMARKNDHKI-UHFFFAOYSA-N 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 231100000315 carcinogenic Toxicity 0.000 description 1
- 238000012754 cardiac puncture Methods 0.000 description 1
- 239000000679 carrageenan Substances 0.000 description 1
- 235000010418 carrageenan Nutrition 0.000 description 1
- 229920001525 carrageenan Polymers 0.000 description 1
- 229940113118 carrageenan Drugs 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- IQFVPQOLBLOTPF-HKXUKFGYSA-L congo red Chemical compound [Na+].[Na+].C1=CC=CC2=C(N)C(/N=N/C3=CC=C(C=C3)C3=CC=C(C=C3)/N=N/C3=C(C4=CC=CC=C4C(=C3)S([O-])(=O)=O)N)=CC(S([O-])(=O)=O)=C21 IQFVPQOLBLOTPF-HKXUKFGYSA-L 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- FDPIMTJIUBPUKL-UHFFFAOYSA-N dimethylacetone Natural products CCC(=O)CC FDPIMTJIUBPUKL-UHFFFAOYSA-N 0.000 description 1
- 150000002009 diols Chemical class 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 208000030172 endocrine system disease Diseases 0.000 description 1
- IRXSLJNXXZKURP-UHFFFAOYSA-N fluorenylmethyloxycarbonyl chloride Chemical compound C1=CC=C2C(COC(=O)Cl)C3=CC=CC=C3C2=C1 IRXSLJNXXZKURP-UHFFFAOYSA-N 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 229940074045 glyceryl distearate Drugs 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- 230000026030 halogenation Effects 0.000 description 1
- 238000005658 halogenation reaction Methods 0.000 description 1
- 239000012456 homogeneous solution Substances 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 238000010907 mechanical stirring Methods 0.000 description 1
- DODCBMODXGJOKD-RGMNGODLSA-N methyl (2s)-2-amino-4-methylpentanoate;hydrochloride Chemical compound Cl.COC(=O)[C@@H](N)CC(C)C DODCBMODXGJOKD-RGMNGODLSA-N 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- MGJXBDMLVWIYOQ-UHFFFAOYSA-N methylazanide Chemical compound [NH-]C MGJXBDMLVWIYOQ-UHFFFAOYSA-N 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 238000006396 nitration reaction Methods 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 239000003495 polar organic solvent Substances 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920000137 polyphosphoric acid Polymers 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- ZLMJMSJWJFRBEC-BJUDXGSMSA-N potassium-38 Chemical compound [38K] ZLMJMSJWJFRBEC-BJUDXGSMSA-N 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- 239000002599 prostaglandin synthase inhibitor Substances 0.000 description 1
- 238000002731 protein assay Methods 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- 239000012066 reaction slurry Substances 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 239000004576 sand Substances 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- ODZPKZBBUMBTMG-UHFFFAOYSA-N sodium amide Chemical compound [NH2-].[Na+] ODZPKZBBUMBTMG-UHFFFAOYSA-N 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- RNVYQYLELCKWAN-UHFFFAOYSA-N solketal Chemical compound CC1(C)OCC(CO)O1 RNVYQYLELCKWAN-UHFFFAOYSA-N 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 238000013222 sprague-dawley male rat Methods 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- ILWRPSCZWQJDMK-UHFFFAOYSA-N triethylazanium;chloride Chemical compound Cl.CCN(CC)CC ILWRPSCZWQJDMK-UHFFFAOYSA-N 0.000 description 1
- 201000008827 tuberculosis Diseases 0.000 description 1
- 238000003828 vacuum filtration Methods 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 229940074411 xylene Drugs 0.000 description 1
- UHVMMEOXYDMDKI-JKYCWFKZSA-L zinc;1-(5-cyanopyridin-2-yl)-3-[(1s,2s)-2-(6-fluoro-2-hydroxy-3-propanoylphenyl)cyclopropyl]urea;diacetate Chemical compound [Zn+2].CC([O-])=O.CC([O-])=O.CCC(=O)C1=CC=C(F)C([C@H]2[C@H](C2)NC(=O)NC=2N=CC(=CC=2)C#N)=C1O UHVMMEOXYDMDKI-JKYCWFKZSA-L 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/27—Esters, e.g. nitroglycerine, selenocyanates of carbamic or thiocarbamic acids, meprobamate, carbachol, neostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/195—Carboxylic acids, e.g. valproic acid having an amino group
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/215—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids
- A61K31/22—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids of acyclic acids, e.g. pravastatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/45—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups
- C07C233/46—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom
- C07C233/51—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom having the carbon atom of the carboxamide group bound to an acyclic carbon atom of a carbon skeleton containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/16—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of hydrocarbon radicals substituted by singly-bound oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/22—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of hydrocarbon radicals substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2603/00—Systems containing at least three condensed rings
- C07C2603/02—Ortho- or ortho- and peri-condensed systems
- C07C2603/04—Ortho- or ortho- and peri-condensed systems containing three rings
- C07C2603/06—Ortho- or ortho- and peri-condensed systems containing three rings containing at least one ring with less than six ring members
- C07C2603/10—Ortho- or ortho- and peri-condensed systems containing three rings containing at least one ring with less than six ring members containing five-membered rings
- C07C2603/12—Ortho- or ortho- and peri-condensed systems containing three rings containing at least one ring with less than six ring members containing five-membered rings only one five-membered ring
- C07C2603/18—Fluorenes; Hydrogenated fluorenes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2603/00—Systems containing at least three condensed rings
- C07C2603/02—Ortho- or ortho- and peri-condensed systems
- C07C2603/40—Ortho- or ortho- and peri-condensed systems containing four condensed rings
Definitions
- This invention relates to methods for treating inflammatory conditions and to compounds and pharmaceutical compositions suitable for
- inflammatory conditions such as atopic dermatitis, contact dermatitis, psoriasis, rheumatoid arthritis, glomerulonephritis, osteoarthritis, lupus erythematosis, scleroderma, asthma and irritable bowel disease has, in the past, involved the use of agents such as aspirin-like nonsteroidal anti-inflammatory agents,
- glucocorticoids methotrexate and cyclophosphamide. Unfortunately these agents generally produce
- nonsteroidal anti-inflammatory drugs often cause gastrointestinal and renal side effects.
- Glucocorticoids suppress the immune system, thus producing opportunistic infection and
- Methotrexate has been associated with patient death
- cyclophosphamide has
- the invention also relates to compounds and pharmaceutical
- compositions suitable for use in such a method are provided.
- the present invention relates to a method of treating an inflammatory condition comprising administering to an animal in need of such treatment at least one compound of Formula I:
- X is methylene, ethylene, ethyleneoxy, or oxygen
- Iipophilic amino acid and Y is -CO 2 H, -CH 2 OH,
- R 1 and R 7 are hydrogen, alkyl, or aryl;
- R 3 and R 4 are, independently, hydrogen, alkyl or aryl
- a and B are, independently, hydrogen, fused phenyl, alkyl, aryl, alkaryl, aralkyl, alkoxy, alkoxyalkyl, halogen, or nitro;
- the present invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising the compound of Formula I (above) in an amount sufficient to produce an anti-inflammatory effect, together with a pharmaceutically acceptable carrier.
- the present invention relates, generally, to compounds of
- Formula I wherein X, Q, R 3 , R 4 , A and B are defined as set forth above, providing that when R 3 , R 4 , A and B are hydrogen and Y is -C0 7 H (or salt thereof), X is not oxygen, and further providing that when A or B are, independently, hydrogen or halogen, R 3 and R 4 are hydrogen, X is oxygen, and Y is -CO 2 H (or salt thereof), C' is not an aromatic amino acid residue.
- the invention does, however, include N-[9H-(fluoren- 9-ylmethoxy)carbonyl]-L-tert-leucine and N-[(9H- fluoren-9-ylmethoxy)carbonyl]-L-neopentylglycine.
- X is methylene, ethylene, ethyleneoxy, or oxygen
- R 1 and R 3 are hydrogen, alkyl (advantageously, C 1-4 ) or aryl (advantageously, C 6-12 );
- R 3 and R 4 are, independently, hydrogen, alkyl (advantageously, C 1-4 ) or aryl (advantageously, C 6-12 ); and
- a and B are, independently, hydrogen, fused phenol, alkyl (advantageously, C 1-9 ), aryl (advantageously, C 6-12 ), alkaryl (advantageously, (C 1-9 )alk(C 6-12 )aryl), aralkyl (advantageously,
- hydrocarbons can be unsubstituted or substituted with a C 1-4 alkyl group.
- Iipophilic amino acid as used herein includes within its scope amino acids, the residues of which do not contain free hydroxy groups, free thiol groups, or basic nitrogen atoms.
- compositions and methods to which the invention relates are provided.
- one that is preferred for use in the present method is the compound N-[(9H-fluoren-9-ylmethoxy)carbonyl]- L-leucine.
- N-[(9H-fluoren-9- ylmethoxy)carbonyl]-L-norleucine, and S-benzyl- ⁇ , ⁇ - dimethyl-N-[9H-(fluoren-9-ylmethoxy)carbonyl]-D- cystein are also known compounds that are preferred for use in the present method.
- novel compounds of Formula I that are preferred for use in the present method are those wherein: i) R 3 , R 4 , A and B are hydrogen, X is methylene, and Q is a Iipophilic amino acid; and ii) R 3 and R 4 are hydrogen, X is oxygen, A and/or B each represents at least one alkyl substituent, and Q is a Iipophilic amino acid.
- novel compounds of Formula I that are the most preferred for use in the present method are those wherein: i) R 3 and R 4 are hydrogen, X is oxygen, A is a methyl group located in the 4 position of the fluorene ring, B is
- R 3 and R 4 are hydrogen, X is oxygen, A is a methyl group located in the 4 position of the fluorene ring, B is hydrogen, and Q is the amino acid homophenylalanine; and iii) R 3 and R 4 are hydrogen, X is oxygen, A is a methyl group located in position 2 of the fluorene ring, B is a methyl group located in position 7 of the fluorene ring, and Q is the amino acid leucine.
- 9H-fluorene can be condensed directly with formaldehyde in the presence of a strong base such as sodium hydride or sodium amide to give the 9-methanol derivative.
- a strong base such as sodium hydride or sodium amide
- Compounds in which the alpha carbon atom is substituted may be prepared by reaction between the selected 9H-fluorene and an aldehyde other than formaldehyde or a ketone, such as acetone or acetophenone, in the presence of a strong base.
- 9H-Fluoren-9-ylmethanols are converted to 9H-fluoren-9-ylmethanol haloformates, carbonates, thiocarbonates, imidylcarbonates or other formate derivatives bearing a grouping ("leaving group") that is readily displaced by a nucleophilic nitrogen of an alpha amino acid.
- the resulting carbonyl derivatives of an activated 9H-fluoren-9-ylmethanol are condensed with an alpha aminocarboxylic acid to form a 9H-fluoren-9-ylmethoxycarbonyl derivative of the general Formula I.
- reaction may be effected in a polar organic solvent such as dioxane, tetrahydrofuran, dimethylformamide or pyridine under alkaline conditions (preferably mild) at a low temperature, for example from 0°C to 25°C during a period of from about 2 to 3 hours.
- a polar organic solvent such as dioxane, tetrahydrofuran, dimethylformamide or pyridine under alkaline conditions (preferably mild) at a low temperature, for example from 0°C to 25°C during a period of from about 2 to 3 hours.
- a preferred solvent is a mixture of dioxane and water.
- the N-[(9H-fluoren-9-ylmethoxy)carbonyl]- amino acid precipitates from solution and may be purified, for example, by recrystallization.
- Utilization of other "leaving groups” may require somewhat elevated temperatures, for example, 25°C to 50°C and longer reaction times, for example, 8 to 12 hours.
- Typical salts include the alkali metal or alkaline earth salts, although it will be appreciated that other nontoxic salts can also be used.
- compounds suitable for use in the present method this invention are administered as sodium, potassium, ammonium, choline or
- the compounds of this invention can be present as D or L optical isomers or, in some cases, as
- the compounds of Formula I include all isomers of such compounds, whether separated or mixtures thereof.
- a compound of Formula I as an anti-inflammatory agent can be demonstrated in animals, such as mice, for example, by measuring the ability of the compound to inhibit edema caused by a variety of inflammatory agents that are generally accepted as producing irritation by differing mechanisms.
- inflammatory agents typically include tetradecanoylphorbolacetate, arachidonic acid, xylene, capsaicin, oxazolone, carrageenan and the like.
- the reverse passive Arthus test offers another measure of the compound's utility in
- Test compounds are typically administered intraperitoneally or topically.
- the test compound can be given in dimethyl sulfoxide or in 0.5% methylcellulose 30 minutes prior to
- test compound can be dissolved in, for example, acetone, ethanol or dimethyl sulfoxide and applied about 15 minutes prior to application of the irritant. Results can be
- non-steroidal anti-inflammatory agents operate by a single mechanism (cyclo-oxygenase inhibitors), thus, they are highly active in a single assay (steroids are usually active in most, if not all, screens but have side effects that prohibit their widespread use).
- the compounds of Formula I are highly active in almost all of the inflammatory screens and are also highly active in the reverse passive artus assay and in adjuvant arthritis, which are
- the compounds of Formula I have the steroid-like spectrum of activity but lack steroid-like toxicity.
- compositions of the present invention comprise, as an active ingredient, at least one compound acid of Formula I (see above), together with a pharmaceutically acceptable carrier.
- the active ingredient is present in the composition in an amount sufficient to produce an
- composition of the invention can be formulated so as to be suitable, for example, for oral, nasal, parenteral, topical, transdermal or rectal administration.
- compositions can also be formulated so as to be suitable for veternary use.
- Compounds (known and novel) that are preferred for use in the pharmaceutical composition of the present invention include those of Formula I wherein R 3 and R 4 are hydrogen, A and B are hydrogen or (C 1-4 ) alkyl, X is oxygen, and Q is a Iipophilic amino acid; compounds that are more preferred are those where A and B are alkyl; compounds that are the most preferred are those where A is a methyl group located in the 2 position of the fluorene ring and B is a methyl group located in the 7 position of the fluorene ring, and Q is leucine, isoleucine or nor-leucine.
- the pharmaceutical composition of the invention includes the active ingredient of Formula I in a quantity selected from 25 mg to 500 mg, advantageously, from about 50 mg to 250 mg, per dosage unit, depending on the route of
- compositions of the invention may be, for example, in solid or liquid form.
- carrier carriers are lactose, magnesium stearate, terra alba, sucrose, talc, stearic acid, gelatin, agar, pectin or acacia.
- the amount of solid carrier present in the composition will vary greatly but preferably will be from about 25 mg to 1 g.
- liquid carriers are syrup, peanut oil, olive oil, sesame oil, propylene glycol,
- the carrier or diluent may include a time delay material well known to the art such as, for example, glyceryl monostearate or glyceryl distearate alone or with a wax.
- the pharmaceutical composition of the invention can be present in dosage unit form.
- the composition can take the form of a tablet (preferrably enteric coated), capsule (preferrably enteric coated), powder, troche, lozenge, inhalant, syrup, emulsion, gel, ointment, cream, lotion transdermal patch, suppository, sterile injectable liquid as well as a liquid suspension or solution.
- the pharmaceutical compositions of the present invention are prepared by conventional techniques such as by mixing, granulating and compressing or dissolving the ingredients as may be appropriate for the desired preparation.
- the method of treating an inflammatory condition according to this invention comprises administering to a subject in need of such treatment an amount of at least one compound of Formula I (see above) sufficient to produce an anti-inflammatory effect.
- the compounds of Formula I can be administered orally, nasally, topically,
- the active ingredient of Formula I (see above) will normally be administered in a daily dosage regimen selected from about 100 mg to 1 g, most preferably from about 200 mg to about 500 mg.
- equal doses will be administered, preferably, between one time per day to one time per week.
- the frequency of administration and the amount of active ingredient to be administered to effect treatment of a particular inflammatory condition can readily be determined by one skilled in the art.
- an aerosol dispensing system wherein the active medicament is incorporated with Freon ®
- aerosol container fluorohydrocarbon or other inert propellant in an aerosol container is of particular applicability.
- Such an aerosol system will deliver a metered dose of about 100 meg to about 650 meg, administered once or twice at a time as needed.
- Phenylalanine (27.25 g, 0.165 mole) was dissolved in a solution of sodium carbonate (31.8 g, 0.3 mole) in 320 ml of water. This mixture was added to a solution of 9-fluorenylmethylsuccinimidyl carbonate (50.8 g, 0.15 mole) dissolved in a minimum amount of dioxane (approximately 90 ml being
- Test compounds (compounds of Formula I where X is oxygen, R 3 , R 4 , A and B are hydrogen and Q is as indicated in Table 1) were administered intraperitoneally (100 mg/kg) or topically as follows.
- Test compounds (compounds of Formula I where X is oxygen, R 3 , R 4 , A and B are hydrogen and Q is as indicated in Table 1) were administered intraperitoneally (100 mg/kg) or topically as follows.
- test compound was dissolved in dimethyl sulfoxide or 0.5% methylcellulose and 100 ⁇ l was injected 30 minutes prior to irritant (100 mg/kg, i.p.).
- test compound was dissolved in either acetone, ethanol or dimethyl sulfoxide and 5 ⁇ l (100 ⁇ g) applied to the upper surface (1 cm') of the ear and an additional 5 ⁇ l (100 ⁇ g) applied to the lower surface (1 cm 2 ) of the ear fifteen minutes prior to application of the irritant.
- a solution of the irritant
- tetradecanoylphorbolacetate 200 ⁇ g/ml was added to the surface of the ear, 5 ⁇ l added to the upper surface and 5 ⁇ l to the lower surface. After three hours, the thickness of the ear was measured to 0.01 mm by a micrometer with loose drag positioned at the lateral-most edge of the mid-point of the pinna. Data were calculated as the inhibition by the test compound of increased ear thickness compared to control animals receiving only the irritant. The results are reported in Table 1.
- Formula I where X is oxygen, R 3 R 4 , A and B are hydrogen and Q is as indicated in Table 2) were administered intraperitoneally (100 mg/kg) as follows.
- test compound was dissolved in DMSO or 0 . 5%
- methylcellulose and 100 ⁇ l was injected 30 minutes prior to i.p. administration of 100 mg/kg of arachidonic acid.
- Test compounds (compounds of Formula I where X is oxygen, R 3 , R 4 , A and B are hydrogen and Q is as indicated in Table 3) were administered intraperitoneally (100 mg/kg) or topically as follows.
- Test compounds (compounds of Formula I where X is oxygen, R 3 , R 4 , A and B are hydrogen and Q is as indicated in Table 3) were administered intraperitoneally (100 mg/kg) or topically as follows.
- test compound was dissolved in
- test compound was dissolved in either acetone, ethanol or dimethyl sulfoxide and 5 ⁇ l (10 ⁇ g) applied to the upper surface (1 cm 2 ) of the ear and an additional 5 ⁇ l (10 ⁇ g) applied to the lower surface (1 cm 2 ) of the ear fifteen minutes prior to application of the irritant.
- the irritant, xylene was added to the surface of the ear, 20 ⁇ l added to the upper surface and 20 ⁇ l to the lower surface. After two hours, the thickness of the ear was measured to 0.01 mm by a micrometer with loose drag positioned at the lateral-most edge of the midpoint of the pinna. Data were calculated as the inhibition by the test compound of increased ear thickness compared to that of control animals receiving only the irritant. The results are reported in Table 3.
- Test compounds (compounds of Formula I where X is oxygen, R 3 , R 4 , A and B are hydrogen and Q is as indicated in Table 4) were administered intraperitoneally (100 mg/kg) as follows.
- the test compound was dissolved in DMSO or 0.5% methylcellulose and 100 ⁇ l was injected 30 minutes prior to irritant.
- the irritant, capsaicin, 25 mg/ml was added to the ear, 5 ⁇ l added to the upper surface and 5 ⁇ l to the lower surface. After thirty minutes, the thickness of the ear was measured to 0.01 mm by a micrometer with loose drag positioned at the lateral-most edge of the mid-point of the pinna. Data were calculated as the
- methylcellulose and 100 ⁇ l (100 mg/kg) was injected 30 minutes prior to irritant.
- the irritant 3% oxazolone in acetone, was added to the surface of the ear, 5 ⁇ l added to the upper surface and 5 ⁇ l to the lower surface. After twenty four hours, the thickness of the ear was measured to 0.01 mm by a micrometer with loose drag positioned at the
- Test compounds (compounds of Formula I where X is oxygen, R 3 , R 4 , A and B are hydrogen and Q is as indicated in Table 6) were dissolved in dimethyl sulfoxide and 200 ⁇ l of this solution (100 mg/kg) were injected intraperitoneally one hour before administration of the antigen .
- the animals were anesthetized inhalationally with isoflurane and then were injected through the penile vein with 1 ml of a solution of 2.5 mg of Evan's blue dye and 5.0 mg of human serum albumin in 1 ml of saline.
- N-[(9H-Fluoren-9-ylmethoxy)carbonyl]- L-leucine methyl ester NPC 15326)
- N-[(9H-Fluoren-9-ylmethoxy)carbonyl]- L-leucine ethyl ester NPC 15327)
- N-[(9H-Fluoren-9-ylmothoxy)carbonyl]- L-leucine benzyl ester NPC 15328
- N-[3-(9H-Fluoren-9-yl)propionyl]-L-leucine NPC 15476)
- reaction was monitored by TLC (silica, 25% ethyl acetate in hexane).
- the reaction mixture was stirred at room temperature for 3 hr. The excess of phosgene was removed by argon bubbling. The solvent was evaporated to afford a slightly yellow oil.
- a solution of the oil in 5 mL of dioxane was charged with a solution of L-leucine (0.53 g, 4 mmol) in 14 mL of 10% aqueous solution of potassium carbonate and 7 mL of dioxane at room temperature.
- the reaction mixture was stirred overnight and diluted with water (100 mL). The water layer was extracted with ethyl acetate (5x30 mL) then acidified to pH 2 with HCl.
- N-[(9H-Fluoren-9-ylethoxy)carbonyl]- L-leucine N-[(9H-Fluoren-9-ylethoxy)carbonyl]- L-leucine (NPC 15521)
- N-[(9H-Fluoren-9-ylmethoxy)carbonyl]- L-leucine t-butyl ester NPC 15527
- N-[(9H-Fluoren-9-ylmethoxy)carbonyl] L-leucine amide NPC 15528
- N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-L-leucine methylamide NPC 15529
- the reaction mixture was stirred for two days, diluted with 150 mL of water and extracted with ethyl acetate (5x25 mL). The water layer was acidified to pH 1. The precipitated oil was extracted with ethyl acetate (3x50 mL), the organic solutions were washed with 1N HCl (3x20 mL), water, brine, dried over magnesium sulfate and evaporated to give an oil which was solidified by stirring in an ether-hexane mixture. The compound was purified by a column chromatography on silica using a solution of
- hydroxylammonium chloride (32.2 g, 464.0 mmol) in of water (108 mL) was charged with 6N sodium hydroxide (77 mL) at 5-10°C.
- the resulting hydroxylamine solution was immediately added to the cupric sulfate solution.
- the resulting solution at 5-10°C was charged with the diazonium solution at the same temperature during 40-50 min (the rate of addition is about 10 cc per minute) with a vigorous stirring. Stirring was continued for 5 min then heated to 70°C and acidified with conc. HCl. The mixture was allowed to stand overnight. The precipitate was filtered off, washed with water and dissolved in 400 mL of 10% sodium bicarbonate solution. This solution was treated with Norit, then filtered and acidified with 6N HCl. The precipitated product weighed 40.4 g (90.4 %).
- reaction mixture was cooled down to 60°C, diluted with water (150 mL) then cooled down to room
- the reaction mixture was stirred for 4 hr at room temperature. The excess of the phosgene was removed by bubbling argon. The solvents were removed under reduced pressure, the residue was dissolved in 10 mL of dioxane and the solution was added to a solution of L-leucine (1.71 g, 13.0 mmol) in 25.0 mL of dioxane and 49.0 mL of 10% potassium carbonate solution at room temperature. The reaction mixture was stirred overnight, the dioxane was removed under reduced pressure and the residue was diluted with 150 mL of water.
- 1,3-dioxolane (9.0 g, 32 mmol) in 350ml acetone at 0°C was slowly charged with 350 mL of Jone's reagent (the reagent was made by dissolving 16 g of chromium trioxide and 64 mL of concentrated sulfuric acid in 400 mL of water).
- the temperature raised to room temperature after all the reagent was added.
- the reaction mixture was monitored by TLC (silica, 25% EtOAc in hexane). The reaction was completed within 5 hr.
- the product was extracted with EtOAc and the organic layer was washed thoroughly with water (x6), until aqueous washings were clear and colorless.
- N- ⁇ [9H-(1-Methoxymethylfluoren-9- yl)methoxy]carbonyl ⁇ -L-leucine NPC 15673
- N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-L- neopentylglycine quarter hydrate NPC 15676
- N- ⁇ [9H-(1-Methylfluoren-9-yl)methoxy]carbonyl ⁇ -L- tert-leucine NPC 15952
- N- ⁇ 9H-[3-(1-Methylfluoren-9-yl)propionyl] ⁇ - L-homophenylalanine NPC-1597
- N- ⁇ 9H-[3-(4-Methylfluoren-9-yl)propionyl] ⁇ -L- leucine quarter hydrate NPC 15975
- N- ⁇ 9H-[3-(1-MethyIfluoren-9-yl)propionyl] ⁇ - L-norleucine, quarter hydrate NPC 15976
- the compound 3-(1-methylfluoren-9- yl) propionyl chloride (its preparation was described in Example 32; 4.0 g, 14.8 mmol) in 80 mL dioxane was charged with a solution of L-norleucine (4.0 g, 30.5 mmol) in 20 mL of 10% aqueous solution of sodium carbonate at room temperature. The reaction mixture was stirred for two hours. Most of the dioxane was removed under reduced pressure.
- Test compounds were administered intraperitoneally or topically as follows. For intraperitoneal administration, the test compound v/as dissolved in dimethyl sulfoxide or 0.5% methylcellulose and 100 microliters was
- micrograms applied to the upper surface and an additional 5 microliters applied to the lower surface of the ear fifteen minutes prior to
- mice 25-30 g body weight, six
- test compound was dissolved is dimethyl sulfoxide or 0.5% methylcellulose and 100 ⁇ L of the solution was injected intraperitoneally 30 minutes prior to the administration of 100 mg/kg of
- arachidponic acid A solution of this irritant, 100 mg/mL in ethanol, was applied to the surface of the ear, 5 ⁇ L to the upper surface and 5 uL to the lower surface. After sixty minutes, the thickness of the ear was measured ot 0.01 mm by a micrometer with the loose drag positioned at the lateral-most edge of the mid-point of the pinna. Data were calculated as the percent inhibityion by the test compound of increased ear thickness compared to control animals recieveing only the irritant. In general, %
- CF-1 mice 25 -30 g body weight, six animals per group, were used.
- the requisite amount of the test compound was dissolved is dimethyl sulfoxide or 0.5% methylcellulose and 100 ⁇ L of the solution was injected intraperitoneally 30 minutes prior to the administration the irritant.
- the irritant xylene was applied to the surface of the ear, 20 ⁇ L to the upper surface and 20 ⁇ L to the lower surface. After two hours, the thickness of the ear was measured to 0.01 mm by a micrometer with the loose drag
- mice 25-30 g body weight, five to six animals per group were used. The mice were
- test compounds were administered
- test compound was dissolved in dimethyl sulfoxide or 0.5%
- irritant 3% oxazolone in acetone
- 5 ⁇ L added to the upper surface
- 5 ⁇ L added to the lower surface.
- the thickness of the ear was measured to 0.01 mm by a micrometer with loose drag, positioned at the lateral-most edge of the mid-point of the pinna. Data were calculated as the inhibition of increased ear thickness compared to control animals' receiving only the irritant. In general, %
- Adjuvant Arthritis Male Sprague Dawley rats, 150 - 200 g, were anesthetized with isoflurane. Drug was
- the rat was then injected in the distal third of the tail with 0.5 mL of saline or 0.5 mL of well-sonicated Freund's complete adjuvant containing 1 mg/mL Mycobacterium
- each rat was weighed and dosed with vehicle or drug suspension as before, but without anesthesia. On day 3, each rat was weighed and anesthetized. Blood was drawn by cardiac puncture into 0.2 mL of EDT ⁇ solution. Blood samples were microcentrifuged for 30 seconds. Then fibrinogen was converted into fibrin using sodium sulfite and the resulting fibrin was assayed using a Lowry protein assay to estimate initial fibrinogen levels.
- Percent inhibition by test compound was determined by subtracting fibrinogen level in non-Freund's adjuvant-injected rats from fibrinogen levels in rats injected with adjuvant alone and those rats injected with adjuvant plus test compound, and dividing the resultant fibrinogen increases in drug treated animals by non-drug treated animals and multiplying by 100.
- N-[9H-(2,3-Denzofluoren-9-ylmethoxy)carbonyl]-L-loucinc NPC 15510
- N-((9H-Fluoren-9-ylmethoxy)carbonyl]-L-leucino amide NPC 15528
Abstract
Description


























Claims



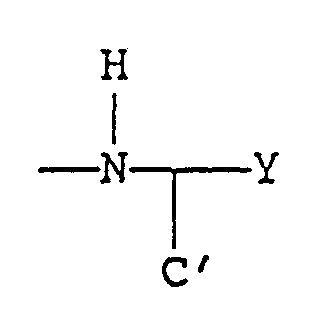




Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002083134A CA2083134A1 (en) | 1990-05-29 | 1991-05-17 | Methods for treating inflammation and compounds and compositions suitable for use therein |
| NO92924517A NO924517L (en) | 1990-05-29 | 1992-11-24 | METHODS FOR TREATMENT OF INFLAMMATION AND COMPOUNDS AND PREPARATIONS SUITABLE FOR USE THEREOF |
| FI925327A FI925327A0 (en) | 1990-05-29 | 1992-11-24 | FOERFARANDE FOER VAORD AV INFLAMMATION OCH I DESSA ANVAENDBARA FOERENINGAR OCH KOMPOSITIONER |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US07/529,356 US5079260A (en) | 1989-06-22 | 1990-05-29 | Method for treating inflammation and compounds and compositions suitable for use therein |
| US529,356 | 1990-05-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1991018596A1 true WO1991018596A1 (en) | 1991-12-12 |
Family
ID=24109577
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1991/003319 WO1991018596A1 (en) | 1990-05-29 | 1991-05-17 | Methods for treating inflammation and compounds and compositions suitable for use therein |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US5079260A (en) |
| EP (1) | EP0531443A4 (en) |
| JP (1) | JPH05509299A (en) |
| AU (1) | AU7951591A (en) |
| CA (1) | CA2083134A1 (en) |
| FI (1) | FI925327A0 (en) |
| IE (1) | IE911823A1 (en) |
| IL (1) | IL98223A0 (en) |
| NZ (1) | NZ238272A (en) |
| WO (1) | WO1991018596A1 (en) |
| ZA (1) | ZA914102B (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996015105A1 (en) * | 1994-11-15 | 1996-05-23 | Italfarmaco S.P.A. | Fluorenyl-hydroxamic derivatives endowed with immunosuppressive and anti-inflammatory activity |
| WO2002000611A2 (en) * | 2000-06-29 | 2002-01-03 | Institut National De La Sante Et De La Recherche Medicale (Inserm) | Fmoc-l-leucine and derivatives thereof as ppar-gamma agonists |
| WO2004106363A2 (en) * | 2003-05-30 | 2004-12-09 | Css-Albachem Limited | A tag for purification of peptides |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5668230A (en) * | 1991-07-23 | 1997-09-16 | Phillips Petroleum Company | Olefin polymerization |
| AU3239393A (en) * | 1991-12-12 | 1993-07-19 | Scios Nova Inc. | Fluorenyl derivatives and their use as anti-inflammatory agents |
| US5472973A (en) * | 1991-12-12 | 1995-12-05 | Scios Nova Inc. | Fluorenyl derivatives as anti-inflammatory agents |
| US5788982A (en) * | 1995-06-16 | 1998-08-04 | Nadoolman; Wolffe | Method and composition for treating oral pain using capsaicin |
| DE60034667T2 (en) | 1999-02-01 | 2008-03-13 | Dermal Research Laboratories, Inc. | PHARMACEUTICAL COMPOSITION OF COMPLEX CARBOHYDRATES AND THEIR USE |
| WO2001009117A1 (en) * | 1999-07-29 | 2001-02-08 | Allellix Neuroscience Inc. | Tricyclic compounds as glycine transport inhibitors |
| US7879824B2 (en) * | 2001-07-31 | 2011-02-01 | Dermal Research Laboratories, Inc. | Methods of preventing or treating diseases and conditions using complex carbohydrates |
| JP2004530632A (en) * | 2000-08-18 | 2004-10-07 | ジェネンテック・インコーポレーテッド | Integrin receptor inhibitor |
| CN100412060C (en) * | 2005-01-27 | 2008-08-20 | 中国科学院大连化学物理研究所 | Prepn of-2-(N-carbazolyl)-ethoxy carbohydrazide |
| DE102005062741A1 (en) * | 2005-12-22 | 2007-06-28 | Bayer Schering Pharma Ag | Fluorenes and carbazoles as ligands of the EP2 receptor |
| CN101328136B (en) * | 2008-07-23 | 2011-02-09 | 中国科学院广州化学研究所 | Aminoacid acidamide compounds and preparation thereof |
| CN108774158A (en) * | 2018-06-20 | 2018-11-09 | 南京肽业生物科技有限公司 | A kind of reaction of Fmoc and hydrophobic amino acid |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3835175A (en) * | 1971-03-15 | 1974-09-10 | Research Corp | 9-fluorenylmethanol haloformates, carbonates and thiocarbonates |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2877269A (en) * | 1956-04-17 | 1959-03-10 | Wm S Merrell Co | Guanyl substituted triphenylethanes, triphenylethylenes and benzalfluorenes |
| US3845097A (en) * | 1969-09-06 | 1974-10-29 | Ajinomoto Kk | N-substituted amino acids and novel ester |
| NL7013043A (en) * | 1969-09-06 | 1971-03-09 | ||
| US3919291A (en) * | 1969-09-06 | 1975-11-11 | Ajinomoto Kk | N-ethylcarbaminomethylisoleucine |
| US3906031A (en) * | 1971-03-15 | 1975-09-16 | Research Corp | Novel 9-fluorenylmethoxycarbonyl compounds |
| EP0129075A3 (en) * | 1983-05-20 | 1985-08-28 | F. HOFFMANN-LA ROCHE & CO. Aktiengesellschaft | Protected amino acid derivatives and their preparation |
| EP0288965A2 (en) * | 1987-04-29 | 1988-11-02 | Hoechst Aktiengesellschaft | Peptides with a phospholipase A2 inhibiting activity |
| JPH04506350A (en) * | 1989-06-22 | 1992-11-05 | ノバ ファーマスーティカル コーポレイション | Pharmaceutical compositions for treating inflammation and methods for treating inflammation |
-
1990
- 1990-05-29 US US07/529,356 patent/US5079260A/en not_active Expired - Lifetime
-
1991
- 1991-05-17 EP EP19910911551 patent/EP0531443A4/en not_active Withdrawn
- 1991-05-17 CA CA002083134A patent/CA2083134A1/en not_active Abandoned
- 1991-05-17 JP JP3510199A patent/JPH05509299A/en active Pending
- 1991-05-17 WO PCT/US1991/003319 patent/WO1991018596A1/en not_active Application Discontinuation
- 1991-05-17 AU AU79515/91A patent/AU7951591A/en not_active Abandoned
- 1991-05-22 IL IL98223A patent/IL98223A0/en unknown
- 1991-05-27 NZ NZ238272A patent/NZ238272A/en unknown
- 1991-05-28 IE IE182391A patent/IE911823A1/en unknown
- 1991-05-29 ZA ZA914102A patent/ZA914102B/en unknown
-
1992
- 1992-11-24 FI FI925327A patent/FI925327A0/en not_active Application Discontinuation
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3835175A (en) * | 1971-03-15 | 1974-09-10 | Research Corp | 9-fluorenylmethanol haloformates, carbonates and thiocarbonates |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996015105A1 (en) * | 1994-11-15 | 1996-05-23 | Italfarmaco S.P.A. | Fluorenyl-hydroxamic derivatives endowed with immunosuppressive and anti-inflammatory activity |
| WO2002000611A2 (en) * | 2000-06-29 | 2002-01-03 | Institut National De La Sante Et De La Recherche Medicale (Inserm) | Fmoc-l-leucine and derivatives thereof as ppar-gamma agonists |
| WO2002000611A3 (en) * | 2000-06-29 | 2002-05-30 | Ass Pour Le Dev De La Rech | Fmoc-l-leucine and derivatives thereof as ppar-gamma agonists |
| WO2004106363A2 (en) * | 2003-05-30 | 2004-12-09 | Css-Albachem Limited | A tag for purification of peptides |
| WO2004106363A3 (en) * | 2003-05-30 | 2005-05-26 | Css Albachem Ltd | A tag for purification of peptides |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0531443A4 (en) | 1993-04-21 |
| ZA914102B (en) | 1993-02-24 |
| FI925327A (en) | 1992-11-24 |
| NZ238272A (en) | 1994-03-25 |
| JPH05509299A (en) | 1993-12-22 |
| IE911823A1 (en) | 1991-12-04 |
| FI925327A0 (en) | 1992-11-24 |
| IL98223A0 (en) | 1992-06-21 |
| US5079260A (en) | 1992-01-07 |
| EP0531443A1 (en) | 1993-03-17 |
| AU7951591A (en) | 1991-12-31 |
| CA2083134A1 (en) | 1991-11-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO1991018596A1 (en) | Methods for treating inflammation and compounds and compositions suitable for use therein | |
| EP0462884B1 (en) | Trh derivatives, their preparations and pharmaceutical compositions containing them | |
| HU215437B (en) | Nitric esters having an inflammatory and anti-platelet aggregation activity and process for preparing them | |
| JPH05310664A (en) | Biaryl substituted 4-aminobutyric acid amide | |
| CA1101846A (en) | Derivatives of 4-hydroxy-phenylglycine | |
| IT9021075A1 (en) | BENZOIC ACID DERIVATIVES SUBSTITUTED FOR CARDIOVASCULAR ACTIVITY | |
| US4134991A (en) | Derivatives of 2-(3-phenyl-2-aminopropionyloxy)-acetic acid | |
| EP0721945B1 (en) | Benzolactam derivative | |
| EP0254354B1 (en) | Pharmaceutically useful derivatives of thiazolidine-4-carboxylic acid | |
| EP0177356B1 (en) | Method for treatment of antidiuresis | |
| US5472973A (en) | Fluorenyl derivatives as anti-inflammatory agents | |
| PT90254B (en) | METHOD FOR PREPARING AMIDES OF CYCENOMETILENIC ACID-1,2-DICARBOXYL ACIDS WITH THERAPEUTIC ACTIVITY AND OF PHARMACEUTICAL COMPOSITIONS CONTAINING THEM | |
| US5519043A (en) | Fluorenyl derivatives | |
| US3676463A (en) | Oxobenzofuran carboxamides | |
| EP0309262B1 (en) | Novel salicylates, their salts, pharmaceutical compositions containing them and process for preparing same | |
| EP0124925B1 (en) | Derivatives of d-2-(6-methoxy-2-naphthyl)-propionic acid having therapeutical activity, process for their preparation and pharmaceutical compositions containing them | |
| SE434835B (en) | N- (1-METHYL-2-PYRROLIDINYLMETHYL) -2,3-DIMETOXY-5-METHYLSULPHAMOYL-BENZAMIDE, ITS PREPARATION AND PHARMACOLOGICAL COMPOSITION CONTAINING THIS NEW BENZAMIDE | |
| WO1990015602A1 (en) | Pharmaceutical compositions and methods for treating inflammation | |
| EP0430738A2 (en) | Use of an imidazopyridine for the manufacture of anesthetic medicaments | |
| KR930006195B1 (en) | Buteroic acid amides, their salts, pharmacetutical compositions containing them and process for preparing same | |
| IE58497B1 (en) | New process for the preparation of derivatives of 4h-1, 2,4-triazole, the new triazoles so obtained, their use as medicaments and the pharmaceutical compositions containing them | |
| US4795758A (en) | 5-[2-(pyrrolidin-1-yl)ethoxy]-p-cymene derivatives, the process for the preparation of the said derivatives and drugs in which the said derivatives are present | |
| SE460969B (en) | APOVINCAMIC ACID DERIVATIVES, PROCEDURES FOR PREPARING THEREOF AND PHARMACEUTICAL COMPOSITIONS CONTAINING THESE DERIVATIVES | |
| KR880001007B1 (en) | Process for the preparation of bicyclic compound | |
| CH660480A5 (en) | D-2- (6-METHOXY-2-NAFTIL) PROPIONIC DERIVATIVES WITH THERAPEUTIC ACTIVITY, PROCEDURE FOR THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU BB BG BR CA FI HU JP KP KR LK MC MG MW NO PL RO SD SU |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE BF BJ CF CG CH CI CM DE DK ES FR GA GB GR IT LU ML MR NL SE SN TD TG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2083134 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 925327 Country of ref document: FI |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1991911551 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1991911551 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1991911551 Country of ref document: EP |