WO1995009843A1 - Hiv protease inhibitors - Google Patents
Hiv protease inhibitors Download PDFInfo
- Publication number
- WO1995009843A1 WO1995009843A1 PCT/US1994/011307 US9411307W WO9509843A1 WO 1995009843 A1 WO1995009843 A1 WO 1995009843A1 US 9411307 W US9411307 W US 9411307W WO 9509843 A1 WO9509843 A1 WO 9509843A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- substituted
- alkyl
- pharmaceutically acceptable
- acceptable salt
- Prior art date
Links
- 239000004030 hiv protease inhibitor Substances 0.000 title abstract description 4
- 150000001875 compounds Chemical class 0.000 claims abstract description 471
- 108010010369 HIV Protease Proteins 0.000 claims abstract description 9
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 5
- 238000000034 method Methods 0.000 claims description 144
- -1 mercapto, hydroxyl Chemical group 0.000 claims description 139
- 229910052739 hydrogen Inorganic materials 0.000 claims description 111
- 239000001257 hydrogen Substances 0.000 claims description 111
- 150000003839 salts Chemical class 0.000 claims description 111
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 claims description 104
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 claims description 82
- 125000000217 alkyl group Chemical group 0.000 claims description 81
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 78
- 125000003118 aryl group Chemical group 0.000 claims description 63
- 125000000623 heterocyclic group Chemical group 0.000 claims description 63
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 62
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 61
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 56
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 50
- 125000003545 alkoxy group Chemical group 0.000 claims description 50
- 229920006395 saturated elastomer Polymers 0.000 claims description 39
- 229910052757 nitrogen Inorganic materials 0.000 claims description 34
- 150000003568 thioethers Chemical class 0.000 claims description 31
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 30
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 27
- 125000004104 aryloxy group Chemical group 0.000 claims description 26
- 125000005843 halogen group Chemical group 0.000 claims description 25
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 24
- 125000003282 alkyl amino group Chemical group 0.000 claims description 22
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 21
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 21
- 125000001544 thienyl group Chemical group 0.000 claims description 20
- 229910052799 carbon Inorganic materials 0.000 claims description 17
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 16
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 16
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 15
- 125000005115 alkyl carbamoyl group Chemical group 0.000 claims description 15
- 229910052736 halogen Inorganic materials 0.000 claims description 15
- 150000002367 halogens Chemical class 0.000 claims description 15
- 125000003396 thiol group Chemical group [H]S* 0.000 claims description 15
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 14
- 229910052760 oxygen Inorganic materials 0.000 claims description 14
- 239000001301 oxygen Substances 0.000 claims description 14
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 13
- 125000002950 monocyclic group Chemical group 0.000 claims description 13
- 229940002612 prodrug Drugs 0.000 claims description 12
- 239000000651 prodrug Substances 0.000 claims description 12
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 12
- 125000006700 (C1-C6) alkylthio group Chemical group 0.000 claims description 11
- 125000002252 acyl group Chemical group 0.000 claims description 11
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 11
- 125000004076 pyridyl group Chemical group 0.000 claims description 11
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 claims description 11
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 10
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 9
- 125000003367 polycyclic group Chemical group 0.000 claims description 8
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 claims description 8
- 125000004414 alkyl thio group Chemical group 0.000 claims description 7
- 125000001624 naphthyl group Chemical group 0.000 claims description 7
- 125000006413 ring segment Chemical group 0.000 claims description 7
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 6
- 125000004652 decahydroisoquinolinyl group Chemical group C1(NCCC2CCCCC12)* 0.000 claims description 6
- 229910052717 sulfur Inorganic materials 0.000 claims description 6
- 125000005000 thioaryl group Chemical group 0.000 claims description 6
- 125000004760 (C1-C4) alkylsulfonylamino group Chemical group 0.000 claims description 5
- 125000001153 fluoro group Chemical group F* 0.000 claims description 5
- 239000011593 sulfur Substances 0.000 claims description 5
- 125000004769 (C1-C4) alkylsulfonyl group Chemical group 0.000 claims description 4
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical compound [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 claims description 4
- 125000002619 bicyclic group Chemical group 0.000 claims description 4
- 125000001246 bromo group Chemical group Br* 0.000 claims description 4
- 230000002401 inhibitory effect Effects 0.000 claims description 4
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 4
- 229910052711 selenium Inorganic materials 0.000 claims description 4
- 239000011669 selenium Substances 0.000 claims description 4
- 125000003003 spiro group Chemical group 0.000 claims description 4
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 3
- 125000004390 alkyl sulfonyl group Chemical group 0.000 claims description 2
- 239000003937 drug carrier Substances 0.000 claims description 2
- 150000002431 hydrogen Chemical class 0.000 claims 30
- QAGYKUNXZHXKMR-UHFFFAOYSA-N CPD000469186 Natural products CC1=C(O)C=CC=C1C(=O)NC(C(O)CN1C(CC2CCCCC2C1)C(=O)NC(C)(C)C)CSC1=CC=CC=C1 QAGYKUNXZHXKMR-UHFFFAOYSA-N 0.000 claims 1
- 241000725303 Human immunodeficiency virus Species 0.000 abstract description 17
- 208000030507 AIDS Diseases 0.000 abstract description 11
- 238000003786 synthesis reaction Methods 0.000 abstract description 4
- 239000003443 antiviral agent Substances 0.000 abstract description 3
- 230000010076 replication Effects 0.000 abstract description 3
- 239000004480 active ingredient Substances 0.000 abstract 1
- 230000004071 biological effect Effects 0.000 abstract 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 387
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 363
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 264
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 232
- 238000002360 preparation method Methods 0.000 description 224
- 238000006243 chemical reaction Methods 0.000 description 181
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 156
- 239000000243 solution Substances 0.000 description 149
- 238000005160 1H NMR spectroscopy Methods 0.000 description 147
- 239000007787 solid Substances 0.000 description 123
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 117
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 107
- 229910001868 water Inorganic materials 0.000 description 107
- 239000003480 eluent Substances 0.000 description 106
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 106
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 105
- 230000002829 reductive effect Effects 0.000 description 105
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 96
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 91
- 239000011541 reaction mixture Substances 0.000 description 91
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 79
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 78
- 235000019439 ethyl acetate Nutrition 0.000 description 77
- 238000004458 analytical method Methods 0.000 description 75
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 71
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 68
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 65
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 62
- 238000004587 chromatography analysis Methods 0.000 description 62
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 54
- 239000006260 foam Substances 0.000 description 54
- 239000002904 solvent Substances 0.000 description 51
- 239000010410 layer Substances 0.000 description 50
- 239000000376 reactant Substances 0.000 description 50
- 239000000203 mixture Substances 0.000 description 49
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 46
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 42
- 238000003818 flash chromatography Methods 0.000 description 42
- 229910052938 sodium sulfate Inorganic materials 0.000 description 42
- 235000011152 sodium sulphate Nutrition 0.000 description 42
- 239000002253 acid Substances 0.000 description 41
- 229940086542 triethylamine Drugs 0.000 description 39
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 34
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 34
- 239000012267 brine Substances 0.000 description 32
- 229960000583 acetic acid Drugs 0.000 description 31
- 239000012044 organic layer Substances 0.000 description 29
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 28
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 27
- 239000013058 crude material Substances 0.000 description 26
- 238000004896 high resolution mass spectrometry Methods 0.000 description 26
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 26
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 24
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 23
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 21
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 21
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 21
- 150000001408 amides Chemical class 0.000 description 20
- 239000000010 aprotic solvent Substances 0.000 description 20
- 239000012043 crude product Substances 0.000 description 20
- 238000010265 fast atom bombardment Methods 0.000 description 20
- 125000004432 carbon atom Chemical group C* 0.000 description 19
- 239000000463 material Substances 0.000 description 19
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 19
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 18
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 18
- 229960004132 diethyl ether Drugs 0.000 description 18
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical class CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 17
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 16
- 239000002585 base Substances 0.000 description 16
- 238000004809 thin layer chromatography Methods 0.000 description 16
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 15
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 15
- 239000003054 catalyst Substances 0.000 description 15
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 14
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 14
- 238000001953 recrystallisation Methods 0.000 description 14
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 13
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 13
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 13
- 239000000908 ammonium hydroxide Substances 0.000 description 13
- 239000003921 oil Substances 0.000 description 13
- 235000019198 oils Nutrition 0.000 description 13
- 229910000027 potassium carbonate Inorganic materials 0.000 description 13
- 235000011181 potassium carbonates Nutrition 0.000 description 13
- 239000002516 radical scavenger Substances 0.000 description 13
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 12
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 12
- 239000003153 chemical reaction reagent Substances 0.000 description 12
- 239000003795 chemical substances by application Substances 0.000 description 12
- 239000012298 atmosphere Substances 0.000 description 11
- 230000015572 biosynthetic process Effects 0.000 description 11
- 239000000706 filtrate Substances 0.000 description 11
- 238000000746 purification Methods 0.000 description 11
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 10
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 10
- 238000001914 filtration Methods 0.000 description 10
- 239000007789 gas Substances 0.000 description 10
- 239000002244 precipitate Substances 0.000 description 10
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 9
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 9
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 9
- 239000003638 chemical reducing agent Substances 0.000 description 9
- 239000000460 chlorine Substances 0.000 description 9
- 230000000694 effects Effects 0.000 description 9
- 150000001412 amines Chemical class 0.000 description 8
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 8
- 239000002002 slurry Substances 0.000 description 8
- 239000012279 sodium borohydride Substances 0.000 description 8
- 229910000033 sodium borohydride Inorganic materials 0.000 description 8
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 8
- 125000001424 substituent group Chemical group 0.000 description 8
- YXHKONLOYHBTNS-UHFFFAOYSA-N Diazomethane Chemical compound C=[N+]=[N-] YXHKONLOYHBTNS-UHFFFAOYSA-N 0.000 description 7
- 238000004440 column chromatography Methods 0.000 description 7
- 238000001816 cooling Methods 0.000 description 7
- 238000005859 coupling reaction Methods 0.000 description 7
- 239000000284 extract Substances 0.000 description 7
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 7
- 230000001737 promoting effect Effects 0.000 description 7
- 230000001177 retroviral effect Effects 0.000 description 7
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 7
- 235000017557 sodium bicarbonate Nutrition 0.000 description 7
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 description 6
- IJFXRHURBJZNAO-UHFFFAOYSA-N 3-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=CC(O)=C1 IJFXRHURBJZNAO-UHFFFAOYSA-N 0.000 description 6
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 6
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 6
- 125000002837 carbocyclic group Chemical group 0.000 description 6
- 210000004027 cell Anatomy 0.000 description 6
- GKIRPKYJQBWNGO-OCEACIFDSA-N clomifene Chemical compound C1=CC(OCCN(CC)CC)=CC=C1C(\C=1C=CC=CC=1)=C(\Cl)C1=CC=CC=C1 GKIRPKYJQBWNGO-OCEACIFDSA-N 0.000 description 6
- 230000008878 coupling Effects 0.000 description 6
- 238000010168 coupling process Methods 0.000 description 6
- 238000010992 reflux Methods 0.000 description 6
- 239000000741 silica gel Substances 0.000 description 6
- 229910002027 silica gel Inorganic materials 0.000 description 6
- 150000003573 thiols Chemical group 0.000 description 6
- BYHMLZGICSEKIY-UHFFFAOYSA-N 3-amino-2-methylbenzoic acid Chemical compound CC1=C(N)C=CC=C1C(O)=O BYHMLZGICSEKIY-UHFFFAOYSA-N 0.000 description 5
- 208000031886 HIV Infections Diseases 0.000 description 5
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical group C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 5
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 5
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 5
- 150000007513 acids Chemical class 0.000 description 5
- 230000001476 alcoholic effect Effects 0.000 description 5
- 150000001298 alcohols Chemical class 0.000 description 5
- 150000008064 anhydrides Chemical class 0.000 description 5
- 150000001450 anions Chemical class 0.000 description 5
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 5
- 238000002425 crystallisation Methods 0.000 description 5
- 230000008025 crystallization Effects 0.000 description 5
- 238000010511 deprotection reaction Methods 0.000 description 5
- 150000002170 ethers Chemical class 0.000 description 5
- 229940052303 ethers for general anesthesia Drugs 0.000 description 5
- 125000001786 isothiazolyl group Chemical group 0.000 description 5
- 229940098779 methanesulfonic acid Drugs 0.000 description 5
- 150000007524 organic acids Chemical class 0.000 description 5
- 239000012074 organic phase Substances 0.000 description 5
- 150000002924 oxiranes Chemical class 0.000 description 5
- 239000000377 silicon dioxide Substances 0.000 description 5
- 239000011734 sodium Substances 0.000 description 5
- 229910052708 sodium Inorganic materials 0.000 description 5
- 239000012312 sodium hydride Substances 0.000 description 5
- 229910000104 sodium hydride Inorganic materials 0.000 description 5
- 125000000547 substituted alkyl group Chemical group 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- YBRBMKDOPFTVDT-UHFFFAOYSA-N tert-butylamine Chemical compound CC(C)(C)N YBRBMKDOPFTVDT-UHFFFAOYSA-N 0.000 description 5
- PJUPKRYGDFTMTM-UHFFFAOYSA-N 1-hydroxybenzotriazole;hydrate Chemical compound O.C1=CC=C2N(O)N=NC2=C1 PJUPKRYGDFTMTM-UHFFFAOYSA-N 0.000 description 4
- HBMCQTHGYMTCOF-UHFFFAOYSA-N 4-hydroxyphenyl acetate Chemical compound CC(=O)OC1=CC=C(O)C=C1 HBMCQTHGYMTCOF-UHFFFAOYSA-N 0.000 description 4
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 4
- 238000005727 Friedel-Crafts reaction Methods 0.000 description 4
- 208000037357 HIV infectious disease Diseases 0.000 description 4
- 238000005481 NMR spectroscopy Methods 0.000 description 4
- 235000019502 Orange oil Nutrition 0.000 description 4
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 4
- 241000700605 Viruses Species 0.000 description 4
- 230000004913 activation Effects 0.000 description 4
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 4
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 4
- 239000007810 chemical reaction solvent Substances 0.000 description 4
- 239000010779 crude oil Substances 0.000 description 4
- 238000010908 decantation Methods 0.000 description 4
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 4
- USIUVYZYUHIAEV-UHFFFAOYSA-N diphenyl ether Chemical compound C=1C=CC=CC=1OC1=CC=CC=C1 USIUVYZYUHIAEV-UHFFFAOYSA-N 0.000 description 4
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 4
- 238000001704 evaporation Methods 0.000 description 4
- 230000008020 evaporation Effects 0.000 description 4
- 238000000605 extraction Methods 0.000 description 4
- 125000005842 heteroatom Chemical group 0.000 description 4
- 150000002391 heterocyclic compounds Chemical class 0.000 description 4
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 description 4
- 229910052500 inorganic mineral Inorganic materials 0.000 description 4
- 150000002596 lactones Chemical group 0.000 description 4
- 229910052744 lithium Inorganic materials 0.000 description 4
- WGOPGODQLGJZGL-UHFFFAOYSA-N lithium;butane Chemical compound [Li+].CC[CH-]C WGOPGODQLGJZGL-UHFFFAOYSA-N 0.000 description 4
- 235000010755 mineral Nutrition 0.000 description 4
- 239000011707 mineral Substances 0.000 description 4
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 4
- 239000010502 orange oil Substances 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 238000012545 processing Methods 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N pyridine Substances C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 4
- 239000011369 resultant mixture Substances 0.000 description 4
- JPJALAQPGMAKDF-UHFFFAOYSA-N selenium dioxide Chemical compound O=[Se]=O JPJALAQPGMAKDF-UHFFFAOYSA-N 0.000 description 4
- WBHQBSYUUJJSRZ-UHFFFAOYSA-M sodium bisulfate Chemical compound [Na+].OS([O-])(=O)=O WBHQBSYUUJJSRZ-UHFFFAOYSA-M 0.000 description 4
- 229910000342 sodium bisulfate Inorganic materials 0.000 description 4
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical group [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 4
- 235000010288 sodium nitrite Nutrition 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- CSRZQMIRAZTJOY-UHFFFAOYSA-N trimethylsilyl iodide Chemical compound C[Si](C)(C)I CSRZQMIRAZTJOY-UHFFFAOYSA-N 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 description 3
- XBNGYFFABRKICK-UHFFFAOYSA-N 2,3,4,5,6-pentafluorophenol Chemical compound OC1=C(F)C(F)=C(F)C(F)=C1F XBNGYFFABRKICK-UHFFFAOYSA-N 0.000 description 3
- RIERSGULWXEJKL-UHFFFAOYSA-N 3-hydroxy-2-methylbenzoic acid Chemical compound CC1=C(O)C=CC=C1C(O)=O RIERSGULWXEJKL-UHFFFAOYSA-N 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- 108091005804 Peptidases Proteins 0.000 description 3
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 3
- 239000004365 Protease Substances 0.000 description 3
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Natural products C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 3
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 3
- 150000001299 aldehydes Chemical class 0.000 description 3
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 3
- 239000001273 butane Substances 0.000 description 3
- 150000001717 carbocyclic compounds Chemical class 0.000 description 3
- 125000004230 chromenyl group Chemical group O1C(C=CC2=CC=CC=C12)* 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000012230 colorless oil Substances 0.000 description 3
- 239000006184 cosolvent Substances 0.000 description 3
- 239000012954 diazonium Substances 0.000 description 3
- 150000001989 diazonium salts Chemical class 0.000 description 3
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- 239000002024 ethyl acetate extract Substances 0.000 description 3
- 238000004992 fast atom bombardment mass spectroscopy Methods 0.000 description 3
- 125000003838 furazanyl group Chemical group 0.000 description 3
- 125000002541 furyl group Chemical group 0.000 description 3
- 229940093915 gynecological organic acid Drugs 0.000 description 3
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 3
- 125000002883 imidazolyl group Chemical group 0.000 description 3
- 210000000987 immune system Anatomy 0.000 description 3
- 208000015181 infectious disease Diseases 0.000 description 3
- 238000002329 infrared spectrum Methods 0.000 description 3
- 150000007529 inorganic bases Chemical class 0.000 description 3
- RBTARNINKXHZNM-UHFFFAOYSA-K iron trichloride Chemical compound Cl[Fe](Cl)Cl RBTARNINKXHZNM-UHFFFAOYSA-K 0.000 description 3
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 3
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 3
- 125000000842 isoxazolyl group Chemical group 0.000 description 3
- 150000002576 ketones Chemical class 0.000 description 3
- 238000001819 mass spectrum Methods 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 3
- 125000004433 nitrogen atom Chemical group N* 0.000 description 3
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 3
- ZWLPBLYKEWSWPD-UHFFFAOYSA-N o-toluic acid Chemical compound CC1=CC=CC=C1C(O)=O ZWLPBLYKEWSWPD-UHFFFAOYSA-N 0.000 description 3
- 235000005985 organic acids Nutrition 0.000 description 3
- MUMZUERVLWJKNR-UHFFFAOYSA-N oxoplatinum Chemical compound [Pt]=O MUMZUERVLWJKNR-UHFFFAOYSA-N 0.000 description 3
- IZUPBVBPLAPZRR-UHFFFAOYSA-N pentachloro-phenol Natural products OC1=C(Cl)C(Cl)=C(Cl)C(Cl)=C1Cl IZUPBVBPLAPZRR-UHFFFAOYSA-N 0.000 description 3
- 229910003446 platinum oxide Inorganic materials 0.000 description 3
- 239000011591 potassium Substances 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 108090000765 processed proteins & peptides Proteins 0.000 description 3
- 102000004196 processed proteins & peptides Human genes 0.000 description 3
- 125000006239 protecting group Chemical group 0.000 description 3
- 125000003373 pyrazinyl group Chemical group 0.000 description 3
- 125000003226 pyrazolyl group Chemical group 0.000 description 3
- 125000000714 pyrimidinyl group Chemical group 0.000 description 3
- 125000000168 pyrrolyl group Chemical group 0.000 description 3
- RAYMXZBXQCGRGX-UHFFFAOYSA-N quinoline-5-carboxylic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=CC2=N1 RAYMXZBXQCGRGX-UHFFFAOYSA-N 0.000 description 3
- 238000001228 spectrum Methods 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 3
- 125000000335 thiazolyl group Chemical group 0.000 description 3
- RMVRSNDYEFQCLF-UHFFFAOYSA-N thiophenol Chemical compound SC1=CC=CC=C1 RMVRSNDYEFQCLF-UHFFFAOYSA-N 0.000 description 3
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 3
- 125000004306 triazinyl group Chemical group 0.000 description 3
- 229910052720 vanadium Inorganic materials 0.000 description 3
- 125000001834 xanthenyl group Chemical group C1=CC=CC=2OC3=CC=CC=C3C(C12)* 0.000 description 3
- LNETULKMXZVUST-UHFFFAOYSA-N 1-naphthoic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=CC2=C1 LNETULKMXZVUST-UHFFFAOYSA-N 0.000 description 2
- DJRFJAVPROZZFL-UHFFFAOYSA-N 1975-52-6 Chemical compound CC1=CC=C([N+]([O-])=O)C=C1C(O)=O DJRFJAVPROZZFL-UHFFFAOYSA-N 0.000 description 2
- QAOJBHRZQQDFHA-UHFFFAOYSA-N 2,3-dichlorobenzoic acid Chemical compound OC(=O)C1=CC=CC(Cl)=C1Cl QAOJBHRZQQDFHA-UHFFFAOYSA-N 0.000 description 2
- RFCQDOVPMUSZMN-UHFFFAOYSA-N 2-Naphthalenethiol Chemical compound C1=CC=CC2=CC(S)=CC=C21 RFCQDOVPMUSZMN-UHFFFAOYSA-N 0.000 description 2
- HDECRAPHCDXMIJ-UHFFFAOYSA-N 2-methylbenzenesulfonyl chloride Chemical compound CC1=CC=CC=C1S(Cl)(=O)=O HDECRAPHCDXMIJ-UHFFFAOYSA-N 0.000 description 2
- YOETUEMZNOLGDB-UHFFFAOYSA-N 2-methylpropyl carbonochloridate Chemical compound CC(C)COC(Cl)=O YOETUEMZNOLGDB-UHFFFAOYSA-N 0.000 description 2
- NURQLCJSMXZBPC-UHFFFAOYSA-N 3,4-dimethylpyridine Chemical compound CC1=CC=NC=C1C NURQLCJSMXZBPC-UHFFFAOYSA-N 0.000 description 2
- RKBQPUFPJRCJKE-UHFFFAOYSA-N 3,5-diamino-2-methylbenzoic acid Chemical compound CC1=C(N)C=C(N)C=C1C(O)=O RKBQPUFPJRCJKE-UHFFFAOYSA-N 0.000 description 2
- AJHPGXZOIAYYDW-UHFFFAOYSA-N 3-(2-cyanophenyl)-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound CC(C)(C)OC(=O)NC(C(O)=O)CC1=CC=CC=C1C#N AJHPGXZOIAYYDW-UHFFFAOYSA-N 0.000 description 2
- KLDLRDSRCMJKGM-UHFFFAOYSA-N 3-[chloro-(2-oxo-1,3-oxazolidin-3-yl)phosphoryl]-1,3-oxazolidin-2-one Chemical compound C1COC(=O)N1P(=O)(Cl)N1CCOC1=O KLDLRDSRCMJKGM-UHFFFAOYSA-N 0.000 description 2
- IQMIVFNEHPKEAI-UHFFFAOYSA-N 3-amino-2-chlorobenzoic acid Chemical compound NC1=CC=CC(C(O)=O)=C1Cl IQMIVFNEHPKEAI-UHFFFAOYSA-N 0.000 description 2
- JMYLVZGGEKBZAB-UHFFFAOYSA-N 3-methoxy-2-methyl-n-phenylbenzamide Chemical compound COC1=CC=CC(C(=O)NC=2C=CC=CC=2)=C1C JMYLVZGGEKBZAB-UHFFFAOYSA-N 0.000 description 2
- OAAVWHUTJIJKOU-UHFFFAOYSA-N 3-methoxy-n-phenylbenzamide Chemical compound COC1=CC=CC(C(=O)NC=2C=CC=CC=2)=C1 OAAVWHUTJIJKOU-UHFFFAOYSA-N 0.000 description 2
- RUQIUASLAXJZIE-UHFFFAOYSA-N 3-methoxybenzoyl chloride Chemical compound COC1=CC=CC(C(Cl)=O)=C1 RUQIUASLAXJZIE-UHFFFAOYSA-N 0.000 description 2
- XVMSFILGAMDHEY-UHFFFAOYSA-N 6-(4-aminophenyl)sulfonylpyridin-3-amine Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=N1 XVMSFILGAMDHEY-UHFFFAOYSA-N 0.000 description 2
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical class OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- BOWHMVWINBRMRI-UHFFFAOYSA-N Clausine P Chemical compound N1C2=C(OC)C=CC=C2C2=C1C=C(OC)C(C)=C2 BOWHMVWINBRMRI-UHFFFAOYSA-N 0.000 description 2
- GKQLYSROISKDLL-UHFFFAOYSA-N EEDQ Chemical compound C1=CC=C2N(C(=O)OCC)C(OCC)C=CC2=C1 GKQLYSROISKDLL-UHFFFAOYSA-N 0.000 description 2
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- 229910021578 Iron(III) chloride Inorganic materials 0.000 description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 2
- 239000012448 Lithium borohydride Substances 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- IOVCWXUNBOPUCH-UHFFFAOYSA-N Nitrous acid Chemical compound ON=O IOVCWXUNBOPUCH-UHFFFAOYSA-N 0.000 description 2
- 101150101537 Olah gene Proteins 0.000 description 2
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 241000288906 Primates Species 0.000 description 2
- NBBJYMSMWIIQGU-UHFFFAOYSA-N Propionic aldehyde Chemical compound CCC=O NBBJYMSMWIIQGU-UHFFFAOYSA-N 0.000 description 2
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- 101710172711 Structural protein Proteins 0.000 description 2
- JEDZLBFUGJTJGQ-UHFFFAOYSA-N [Na].COCCO[AlH]OCCOC Chemical compound [Na].COCCO[AlH]OCCOC JEDZLBFUGJTJGQ-UHFFFAOYSA-N 0.000 description 2
- CSCPPACGZOOCGX-WFGJKAKNSA-N acetone d6 Chemical compound [2H]C([2H])([2H])C(=O)C([2H])([2H])[2H] CSCPPACGZOOCGX-WFGJKAKNSA-N 0.000 description 2
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- 150000001262 acyl bromides Chemical class 0.000 description 2
- 150000001263 acyl chlorides Chemical class 0.000 description 2
- 150000003973 alkyl amines Chemical class 0.000 description 2
- 229940024606 amino acid Drugs 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 2
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 2
- 125000003180 beta-lactone group Chemical group 0.000 description 2
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 2
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 2
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical group BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 2
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 2
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 2
- 239000003610 charcoal Substances 0.000 description 2
- 125000003636 chemical group Chemical group 0.000 description 2
- 150000008280 chlorinated hydrocarbons Chemical class 0.000 description 2
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 2
- 239000007822 coupling agent Substances 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 238000002451 electron ionisation mass spectrometry Methods 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 239000012362 glacial acetic acid Substances 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 2
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 2
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 2
- 125000001041 indolyl group Chemical group 0.000 description 2
- 239000012442 inert solvent Substances 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 125000002346 iodo group Chemical group I* 0.000 description 2
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- ZIPLFLRGHZAXSJ-UHFFFAOYSA-N isoquinoline-5-carboxylic acid Chemical compound N1=CC=C2C(C(=O)O)=CC=CC2=C1 ZIPLFLRGHZAXSJ-UHFFFAOYSA-N 0.000 description 2
- 125000005956 isoquinolyl group Chemical group 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical compound COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 2
- KRKPYFLIYNGWTE-UHFFFAOYSA-N n,o-dimethylhydroxylamine Chemical compound CNOC KRKPYFLIYNGWTE-UHFFFAOYSA-N 0.000 description 2
- DUWWHGPELOTTOE-UHFFFAOYSA-N n-(5-chloro-2,4-dimethoxyphenyl)-3-oxobutanamide Chemical compound COC1=CC(OC)=C(NC(=O)CC(C)=O)C=C1Cl DUWWHGPELOTTOE-UHFFFAOYSA-N 0.000 description 2
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 2
- BAULSHLTGVOYKM-UHFFFAOYSA-N n-butylbenzamide Chemical compound CCCCNC(=O)C1=CC=CC=C1 BAULSHLTGVOYKM-UHFFFAOYSA-N 0.000 description 2
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 2
- 125000000018 nitroso group Chemical group N(=O)* 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 2
- 239000007800 oxidant agent Substances 0.000 description 2
- 125000005327 perimidinyl group Chemical group N1C(=NC2=CC=CC3=CC=CC1=C23)* 0.000 description 2
- 125000004934 phenanthridinyl group Chemical group C1(=CC=CC2=NC=C3C=CC=CC3=C12)* 0.000 description 2
- 125000004625 phenanthrolinyl group Chemical group N1=C(C=CC2=CC=C3C=CC=NC3=C12)* 0.000 description 2
- 125000001791 phenazinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3N=C12)* 0.000 description 2
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 2
- 125000001644 phenoxazinyl group Chemical group C1(=CC=CC=2OC3=CC=CC=C3NC12)* 0.000 description 2
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 2
- HXEACLLIILLPRG-UHFFFAOYSA-N pipecolic acid Chemical compound OC(=O)C1CCCCN1 HXEACLLIILLPRG-UHFFFAOYSA-N 0.000 description 2
- 229920001184 polypeptide Polymers 0.000 description 2
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 description 2
- 229910000105 potassium hydride Inorganic materials 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 235000019260 propionic acid Nutrition 0.000 description 2
- 239000003586 protic polar solvent Substances 0.000 description 2
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 2
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 2
- 125000002098 pyridazinyl group Chemical group 0.000 description 2
- 238000010791 quenching Methods 0.000 description 2
- 230000000171 quenching effect Effects 0.000 description 2
- LOAUVZALPPNFOQ-UHFFFAOYSA-N quinaldic acid Chemical compound C1=CC=CC2=NC(C(=O)O)=CC=C21 LOAUVZALPPNFOQ-UHFFFAOYSA-N 0.000 description 2
- VQMSRUREDGBWKT-UHFFFAOYSA-N quinoline-4-carboxylic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=NC2=C1 VQMSRUREDGBWKT-UHFFFAOYSA-N 0.000 description 2
- 125000005493 quinolyl group Chemical group 0.000 description 2
- 238000010839 reverse transcription Methods 0.000 description 2
- 229910052703 rhodium Inorganic materials 0.000 description 2
- 239000010948 rhodium Substances 0.000 description 2
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 2
- 239000012047 saturated solution Substances 0.000 description 2
- 150000003335 secondary amines Chemical class 0.000 description 2
- 229960001153 serine Drugs 0.000 description 2
- 239000012419 sodium bis(2-methoxyethoxy)aluminum hydride Substances 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 125000000446 sulfanediyl group Chemical group *S* 0.000 description 2
- 208000011580 syndromic disease Diseases 0.000 description 2
- PUGUQINMNYINPK-UHFFFAOYSA-N tert-butyl 4-(2-chloroacetyl)piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCN(C(=O)CCl)CC1 PUGUQINMNYINPK-UHFFFAOYSA-N 0.000 description 2
- 150000003512 tertiary amines Chemical class 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- 125000004627 thianthrenyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3SC12)* 0.000 description 2
- 150000007970 thio esters Chemical class 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 125000005270 trialkylamine group Chemical group 0.000 description 2
- HFFLGKNGCAIQMO-UHFFFAOYSA-N trichloroacetaldehyde Chemical compound ClC(Cl)(Cl)C=O HFFLGKNGCAIQMO-UHFFFAOYSA-N 0.000 description 2
- FAQYAMRNWDIXMY-UHFFFAOYSA-N trichloroborane Chemical compound ClB(Cl)Cl FAQYAMRNWDIXMY-UHFFFAOYSA-N 0.000 description 2
- 238000002211 ultraviolet spectrum Methods 0.000 description 2
- 241001430294 unidentified retrovirus Species 0.000 description 2
- 230000003612 virological effect Effects 0.000 description 2
- 125000004933 β-carbolinyl group Chemical group C1(=NC=CC=2C3=CC=CC=C3NC12)* 0.000 description 2
- KOJVNDPZLOBKMK-UHFFFAOYSA-N (3-iodo-4-methylphenyl)methanol Chemical compound CC1=CC=C(CO)C=C1I KOJVNDPZLOBKMK-UHFFFAOYSA-N 0.000 description 1
- UOCLXMDMGBRAIB-UHFFFAOYSA-N 1,1,1-trichloroethane Chemical compound CC(Cl)(Cl)Cl UOCLXMDMGBRAIB-UHFFFAOYSA-N 0.000 description 1
- KEBVOFVJYCXIBZ-UHFFFAOYSA-N 1,2,3,4-tetrahydroquinoline-5-carboxylic acid Chemical compound N1CCCC2=C1C=CC=C2C(=O)O KEBVOFVJYCXIBZ-UHFFFAOYSA-N 0.000 description 1
- RBACIKXCRWGCBB-UHFFFAOYSA-N 1,2-Epoxybutane Chemical compound CCC1CO1 RBACIKXCRWGCBB-UHFFFAOYSA-N 0.000 description 1
- WMXFNCKPYCAIQW-UHFFFAOYSA-N 1,2-dimethoxy-3-methylbenzene Chemical compound COC1=CC=CC(C)=C1OC WMXFNCKPYCAIQW-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- BHKKSKOHRFHHIN-MRVPVSSYSA-N 1-[[2-[(1R)-1-aminoethyl]-4-chlorophenyl]methyl]-2-sulfanylidene-5H-pyrrolo[3,2-d]pyrimidin-4-one Chemical compound N[C@H](C)C1=C(CN2C(NC(C3=C2C=CN3)=O)=S)C=CC(=C1)Cl BHKKSKOHRFHHIN-MRVPVSSYSA-N 0.000 description 1
- IUSDBEVKVROXPY-UHFFFAOYSA-N 1-chloro-3,4-dimethoxy-2-methylbenzene Chemical compound COC1=CC=C(Cl)C(C)=C1OC IUSDBEVKVROXPY-UHFFFAOYSA-N 0.000 description 1
- ZQXCQTAELHSNAT-UHFFFAOYSA-N 1-chloro-3-nitro-5-(trifluoromethyl)benzene Chemical compound [O-][N+](=O)C1=CC(Cl)=CC(C(F)(F)F)=C1 ZQXCQTAELHSNAT-UHFFFAOYSA-N 0.000 description 1
- HNEGJTWNOOWEMH-UHFFFAOYSA-N 1-fluoropropane Chemical group [CH2]CCF HNEGJTWNOOWEMH-UHFFFAOYSA-N 0.000 description 1
- NFKAWBGFIMBUMB-UHFFFAOYSA-N 1-phenylpentan-2-one Chemical compound CCCC(=O)CC1=CC=CC=C1 NFKAWBGFIMBUMB-UHFFFAOYSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- YPQAFWHSMWWPLX-UHFFFAOYSA-N 1975-50-4 Chemical compound CC1=C(C(O)=O)C=CC=C1[N+]([O-])=O YPQAFWHSMWWPLX-UHFFFAOYSA-N 0.000 description 1
- ROGHUJUFCRFUSO-UHFFFAOYSA-N 1h-indole-4-carboxylic acid Chemical compound OC(=O)C1=CC=CC2=C1C=CN2 ROGHUJUFCRFUSO-UHFFFAOYSA-N 0.000 description 1
- 125000000453 2,2,2-trichloroethyl group Chemical group [H]C([H])(*)C(Cl)(Cl)Cl 0.000 description 1
- NSEBBFDHPSOJLT-UHFFFAOYSA-N 2,3,5-trimethoxybenzaldehyde Chemical compound COC1=CC(OC)=C(OC)C(C=O)=C1 NSEBBFDHPSOJLT-UHFFFAOYSA-N 0.000 description 1
- LBXGBNHUNHWYRM-UHFFFAOYSA-N 2,3-dimethoxybenzonitrile Chemical compound COC1=CC=CC(C#N)=C1OC LBXGBNHUNHWYRM-UHFFFAOYSA-N 0.000 description 1
- HCSBTDBGTNZOAB-UHFFFAOYSA-N 2,3-dinitrobenzoic acid Chemical compound OC(=O)C1=CC=CC([N+]([O-])=O)=C1[N+]([O-])=O HCSBTDBGTNZOAB-UHFFFAOYSA-N 0.000 description 1
- LHJFBVDNRPBOOW-UHFFFAOYSA-N 2,6-dichloro-3-hydroxybenzoic acid Chemical compound OC(=O)C1=C(Cl)C=CC(O)=C1Cl LHJFBVDNRPBOOW-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- OBSIQMZKFXFYLV-UHFFFAOYSA-N 2-amino-3-phenylpropanamide Chemical compound NC(=O)C(N)CC1=CC=CC=C1 OBSIQMZKFXFYLV-UHFFFAOYSA-N 0.000 description 1
- 125000005999 2-bromoethyl group Chemical group 0.000 description 1
- IURVFJPLQYDRHQ-UHFFFAOYSA-N 2-butyl-3-hydroxybenzoic acid Chemical compound CCCCC1=C(O)C=CC=C1C(O)=O IURVFJPLQYDRHQ-UHFFFAOYSA-N 0.000 description 1
- FMYQQHXWBCLWIE-UHFFFAOYSA-N 2-butyl-3-methoxy-n-phenylbenzamide Chemical compound CCCCC1=C(OC)C=CC=C1C(=O)NC1=CC=CC=C1 FMYQQHXWBCLWIE-UHFFFAOYSA-N 0.000 description 1
- ONYBTYIVIZTLHT-UHFFFAOYSA-N 2-chloro-3,5-dihydroxybenzoic acid Chemical compound OC(=O)C1=CC(O)=CC(O)=C1Cl ONYBTYIVIZTLHT-UHFFFAOYSA-N 0.000 description 1
- HMEJVKGZESKXFC-UHFFFAOYSA-N 2-chloro-3-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=CC(O)=C1Cl HMEJVKGZESKXFC-UHFFFAOYSA-N 0.000 description 1
- IBRSSZOHCGUTHI-UHFFFAOYSA-N 2-chloropyridine-3-carboxylic acid Chemical compound OC(=O)C1=CC=CN=C1Cl IBRSSZOHCGUTHI-UHFFFAOYSA-N 0.000 description 1
- RCGJBBOBRZDMTL-UHFFFAOYSA-N 2-ethyl-3-hydroxybenzoic acid Chemical compound CCC1=C(O)C=CC=C1C(O)=O RCGJBBOBRZDMTL-UHFFFAOYSA-N 0.000 description 1
- FZERVMGFXOOUEB-UHFFFAOYSA-N 2-ethyl-3-methoxy-n-phenylbenzamide Chemical compound CCC1=C(OC)C=CC=C1C(=O)NC1=CC=CC=C1 FZERVMGFXOOUEB-UHFFFAOYSA-N 0.000 description 1
- VKSRITXZTWTEEW-UHFFFAOYSA-N 2-fluoro-3-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=CC(O)=C1F VKSRITXZTWTEEW-UHFFFAOYSA-N 0.000 description 1
- UEKWKQSBJDJVCK-UHFFFAOYSA-N 2-fluoro-3-methoxy-n-phenylbenzamide Chemical compound COC1=CC=CC(C(=O)NC=2C=CC=CC=2)=C1F UEKWKQSBJDJVCK-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- PZUXUOZSSYKAMX-UHFFFAOYSA-N 2-iodo-3-methylbenzoic acid Chemical compound CC1=CC=CC(C(O)=O)=C1I PZUXUOZSSYKAMX-UHFFFAOYSA-N 0.000 description 1
- CDVNZMKTJIBBBV-UHFFFAOYSA-N 2-methyl-3,5-dinitrobenzoic acid Chemical compound CC1=C(C(O)=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O CDVNZMKTJIBBBV-UHFFFAOYSA-N 0.000 description 1
- BNYCIDYVYCUCBU-UHFFFAOYSA-N 2-methyl-3-(methylamino)benzoic acid Chemical compound CNC1=CC=CC(C(O)=O)=C1C BNYCIDYVYCUCBU-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M 2-methylbenzenesulfonate Chemical compound CC1=CC=CC=C1S([O-])(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- ZJSQZQMVXKZAGW-UHFFFAOYSA-N 2H-benzotriazol-4-ol hydrate Chemical group O.OC1=CC=CC2=C1N=NN2 ZJSQZQMVXKZAGW-UHFFFAOYSA-N 0.000 description 1
- ZDTVOKFHNGKTNZ-UHFFFAOYSA-N 3,4-dihydroxy-2-methylbenzoic acid Chemical compound CC1=C(O)C(O)=CC=C1C(O)=O ZDTVOKFHNGKTNZ-UHFFFAOYSA-N 0.000 description 1
- OUEJXYZKZCQELU-UHFFFAOYSA-N 3,4-dimethoxy-2-methylbenzoic acid Chemical compound COC1=CC=C(C(O)=O)C(C)=C1OC OUEJXYZKZCQELU-UHFFFAOYSA-N 0.000 description 1
- 125000002774 3,4-dimethoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C(OC([H])([H])[H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 1
- FIUFLISGGHNPSM-UHFFFAOYSA-N 3-(4-methoxyphenyl)propanoic acid Chemical compound COC1=CC=C(CCC(O)=O)C=C1 FIUFLISGGHNPSM-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- ABFYEILPZWAIBN-UHFFFAOYSA-N 3-(iminomethylideneamino)-n,n-dimethylpropan-1-amine;hydrochloride Chemical compound Cl.CN(C)CCCN=C=N ABFYEILPZWAIBN-UHFFFAOYSA-N 0.000 description 1
- ONMOULMPIIOVTQ-UHFFFAOYSA-M 3-Nitrobenzene sulphonate Chemical compound [O-][N+](=O)C1=CC=CC(S([O-])(=O)=O)=C1 ONMOULMPIIOVTQ-UHFFFAOYSA-M 0.000 description 1
- PAGROCHVTXMVTP-UHFFFAOYSA-N 3-amino-2-bromobenzoic acid Chemical compound NC1=CC=CC(C(O)=O)=C1Br PAGROCHVTXMVTP-UHFFFAOYSA-N 0.000 description 1
- BYHMLZGICSEKIY-UHFFFAOYSA-M 3-amino-2-methylbenzoate Chemical compound CC1=C(N)C=CC=C1C([O-])=O BYHMLZGICSEKIY-UHFFFAOYSA-M 0.000 description 1
- XFDUHJPVQKIXHO-UHFFFAOYSA-N 3-aminobenzoic acid Chemical compound NC1=CC=CC(C(O)=O)=C1 XFDUHJPVQKIXHO-UHFFFAOYSA-N 0.000 description 1
- XMKZAIHFVHJGPV-UHFFFAOYSA-N 3-fluoro-2-methylbenzoic acid Chemical compound CC1=C(F)C=CC=C1C(O)=O XMKZAIHFVHJGPV-UHFFFAOYSA-N 0.000 description 1
- OYAQDECKBQDDBJ-UHFFFAOYSA-N 3-hydroxy-2,6-dimethylbenzoic acid Chemical compound CC1=CC=C(O)C(C)=C1C(O)=O OYAQDECKBQDDBJ-UHFFFAOYSA-N 0.000 description 1
- JLNDMDZMWLVUGD-UHFFFAOYSA-N 3-hydroxy-2-propan-2-ylbenzoic acid Chemical compound CC(C)C1=C(O)C=CC=C1C(O)=O JLNDMDZMWLVUGD-UHFFFAOYSA-N 0.000 description 1
- OSMPVDKPQINFEZ-UHFFFAOYSA-N 3-hydroxy-2-propylbenzoic acid Chemical compound CCCC1=C(O)C=CC=C1C(O)=O OSMPVDKPQINFEZ-UHFFFAOYSA-N 0.000 description 1
- QOXOZONBQWIKDA-UHFFFAOYSA-N 3-hydroxypropyl Chemical group [CH2]CCO QOXOZONBQWIKDA-UHFFFAOYSA-N 0.000 description 1
- JPCISVSOTKMFPG-UHFFFAOYSA-N 3-methoxy-2-methylbenzoic acid Chemical compound COC1=CC=CC(C(O)=O)=C1C JPCISVSOTKMFPG-UHFFFAOYSA-N 0.000 description 1
- XRWDUUFEQGNHCY-UHFFFAOYSA-N 3-methoxy-2-propan-2-ylbenzonitrile Chemical compound COC1=CC=CC(C#N)=C1C(C)C XRWDUUFEQGNHCY-UHFFFAOYSA-N 0.000 description 1
- BPIHQPBYRGOHCJ-UHFFFAOYSA-N 3-methoxy-n-phenyl-2-propylbenzamide Chemical compound CCCC1=C(OC)C=CC=C1C(=O)NC1=CC=CC=C1 BPIHQPBYRGOHCJ-UHFFFAOYSA-N 0.000 description 1
- QMYGFTJCQFEDST-UHFFFAOYSA-N 3-methoxybutyl acetate Chemical group COC(C)CCOC(C)=O QMYGFTJCQFEDST-UHFFFAOYSA-N 0.000 description 1
- OSMAGAVKVRGYGR-UHFFFAOYSA-N 3-methylpyridine-4-carboxylic acid Chemical compound CC1=CN=CC=C1C(O)=O OSMAGAVKVRGYGR-UHFFFAOYSA-N 0.000 description 1
- SILCYILEJNVSKZ-UHFFFAOYSA-N 3-phenyl-2-(phenylmethoxycarbonylamino)propanethioic s-acid Chemical compound C=1C=CC=CC=1COC(=O)NC(C(=O)S)CC1=CC=CC=C1 SILCYILEJNVSKZ-UHFFFAOYSA-N 0.000 description 1
- 125000002103 4,4'-dimethoxytriphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)(C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H])C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H] 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- MBDUKNCPOPMRJQ-UHFFFAOYSA-N 4-amino-2-chlorobenzoic acid Chemical compound NC1=CC=C(C(O)=O)C(Cl)=C1 MBDUKNCPOPMRJQ-UHFFFAOYSA-N 0.000 description 1
- PXACTUVBBMDKRW-UHFFFAOYSA-N 4-bromobenzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=C(Br)C=C1 PXACTUVBBMDKRW-UHFFFAOYSA-N 0.000 description 1
- SXIFAEWFOJETOA-UHFFFAOYSA-N 4-hydroxy-butyl Chemical group [CH2]CCCO SXIFAEWFOJETOA-UHFFFAOYSA-N 0.000 description 1
- OBKXEAXTFZPCHS-UHFFFAOYSA-N 4-phenylbutyric acid Chemical compound OC(=O)CCCC1=CC=CC=C1 OBKXEAXTFZPCHS-UHFFFAOYSA-N 0.000 description 1
- VATJFXXKGNSLIW-UHFFFAOYSA-N 5-(hydroxymethyl)-2-methylbenzoic acid Chemical compound CC1=CC=C(CO)C=C1C(O)=O VATJFXXKGNSLIW-UHFFFAOYSA-N 0.000 description 1
- FSXVZWAWYKMFMX-UHFFFAOYSA-N 5-amino-2-methylbenzoic acid Chemical compound CC1=CC=C(N)C=C1C(O)=O FSXVZWAWYKMFMX-UHFFFAOYSA-N 0.000 description 1
- DTVYNUOOZIKEEX-UHFFFAOYSA-N 5-aminoisoquinoline Chemical compound N1=CC=C2C(N)=CC=CC2=C1 DTVYNUOOZIKEEX-UHFFFAOYSA-N 0.000 description 1
- ZIOYQUNKXJQXQY-UHFFFAOYSA-N 5-hydroxy-2-methylbenzoic acid Chemical compound CC1=CC=C(O)C=C1C(O)=O ZIOYQUNKXJQXQY-UHFFFAOYSA-N 0.000 description 1
- MEXUTNIFSHFQRG-UHFFFAOYSA-N 6,7,12,13-tetrahydro-5h-indolo[2,3-a]pyrrolo[3,4-c]carbazol-5-one Chemical compound C12=C3C=CC=C[C]3NC2=C2NC3=CC=C[CH]C3=C2C2=C1C(=O)NC2 MEXUTNIFSHFQRG-UHFFFAOYSA-N 0.000 description 1
- ILDXWVHMDHODDX-UHFFFAOYSA-N 6-methyl-1,2,3,4-tetrahydroquinoline-5-carboxylic acid Chemical compound N1CCCC2=C(C(O)=O)C(C)=CC=C21 ILDXWVHMDHODDX-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- ONMOULMPIIOVTQ-UHFFFAOYSA-N 98-47-5 Chemical compound OS(=O)(=O)C1=CC=CC([N+]([O-])=O)=C1 ONMOULMPIIOVTQ-UHFFFAOYSA-N 0.000 description 1
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- ZNSMNVMLTJELDZ-UHFFFAOYSA-N Bis(2-chloroethyl)ether Chemical compound ClCCOCCCl ZNSMNVMLTJELDZ-UHFFFAOYSA-N 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- QRLQEUAUTJAPFJ-UHFFFAOYSA-N C1=CC=CC2=C(NC(C)(C)C)C(C)=CC=C21 Chemical compound C1=CC=CC2=C(NC(C)(C)C)C(C)=CC=C21 QRLQEUAUTJAPFJ-UHFFFAOYSA-N 0.000 description 1
- 210000004366 CD4-positive T-lymphocyte Anatomy 0.000 description 1
- CKDWPUIZGOQOOM-UHFFFAOYSA-N Carbamyl chloride Chemical compound NC(Cl)=O CKDWPUIZGOQOOM-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- BYMMIQCVDHHYGG-UHFFFAOYSA-N Cl.OP(O)(O)=O Chemical compound Cl.OP(O)(O)=O BYMMIQCVDHHYGG-UHFFFAOYSA-N 0.000 description 1
- 229920000742 Cotton Polymers 0.000 description 1
- XFXPMWWXUTWYJX-UHFFFAOYSA-N Cyanide Chemical compound N#[C-] XFXPMWWXUTWYJX-UHFFFAOYSA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical group FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 241000713340 Human immunodeficiency virus 2 Species 0.000 description 1
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-L L-tartrate(2-) Chemical compound [O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O FEWJPZIEWOKRBE-JCYAYHJZSA-L 0.000 description 1
- 125000002977 L-tyrosinyl radical cation group Chemical group 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- VZUNGTLZRAYYDE-UHFFFAOYSA-N N-methyl-N'-nitro-N-nitrosoguanidine Chemical compound O=NN(C)C(=N)N[N+]([O-])=O VZUNGTLZRAYYDE-UHFFFAOYSA-N 0.000 description 1
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 1
- WWNNZCOKKKDOPX-UHFFFAOYSA-N N-methylnicotinate Chemical compound C[N+]1=CC=CC(C([O-])=O)=C1 WWNNZCOKKKDOPX-UHFFFAOYSA-N 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- JCXJVPUVTGWSNB-UHFFFAOYSA-N Nitrogen dioxide Chemical compound O=[N]=O JCXJVPUVTGWSNB-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical compound ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 206010035664 Pneumonia Diseases 0.000 description 1
- 108010076039 Polyproteins Proteins 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 241000220317 Rosa Species 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- 238000005614 Skraup synthesis reaction Methods 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical compound OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- ZZXDRXVIRVJQBT-UHFFFAOYSA-M Xylenesulfonate Chemical compound CC1=CC=CC(S([O-])(=O)=O)=C1C ZZXDRXVIRVJQBT-UHFFFAOYSA-M 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- XAKBSHICSHRJCL-UHFFFAOYSA-N [CH2]C(=O)C1=CC=CC=C1 Chemical group [CH2]C(=O)C1=CC=CC=C1 XAKBSHICSHRJCL-UHFFFAOYSA-N 0.000 description 1
- VYWQTJWGWLKBQA-UHFFFAOYSA-N [amino(hydroxy)methylidene]azanium;chloride Chemical compound Cl.NC(N)=O VYWQTJWGWLKBQA-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- IPBVNPXQWQGGJP-UHFFFAOYSA-N acetic acid phenyl ester Natural products CC(=O)OC1=CC=CC=C1 IPBVNPXQWQGGJP-UHFFFAOYSA-N 0.000 description 1
- ZKRBPKMEQZUFMP-UHFFFAOYSA-N acetonitrile;1,1,1-trichloroethane Chemical compound CC#N.CC(Cl)(Cl)Cl ZKRBPKMEQZUFMP-UHFFFAOYSA-N 0.000 description 1
- YTIVTFGABIZHHX-UHFFFAOYSA-L acetylenedicarboxylate(2-) Chemical compound [O-]C(=O)C#CC([O-])=O YTIVTFGABIZHHX-UHFFFAOYSA-L 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000001266 acyl halides Chemical class 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910001854 alkali hydroxide Inorganic materials 0.000 description 1
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 125000004656 alkyl sulfonylamino group Chemical group 0.000 description 1
- 230000002152 alkylating effect Effects 0.000 description 1
- 125000005263 alkylenediamine group Chemical group 0.000 description 1
- BHELZAPQIKSEDF-UHFFFAOYSA-N allyl bromide Chemical compound BrCC=C BHELZAPQIKSEDF-UHFFFAOYSA-N 0.000 description 1
- 125000000266 alpha-aminoacyl group Chemical group 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- XXMYKMWELWQWHW-UHFFFAOYSA-N amino 2-methylbenzoate Chemical compound CC1=CC=CC=C1C(=O)ON XXMYKMWELWQWHW-UHFFFAOYSA-N 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- 150000001448 anilines Chemical class 0.000 description 1
- 125000005428 anthryl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C3C(*)=C([H])C([H])=C([H])C3=C([H])C2=C1[H] 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000036436 anti-hiv Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 125000002785 azepinyl group Chemical group 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000004619 benzopyranyl group Chemical group O1C(C=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 239000012455 biphasic mixture Substances 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 229910000085 borane Inorganic materials 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- KMGBZBJJOKUPIA-UHFFFAOYSA-N butyl iodide Chemical compound CCCCI KMGBZBJJOKUPIA-UHFFFAOYSA-N 0.000 description 1
- 125000004744 butyloxycarbonyl group Chemical group 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 1
- 239000000920 calcium hydroxide Substances 0.000 description 1
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 150000001718 carbodiimides Chemical class 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 229950005499 carbon tetrachloride Drugs 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 230000021523 carboxylation Effects 0.000 description 1
- 238000006473 carboxylation reaction Methods 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- UOCJDOLVGGIYIQ-PBFPGSCMSA-N cefatrizine Chemical group S([C@@H]1[C@@H](C(N1C=1C(O)=O)=O)NC(=O)[C@H](N)C=2C=CC(O)=CC=2)CC=1CSC=1C=NNN=1 UOCJDOLVGGIYIQ-PBFPGSCMSA-N 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- KXZJHVJKXJLBKO-UHFFFAOYSA-N chembl1408157 Chemical compound N=1C2=CC=CC=C2C(C(=O)O)=CC=1C1=CC=C(O)C=C1 KXZJHVJKXJLBKO-UHFFFAOYSA-N 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- KVSASDOGYIBWTA-UHFFFAOYSA-N chloro benzoate Chemical compound ClOC(=O)C1=CC=CC=C1 KVSASDOGYIBWTA-UHFFFAOYSA-N 0.000 description 1
- FZFAMSAMCHXGEF-UHFFFAOYSA-N chloro formate Chemical compound ClOC=O FZFAMSAMCHXGEF-UHFFFAOYSA-N 0.000 description 1
- 239000002026 chloroform extract Substances 0.000 description 1
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 125000000490 cinnamyl group Chemical group C(C=CC1=CC=CC=C1)* 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- DOBRDRYODQBAMW-UHFFFAOYSA-N copper(i) cyanide Chemical compound [Cu+].N#[C-] DOBRDRYODQBAMW-UHFFFAOYSA-N 0.000 description 1
- 125000004093 cyano group Chemical group *C#N 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 230000002498 deadly effect Effects 0.000 description 1
- GHVNFZFCNZKVNT-UHFFFAOYSA-N decanoic acid Chemical compound CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 description 1
- 210000001787 dendrite Anatomy 0.000 description 1
- 230000005595 deprotonation Effects 0.000 description 1
- 238000010537 deprotonation reaction Methods 0.000 description 1
- 238000003795 desorption Methods 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 238000006193 diazotization reaction Methods 0.000 description 1
- ZFTFAPZRGNKQPU-UHFFFAOYSA-N dicarbonic acid Chemical compound OC(=O)OC(O)=O ZFTFAPZRGNKQPU-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- NKDDWNXOKDWJAK-UHFFFAOYSA-N dimethoxymethane Chemical compound COCOC NKDDWNXOKDWJAK-UHFFFAOYSA-N 0.000 description 1
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 1
- ZZVUWRFHKOJYTH-UHFFFAOYSA-N diphenhydramine Chemical group C=1C=CC=CC=1C(OCCN(C)C)C1=CC=CC=C1 ZZVUWRFHKOJYTH-UHFFFAOYSA-N 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 230000005520 electrodynamics Effects 0.000 description 1
- 238000007336 electrophilic substitution reaction Methods 0.000 description 1
- 238000000921 elemental analysis Methods 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- DQYBDCGIPTYXML-UHFFFAOYSA-N ethoxyethane;hydrate Chemical compound O.CCOCC DQYBDCGIPTYXML-UHFFFAOYSA-N 0.000 description 1
- FCZCIXQGZOUIDN-UHFFFAOYSA-N ethyl 2-diethoxyphosphinothioyloxyacetate Chemical compound CCOC(=O)COP(=S)(OCC)OCC FCZCIXQGZOUIDN-UHFFFAOYSA-N 0.000 description 1
- 125000006125 ethylsulfonyl group Chemical group 0.000 description 1
- 125000006351 ethylthiomethyl group Chemical group [H]C([H])([H])C([H])([H])SC([H])([H])* 0.000 description 1
- 239000011737 fluorine Chemical group 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical compound [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 230000002140 halogenating effect Effects 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- KKLGDUSGQMHBPB-UHFFFAOYSA-N hex-2-ynedioic acid Chemical compound OC(=O)CCC#CC(O)=O KKLGDUSGQMHBPB-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-M hexanoate Chemical compound CCCCCC([O-])=O FUZZWVXGSFPDMH-UHFFFAOYSA-M 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical compound [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- 229960000443 hydrochloric acid Drugs 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- USZLCYNVCCDPLQ-UHFFFAOYSA-N hydron;n-methoxymethanamine;chloride Chemical compound Cl.CNOC USZLCYNVCCDPLQ-UHFFFAOYSA-N 0.000 description 1
- BNTRVUUJBGBGLZ-UHFFFAOYSA-N hydron;pyridine-4-carbonyl chloride;chloride Chemical compound Cl.ClC(=O)C1=CC=NC=C1 BNTRVUUJBGBGLZ-UHFFFAOYSA-N 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 150000002460 imidazoles Chemical class 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 239000002955 immunomodulating agent Substances 0.000 description 1
- 229940121354 immunomodulator Drugs 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 229910052740 iodine Chemical group 0.000 description 1
- 239000011630 iodine Chemical group 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- FMKOJHQHASLBPH-UHFFFAOYSA-N isopropyl iodide Chemical compound CC(C)I FMKOJHQHASLBPH-UHFFFAOYSA-N 0.000 description 1
- 125000005928 isopropyloxycarbonyl group Chemical group [H]C([H])([H])C([H])(OC(*)=O)C([H])([H])[H] 0.000 description 1
- XVJDHUCBVHTPGE-UHFFFAOYSA-N isoquinoline-5-carbonitrile Chemical compound N1=CC=C2C(C#N)=CC=CC2=C1 XVJDHUCBVHTPGE-UHFFFAOYSA-N 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000004628 isothiazolidinyl group Chemical group S1N(CCC1)* 0.000 description 1
- 125000003965 isoxazolidinyl group Chemical group 0.000 description 1
- HXEACLLIILLPRG-RXMQYKEDSA-N l-pipecolic acid Natural products OC(=O)[C@H]1CCCCN1 HXEACLLIILLPRG-RXMQYKEDSA-N 0.000 description 1
- 231100001231 less toxic Toxicity 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- AFRJJFRNGGLMDW-UHFFFAOYSA-N lithium amide Chemical compound [Li+].[NH2-] AFRJJFRNGGLMDW-UHFFFAOYSA-N 0.000 description 1
- 210000002751 lymph Anatomy 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- OHZZTXYKLXZFSZ-UHFFFAOYSA-I manganese(3+) 5,10,15-tris(1-methylpyridin-1-ium-4-yl)-20-(1-methylpyridin-4-ylidene)porphyrin-22-ide pentachloride Chemical compound [Cl-].[Cl-].[Cl-].[Cl-].[Cl-].[Mn+3].C1=CN(C)C=CC1=C1C(C=C2)=NC2=C(C=2C=C[N+](C)=CC=2)C([N-]2)=CC=C2C(C=2C=C[N+](C)=CC=2)=C(C=C2)N=C2C(C=2C=C[N+](C)=CC=2)=C2N=C1C=C2 OHZZTXYKLXZFSZ-UHFFFAOYSA-I 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 125000005341 metaphosphate group Chemical group 0.000 description 1
- IZDROVVXIHRYMH-UHFFFAOYSA-N methanesulfonic anhydride Chemical compound CS(=O)(=O)OS(C)(=O)=O IZDROVVXIHRYMH-UHFFFAOYSA-N 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- GSYSFVSGPABNNL-UHFFFAOYSA-N methyl 2-dimethoxyphosphoryl-2-(phenylmethoxycarbonylamino)acetate Chemical group COC(=O)C(P(=O)(OC)OC)NC(=O)OCC1=CC=CC=C1 GSYSFVSGPABNNL-UHFFFAOYSA-N 0.000 description 1
- IZYBEMGNIUSSAX-UHFFFAOYSA-N methyl benzenecarboperoxoate Chemical compound COOC(=O)C1=CC=CC=C1 IZYBEMGNIUSSAX-UHFFFAOYSA-N 0.000 description 1
- 229940095102 methyl benzoate Drugs 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- GRVDJDISBSALJP-UHFFFAOYSA-N methyloxidanyl Chemical group [O]C GRVDJDISBSALJP-UHFFFAOYSA-N 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 125000004092 methylthiomethyl group Chemical group [H]C([H])([H])SC([H])([H])* 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- UHAAFJWANJYDIS-UHFFFAOYSA-N n,n'-diethylmethanediimine Chemical compound CCN=C=NCC UHAAFJWANJYDIS-UHFFFAOYSA-N 0.000 description 1
- WEVSWKCMKXOKBQ-UHFFFAOYSA-N n,n,2,4,6-pentamethylbenzamide Chemical compound CN(C)C(=O)C1=C(C)C=C(C)C=C1C WEVSWKCMKXOKBQ-UHFFFAOYSA-N 0.000 description 1
- OFCCYDUUBNUJIB-UHFFFAOYSA-N n,n-diethylcarbamoyl chloride Chemical compound CCN(CC)C(Cl)=O OFCCYDUUBNUJIB-UHFFFAOYSA-N 0.000 description 1
- RLKHFSNWQCZBDC-UHFFFAOYSA-N n-(benzenesulfonyl)-n-fluorobenzenesulfonamide Chemical compound C=1C=CC=CC=1S(=O)(=O)N(F)S(=O)(=O)C1=CC=CC=C1 RLKHFSNWQCZBDC-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- SHVJWMYCHXFRMF-UHFFFAOYSA-N n-tert-butyl-2-methylbenzamide Chemical compound CC1=CC=CC=C1C(=O)NC(C)(C)C SHVJWMYCHXFRMF-UHFFFAOYSA-N 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- 125000005029 naphthylthio group Chemical group C1(=CC=CC2=CC=CC=C12)S* 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000000627 niacin group Chemical group 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-M octanoate Chemical compound CCCCCCCC([O-])=O WWZKQHOCKIZLMA-UHFFFAOYSA-M 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 229940042443 other antivirals in atc Drugs 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- FJCFFCXMEXZEIM-UHFFFAOYSA-N oxiniacic acid Chemical compound OC(=O)C1=CC=C[N+]([O-])=C1 FJCFFCXMEXZEIM-UHFFFAOYSA-N 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 125000006503 p-nitrobenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1[N+]([O-])=O)C([H])([H])* 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 210000001428 peripheral nervous system Anatomy 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 125000005561 phenanthryl group Chemical group 0.000 description 1
- DYUMLJSJISTVPV-UHFFFAOYSA-N phenyl propanoate Chemical compound CCC(=O)OC1=CC=CC=C1 DYUMLJSJISTVPV-UHFFFAOYSA-N 0.000 description 1
- 229940049953 phenylacetate Drugs 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 229950009215 phenylbutanoic acid Drugs 0.000 description 1
- 125000003356 phenylsulfanyl group Chemical group [*]SC1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- UHZYTMXLRWXGPK-UHFFFAOYSA-N phosphorus pentachloride Chemical compound ClP(Cl)(Cl)(Cl)Cl UHZYTMXLRWXGPK-UHFFFAOYSA-N 0.000 description 1
- IPNPIHIZVLFAFP-UHFFFAOYSA-N phosphorus tribromide Chemical compound BrP(Br)Br IPNPIHIZVLFAFP-UHFFFAOYSA-N 0.000 description 1
- FAIAAWCVCHQXDN-UHFFFAOYSA-N phosphorus trichloride Chemical compound ClP(Cl)Cl FAIAAWCVCHQXDN-UHFFFAOYSA-N 0.000 description 1
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical group [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 description 1
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 150000004885 piperazines Chemical class 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 1
- 108700004029 pol Genes Proteins 0.000 description 1
- 101150088264 pol gene Proteins 0.000 description 1
- 230000001323 posttranslational effect Effects 0.000 description 1
- 239000011736 potassium bicarbonate Substances 0.000 description 1
- 235000015497 potassium bicarbonate Nutrition 0.000 description 1
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- 239000012286 potassium permanganate Substances 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 230000001566 pro-viral effect Effects 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- KCXFHTAICRTXLI-UHFFFAOYSA-N propane-1-sulfonic acid Chemical compound CCCS(O)(=O)=O KCXFHTAICRTXLI-UHFFFAOYSA-N 0.000 description 1
- QLNJFJADRCOGBJ-UHFFFAOYSA-N propionamide Chemical compound CCC(N)=O QLNJFJADRCOGBJ-UHFFFAOYSA-N 0.000 description 1
- 125000004742 propyloxycarbonyl group Chemical group 0.000 description 1
- UORVCLMRJXCDCP-UHFFFAOYSA-M propynoate Chemical compound [O-]C(=O)C#C UORVCLMRJXCDCP-UHFFFAOYSA-M 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- NIPZZXUFJPQHNH-UHFFFAOYSA-N pyrazine-2-carboxylic acid Chemical compound OC(=O)C1=CN=CC=N1 NIPZZXUFJPQHNH-UHFFFAOYSA-N 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 238000006268 reductive amination reaction Methods 0.000 description 1
- 238000009877 rendering Methods 0.000 description 1
- 230000003362 replicative effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 229910052702 rhenium Inorganic materials 0.000 description 1
- WUAPFZMCVAUBPE-UHFFFAOYSA-N rhenium atom Chemical compound [Re] WUAPFZMCVAUBPE-UHFFFAOYSA-N 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 229940116351 sebacate Drugs 0.000 description 1
- CXMXRPHRNRROMY-UHFFFAOYSA-L sebacate(2-) Chemical compound [O-]C(=O)CCCCCCCCC([O-])=O CXMXRPHRNRROMY-UHFFFAOYSA-L 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 159000000000 sodium salts Chemical group 0.000 description 1
- LJRGBERXYNQPJI-UHFFFAOYSA-M sodium;3-nitrobenzenesulfonate Chemical compound [Na+].[O-][N+](=O)C1=CC=CC(S([O-])(=O)=O)=C1 LJRGBERXYNQPJI-UHFFFAOYSA-M 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- TYFQFVWCELRYAO-UHFFFAOYSA-L suberate(2-) Chemical compound [O-]C(=O)CCCCCCC([O-])=O TYFQFVWCELRYAO-UHFFFAOYSA-L 0.000 description 1
- 125000003107 substituted aryl group Chemical group 0.000 description 1
- 125000005346 substituted cycloalkyl group Chemical group 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- PXQLVRUNWNTZOS-UHFFFAOYSA-N sulfanyl Chemical compound [SH] PXQLVRUNWNTZOS-UHFFFAOYSA-N 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 125000001273 sulfonato group Chemical group [O-]S(*)(=O)=O 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 229940032330 sulfuric acid Drugs 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- YZUAOVCUGSBIPP-UHFFFAOYSA-N tert-butyl N-[1-([1,2,4]triazolo[4,3-a]pyridin-3-yl)ethyl]carbamate Chemical compound C1=CC=CN2C(C(NC(=O)OC(C)(C)C)C)=NN=C21 YZUAOVCUGSBIPP-UHFFFAOYSA-N 0.000 description 1
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical group C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000003698 tetramethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000006090 thiamorpholinyl sulfone group Chemical group 0.000 description 1
- 125000006089 thiamorpholinyl sulfoxide group Chemical group 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- HFRXJVQOXRXOPP-UHFFFAOYSA-N thionyl bromide Chemical compound BrS(Br)=O HFRXJVQOXRXOPP-UHFFFAOYSA-N 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 229940029273 trichloroacetaldehyde Drugs 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- KBMBVTRWEAAZEY-UHFFFAOYSA-N trisulfane Chemical compound SSS KBMBVTRWEAAZEY-UHFFFAOYSA-N 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 229960005486 vaccine Drugs 0.000 description 1
- 210000002845 virion Anatomy 0.000 description 1
- 229940071104 xylenesulfonate Drugs 0.000 description 1
- HBOMLICNUCNMMY-XLPZGREQSA-N zidovudine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](N=[N+]=[N-])C1 HBOMLICNUCNMMY-XLPZGREQSA-N 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/28—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a non-condensed six-membered aromatic ring of the carbon skeleton
- C07C237/42—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a non-condensed six-membered aromatic ring of the carbon skeleton having nitrogen atoms of amino groups bound to the carbon skeleton of the acid part, further acylated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/50—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton
- C07C323/51—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton
- C07C323/60—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton with the carbon atom of at least one of the carboxyl groups bound to nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/16—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/08—Indoles; Hydrogenated indoles with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/60—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/36—Radicals substituted by singly-bound nitrogen atoms
- C07D213/40—Acylated substituent nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/48—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/48—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
- C07D215/50—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/02—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with only hydrogen atoms or radicals containing only carbon and hydrogen atoms, directly attached to carbon atoms of the nitrogen-containing ring; Alkylene-bis-isoquinolines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/02—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with only hydrogen atoms or radicals containing only carbon and hydrogen atoms, directly attached to carbon atoms of the nitrogen-containing ring; Alkylene-bis-isoquinolines
- C07D217/06—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with only hydrogen atoms or radicals containing only carbon and hydrogen atoms, directly attached to carbon atoms of the nitrogen-containing ring; Alkylene-bis-isoquinolines with the ring nitrogen atom acylated by carboxylic or carbonic acids, or with sulfur or nitrogen analogues thereof, e.g. carbamates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/22—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the nitrogen-containing ring
- C07D217/26—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/553—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having one nitrogen atom as the only ring hetero atom
- C07F9/576—Six-membered rings
- C07F9/62—Isoquinoline or hydrogenated isoquinoline ring systems
Definitions
- This invention relates to a novel series of chemical
- AIDS Acquired Immune Deficiency Syndrome
- HTLV-III human T-lymphotropic retrovirus III
- HIV is a member of the class of viruses known as
- retroviruses The retroviral genome is composed of RNA which is converted to DNA by reverse transcription. This retroviral DNA is then stably integrated into a host cell's chromosome and,
- HIV appears to have a particular affinity for the human T-4 lymphocyte cell which plays a vital role in the body's immune system. HIV infection of these white blood cells depletes this white cell population. Eventually, the immune system is rendered inoperative and ineffective against various opportunistic diseases such as, among others, pneumocystic carini pneumonia, Karposis sarcoma, and cancer of the lymph system.
- the drug azidothymidine has been found effective for inhibiting the reverse transcription of the retroviral genome of the HIV virus, thus giving a measure of control, though not a cure, for patients afflicted with AIDS.
- the search continues for drugs that can cure or at least provide an improved measure of control of the deadly HIV virus.
- Retroviral replication routinely features post-translational processing of polyproteins. This processing is accomplished by virally encoded HIV protease enzyme. This yields mature polypeptides that will subsequently aid in the formation and function of infectious virus. If this molecular processing is stifled, then the normal production of HIV is terminated.
- inhibitors of HIV protease may function as anti-HIV viral agents.
- HIV protease is one of the translated products from the HIV structural protein pol gene. This retroviral protease
- HIV protease inhibitors may have the advantages of being more readily available, longer lived in virus, and less toxic than currently available drugs, possibly due to their specificity for the retroviral protease.
- a novel class of chemical compounds that can inhibit and/or block the activity of the HIV protease, which halts the proliferation of HIV virus, pharmaceutical compositions containing these compounds, and the use of the compounds as inhibitors of the HIV protease.
- the present invention relates to compounds falling within formula (1) below, and pharmaceutically acceptable salts thereof, that inhibit the protease encoded by human immunodeficiency virus (HIV) type 1 (HIV-1) or type 2 (HIV-2). These compounds are useful in the treatment of infection by HIV and the treatment of the acquired immune deficiency syndrome (AIDS).
- HIV human immunodeficiency virus
- the compounds, their pharmaceutically acceptable salts, and the pharmaceutical compositions of the present invention can be used alone or in combination with other antivirals, immunomodulators, antibiotics or vaccines.
- Compounds of the present invention can also be used as prodrugs. Methods of treating AIDS, methods of treating HIV infection and methods of inhibiting HIV protease are disclosed.
- the compounds of the present invention are of the formula (1) :
- Q 1 and Q 2 are independently selected from hydrogen and substituted and unsubstituted alkyl and aryl, and Q 1 and Q 2 may form a ring with G, Q 3 is selected from mercapto and substituted and unsubstituted alkoxyl, aryloxyl, thioether, amino, alkyl,
- Q 4 -Q 8 are independently selected from hydrogen, hydroxyl, mercapto, nitro, halogen, -O-J, wherein J is a substituted or unsubstituted hydrolyzable group, and substituted and
- Y and G are independently selected from oxygen, -NH,
- D is carbon or nitrogen, and wherein D is singly bonded to each of the adjacent ring atoms,
- E is carbon or nitrogen
- Q 9 is selected from hydrogen, halogen, hydroxyl, mercapto, and substituted and unsubstituted alkoxyl, aryloxyl, thioether, amino, alkyl, and aryl, wherein Q 9 may form part of a ring,
- A is a carbocycle or heterocycle, which is optionally further substituted
- B is a carbocycle or heterocycle, which is optionally further substituted
- the invention more particularly relates -to preferred
- Q 1 and Q 2 are substituted or unsubstituted alkyl and the other is as defined above,
- Q 3 is selected from thioether and aryl
- Q 4 -Q 8 are independently selected from hydrogen, hydroxyl, halogen, -O-J, wherein J is a substituted or unsubstituted
- hydrolyzable group and substituted and unsubstituted alkoxyl, amino, alkyl, and L 6 C(O)L 4 , wherein L 6 is a single bond, -O or -N, and further wherein L. is preferably alkyl, hydroxyl, alkoxyl or hydrogen, and further wherein any one or more of Q 4 -Q 8 may form part of a ring,
- Y and G are each oxygen
- D is nitrogen, and wherein D is singly bonded to each of the adjacent ring atoms,
- E is carbon or nitrogen
- A is a carbocycle or heterocycle that is an aromatic or partially saturated, 5-7 membered mono-ring, which is optionally further substituted
- B is a heterocycle that is a saturated or partially saturated, 8-12 membered poly-ring, which is optionally further substituted
- the invention even more particularly relates to compounds of the formula (1) wherein:
- one of Q 1 and Q 2 is substituted or unsubstituted alkyl, preferably t-butyl, and the other is hydrogen,
- Q 3 is selected from thioaryl and aryl, preferably thiophenyl and phenyl,
- Q 4 is alkyl, preferably methyl
- Q 5 is hydroxyl or -O-J, wherein J is a hydrolyzable group, or substituted or unsubstituted alkoxyl or amino,
- Q 6 -Q 8 are independently selected from hydrogen, hydroxyl, halogen, -O-J, wherein J is a substituted or unsubstituted
- hydrolyzable group and substituted and unsubstituted alkoxyl, amino, alkyl, and L 6 C(O)L 4 , wherein L 6 is a single bond, -O or -N, and further wherein L 4 is preferably alkyl, hydroxyl, alkoxyl or hydrogen, and further wherein any one or more of Q 6 - Q 8 may form part of a ring,
- Y and G are each oxygen
- D is nitrogen, and wherein D is singly bonded to each of the adjacent ring atoms,
- Q 9 is hydrogen
- A is a carbocycle that is an aromatic, 5-6 membered monocyclic ring, preferably phenyl, which is optionally further substituted,
- B is a heterocycle that is a saturated, 6-14 membered monocyclic or polycyclic ring, which is optionally further
- M 1 and M 2 are independently selected from hydrogen, mercapto, hydroxyl, and substituted and unsubstituted thioether, alkyl, alkoxyl, aryloxyl, amino, five membered heterocycle and carbocycle, sulfinyl, sulfonyl, and acyl, and wherein M 1 and M 2 optionally form a ring having up to 10 members, wherein preferably M 1 and M 2 independently have from zero to eight non-hydrogen atoms;
- Preferred compounds of the formula (1) include those wherein: one of Q 1 and Q 2 is tertiary alkyl, preferably t-butyl, and the other is hydrogen, Q 3 is thiophenyl, phenyl, naphthyl, or thionaphthyl,
- Q 5 is hydroxyl, amino, or -O-J, wherein J is a substituted or unsubstituted hydrolyzable group
- Q 6 -Q 8 are independently selected from hydrogen, hydroxyl, halogen, -O-J, wherein J is a substituted or unsubstituted
- hydrolyzable group and substituted and unsubstituted alkoxyl, amino, alkyl, and L 6 C(O)L 4 , wherein L 6 is a single bond, -O or -N, and further wherein L. is preferably alkyl, hydroxyl, alkoxyl or hydrogen, and further wherein any one or more of Q 6 - Q 8 may form part of a ring,
- Y and G are each oxygen
- D is nitrogen, and wherein D is singly bonded to each of the adjacent ring atoms,
- A is phenyl, which is optionally further substituted
- B is a heterocycle that is a saturated, 9-10 membered bi-ring, preferably decahydroisoquinolinyl or octahydrothieno[3,2-c]pyridinyl,
- the compounds have the formula 1 (A) :
- Z is a group having the structure:
- a is 1, 2, 3, 4, or 5;
- b is 1, or 2;
- c is 1, or 2;
- d is 1, 2, 3, or 4;
- each R is independently hydrogen, hydroxy, thiol, halo, amino, C 1 -C 4 alkylamino, di(C 1 -C 4 )alkylamino, nitro, carboxy, C 1 -
- alkoxycarbonyl carbamoyl, N-(C 1 -C 4 )alkylcarbamoyl, C 1 -C 4 alkylsulfonyl, N,N-di(C 1 -C 4 )alkylcarbamoyl, or C 1 -C 4 alkylsulfonylamino;
- a 1 and A 2 are independently -CH 2 - or -N(R 8 )-;
- a 3 and A 4 are independently -CH- or -N-;
- a 5 and A 6 are independently -CH 2 - or -N(R 9 )-;
- a 7 and A 8 are independently -CH- or -N-;
- R 8 is hydrogen or C 1 -C 4 alkyl
- R 9 is hydrogen or C 1 -C 4 alkyl
- R 1 is aryl, or -S-aryl
- X is a group having the structure:
- R is hydrogen, C 1 -C 4 alkyl, or -CH 2 -pyridyl;
- R 3 is a group having the structure:
- R 4 at each occurrence is independently hydrogen, C 1 -C 6 alkyl or hydroxy(C 1 -C 4 )alkyl;
- R 5 and R 6 are independently selected from hydrogen, hydroxy, C 1 -C 6 alkyl, C 1 -C 6 alkoxy, or hydroxy (C 1 -C 4 ) alkyl; with the provisos that :
- a 5 and A 6 cannot both be -N(R 9 )-;
- R 1 is aryl, or -S-aryl; X 1 is a group having the formula: , or
- T 2 is hydrogen, halo, or C 1 -C 4 alkyl
- R 3 is a group having the structure:
- p 4 or 5;
- R 4 at each occurrence is independently hydrogen
- R and R are independently selected from hydrogen, hydroxy,
- Z is a group having the structure :
- a is 1, 2, 3, 4, or 5;
- b is 1, or 2;
- c is 1, or 2;
- d is 1, 2, 3, or 4;
- each R 7 is independently hydrogen, hydroxy, thiol, halo, amino, C 1 -C 4 alkylamino, di(C 1 -C 4 )alkylamino, nitro, carboxy, C 1 -
- alkoxycarbonyl carbamoyl, N-(C 1 -C 4 )alkylcarbamoyl, C -C.
- alkylsulfonyl N,N-di(C 1 -C 4 )alkylcarbamoyl, or C 1 -C 4
- a 1 and A 2 are independently -CH 2 - or -N(R 8 )-;
- a 3 and A 4 are independently -CH- or -N-;
- a 5 and A 6 are independently -CH 2 - or -N(R 9 )-;
- a 7 and A 8 are independently -CH- or -N-;
- R 8 is hydrogen or C 1 -C 4 alkyl
- R 9 is hydrogen or C 1 -C 4 alkyl
- T 2 is hydrogen, or C 1 -C 4 alkyl
- a 5 and A 6 cannot both be -N(R 9 )-;
- Preferred species of the formula (1) are:
- the present invention further provides pharmaceutical formulations comprising an effective amount of a compound of formula (1) or a pharmaceutically acceptable salt thereof, in combination with a pharmaceutically acceptable carrier, such as a diluent or excipient.
- a pharmaceutically acceptable carrier such as a diluent or excipient.
- the present invention further provides a method of treating AIDS comprising administering to a host or patient, such as a primate, an effective amount of a compound of the present
- the present invention further provides a method of inhibiting HIV replication comprising administering to an HIV infected cell, a cell susceptible to HIV infection or a host or patient, such as a primate, an effective amount of a compound of the present invention.
- the present invention provides new compounds falling within formula (1), as described above, that are useful for treating HIV infection and/or AIDS.
- Compounds of the formula (1) may be prodrugs.
- compounds wherein at least one of Q 4 -Q 8 is -O-J, as defined above, may be used as prodrugs, which can serve to improve the
- the preparation of the prodrugs may be carried out by reacting a compound of the formula (1), wherein at least one of
- Q 4 -Q 8 is -O-H, with, for example, an activated amino acyl, phosphoryl or hemisuccinyl derivative.
- alkyl refers to straight or branched
- C 1 -C 8 alkyl represents a
- C 1 -C 6 alkyl groups include methyl, ethyl, n-propyl, isopropyl, butyl, isobutyl, sec-butyl, t-butyl, pentyl, neo-pentyl, hexyl, isohexyl, and the like.
- the term "C 1 -C 6 alkyl” includes within its definition the term "C 1 -C 4 alkyl".
- cycloalkyl represents a saturated or partially saturated, mono- or poly-carbocylic ring, preferably having 5-14 ring carbon atoms.
- exemplary cycloalkyls include monocyclic rings having from 3-7, preferably 3-6, carbon atoms, such as
- cyclopropyl cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and the like.
- An exemplary cycloalkyl is a C 5 -C 7 cycloalkyl, which is a saturated hydrocarbon ring structure containing from five to seven carbon atoms.
- alkoxyl represents -O-alkyl.
- An example of an alkoxyl is a C 1 -C 6 alkoxyl, which represents a straight or
- C 1 -C 9 alkoxyl groups include methoxyl, ethoxyl, propoxyl, isopropoxyl, butoxyl, sec-butoxyl, t-butoxyl, pentoxyl, hexoxyl, and the like.
- C 1 -C 6 alkoxyl includes within its definition a C 1 -C 4 alkoxyl.
- aryl refers to a carbocyclic or heterocyclic, aromatic, 5-14 membered monocyclic or polycyclic ring.
- exemplary aryls include phenyl, naphthyl, anthryl,
- beta-carbolinyl phenanthridinyl, acridinyl, perimidinyl
- phenanthrolinyl phenazinyl, isothiazolyl, phenothiazinyl, and phenoxazinyl.
- aryloxyl represents -O-aryl
- hydrolyzable group is a group, which .when bonded to an oxygen, forms an ester, which can be hydrolyzed in vivo to a hydroxyl group.
- exemplary hydrolyzable groups which are
- acyl function optionally substituted, include acyl function, sulfonate function and phosphate function.
- hydrolyzable groups include blocked or unblocked amino acid residue, a hemisuccinate residue, and a nicotinate residue.
- halogen represents chlorine, fluorine, bromine or iodine.
- halo represents chloro, fluoro, bromo or iodo.
- carbocycle represents an aromatic or a saturated or a partially saturated 5-14 membered monocyclic or polycyclic ring, such as a 5- to 7-membered monocyclic or 7- to 10-membered bicyclic ring, wherein all the ring members are carbon atoms.
- heterocycle represents an aromatic or a saturated or a partially saturated, 5-14 membered, monocylic or polycyclic ring, such as a 5- to 7-membered monocyclic or 7- to 10-membered bicyclic ring, having from one to three heteroatoms selected from nitrogen, oxygen and sulfur, and wherein any nitrogen and sulfur heteroatoms may optionally be oxidized, and any nitrogen
- heteroatom may optionally be quaternized.
- the heterocyclic ring may be attached at any suitable heteroatom or carbon atom.
- heterocycles examples include decahydroisoquinolinyl, octahydro-thieno [3, 2-c] pyridinyl, piperidinyl, piperazinyl, azepinyl, pyrrolyl, pyrrolidinyl, pyrazolyl, pyrazolidinyl, imidazolyl, isobenzofuranyl, furazanyl, imidazolinyl,
- quinolinyl chromenyl, xanthenyl, isoquinolinyl, benzimidazolyl, thiadiazolyl, benzopyranyl, benzothiazolyl, benzoazolyl, furyl, tetrahydrofuryl, tetrahydropyranyl, thienyl, benzothienyl, benzo[b]thienyl, naphtho[2,3-b]thienyl, thiamorpholinyl,
- thiamorpholinylsulfoxide thiamorpholinylsulfone, oxadiazolyl, triazolyl, tetrahydroquinolinyl, tetrahydrisoquinolinyl,
- phenoxathienyl indolizinyl, isoindolyl, indazolyl, purinyl, isoquinolyl, quinolyl, phthalazinyl, naphthyridinyl,
- quinoxyalinyl quinzolinyl, tetrahydroquinolinyl, cinnolinyl, pteridinyl, carbazolyl, beta-carbolinyl, phenanthridinyl,
- acridinyl acridinyl, perimidinyl, phenanthrolinyl, phenazinyl, isothiazolyl, phenothiazinyl, and phenoxazinyl.
- thioether includes S-aryl, such as phenylthio and naphthylthio; S-heterocycle where the heterocycle is saturated or partially saturated; S-(C 5 -C 7 -cycloalkyl; and S-alkyl, such as C 1 -C 6 alkylthio.
- the -aryl, the -heterocycle, the -cycloalkyl and the -alkyl can optionally be substituted.
- An example of a thioether is "C 1 -C 6 alkylthio", which represents a straight or branched alkyl chain having from one to six carbon atoms attached to a sulfur atom.
- Exemplary C 1 -C 6 alkylthio groups include methylthio, ethylthio, propylthio, isopropylthio,
- mercapto represents -SH.
- amino represents -NL 1 L 2 , wherein L 1 and L 2 are preferably independently selected from oxygen, carbocycle, heterocycle, alkyl, sulfonyl and hydrogen; or NC(O)L 3 , wherein L 3 is preferably alkyl, alkoxyl, hydrogen or -NL 1 L 2 .
- the aryl, alkyl and alkoxyl groups can optionally be substituted.
- An example of an amino is C 1 -C 4 alkylamino, which represents a straight or branched alkyl chain having from one to four carbon atoms attached to an amino group.
- Exemplary C 1 -C 4 alkylamino groups include methylamino, ethylamino, propylamino, isopropylamino, butylamino, sec-butylamino, and the like.
- Another example of an amino is di (C 1 -C 4 ) alkylamino, which represents two straight or branched alkyl chains, each having from one to four carbon atoms attached to a common amino group.
- Exemplary di(C 1 -C 4 )alkylamino groups include dimethylamino, ethylmethylamino, methylpropylamino, ethylisopropylamino, butylmethylamino, sec-butylethylamino, and the like.
- An example of an amino is C 1 -C 4 alkylsulfonylamino, which has a straight or branched alkyl chain having from one to four carbon atoms attached to a sulfonylammo moiety.
- Exemplary C 1 -C 4 alkylsulfonylamino groups include methylsulfonylamino, ethylsulfonylamino, propylsulfonylamino, isopropylsulfonylamino, butylsulfonylammo, sec-butylsulfonylamino, t-butylsulfonylamino, and the like.
- acyl represents L 6 C(O)L 4 , wherein L 6 is a single bond, -O or -N, and further wherein L is preferably alkyl, amino, hydroxyl, alkoxyl or hydrogen.
- the alkyl and alkoxyl groups can optionally be substituted.
- An exemplary acyl is a C 1 -C 4
- alkoxycarbonyl which is a straight or branched alkoxyl chain having from one to four carbon atoms attached to a carbonyl moiety.
- exemplary C 1 -C 4 alkoxycarbonyl groups include
- exemplary acyl is a carboxy wherein L 6 is a single bond and L 4 is alkoxyl, hydrogen, or hydroxyl.
- a further exemplary acyl is N- (C 1 -C 4 )alkylcarbamoyl (L 6 is a single bond and L 4 is an amino), which is a straight or branched alkyl chain having from one to four carbon atoms attached to the nitrogen atom of a carbamoyl moiety.
- N-(C 1 -C 4 )alkylcarbamoyl groups include N-methylcarbamoyl, N-ethylcarbamoyl, N-propylcarbamoyl, N-isopropylcarbamoyl, N-butylcarbamoyl, and N-t-butylcarbamoyl, and the like.
- Yet another exemplary acyl is N,N-di(C 1 -C 4 )alkylcarbamoyl, which has two straight or branched alkyl chains, each having from one to four carbon atoms attached to the nitrogen atom of a carbamoyl moiety.
- N,N-di(C 1 -C 4 )alkylcarbamoyl groups include N,N-dimethylcarbamoyl, N,N- ethylmethylcarbamoyl, N,N-methylpropylcarbamoyl,
- alkyl preferably alkyl, amino, aryl, cycloalkyl or heterocycle.
- the alkyl, aryl, cycloalkyl and heterocycle can all optionally be substituted.
- sulfonyl represents -SO 2 -L 5 , wherein L 5 is preferably alkyl, aryl, cycloalkyl, heterocycle or amino.
- L 5 is preferably alkyl, aryl, cycloalkyl, heterocycle or amino.
- the alkyl, aryl, cycloalkyl and heterocycle can all optionally be substituted.
- An example of a sulfonyl is a C 1 -C 4 alkylsulfonyl, which is a straight or branched alkyl chain having from one to four carbon atoms attached to a sulfonyl moiety.
- Exemplary C 1 -C 4 alkylsulfonyl groups include methylsulfonyl, ethylsulfonyl, propylsulfonyl, isopropylsulfonyl, butylsulfonyl, sec-butylsulfonyl, t-butylsulfonyl and the like.
- all chemical groups may be substituted or unsubstituted as long as the valences of such groups allow such substitutions, even if the definitions of the chemical groups do not explicitly state that the groups are substituted or unsubstituted.
- a group is simply defined as alkyl, it may be substituted or unsubstituted alkyl.
- substituents for alkyl and aryl include mercapto, thioether, nitro (NO 2 ), amino, aryloxyl, halogen, hydroxyl, alkoxyl, and acyl, as well as aryl, cycloalkyl and saturated and partially saturated heterocycles.
- substituents for heterocycle and cycloalkyl include those listed above for alkyl and aryl, as well as aryl and alkyl.
- Exemplary substituted aryls include a phenyl or naphthyl ring substituted with one or more substituents, preferably one to three substituents, independently selected from halo, hydroxy,
- C 1 -C 4 morpholino (C 1 -C 4 )alkoxy carbonyl, pyridyl (C 1 -C 4 )alkoxycarbonyl, halo (C 1 -C 4 )alkyl, C 1 -C 4 alkyl, C 1 -C 4 alkoxy, carboxy, C 1 -C 4 alkoxycarbonyl, carbamoyl, N-(C 1 -C 4 )alkylcarbamoyl, amino,
- R 7 is hydroxy, C 1 -C 4 alkoxy, carboxy, C 1 -C 4 alkoxycarbonyl, amino, carbamoyl, C 1 -C 4 alkylamino or di(C 1 -C 4 )alkylamino.
- Another substituted alkyl is halo(C 1 -C 4 )alkyl, which
- Exemplary halo(C 1 -C 4 )alkyl groups include chloromethyl, 2-bromoethyl, 1-chloroisopropyl, 3-fluoropropyl, 2,3-dibromobutyl, 3-chloroisobutyl, iodo-t-butyl, trifluoromethyl and the like.
- hydroxy(C 1 -C 4 )alkyl which represents a straight or branched alkyl chain having from one to four carbon atoms with a hydroxy group attached to it.
- exemplary hydroxy(C 1 -C 4 )alkyl groups include hydroxymethyl, 2-hydroxyethyl, 3-hydroxypropyl, 2-hydroxyisopropyl, 4-hydroxybutyl and the like.
- C 1 -C 4 alkylthio(C 1 -C 4 )alkyl is a straight or branched C 1 -C 4 alkyl group with a C 1 -C 4 alkylthio group attached to it.
- Exemplary C 1 -C 4 alkylthio (C 1 -C 4 )alkyl groups include methylthiomethyl, ethylthiomethyl, propylthiopropyl, sec-butylthiomethyl, and the like.
- heterocycle (C 1 -C 4 )alkyl which is a straight or branched alkyl chain having from one to four carbon atoms with a heterocycle attached to it.
- Exemplary heterocycle(C 1 -C 4 )alkyls include pyrrolylmethyl, quinolinylmethyl, 1-indolylethyl, 2-furylethyl, 3-thien-2-ylpropyl, 1-imidazolylisopropyl, 4-thiazolylbutyl and the like.
- aryl(C 1 -C 4 )alkyl which is a straight or branched alkyl chain having from one to four carbon atoms with an aryl group attached to it.
- exemplary aryl (C 1 -C 4 )alkyl groups include phenylmethyl, 2-phenylethyl,
- the heterocycle can, for example, be substituted with 1, 2 or 3 substituents independently selected from halo, halo(C 1 -C 4 )alkyl, C 1 -C 4 alkyl, C 1 -C 4 alkoxy, carboxy, C 1 -C 4
- alkoxycarbonyl carbamoyl, N-(C 1 -C 4 )alkylcarbamoyl, amino,
- C 1 -C 4 alkylamino di(C 1 -C 4 )alkylamino or a group having the structure -(CH 2 ) a -R 7 where a is 1, 2, 3 or 4 and R 7 is hydroxy, C 1 -C 4 alkoxy, carboxy, C 1 -C 4 alkoxycarbonyl, amino, carbamoyl, C 1 -C 4 alkylamino or di(C 1 -C 4 )alkylamino.
- substituted heterocycles include 3-N-t-butyl carboxamide decahydroisoquinolinyl, 6-N-t-butyl carboxamide octahydro-thieno[3,2-c]pyridinyl, 3-methylimidazolyl,
- 6-methoxybenzimidazolyl 4-hydroxyfuryl, 4-methylisoquinolinyl, 6,8-dibromoquinolinyl, 4,8-dimethylnaphthyl, 2-methyl-1,2,3,4-tetrahydroisoquinolinyl, N-methyl-quinolin-2-yl,
- Exemplary heterocyclic ring systems represented by A or B include (1) 5-membered monocyclic ring groups such as thienyl, pyrrolyl, imidazolyl, pyrazolyl, furyl, isothiazolyl, furazanyl, isoxazolyl, thiazolyl and the like; (2) 6-membered monocyclic groups such as pyridyl, pyrazinyl, pyrimidinyl, pyridazinly, triazinyl and the like; and (3) polycyclic heterocyclic rings groups, such as decahydroisoquinolinyl, octahydro-thieno [3,2-c] pyridinyl, benzo[b]thienyl, naphtho[2,3-b]thianthrenyl,
- a cycloalkyl may be optionally substituted with 1, 2 or 3 substituents independently selected from halo, halo (C. -C.) alkyl, C 1 -C 4 alkyl, C 1 -C 4 alkoxy, carboxy, C 1 -C 4 alkoxycarbonyl, carbamoyl, N-(C 1 -C 4 )alkylcarbamoyl, amino, C 1 -C 4 alkylamino, di(C 1 -C 4 )alkylamino or a group having the structure -(CH 2 ) a -R 7 where a is 1, 2, 3 or 4 and R 7 is hydroxy, C 1 -C 4 alkoxy, carboxy, C 1 -C 4 alkoxycarbonyl, amino, carbamoyl, C 1 -C 4 alkylamino or di(C 1 -C 4 )alkylamino.
- substituents independently selected from halo, halo (C. -C
- Exemplary substituted hydrolyzable groups include N-benzyl glycyl, N-Cbz-L-valyl, and N-methyl nicotinate.
- the compounds of the present invention have at least two asymmetric centers denoted by an asterisk in the formula (1) below:
- the compounds of the present invention can occur in any of the possible
- stereoisomers which can be optically active or racemic, or can be used alone as essentially pure stereisomers, i.e., at least 95% pure. All asymmetric forms, individual stereoisomers and
- the individual stereoisomers may be prepared from their respective precursors by the procedures described above, by resolving the racemic mixtures, or by separating the
- the resolution can be carried out in the presence of a resolving agent, by chromatography or by repeated crystallization or by some combination of these techniques which are known in the art. Further details regarding resolutions can be found in Jacques et al., Enantiomers, Racemates, and
- the compounds of the present invention are substantially pure, i.e, over 50% pure. More preferably, the compounds are at least 75% pure. Even more preferably, the compounds are more than 90% pure. Even more preferably, the compounds are at least 95% pure, more preferably, at least 97% pure, and most preferably at least 99% pure.
- the invention includes the
- a compound of this invention may possess a
- pharmaceutically acceptable salt refers to salts of the compounds of the above formula which are substantially non-toxic to living organisms.
- pharmaceutically acceptable salts include those salts prepared by reaction of the compounds of the present invention with a mineral or organic acid or an inorganic base.
- the reactants are generally combined in a mutual solvent such as diethylether or benzene, for acid addition salts, or water or alcohols for base addition salts.
- the salts normally precipitate out of solution within about one hour to about ten days and can be isolated by filtration or other conventional methods. Such salts are known as acid addition and base addition salts.
- Acids that may be employed to form acid addition salts are inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, phosphoric acid, and the like, and organic acids such as p-toluenesulfonic, methanesulfonic acid, oxalic acid, p-bromophenylsulfonic acid, carbonic acid, succinic acid, citric acid, benzoic acid, acetic acid, and the like.
- inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, phosphoric acid, and the like
- organic acids such as p-toluenesulfonic, methanesulfonic acid, oxalic acid, p-bromophenylsulfonic acid, carbonic acid, succinic acid, citric acid, benzoic acid, acetic acid, and the like.
- sulfate pyrosulfate, bisulfate, sulfite, bisulfite, phosphate, monohydrogenphosphate, dihydrogenphosphate, metaphosphate, pyrophosphate, chloride, bromide, iodide, acetate, propionate, decanoate, caprylate, acrylate, formate, isobutyrate, caproate, heptanoate, propiolate, oxalate, malonate, succinate, suberate, sebacate, fumarate, maleate, butyne-1,4-dioate, hexyne-1,6-dioate, benzoate, chlorobenzoate, methylbenzoate, dinitrobenzoate, hydroxybenzoate, methoxybenzoate, phthalate, sulfonate,
- xylenesulfonate phenylacetate, phenylpropionate, phenylbutyrate, citrate, lactate, g-hydroxybutyrate, glycollate, tartrate, methane-sulfonate, propanesulfonate, naphthalene-1-sulfonate, napththalene-2-sulfonate, mandelate and the like.
- Preferred pharmaceutically acceptable acid addition salts are those formed with mineral acids such as hydrochloric acid and hydrobromic acid, and those formed with organic acids such as maleic acid and methanesulfonic acid.
- Base addition salts include those derived from inorganic and organic bases, such as ammonium or alkali or alkaline earth metal hydroxides, carbonates, bicarbonates, and the like.
- bases useful in preparing the salts of this invention thus include sodium hydroxide, potassium hydroxide, ammonium hydroxide, potassium carbonate, sodium carbonate, sodium bicarbonate, potassium bicarbonate, calcium hydroxide, calcium carbonate and the like.
- the potassium and sodium salt forms are particularly preferred.
- any salt of this invention is not of a critical nature, so long as the salt as a whole is pharmacologically acceptable and as long as the counterion does not contribute undesired qualities to the salt as a whole.
- Z is a group having the structure:
- R 2 is hydrogen, hydroxy, C 1 -C 4 alkyl, halo, amino, nitro, or trifluoromethyl; a is 1, 2, or 3;
- R 3 is -C(O)NR 4 R 4 ;
- R 2 is hydrogen, methyl, ethyl, propyl, chloro, fluoro, hydroxy, or amino;
- X is , or ;
- R is -CH 2 -pyridyl
- R 1 is phenyl or -S-phenyl
- R 3 is -C(O)NH(R 4 );
- Z is R 2a is methyl, ethyl, or propyl
- R 2b is hydrogen, hydroxy, or amino
- R 2c is hydrogen, hydroxy, or amino
- R is -C(O)NH(t-butyl);
- X is ;
- T 2 i.s hydrogen or methyl
- Z 1 is a group having the structure:
- R 7 is hydrogen, C 1 -C 4 alkyl, halo, nitro, amino, hydroxy; a is 1, 2, or 3;
- R 7 is hydrogen, methyl, ethyl, hydroxy, amino, chloro
- R is -S-phenyl, or -S-naphth-2-yl
- R 3 is -C(O)NR 4 R 4 ;
- Z 1 is R 7 a is hydrogen, methyl, ethyl, chloro, bromo, or fluoro; R 7 b is hydrogen, hydroxy, chloro, or ammo;
- R 7 c is hydrogen, hydroxy, or amino
- R 3 is -C(O)NH(t-butyl);
- Preferred compounds are:
- Each of the above five formulae has five assymetric centers and thus defines a compound selected from the group of 32
- Preferred stereisomers of these compounds are :
- the compounds of formula 1 can be prepared according to the following Reaction I.
- Reaction I is a standard coupling reaction commonly employed in the synthesis of amides or peptides which is carried out by reacting an appropriately substituted amine of formula IA, with an appropriately substituted carboxylic acid reactant of formula IB, in an aprotic solvent or mixture of solvents.
- the reaction is typically carried out in the presence or absence of a promoting agent, preferably in the presence of a promoting agent, and in the presence of a coupling reagent.
- Typical aprotic solvents for this reaction are tetrahydrofuran and dimethylformamide, or a mixture of such solvents.
- the reaction is typically carried out at a temperature from about -30°C to about 25°C.
- the amine reactant is generally employed in equimolar proportions relative to the carboxylic acid reactant, in the presence of an equimolar quantity to a slight excess of the coupling reagent.
- Typical coupling reagents include the carbodiimides such as
- DCC dicyclohexylcarbodiimide
- N,N'-diethylcarbodiimide the imidazoles such as carbonyldiimidazole
- reagents such as bis (2-oxo-3-oxazolidinyl)phosphinic chloride (BOP-Cl) or N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline (EEDQ) .
- BOP-Cl bis (2-oxo-3-oxazolidinyl)phosphinic chloride
- EEDQ N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline
- a preferred coupling reagent for this reaction is DCC.
- a promoting agent is preferably included for this reaction; a preferred promoting agent is hydroxybenzotriazole hydrate (HOBT ⁇ H 2 O).
- the compound may be isolated, if desired, by procedures known in the art, for example, the compound may be crystallized and then collected by filtration, or the reaction solvent may be removed by extraction, evaporation or decantation.
- the compound may be further purified, if desired, by common techniques such as crystallization or chromatography over solid supports such as silica gel or alumina.
- the starting compounds of formula IA may be prepared according to the procedures shown in Reaction Scheme A.
- v is an amino-protecting group
- ZZ is halo
- Reaction Scheme A is accomplished by carrying out reactions 1-7 in sequential order. Once a reaction is complete, the intermediate compound may be isolated, if desired, by
- the compound may be crystallized and then collected by filtration, or the reaction solvent may be removed by extraction, evaporation or decantation.
- the intermediate compound may be further purified, if desired, by common techniques such as crystallization or chromatography over solid supports such as silica gel or alumina, before carrying out the next step of the reaction scheme.
- Reaction A.1 is carried out by converting an amino-protected carboxylic acid reactant having the structure:
- the amino-protected carboxylic acid reactant may be reacted with a C 1 -C 6 alkylchloroformate, such as
- isobutylchloroformate preferably in the presence of an acid scavenger.
- Preferred acid scavengers are the trialkylamines, preferably triethylamine.
- the reaction is typically carried out in an aprotic solvent such as ethyl acetate. Solvent choice is not critical so long as the solvent employed is inert to the ongoing reaction, and the reactants are sufficiently solubilized to effect the desired reaction.
- the resulting mixed anhydride reactant is preferably used in Reaction A.2 without further isolation or purification.
- Reaction A.2 is accomplished in two steps. First, a solution of sodium hydroxide, covered with a layer of an ether solvent, preferably diethyl ether, is reacted with a large excess of
- the sodium hydroxide is preferably used as an aqueous solution having about four to six mol/liter of sodium hydroxide.
- the organic layer is dried over a dessicant such as potassium hydroxide.
- This solution is then reacted with the mixed anhydride from Reaction A.1, above, to form the corresponding alpha-diazo carbonyl compound.
- the diazomethane reactant is preferably used in this reaction without isolation or purification.
- the reaction is typically carried out at a temperature of from about -50°C to about -10°C, preferably about -20°C.
- Reaction A.3 the alpha-diazo carbonyl compound prepared in Reaction A.2 is reacted with an acid of the formula H-ZZ where ZZ is halo, typically in an aprotic solvent such as diethylether to form an alpha-halo carbonyl compound.
- a preferred acid reactant is hydrochloric acid which provides the corresponding alpha-chloro carbonyl compound.
- the reaction is typically carried out at a temperature from about -30°C to about 0°C. Solvent choice is not critical so long as the solvent employed is inert to the ongoing reaction and the reactants are sufficiently
- the acid reactant is typically added in the form of an anhydrous gas in small
- reaction can be monitored by thin layer chromatography.
- Reaction A.4 the carbonyl moiety on the compound prepared in Reaction A.3 is reduced using standard conditions known in the art to form the corresponding alpha-chloro hydroxy compound.
- the compound prepared in Reaction A.3 is combined with a reducing agent in a mixture of solvents.
- Typical reducing agents include sodium borohydride, lithium borohydride, zinc borohydride, diisobutylaluminum hydride, and sodium bis (2-methoxy-ethoxy) aluminum hydride.
- a preferred reducing agent is sodium
- borohydride Typical solvent mixtures include a protic and aprotic mixture such as tetrahydrofuran/water. Solvent choice is not critical so long as the solvent employed is inert to the ongoing reaction, and the reactants are sufficiently solubilized to effect the desired reaction.
- the reaction is typically carried out at a temperature from about -10°C, preferably about 0°C.
- the alpha-chloro hydroxy compound may be reacted with a potassium hydroxide/ethanol mixture in an alcoholic solvent such as ethanol.
- the reaction is typically carried out at a
- reaction is carried out at room temperature from about 0°C to about the reflux temperature of the solvent.
- the reaction is carried out at room temperature
- Reaction A.6 the epoxide prepared in Reaction A.5 is reacted with a heterocyclic reactant :
- solvent choice is not critical so long as the solvent employed is inert to the ongoing reaction, and the reactants are sufficiently solubilized to effect the desired reaction.
- Typical solvents for this reaction include the alcohols, preferably isopropanol or ethanol. The reaction is preferably carried out at a temperature of about 80°C.
- Reaction A.7 is a standard amino deprotection reaction using procedures and methods known in the art to afford the
- This amine which is used in Reaction I, above.
- This amine may be reacted without purification, but it is preferably purified first.
- the compounds of formula IA, where Q 3 is -S-aryl are typically prepared by first reacting amino-protected serine with triphenylphosphine and diethylazodicarboxylate (DEAD) in an aprotic solvent at a temperature of from about -80°C to 0°C to form the corresponding beta-lactone.
- the reaction is typically carried out in an ether, such as tetrahydrofuran at a temperature of from about -80°C to -50°C.
- the lactone ring is opened to provide a compound having the structure :
- thioanion compound is preferably formed by reacting the
- thiol with a strong base, such as sodium hydride or potassium hydride.
- a strong base such as sodium hydride or potassium hydride.
- This reaction is typically carried out in an aprotic solvent at a temperature from about 0°C to about 40°C and under an inert atmosphere, such as nitrogen.
- Typical solvents for this reaction include ethers, preferably tetrahydrofuran.
- the compounds of formula IA, where Q 3 is -S-aryl may be prepared using the procedures detailed in Photaki .
- the compounds may be prepared by reacting doubly protected serine (carboxy-protected and amino-protected) with toluenesulfonyl chloride in the presence of dimethylaminopyridine (DMAP) and an acid scavenger such as pyridine in an aprotic solvent such as methylene chloride to form the corresponding toluenesulfonate which may then be reacted with an appropriately substituted thioanion having the structure, -S- aryl.
- DMAP dimethylaminopyridine
- an acid scavenger such as pyridine
- an aprotic solvent such as methylene chloride
- the thioanion compound is preferably formed by reacting the corresponding thiol with a strong base as described above.
- the carboxy-protecting group may be removed from the resulting doubly protected arylthioalanine using conditions known in the art.
- the heterocyclic reactants were typically prepared from the corresponding amino-protected amino acids by acid activation followed by treatment with an alkylamine. This reaction is typically carried out in the presence of an acid scavenger, such as N-methylmorpholine. Removal of the aminoprotecting group using standard chemical deprotecting techniques then provides the desired heterocyclic reactants.
- the [3S- (3R* , 4aR* , 8aR*) ]-decahydroisoquinoline-3-N-t-butylcarboxamide was prepared using 2S-1,2,3,4-tetrahydro-3-isoquinolinecarboxylic acid by the following procedure:
- the piperazine reactants may be prepared by converting an appropriately substituted pyrazine compound to the corresponding piperazine compound using procedures known in the art, preferably using catalytic hydrogenation.
- the hydrogenation may be accomplished by combining the pyrazine reactant with a catalyst under a hydrogen atmosphere in an aprotic solvent at a temperature from about 0°C to about 60°C.
- Suitable catalysts include palladium-on-carbon, platinum metal, platinum oxide and the like.
- a preferred catalyst is platinum oxide.
- Typical solvents for this reaction include tetrahydrofuran, dimethylformamide or a mixture of tetrahydrofuran and dimethylformamide.
- the nitrogen atom on the resultant piperazine reactant may be alkylated using procedures known in the art.
- the piperazine reactant may be reacted with a halo (C 1 -C 4 )alkyl, or halomethylpyridine, such as methyl iodide or chloromethylpyridme.
- Preferred halo substituents include chloro, bromo and iodo.
- the reaction is carried out at temperatures of from about 0°C to 60°C in a mutually inert solvent and in the presence of an acid
- a preferred acid scavenger is potassium carbonate.
- Typical solvents include a mixture of a protic and aprotic
- solvents such as acetonitrile and water. Solvent choice is not critical so long as the solvent employed is inert to the ongoing reaction and the reactants are sufficiently solubilized to effect the desired reaction.
- the alkylated piperazine reactant may be prepared using reductive amination.
- the piperazine reactant prepared above may be reacted with an aldehyde (for example, 3 -pyridine carboxylic aldehyde, ethanal, propanal) or a ketone in the presence of a reducing agent and an acid.
- the reaction is typically carried out in an alcoholic solvent such as methanol, ethanol or isopropanol.
- Typical reducing agents include sodium borohydride, lithium cyanoborohydride, sodium
- cyanoborohydride and the like.
- a preferred reducing agent is sodium cyanoborohydride.
- Typical acids include any protic acid such as hydrochloric acid, sulfuric acid, methanesulfonic acid, or acetic acid.
- a preferred acid is acetic acid.
- V 0 and V 1 are independently hydrogen, C 1 -C 8 alkyl, or hydroxy
- V 2 is hydrogen, an ammo-protecting group, or a group of the formula:
- V 3 is -(CH 2 ) t -V 3 ';
- t 0, 1, 2, 3, or 4;
- V 3' is aryl, -O-aryl, or -S-aryl
- V 4 is hydrogen or an ammo-protectmg group; f, h and j are each independently 0, 1 or 2; g and i are each independently 0 or
- V 5 is -CH 2 -, -CHV 5' -, or -CV 5' V 5' -;
- V 6 is -CH 2 -, -CHV 6' , -CV 6' v 6' -;
- V 7 is -CH 2 -, -CHV 7' , or -CV 7 ' V 7 ' -;
- each of V 5' , V 6' , and V 7' is independently selected from halo, hydroxy, C 1 -C 6 alkyl, halo (C 1 -C 6 ) alkyl, hydroxy (C 1 -C 6 ) alkyl, C 1 -C 6 , alkoxy, C 1 -C 6 alkylthio, amino, or cyano;
- T and W are independently -S-, -S(O)-, -S(O) 2 -, - O-, -NH-, or -(V 9 )-;
- V 9 is C 1 -C 6 alkyl, aryl (C 1 -C 6 ) alkyl, aryl, or acyl;
- f, g, h, i and j must be 2, 3, 4, or 5;
- V 5 is -CV 5' v 5' -
- V 6 must be -CH 2 - or - CHV 6' -
- V 7 must be -CH 2 - or -CHV 7' -;
- V 6 is -CV 6' v 6' -
- V 5 must be -CH 2 - or - CHV 5' -
- V 7 must be -CH 2 - or -CHV 7' -;
- V 7 is -CV 7' v 7' -
- V 5 must be -CH 2 - or - CHV 5' -
- V 6 must be -CH 2 - or -CHV 6' -;
- V 4 , V 3 , V 0 , V 1 , V 5 , T, V 6 , W, V 7 , f, g, h, i, j, are defined as they are above for formula 2, including their
- V 3' , t, V 5' , V 6' , V 7' , and V 9' definitions of V 3' , t, V 5' , V 6' , V 7' , and V 9' ,
- V A is an amino-protecting group
- U in reaction 1-3 above represents the presence of double bonds between, for example, V 5 and V 6 , V 5 and V 5 , or V 7 and V 6 and the like, where g is 0, h is 0 and f is 2, or i is O,
- Reaction Scheme II is accomplished by carrying out reactions 1-3 (or 1-5) in sequential order.
- the intermediate compound may be isolated, if desired, by procedures known in the art, for example, the compound may be crystallized and then collected by filtration, or the reaction solvent may be removed by extraction, evaporation or decantation.
- the intermediate compound may be further purified, if desired, by common techniques such as crystallization or chromatography over solid supports such as silica gel or alumina, before carrying out the next step of the reaction scheme.
- Reaction II.1 is typically carried out by activating the carboxylic acid moiety using, for example, DCC or a mixed
- anhydride such as isobutyl
- reaction followed by reaction with a primary or secondary amine having the formula NV 0 V 1 where V 0 and V 1 are as defined above for formula (2).
- the reaction is typically carried out in a nonpolar aprotic solvent or mixture of solvents in the presence or absence of an acid scavenger at a temperature of from about -20°C to about 25°C to afford the corresponding amide.
- Suitable solvents for this reaction include ethers and chlorinated hydrocarbons, preferably diethyl ether, chloroform, or methylene chloride.
- this reaction is carried out in the presence of an acid scavenger such as a tertiary amine, preferably triethylamine.
- an acid scavenger such as a tertiary amine, preferably triethylamine.
- the amide afforded by this reaction may be isolated or further reacted as shown in Reaction II.2.
- Reaction II.2 is typically carried out by reacting the compound obtained from Reaction II.1 using the procedures detailed in Comprehensive Organic Synthesis, "Heteroatom Manipulation", Barry M. Trost, ed., volume 6, pages 736-746, (1991).
- an appropriately substituted monocyclic ring is reacted with an aldehyde, such as formaldehyde or trichloroacetaldehyde, in the presence of an acid.
- the acid may be used as a solvent.
- Typical acids include hydrochloric acid, hydrobromic acid, sulfuric acid, acetic acid, trifluoroacetic acid, and the like.
- a co-solvent may optionally be added to the reaction mixture.
- co-solvent choice is not critical so long as the co-solvent employed is inert to the ongoing reaction, and the reactants are sufficiently solubilized to effect the desired reaction.
- Typical solvents for this reaction include halogenated solvents such as methylene chloride, trichloroethane, carbontetrachloride, and the like.
- the aldehyde may be produced in situ using for example, dimethoxymethane and a suitable acid.
- reaction II.3 the compound isolated from reaction II.2 is reduced to provide a saturated heterocyclic compound as depicted above.
- Catalytic hydrogenation is a preferred method of
- Typical catalysts include palladium catalysts, rhodium catalysts (for example rhodium on aluminum) and rhenium catalysts.
- Preferred catalysts include palladium-on-carbon.
- solvents for this reaction include the C 1 -C 4 alcohols,
- the reaction is typically carried out under an atmosphere of hydrogen from about 1000 to about 4000 psi at a temperature of from about 25°C to about 150°C. Preferably, the reaction is carried out under an atmosphere of hydrogen from about 2000 to about 3000 psi at a temperature of from about 50°C to 100°C.
- the catalyst is generally employed in a amount ranging from about equimolar proportions to about a
- Reactions II.4 and II.5 may be used to prepare compounds of formula (3) which correspond to compounds of formula (2) where
- V 3 and V 4 are as defined above for formula (2), including their definitions of V 3' and t.
- Reaction II.4 is a standard amino deprotection reaction using procedures and methods known in the art to afford the
- the compound isolated from II.3 may be deprotected using trimethylsilyliodide (TMSI) in an aprotic solvent or mixture of solvents at a
- Typical solvents include methylene chloride, acetonitrile trichloroethane, and the like.
- Reaction II.5 the epoxide prepared in Reaction A.5, above, in which Q 3 of Reaction A.5 is replaced by V 3 , is reacted with the compound isolated from Reaction II.4 in an alcoholic solvent at a temperature of from about 20°C to 100°C.
- Solvent choice is not critical so long as the solvent employed is inert to the ongoing reaction, and the reactants are sufficiently solubilized to effect the desired reaction.
- Typical solvents for this reaction include the alcohols, preferably isopropanol or ethanol.
- the reaction is preferably carried out at a temperature of about 80°C.
- reaction II.5 may optionally be deprotected to provide a compound of formula (3) where V A is hydrogen.
- the epoxide used in Reaction II.5 may be synthesized using Reaction Scheme A above in which Q 3 of Scheme A is replaced by V 3 .
- this reactant may be prepared by further
- carbocyclic or heterocyclic compound for example, carbocyclic or heterocyclic compounds of the formula
- an oxidizing agent such as selenium dioxide or potassium permanganate at temperatures of from about 0°C to 200°C in a mutually inert solvent, such as water or diphenylether.
- a second method for preparing compounds of the formula (IB) involves protecting an appropriately substituted carboxylated carbocyclic or heterocyclic group with a carboxy-protecting group, and then further substituting the carbocyclic or heterocyclic group using procedures known in the art.
- the carboxy-protecting group may then be removed using procedures known in the art to provide the desired carboxylic acid reactant of formula (IB).
- carboxy-protecting groups include methyl, p-nitrobenzyl, p- methylbenzyl, p-methoxybenzyl, 3,4-dimethoxybenzyl, 2,4-dimethoxybenzyl, 2,4,6-trimethoxybenzyl, 2,4,6-trimethylbenzyl, pentamethylbenzyl, 3,4-methylenedioxybenzyl, benzhydryl, 4,4'-dimethoxybenzhydryl, 2,2',4,4'-tetramethoxybenzhydryl, t-butyl, t-amyl, trityl, 4-methoxytrityl, 4,4'-dimethoxy-trityl, 4,4',4'trimethoxytrityl, 2-phenylprop-2-yl, trimethylsilyl, t-butyld
- a preferred procedure for protecting the carboxy moiety involves the acid activation of the carboxy moiety, followed by the formation of an amide.
- the carboxy moiety may be converted to an acyl halide, acyl anhydride, acyl imidazole and the like, preferably in the presence of an acid scavenger to form an activated carboxy moiety.
- an acid scavenger preferably in the presence of an acid scavenger to form an activated carboxy moiety.
- a commercially available acid chloride is typically employed, obviating the need for further acid activation.
- Preferred acid scavengers are the
- trialkylamines preferably triethylamine.
- the reaction is typically carried out in an aprotic solvent such as diethylether, methylene chloride or the like.
- a preferred solvent is methylene chloride.
- Solvent choice is not critical so long as the solvent employed is inert to the ongoing reaction, and the reactants are sufficiently solubilized to effect the desired reaction.
- the activated carboxy moiety is then reacted with an amine, R 11 -NH 2 , for example aniline, in an aprotic solvent to provide an amide reactant
- the amide reactant may be further substituted by ortho deprotonation of the group
- the amide reactant is generally deprotonated twice using two equivalents of a strong base such as n-butyl lithium or sec-butyl lithium relative to the amide reactant, optionally in the presence of a metal coordinating agent such as tetramethylethylenediamine (TMEDA).
- a metal coordinating agent such as tetramethylethylenediamine (TMEDA).
- TEDA tetramethylethylenediamine
- the reaction is typically carried out in an aprotic solvent, preferably an ether such as diethylether, tetrahydrofuran or the like at a temperature from about -78°C to about 25°C.
- the resultant compound may then be hydrolyzed using
- a suitable hydrolysis involves exposing the amide reactant to a strong mineral acid, organic acid, or a mineral acid/organic mixture at a temperature from about 100°C to about 160°C.
- Typical acids which may be used in this reaction include hydrobromic acid, acetic acid, hydrochloric acid and the like.
- a sealed tube may
- a third method for preparing the substituted carboxylic acid reactant of formula (IB) involves diazotization of an aniline, followed by a quenching of the resultant diazonium salt.
- the amino moiety of the aniline reactant is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- Nitrous acid may be produced in si tu by treating sodium nitrite with an aqueous solution of a strong acid such as hydrochloric acid, or sulfuric acid. This reaction is typically carried out at or below 5°C.
- the diazonium salt is then quenched by reaction with suitable reagent to provide the desired substituted aromatic system.
- suitable quenching reagents include water, cyanide, halide, aqueous sulfuric acid, and the like. Typically, the reaction will be heated to facilitate the desired reaction.
- carboxylation may be accomplished using a number of different reagents.
- the carboxylation may be accomplished using a number of different reagents. For example, the
- carbocyclic or heterocyclic reagent may be reacted with phosgene, oxalyl chloride, urea hydrochloride, or N,N-diethylcarbamoyl chloride in the presence of Friedel-Crafts catalysts.
- a variation of this method involves reacting the carbocyclic or heterocyclic reagent with an alkyl thiolchloroformate (RSCOCl), or a carbamoyl chloride (H 2 NCOCl) to provide an amide and a thiol ester,
- RSCOCl alkyl thiolchloroformate
- H 2 NCOCl carbamoyl chloride
- the amide and thiol ester may then be hydrolyzed to provide the desired carboxy group. March, at 491.
- Friedel-Crafts catalysts include the Lewis acids, such as aluminum bromide (AlBr 3 ), aluminum chloride (AlCl 3 ), iron (III) chloride (FeCl 3 ), boron trichloride (BCl 3 ), boron
- the quinoline carboxylic acid reactants may be prepared by reacting an appropriately substituted aniline with glycerol using the Skraup reaction disclosed in Bradford, L. et al., J. Chem. Soc., 1947, p 437.
- 3-amino benzoic acid may be reacted with glycerol in the presence of an oxidizing agent such as m-nitro benzene sulfonic acid or sodium m-nitro benzene sulfonate in a 60-75% aqueous solution of sulfuric acid to provide the desired carboxy-substituted quinoline.
- the reaction is typically carried out at a temperature from about 35°C to reflux temperature for one to six hours, preferably from about 50°C to reflux temperature for two to four hours.
- the resultant reactants may then be reduced or hydrogenated using procedures known in the art. See e.g., March, at 700.
- a preferred procedure involves catalytic hydrogenation, for example by combining the quinoline carboxylic acid reactant with hydrogen gas in the presence of a catalyst.
- a preferred catalyst is palladium-on-carbon.
- Typical solvents suitable for use in this reaction include any organic solvent such as ethyl acetate.
- Solvent choice is not critical so long as the solvent employed is inert to the ongoing reaction.
- the reaction is generally
- the compounds of formula IA, in which Q 3 is replaced by R 1 can be prepared according to the following Reaction Scheme B.
- R is an amino-protecting group
- R 1 , R 3 , and T 2 are as defined above for formula 1(B), including the definition of R 4 , R 5 , R 6 , and p.
- Reaction Scheme B is accomplished by carrying out reactions 1-6 in sequential order. Once a reaction is complete, the intermediate compound may be isolated, if desired by
- the compound may be crystallized and then collected by filtration, or the reaction solvent may be removed by extraction, evaporation or decantation.
- the intermediate compound may be further purified, if desired, by common techniques such as crystallization or chromatography over solid supports such as silica gel or alumina, before carrying out the next step of the reaction scheme.
- reaction B.1 the reaction is typically carried out by activating, that is, converting, a suitably substituted:
- Reaction B.2 the acyl chloride or acyl bromide, prepared in Reaction B.1, typically is reacted with ammonia or a primary or secondary amine having the formula
- R 4 , R 5 , R 6 and p are as defined above for formula 1(B), in a nonpolar aprotic solvent or mixture of solvents in the presence or absence of an acid scavenger to afford the corresponding amide.
- the reaction is typically carried out at a temperature of from about -20°C to about 25°C.
- Typical solvents for this reaction include ethers and chlorinated hydrocarbons, preferably
- this reaction is carried out in the presence of an acid scavenger such as a tertiary amine, preferably triethylamine.
- an acid scavenger such as a tertiary amine, preferably triethylamine.
- Reaction B.3 the amide prepared in Reaction B.2, is reacted with a strong base in the presence of a solubilizing agent to afford the corresponding anion which is then reacted in
- Reaction B.4 with a Weinreb amide to afford a ketone is typically carried out in an aprotic solvent at a temperature of from about -78°C to about 0°C.
- Typical bases used in Reaction B.3 include lithium amide bases and alkyl lithium bases, preferably C 1 -C 4 alkyl lithium bases and lithium di(C 1 -C 6 ) alkylamide bases.
- Typical solubilizing agents for Reaction 3 are tetramethyl (C 1 -C 4 )alkylenediamines, preferably tetramethylethylenediamine.
- Reaction B.4 typically is carried out in an aprotic solvent at a temperature from about -80°C to about -40°C.
- Typical solvents for Reactions B.3 and B.4 include ethers, preferably tetrahydrofuran.
- the anion is generally employed in an amount ranging from about equimolar proportions to about a three molar excess of the anion, preferably in about a two molar excess of the anion relative to the Weinreb amide reactant.
- Reaction B.5 the ketone prepared in Reaction B.3, is reduced to the corresponding alcohol using a suitable reducing agent.
- the reaction is carried out in a protic solvent at a temperature of from about -25°C to about 25°C.
- Typical reducing agents for this reaction include sodium borohydride, lithium borohydride, diisobutylaluminum hydride, and sodium bis (2-methoxyethoxy) aluminum hydride.
- a preferred reducing agent is sodium borohydride.
- Typical protic solvents for this reaction include alcohols, preferably ethanol.
- Reaction B.6 is a standard amino deprotection reaction using procedures and methods known in the art to afford the
- This amine which is used in Reaction I above.
- This amine may be reacted without purification, but it is preferably purified first.
- the Weinreb amide used as a reactant in Reaction B.4 is typically prepared by reacting an amino-protected amino acid with N-methoxy-N-methyl-amine in the presence of a promoting agent, an acid scavenger, and a coupling agent.
- the reaction is typically carried out in an aprotic solvent or mixture of solvents at a temperature of from about -25°C to 25°C.
- a preferred promoting agent for this reaction is HOBT.H 2 O.
- Preferred acid scavengers are the tertiary alkylamines, preferably triethylamine or N-methyl-morpholine.
- a preferred coupling reagent is ethyl
- the thioanion compound is preferably formed by reacting the corresponding thiol with a strong base, such as sodium hydride or potassium hydride. This reaction is typically carried out in an aprotic solvent at a temperature from about 0°C to about 40°C and under an inert atmosphere, such as nitrogen. Typical solvents for this reaction include ethers, preferably tetrahydrofuran.
- the desired amide reactant is then formed by reacting the resulting carboxylic acid reactant with N-methoxy-N-methyl-amine in the presence of a promoting agent, an acid scavenger and a coupling agent
- the compounds of formula (IA), where R 1 replaces Q 3 and where R 1 is -S-aryl may be prepared in Scheme B using the procedures detailed in Photaki, JACS, 85, 1123 (1963), and Sasaki, N.A. et al., Tetrahedron Letters, 28, 6069 (1987).
- the compounds may be prepared by reacting doubly protected serine (carboxy-protected and amino-protected) with toluenesulfonyl chloride in the presence of dimethylaminopyridine (DMAP) and an acid scavenger such as pyridine in an aprotic solvent such as methylene chloride to form the corresponding toluenesulfonate compound which may then be reacted with an appropriately substituted thioanion having the structure, -S-aryl.
- DMAP dimethylaminopyridine
- an acid scavenger such as pyridine
- an aprotic solvent such as methylene chloride
- the thioanion compound is preferably formed by reacting the corresponding thiol with a strong base as described above.
- the carboxy-protecting group may then be removed from the resulting doubly protected arylthioalanine using conditions known in the art.
- an intermediate for making compounds of the present invention is prepared as follows.
- the intermediate has the formula 4 :
- R 1 is aryl, or -S-aryl
- R 10 is hydrogen or an amino-protecting group
- R 0 is C 1 -C 4 alkyl or -CH 2 -pyridyl
- R 3 is a group having the structure
- p 4 or 5 ;
- R 4 at each occurrence is independently hydrogen, C 1 -C 6 alkyl or hydroxy(C 1 -C 4 )alkyl;
- R 5 and R 6 are independently selected from hydrogen, hydroxy, C 1 -C 6 , alkyl, C 1 -C 6 alkoxy, or hydroxy (C 1 -C 4 ) alkyl;
- the intermediate having the formula 4 is typically made by the process comprising: (a) reducing a compound of the formula
- R b is an amino protecting group
- NMR spectra were obtained on a Bruker Corp. 270 MHz instrument or on a General Electric QE-300 300 MHz instrument. The chemical shifts are expressed in delta values (ppm downfield from tetramethylsilane). MS(FD) spectra were taken on a Varian-MAT 731 Spectrometer using carbon dendrite emitters. EIMS spectra were obtained on a CEC 21-110 instrument from Consolidated
- IR (CHCl 3 ) 3600-3100 (br.), 2929, 2865, 1671, 1515, 1455,
- IR (CHCD) 3630, 3412, 3011, 1720, 1502, 1236, 1044 cm -1 .
- IR (CHCI 3 ) 3640, 3022, 2976, 1720, 1502, 1235, 1045 cm -1 .
- the subtitled compound of Preparation 4E (371 mg, .0.77 mmol) was deprotected substantially as detailed in Preparation 1B, using 110 mg of 10% palladium-on-carbon and hydrogen gas in 20 mL of ethanol to provide 260 mg of a white foam.
- reaction mixture was reacted for approximately thirty minutes, concentrated under reduced pressure, redissolved in 500 mL of methylene chloride and then washed sequentially with water, hydrochloric acid (pH 2), saturated sodium bicarbonate, water, 1M potassium hydroxide, and brine, dried over sodium sulfate, and concentrated to provide 68.5 g of a white solid.
- This residue was redissolved in 4 mL of ammonium hydroxide.
- the resultant solution was extracted four times with 10 mL portions of a 10% solution of isopropanol in chloroform.
- the combined organic layers were dried over sodium sulfate, filtered and concentrated under reduced pressure to provide a residue.
- This residue was purified using radial chromatograghy (2 mm plate; gradient eluent of 10-30% methanol in methylene chloride containing 1% ammonium hydroxide) to provide 65 mg of a light yellow foam.
- a solution was prepared containing 50 mg (0.081 mmol) of the subtitled intermediate of Preparation 7A and 1 mL of a 38% aqueous hydrobromic acid solution. in acetic acid.
- the resultant reaction mixture was allowed to react at room temperature for approximately 1 hour and then was concentrated under reduced pressure to provide a residue. This residue was slurried with toluene and then concentrated under reduced pressure to provide 61 mg of the desired subtitled intermediate. This compound was used crude without purification in Example 9.
- the desired subtitled intermediate was prepared substantially in accordance with the procedure detailed in Procedure 2A, using 13.1 mL (127 mmol) of thiophenol, 4.6 g (117 mmol) of a 60% sodium hydride solution and 25.6 g (116 mmol) of L-N (benzyloxycarbonyl)-serine ⁇ -lactone in 450 mL of tetrahydrofuran to provide a residue.
- This residue was purified using flash chromatography (gradient eluent of 0-2% acetic acid in a 4:1 methylene chloride/ethyl acetate mixture) to provide 27.9 g of a white solid.
- the desired subtitled compound was prepared substantially in accordance with the procedure detailed in Procedure 2B, using 12.1 g (37 mmol) of the subtitled compound of Preparation 8A, 5.09 mL (37 mmol) of triethylamine, 7.13 mL (55 mmol) isobutyl
- the desired subtitled compound was prepared substantially in accordance with the procedure detailed in Procedure 2C, using 22.3 g (63 mmol) of the subtitled compound of Preparation 8B and small quantities of hydrochloric acid (gas) in 400 mL of diethylether to provide 21 g of a white solid. This solid was used without further purification.
- the desired subtitled compound was prepared substantially in accordance with the procedure detailed in Procedure 2D, using 21 g (58 mmol) of the subtitled compound of Preparation 8C, and 2.4 g (63 mmol) of sodium borohydride in 300 mL of tetrahydrofuran to provide a residue. This residue was purified using flash
- the desired subtitled compound was prepared substantially in accordance with the procedure detailed in Procedure 2E, using 8.3 g (23 mmol) of the subtitled compound of Preparation 8D, 1.4 g (25 mmol) of potassium hydroxide in 400 mL of ethanol to provide a residue. This residue was purified using flash chromatography (gradient eluent of 0-2% ethyl acetate in methylene chloride) to provide 6.4 g of a white solid.
- the desired subtitled compound was prepared substantially in accordance with the procedure detailed in Procedure 2F, using 6.3 g (19 mmol) of the subtitled compound of Preparation 8E, 5 g (21 mmol) of [3S- (3R* , 4aR*, 8aR*) ]-decahydroisoquinoline-3-N-t-butylcarboxamide in 300 mL of ethanol to provide a residue.
- This residue was purified using flash chromatography (gradient eluent of 0-20% ethyl acetate in methylene chloride) to provide 4.3 g of a white solid.
- the desired subtitled compound was prepared substantially in accordance with the procedure detailed in Procedure 2G using 1 g
- the resulting reaction mixture was reacted for approximately thirty minutes and then diluted with 1N sodium bicarbonate.
- the resultant layers were separated and the organic layer was washed sequentially with water, IM sodium hydroxide and then brine, dried over sodium sulfate, filtered and then reduced to dryness under reduced pressure to provide 31.6 g of an off-white solid.
Abstract
Description
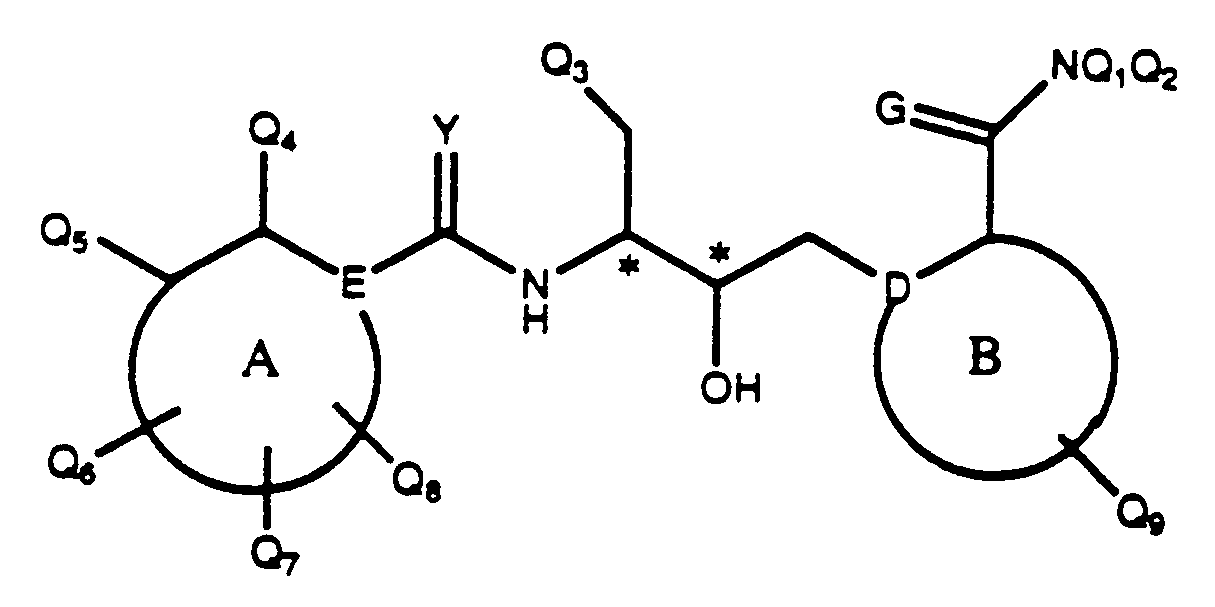































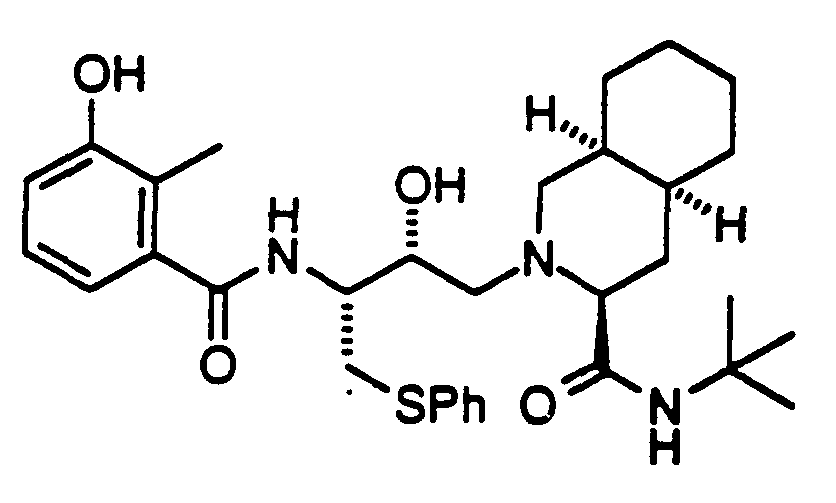
























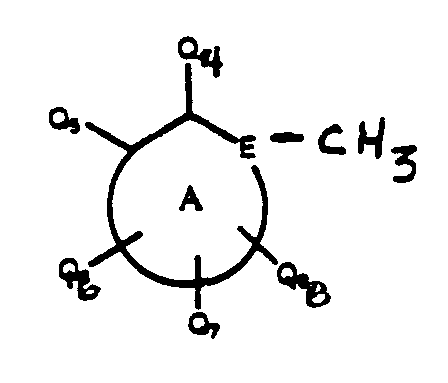

















































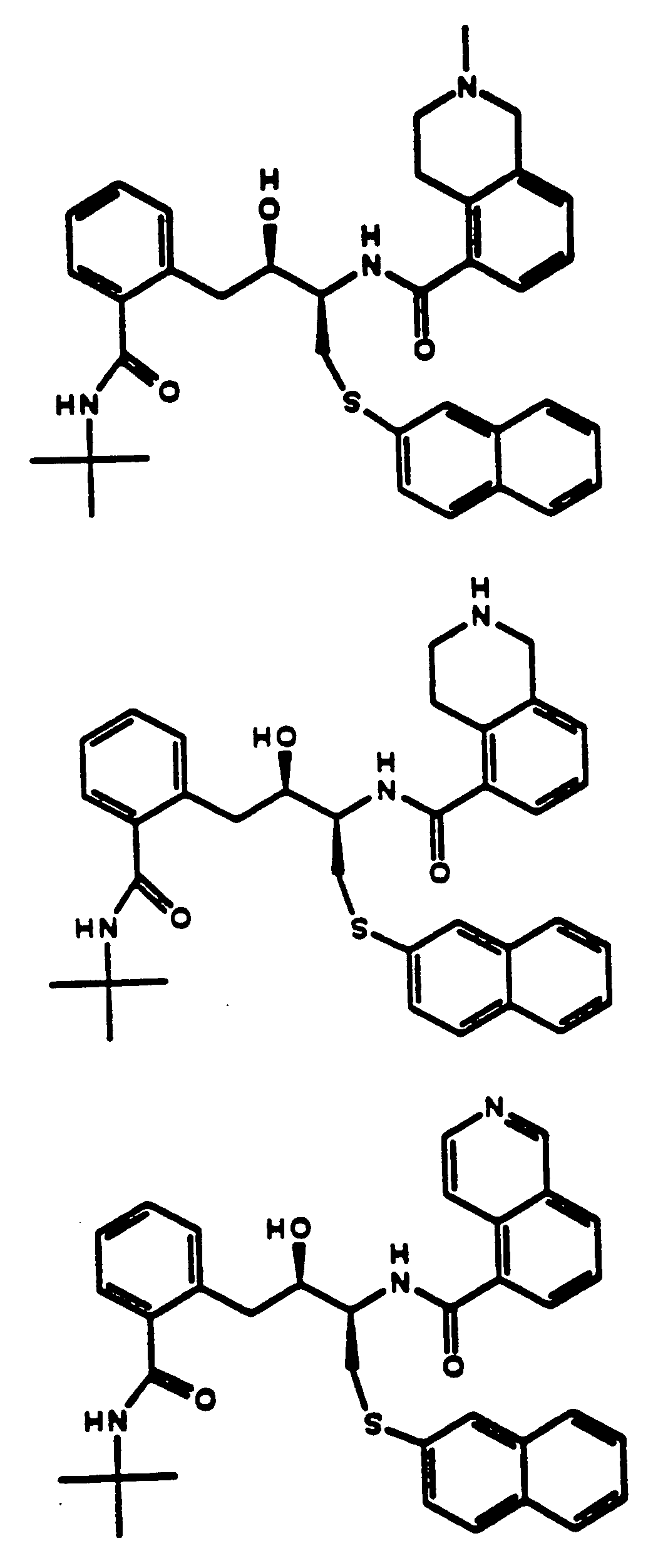



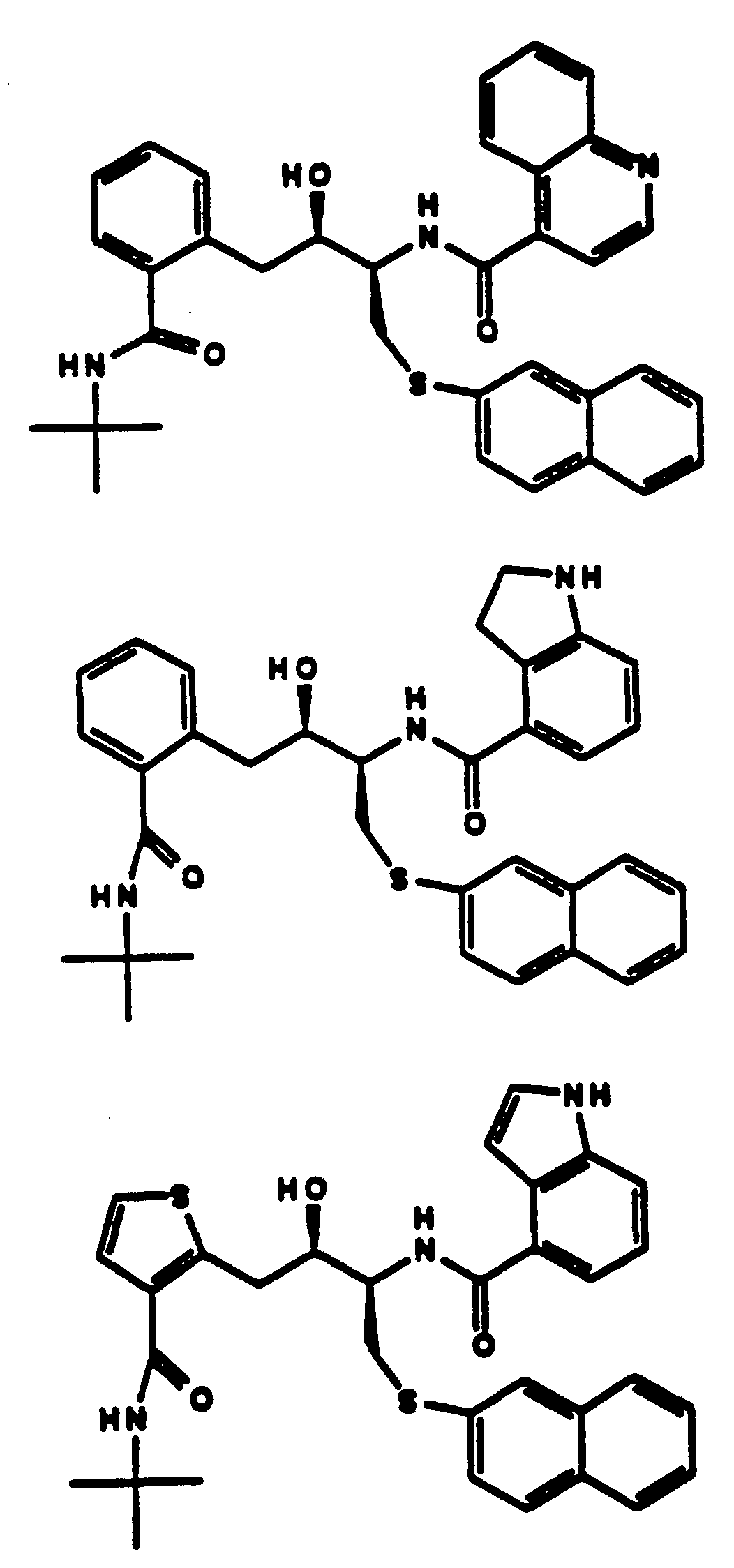










Claims






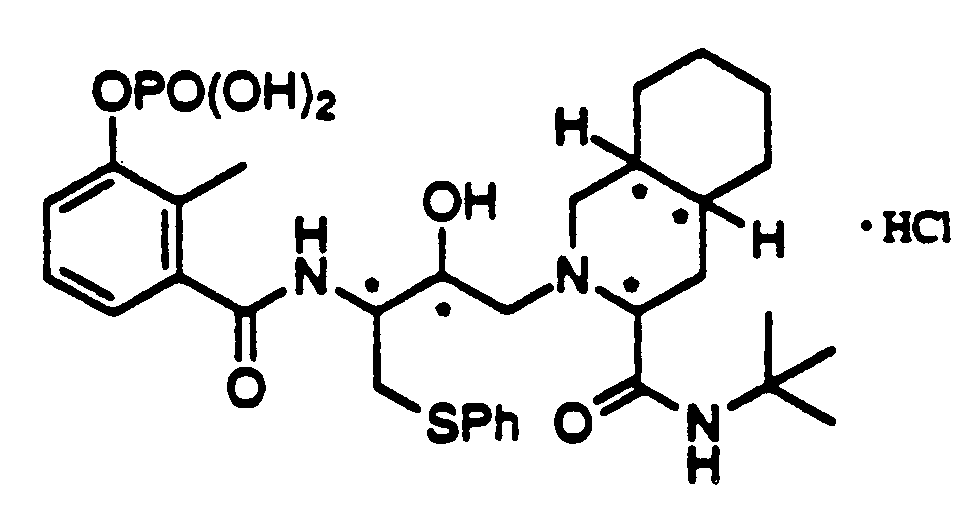





















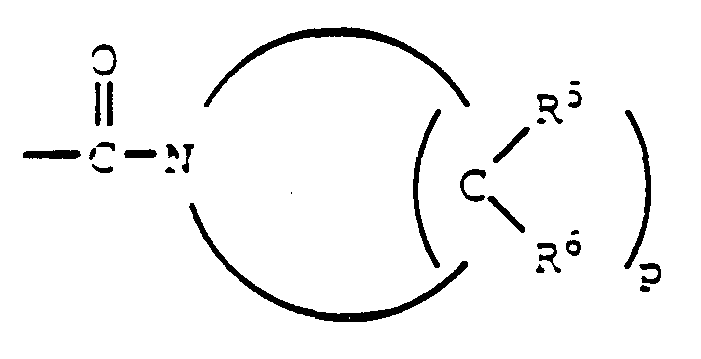


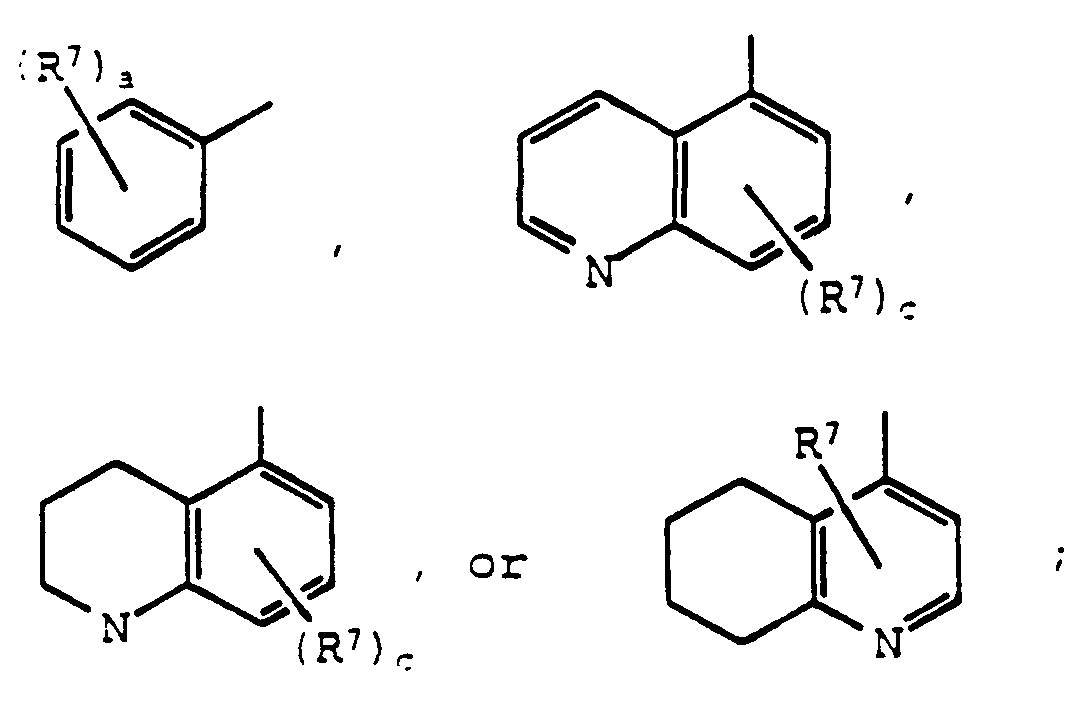


Priority Applications (27)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| UA96041689A UA56984C2 (en) | 1993-10-07 | 1994-07-10 | HIV protease inhibitors, a pharmaceutical composition and a method for inhibition of HIV protease |
| EP94930609A EP0722439B1 (en) | 1993-10-07 | 1994-10-07 | Hiv protease inhibitors |
| HU9600908A HU226814B1 (en) | 1993-10-07 | 1994-10-07 | Hiv protease inhibitors based on decahydroisoquinoline-carboxamide derivatives and use thereof |
| AT94930609T ATE222240T1 (en) | 1993-10-07 | 1994-10-07 | HIV - PROTEASE INHIBITORS |
| TJ96000281A TJ339B (en) | 1993-10-07 | 1994-10-07 | A compound pharmaceutical composition and method for hiv protease inhibitors |
| APAP/P/1996/000844A AP600A (en) | 1993-10-07 | 1994-10-07 | HIV Protease inhibitors. |
| AU79674/94A AU694746B2 (en) | 1993-10-07 | 1994-10-07 | HIV protease inhibitors |
| BR9407782A BR9407782A (en) | 1993-10-07 | 1994-10-07 | HIV protease inhibitors |
| JP7511006A JP2951724B2 (en) | 1993-10-07 | 1994-10-07 | HIV protease inhibitor |
| MD96-0141A MD1507G2 (en) | 1993-10-07 | 1994-10-07 | Inhibitors, pharmaceutical composition on base thereof and process for HIV protease inhibition |
| DK94930609T DK0722439T3 (en) | 1993-10-07 | 1994-10-07 | HIV protease inhibitors |
| HU0900219A HU227885B1 (en) | 1993-10-07 | 1994-10-07 | Hiv protease inhibitor carboxamide-derivatives with hiv protease inhibitor effect |
| SK439-96A SK284115B6 (en) | 1993-10-07 | 1994-10-07 | HIV protease inhibitors |
| RU96109378A RU2139280C1 (en) | 1993-10-07 | 1994-10-07 | Compound, pharmaceutical composition and method of inhibition of hiv-protease activity |
| CA002173328A CA2173328C (en) | 1993-10-07 | 1994-10-07 | Hiv protease inhibitors |
| DE69431193T DE69431193T2 (en) | 1993-10-07 | 1994-10-07 | HIV PROTEASE INHIBITORS |
| RO96-00738A RO119363B1 (en) | 1993-10-07 | 1994-10-07 | Decahydroisoquinoline derivative, pharmaceutical composition and use thereof |
| NZ275633A NZ275633A (en) | 1993-10-07 | 1994-10-07 | N-(3-hetero)cyclyl-2-hydroxy propyl)-(hetero)cyclyl carboxamide derivatives and analogues |
| PL94313871A PL185647B1 (en) | 1993-10-07 | 1994-10-07 | Inhibitors of hiv protease |
| SI9430421T SI0722439T1 (en) | 1993-10-07 | 1994-10-07 | Hiv protease inhibitors |
| EE9600091A EE05399B1 (en) | 1993-10-07 | 1994-10-07 | HIV protease inhibitors |
| SK493-2002A SK284116B6 (en) | 1993-10-07 | 1994-10-07 | Carboaxamide compounds, pharmaceutical composition containing thereof and use thereof |
| KR1019960700897A KR100190517B1 (en) | 1993-10-07 | 1996-02-23 | Hiv protease inhibitors |
| BG100455A BG62567B1 (en) | 1993-10-07 | 1996-03-27 | Hiv-protease inhibitors |
| FI961449A FI114794B (en) | 1993-10-07 | 1996-03-29 | HIV protease inhibitors |
| NO961382A NO307050B1 (en) | 1993-10-07 | 1996-04-03 | HIV protease inhibitors |
| HK98114956A HK1013650A1 (en) | 1993-10-07 | 1998-12-23 | Hiv protease inhibitors |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US13354393A | 1993-10-07 | 1993-10-07 | |
| US13369693A | 1993-10-07 | 1993-10-07 | |
| US08/190,764 US5484926A (en) | 1993-10-07 | 1994-02-02 | HIV protease inhibitors |
| US08/190,764 | 1994-02-02 | ||
| US08/133,543 | 1994-02-02 | ||
| US08/133,696 | 1994-02-02 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1995009843A1 true WO1995009843A1 (en) | 1995-04-13 |
Family
ID=27384442
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1994/011307 WO1995009843A1 (en) | 1993-10-07 | 1994-10-07 | Hiv protease inhibitors |
Country Status (36)
| Country | Link |
|---|---|
| US (14) | US5484926A (en) |
| EP (3) | EP1340744B1 (en) |
| JP (2) | JP2951724B2 (en) |
| KR (1) | KR100190517B1 (en) |
| CN (2) | CN1195737C (en) |
| AP (1) | AP600A (en) |
| AT (3) | ATE286025T1 (en) |
| AU (1) | AU694746B2 (en) |
| BG (1) | BG62567B1 (en) |
| BR (1) | BR9407782A (en) |
| CA (2) | CA2268709C (en) |
| CZ (1) | CZ290417B6 (en) |
| DE (3) | DE69434977T2 (en) |
| DK (2) | DK0722439T3 (en) |
| EE (1) | EE05399B1 (en) |
| ES (3) | ES2287387T3 (en) |
| FI (1) | FI114794B (en) |
| GE (2) | GEP20002209B (en) |
| HK (3) | HK1013650A1 (en) |
| HU (2) | HU227885B1 (en) |
| MD (1) | MD1507G2 (en) |
| MY (1) | MY138860A (en) |
| NO (1) | NO307050B1 (en) |
| NZ (2) | NZ329626A (en) |
| OA (1) | OA10718A (en) |
| PL (1) | PL185647B1 (en) |
| PT (2) | PT722439E (en) |
| RO (1) | RO119363B1 (en) |
| RU (1) | RU2139280C1 (en) |
| SI (2) | SI0722439T1 (en) |
| SK (2) | SK284116B6 (en) |
| TJ (1) | TJ339B (en) |
| TW (1) | TW432049B (en) |
| UA (1) | UA56984C2 (en) |
| UY (1) | UY23840A1 (en) |
| WO (1) | WO1995009843A1 (en) |
Cited By (55)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0741719A1 (en) * | 1994-02-02 | 1996-11-13 | Eli Lilly And Company | Intermediate and process for making |
| EP0744948A1 (en) * | 1994-02-02 | 1996-12-04 | Eli Lilly And Company | Protease inhibitors |
| EP0746320A1 (en) * | 1994-02-02 | 1996-12-11 | Eli Lilly And Company | Hiv protease inhibitors and intermediates |
| WO1997011938A1 (en) * | 1995-09-26 | 1997-04-03 | Japan Tobacco Inc. | Process for producing amide derivatives and intermediate compounds |
| WO1997011937A1 (en) * | 1995-09-26 | 1997-04-03 | Japan Tobacco Inc. | Process for producing amide derivatives and intermediates |
| WO1998040357A2 (en) * | 1997-03-13 | 1998-09-17 | Agouron Pharmaceuticals, Inc. | Hiv protease inhibitors |
| DE19730602A1 (en) * | 1997-07-17 | 1999-01-21 | Clariant Gmbh | 3-Acetoxy-2-methylbenzoic acid chloride and a process for its preparation |
| US5962725A (en) * | 1996-09-05 | 1999-10-05 | Agouron Pharmaceuticals, Inc. | Intermediate compounds useful for making HIV protease inhibitors such as nelfinavir |
| WO1999054297A1 (en) * | 1998-04-16 | 1999-10-28 | Nagase & Company, Ltd. | Process for preparing chloro alcohol derivatives and intermediates |
| US6001851A (en) * | 1997-03-13 | 1999-12-14 | Agouron Pharmaceuticals, Inc. | HIV protease inhibitors |
| US6084107A (en) * | 1997-09-05 | 2000-07-04 | Agouron Pharmaceuticals, Inc. | Intermediates for making HIV-protease inhibitors |
| US6107511A (en) * | 1998-07-29 | 2000-08-22 | Kaneka Corporation | Process for the purification or isolation of (2S,3R)-1-halo-2-hydroxy-3-(protected amino)4-phenylthiobutanes or optical antipodes thereof |
| US6117999A (en) * | 1996-09-05 | 2000-09-12 | Agouron Phramaceuticals, Inc. | Methods of making HIV-protease inhibitors and intermediates for making HIV-protease inhibitors |
| US6197966B1 (en) | 1998-01-16 | 2001-03-06 | Japan Tobacco Inc. | Production method of optically active amino alcohols |
| US6232333B1 (en) | 1996-11-21 | 2001-05-15 | Abbott Laboratories | Pharmaceutical composition |
| US6319946B1 (en) | 1999-02-12 | 2001-11-20 | Vertex Pharmaceuticals Incorporated | Inhibitors of aspartyl protease |
| US6436989B1 (en) | 1997-12-24 | 2002-08-20 | Vertex Pharmaceuticals, Incorporated | Prodrugs of aspartyl protease inhibitors |
| WO2002064553A1 (en) * | 2001-02-14 | 2002-08-22 | Kureha Chemical Industry Company, Limited | Process for preparation of halogenoalcohol derivatives |
| US6613743B2 (en) | 1998-06-19 | 2003-09-02 | Vertex Pharmaceuticals Incorporated | Sulfonamide inhibitors of aspartyl protease |
| US6703403B2 (en) | 1995-06-29 | 2004-03-09 | Abbott Laboratories | Method for improving pharmacokinetics |
| US6846813B2 (en) * | 2000-06-30 | 2005-01-25 | Pharmacia & Upjohn Company | Compounds to treat alzheimer's disease |
| BG64457B1 (en) * | 1996-03-22 | 2005-03-31 | Glaxo Group Limited | Hiv protease inhibitor-containing pharmaceutical form for oral administration |
| US6878728B1 (en) | 1999-06-11 | 2005-04-12 | Vertex Pharmaceutical Incorporated | Inhibitors of aspartyl protease |
| US6953858B2 (en) | 2001-06-11 | 2005-10-11 | Agouron Pharmaceuticals, Inc. | HIV protease inhibitors, compositions containing the same, their pharmaceutical uses and materials for their synthesis |
| WO2006127133A2 (en) | 2005-04-01 | 2006-11-30 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| WO2007087504A2 (en) | 2006-01-25 | 2007-08-02 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| WO2007087505A2 (en) | 2006-01-25 | 2007-08-02 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| WO2007092729A2 (en) | 2006-02-02 | 2007-08-16 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| EP1917958A2 (en) | 2000-01-19 | 2008-05-07 | Abbott Laboratories | Improved HIV protease inhibitors pharmaceutical formulations |
| WO2009151695A1 (en) | 2008-03-13 | 2009-12-17 | Wellstat Therapeutics Corporation | Compounds and method for reducing uric acid |
| WO2010144869A2 (en) | 2009-06-12 | 2010-12-16 | Nektar Therapeutics | Protease inhibitors |
| EP2266946A2 (en) | 2003-02-13 | 2010-12-29 | Wellstat Therapeutics Corporation | Compound For The Treatment Of Metabolic Disorders |
| USRE42889E1 (en) | 1992-08-25 | 2011-11-01 | G.D. Searle Llc | α- and β- amino acid hydroxyethylamino sulfonamides useful as retroviral protease inhibitors |
| US8076514B2 (en) | 2005-07-20 | 2011-12-13 | Eli Lilly And Company | Phenyl compounds and their use in the treatment of Type II diabetes |
| US8106090B2 (en) | 2005-07-20 | 2012-01-31 | Eli Lilly And Company | 1-amino linked compounds |
| US8114901B2 (en) | 2002-07-12 | 2012-02-14 | Eli Lilly And Company | Crystalline 2,5-dione-3-(1-methyl-1H-indol-3-yl)-4-[1-(pyridin-2-ylmethyl)piperidin-4-yl]-1H-indol-3-yl]-1H-pyrrole mono-hydrochloride |
| USRE43596E1 (en) | 1992-08-25 | 2012-08-21 | G.D. Searle Llc | α- and β-amino acid hydroxyethylamino sulfonamides useful as retroviral protease inhibitors |
| US8247436B2 (en) | 2010-03-19 | 2012-08-21 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of CF |
| EP2522367A1 (en) | 2007-03-12 | 2012-11-14 | Nektar Therapeutics | Oligomer-protease inhibitor conjugates |
| US8455497B2 (en) | 1999-06-11 | 2013-06-04 | Vertex Pharmaceuticals Incorporated | Inhibitors of aspartyl protease |
| US8551973B2 (en) | 2008-12-23 | 2013-10-08 | Gilead Pharmasset Llc | Nucleoside analogs |
| US8716263B2 (en) | 2008-12-23 | 2014-05-06 | Gilead Pharmasset Llc | Synthesis of purine nucleosides |
| US8716262B2 (en) | 2008-12-23 | 2014-05-06 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8779144B2 (en) | 2006-03-16 | 2014-07-15 | Evotec (Us) Inc. | Bicycloheteroaryl compounds as P2X7 modulators and uses thereof |
| US8859756B2 (en) | 2010-03-31 | 2014-10-14 | Gilead Pharmasset Llc | Stereoselective synthesis of phosphorus containing actives |
| US9095620B2 (en) | 2008-03-12 | 2015-08-04 | Nektar Therapeutics | Reagents |
| US9227990B2 (en) | 2012-10-29 | 2016-01-05 | Cipla Limited | Antiviral phosphonate analogues and process for preparation thereof |
| US10640457B2 (en) | 2009-12-10 | 2020-05-05 | The Trustees Of Columbia University In The City Of New York | Histone acetyltransferase activators and uses thereof |
| WO2021176369A1 (en) | 2020-03-06 | 2021-09-10 | Pfizer Inc. | Methods of inhibiting sars-cov-2 replication and treating coronavirus disease 2019 |
| WO2021209563A1 (en) | 2020-04-16 | 2021-10-21 | Som Innovation Biotech, S.A. | Compounds for use in the treatment of viral infections by respiratory syndrome-related coronavirus |
| US11426412B2 (en) | 2017-10-18 | 2022-08-30 | Jubilant Epipad LLC | Imidazo-pyridine compounds as PAD inhibitors |
| US11459338B2 (en) | 2017-11-24 | 2022-10-04 | Jubilant Episcribe Llc | Heterocyclic compounds as PRMT5 inhibitors |
| US11529341B2 (en) | 2018-03-13 | 2022-12-20 | Jubilant Prodel LLC | Bicyclic compounds as inhibitors of PD1/PD-L1 interaction/activation |
| US11629135B2 (en) | 2017-11-06 | 2023-04-18 | Jubilant Prodell Llc | Pyrimidine derivatives as inhibitors of PD1/PD-L1 activation |
| US11833156B2 (en) | 2017-09-22 | 2023-12-05 | Jubilant Epipad LLC | Heterocyclic compounds as pad inhibitors |
Families Citing this family (91)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| USH1649H (en) * | 1987-07-31 | 1997-05-06 | Barrish; Joel C. | HIV protease inhibitor combinations |
| US5484926A (en) * | 1993-10-07 | 1996-01-16 | Agouron Pharmaceuticals, Inc. | HIV protease inhibitors |
| US5514801A (en) * | 1992-12-29 | 1996-05-07 | Monsanto Company | Cyclic sulfone containing retroviral protease inhibitors |
| IL110255A (en) * | 1993-07-16 | 1998-12-06 | Merck & Co Inc | Formation and resolution of 2-tert-butylcarboxamido-piperazine |
| US20030207813A1 (en) * | 1996-12-09 | 2003-11-06 | G.D. Searle | Retroviral protease inhibitor combinations |
| UA49803C2 (en) * | 1994-06-03 | 2002-10-15 | Дж.Д. Сьорль Енд Ко | Method for treatment of retrovirus infections |
| US5831117A (en) * | 1995-01-20 | 1998-11-03 | G. D. Searle & Co. | Method of preparing retroviral protease inhibitor intermediates |
| US5914332A (en) * | 1995-12-13 | 1999-06-22 | Abbott Laboratories | Retroviral protease inhibiting compounds |
| DE69731715T2 (en) * | 1996-06-05 | 2005-11-03 | Sega Enterprises, Ltd. | IMAGE PROCESSOR, IMAGE PROCESSING, GAME COMPUTER AND RECORD MEDIUM |
| UA67727C2 (en) * | 1996-09-05 | 2004-07-15 | Агурон Фармасевтікелс, Інк. | Intermediate compounds for preparation of hiv-protease inhibitors and methods for the preparation of hiv-protease inhibitors |
| US5705647A (en) * | 1996-09-05 | 1998-01-06 | Agouron Pharmaceuticals, Inc. | Intermediates for making HIV-protease inhibitors |
| DE19704885C1 (en) * | 1997-02-11 | 1998-06-10 | Clariant Gmbh | Preparation of 3-acetoxy-2-methyl-benzoic acid |
| EP0975601B1 (en) * | 1997-04-10 | 2003-03-12 | F. Hoffmann-La Roche Ag | Process for making a butylthio-isoquinoline and intermediates therefor |
| US6130348A (en) | 1997-04-10 | 2000-10-10 | Hoffmann-La Roche Inc. | Process for a phenylthiobutyl-isoquinoline and intermediates therefor |
| US6123694A (en) * | 1997-05-09 | 2000-09-26 | Paragon Trade Brands | Disposable absorbent article with unitary leg gathers |
| US6124500A (en) * | 1998-03-09 | 2000-09-26 | Rohm And Haas Company | Process for synthesizing benzoic acids |
| FR2776292B1 (en) | 1998-03-20 | 2004-09-10 | Oncopharm | CEPHALOTAXANES SUPPORTING THE SIDE CHAIN AND THEIR SYNTHESIS PROCESS |
| US6538006B1 (en) | 1998-07-08 | 2003-03-25 | Pharmacia Corporation | Retroviral protease inhibitors |
| US7115584B2 (en) * | 1999-01-22 | 2006-10-03 | Emory University | HIV-1 mutations selected for by β-2′,3′-didehydro-2′,3′-dideoxy-5-fluorocytidine |
| US7635690B2 (en) * | 1999-01-22 | 2009-12-22 | Emory University | HIV-1 mutations selected for by β-2′,3′-didehydro-2′,3′-dideoxy-5-fluorocytidine |
| AU3910700A (en) | 1999-03-22 | 2000-10-09 | Immugen Pharmaceuticals, Inc. | Treatment of immune diseases |
| US6566560B2 (en) | 1999-03-22 | 2003-05-20 | Immugen Pharmaceuticals, Inc. | Resorcinolic compounds |
| US6589962B1 (en) | 1999-07-20 | 2003-07-08 | Merck & Co., Inc. | Alpha-hydroxy-gamma-[[(carbocyclic-or heterocyclic-substituted)amino]carbonyl]alkanamide derivatives and uses thereof |
| EP1202626A4 (en) * | 1999-07-20 | 2002-10-30 | Merck & Co Inc | Alpha-hydroxy-gamma-[[(carbocyclic-or heterocyclic-substituted)amino]carbonyl]alkanamide derivatives and uses thereof |
| KR100339831B1 (en) | 1999-08-18 | 2002-06-07 | 김태성 | New ethyl arizidine derivatives and their preparation methods |
| US6403799B1 (en) | 1999-10-21 | 2002-06-11 | Agouron Pharmaceuticals, Inc. | Methods for the preparation of intermediates in the synthesis of HIV-protease inhibitors |
| ATE377011T1 (en) | 1999-11-24 | 2007-11-15 | Merck & Co Inc | GAMMA-HYDROXY-2-(FLUORALKYLAMINOCARBONYL)-1-PIPERAZINE PENTANAMIDE AS AN HIV PROTEASE INHIBITOR |
| US6992081B2 (en) | 2000-03-23 | 2006-01-31 | Elan Pharmaceuticals, Inc. | Compounds to treat Alzheimer's disease |
| CA2401749A1 (en) * | 2000-03-23 | 2001-09-27 | Elan Pharmaceuticals, Inc. | Compounds and methods to treat alzheimer's disease |
| AU2001259817A1 (en) | 2000-05-04 | 2001-11-12 | The Government Of The United States Of America, As Represented By The Secretary, Department Of Health And Human Services, The National Institutes Of Health | Methods of and compounds for inhibiting calpains |
| US20020198207A1 (en) * | 2000-05-18 | 2002-12-26 | Kath John Charles | Novel Hexanoic acid derivatives |
| WO2002002505A2 (en) | 2000-06-30 | 2002-01-10 | Elan Pharmaceuticals, Inc. | Compounds to treat alzheimer's disease |
| EP1666452A2 (en) | 2000-06-30 | 2006-06-07 | Elan Pharmaceuticals, Inc. | Compounds to treat Alzheimer's disease |
| PE20020276A1 (en) | 2000-06-30 | 2002-04-06 | Elan Pharm Inc | SUBSTITUTE AMINE COMPOUNDS AS ß-SECRETASE INHIBITORS FOR THE TREATMENT OF ALZHEIMER |
| US20030096864A1 (en) * | 2000-06-30 | 2003-05-22 | Fang Lawrence Y. | Compounds to treat alzheimer's disease |
| US7425537B2 (en) * | 2000-08-22 | 2008-09-16 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | SH2 domain binding inhibitors |
| WO2002026224A2 (en) * | 2000-09-28 | 2002-04-04 | Immugen Pharmaceuticals, Inc. | Methods and compounds for inhibiting eicosanoid metabolism and platelet aggregation |
| AU2002213429A1 (en) * | 2000-09-28 | 2002-04-08 | Immugen Pharmaceuticals, Inc. | Antiviral methods and compounds |
| IT1318986B1 (en) * | 2000-10-09 | 2003-09-19 | Archimica S P A Ora Clariant L | PROCEDURE FOR THE PREPARATION OF (S) -N-TERBUTYL-1,2,3,4-TETRAIDROISOCHINOLIN-3-CARBOXYAMIDE. |
| WO2002066952A2 (en) * | 2000-10-19 | 2002-08-29 | Target Discovery, Inc | Mass defect labeling for the determination of oligomer sequences |
| MXPA04000140A (en) * | 2001-06-27 | 2004-06-03 | Elan Pharm Inc | Beta-hydroxyamine derivatives useful in treatment of alzheimer's disease. |
| US20070213407A1 (en) * | 2001-06-29 | 2007-09-13 | Elan Pharmaceuticals And Pharmacia & Upjohn Company Llc | Compounds to treat Alzheimer's disease |
| US20030191121A1 (en) * | 2001-08-09 | 2003-10-09 | Miller Ross A. | Piperazine carboxamide intermediates of HIV protease inhibitors and processes for their preparation |
| US20030068655A1 (en) * | 2001-09-12 | 2003-04-10 | Protiveris, Inc. | Microcantilever apparatus and methods for detection of enzymes |
| US6696494B2 (en) | 2001-10-22 | 2004-02-24 | Enanta Pharmaceuticals, Inc. | α-hydroxyarylbutanamine inhibitors of aspartyl protease |
| EP1490090A4 (en) | 2002-02-22 | 2006-09-20 | New River Pharmaceuticals Inc | Active agent delivery systems and methods for protecting and administering active agents |
| US20040067216A1 (en) * | 2002-02-22 | 2004-04-08 | Karki Shyam B. | Hiv protease inhibitors supported on cation exchange resins for oral administration |
| US7157489B2 (en) * | 2002-03-12 | 2007-01-02 | The Board Of Trustees Of The University Of Illinois | HIV protease inhibitors |
| US20030232101A1 (en) * | 2002-03-18 | 2003-12-18 | Immugen Pharmaceuticals, Inc. | Topical formulations of resorcinols and cannibinoids and methods of use |
| DE10212885A1 (en) * | 2002-03-22 | 2003-10-02 | Bayer Ag | Process for the preparation of 3-hydroxy-2-methylbenzoic acid |
| EP1371626A3 (en) * | 2002-06-13 | 2004-07-21 | Bayer Chemicals AG | Process for the preparation of 3-Alkoxy-2-methylbenzoic acids |
| DE10226219A1 (en) * | 2002-06-13 | 2004-01-08 | Bayer Ag | Process for the preparation of 3-acyloxy-2-methylbenzoic acids |
| DE50302717D1 (en) * | 2002-08-21 | 2006-05-11 | Lanxess Deutschland Gmbh | Chiral diphosphorus compounds and their transition metal complexes |
| JP2006525307A (en) * | 2003-05-08 | 2006-11-09 | ファイザー インコーポレイテッド | Intermediates useful for the synthesis of HIV-protease inhibitors and methods for their preparation |
| WO2005004868A1 (en) * | 2003-07-15 | 2005-01-20 | Arigen, Inc. | Anti-coronavirus drug |
| US7211588B2 (en) * | 2003-07-25 | 2007-05-01 | Zentaris Gmbh | N-substituted indolyl-3-glyoxylamides, their use as medicaments and process for their preparation |
| US8377952B2 (en) * | 2003-08-28 | 2013-02-19 | Abbott Laboratories | Solid pharmaceutical dosage formulation |
| US8025899B2 (en) * | 2003-08-28 | 2011-09-27 | Abbott Laboratories | Solid pharmaceutical dosage form |
| RS51799B (en) | 2004-07-27 | 2011-12-31 | Gilead Sciences Inc. | Nucleoside phosphonate conjugates as anti hiv agents |
| US8367832B2 (en) * | 2004-08-23 | 2013-02-05 | Mylan Laboratories Limited | Crystalline forms of Nelfinavir mesylate |
| JP4788958B2 (en) * | 2006-02-28 | 2011-10-05 | 東亞合成株式会社 | Antiviral peptides and uses thereof |
| JP4831410B2 (en) * | 2006-02-28 | 2011-12-07 | 東亞合成株式会社 | Antiviral peptides and antiviral agents |
| US20080161324A1 (en) * | 2006-09-14 | 2008-07-03 | Johansen Lisa M | Compositions and methods for treatment of viral diseases |
| WO2008041087A1 (en) * | 2006-10-06 | 2008-04-10 | Aurobindo Pharma Limited | An improved process for preparation of amorphous nelfinavir mesylate |
| US20110009411A1 (en) * | 2007-06-29 | 2011-01-13 | Gilead Sciences ,Inc. | Therapeutic compositions and the use thereof |
| AP2965A (en) * | 2007-06-29 | 2014-09-30 | Gilead Sciences Inc | Therapeutic compositions and the use thereof |
| US20110105477A1 (en) * | 2007-09-14 | 2011-05-05 | The Regents Of The University Of Michigan | Compositions and methods relating to hiv protease inhibition |
| JP2010540517A (en) * | 2007-09-25 | 2010-12-24 | メルク・シャープ・エンド・ドーム・コーポレイション | HIV protease inhibitor |
| US8173621B2 (en) | 2008-06-11 | 2012-05-08 | Gilead Pharmasset Llc | Nucleoside cyclicphosphates |
| AR074506A1 (en) | 2008-12-09 | 2011-01-19 | Gilead Sciences Inc | TOLL TYPE RECEIVERS MODULATORS |
| SG171308A1 (en) * | 2008-12-11 | 2011-07-28 | Shionogi & Co | Synthesis of carbamoylpyridone hiv integrase inhibitors and intermediates |
| CA2762582A1 (en) | 2009-05-27 | 2010-12-02 | Merck Sharp & Dohme Corp. | Hiv protease inhibitors |
| ES2907862T3 (en) * | 2009-12-10 | 2022-04-26 | Univ Columbia | Histone acetyltransferase activators and uses thereof |
| DE102010004957A1 (en) | 2010-01-14 | 2011-07-21 | Universitätsklinikum Jena, 07743 | Biologically active molecules for influencing virus, bacterial, parasite-infected cells and / or tumor cells and methods for their use |
| US20110223131A1 (en) | 2010-02-24 | 2011-09-15 | Gilead Sciences, Inc. | Antiviral compounds |
| TWI577377B (en) | 2010-09-16 | 2017-04-11 | Viiv醫療保健公司 | Pharmaceutical compositions |
| US9079834B2 (en) | 2010-10-28 | 2015-07-14 | Merck Canada Inc. | HIV protease inhibitors |
| EP2771332B1 (en) | 2011-10-26 | 2016-06-29 | Merck Canada Inc. | Thiophen and thiazol sulfonamid derivatives as HIV protease inhibitors for the treatment of AIDS |
| BR112015005347A2 (en) | 2012-09-11 | 2017-08-08 | Merck Sharp & Dohme Corp E Merck Canada Inc | compound, pharmaceutical composition, and method for treatment or prophylaxis of HIV infection or for treatment, prophylaxis, or delay in the onset of AIDS |
| RU2505286C1 (en) | 2012-12-29 | 2014-01-27 | Открытое Акционерное Общество "Фармасинтез" | Pharmaceutical composition for treating hiv infection, method for preparing it, and method of treating |
| WO2015013835A1 (en) | 2013-07-31 | 2015-02-05 | Merck Sharp & Dohme Corp. | Piperazine derivatives as hiv protease inhibitors |
| RU2543322C1 (en) * | 2013-09-19 | 2015-02-27 | Открытое Акционерное Общество "Фармасинтез" | Pharmaceutical composition for treating hiv-infection, method for preparing and method of using |
| US9737545B2 (en) | 2013-12-19 | 2017-08-22 | Merck Sharp & Dohme Corp. | HIV protease inhibitors |
| EP3082822B1 (en) | 2013-12-19 | 2020-01-15 | Merck Sharp & Dohme Corp. | Hiv protease inhibitors |
| WO2015134366A1 (en) | 2014-03-06 | 2015-09-11 | Merck Sharp & Dohme Corp. | Hiv protease inhibitors |
| US10138255B2 (en) | 2014-03-10 | 2018-11-27 | Merck Sharp & Dohme Corp. | Piperazine derivatives as HIV protease inhibitors |
| WO2016001907A1 (en) | 2014-07-02 | 2016-01-07 | Prendergast Patrick T | Mogroside iv and mogroside v as agonist/stimulator/un-blocking agent for toll-like receptor 4 and adjuvant for use in human/animal vaccine and to stimulate immunity against disease agents. |
| ES2930667T3 (en) | 2014-07-11 | 2022-12-21 | Gilead Sciences Inc | Toll-like receptor modulators for the treatment of HIV |
| EP3212196A4 (en) | 2014-10-29 | 2018-07-11 | Wisconsin Alumni Research Foundation | Boronic acid inhibitors of hiv protease |
| US20180263985A1 (en) | 2015-09-15 | 2018-09-20 | Gilead Sciences, Inc. | Modulators of toll-like receptors for the treatment of hiv |
| US10539397B2 (en) | 2017-04-12 | 2020-01-21 | Wilcox Industries Corp. | Modular underwater torpedo system |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0346847A2 (en) * | 1988-06-13 | 1989-12-20 | F. Hoffmann-La Roche Ag | Amino acid derivatives |
| EP0539192A1 (en) * | 1991-10-23 | 1993-04-28 | Merck & Co. Inc. | HIV Protease inhibitors |
| EP0560268A1 (en) * | 1992-03-13 | 1993-09-15 | Bio-Mega/Boehringer Ingelheim Research Inc. | Substituted pipecolinic acid derivatives as HIV protease inhibitors |
Family Cites Families (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2448392A1 (en) * | 1979-02-12 | 1980-09-05 | Vilbiss Toussaint De | AUTOMATIC DEVICE FOR PROJECTING COATING PRODUCTS |
| US5142056A (en) * | 1989-05-23 | 1992-08-25 | Abbott Laboratories | Retroviral protease inhibiting compounds |
| IL89900A0 (en) * | 1988-04-12 | 1989-12-15 | Merck & Co Inc | Hiv protease inhibitors useful for the treatment of aids and pharmaceutical compositions containing them |
| IL91307A0 (en) * | 1988-08-24 | 1990-03-19 | Merck & Co Inc | Hiv protease inhibitors and pharmaceutical compositions for the treatment of aids containing them |
| EP0361341A3 (en) * | 1988-09-28 | 1991-07-03 | Miles Inc. | Therapeutics for aids based on inhibitors of hiv protease |
| US5063208A (en) * | 1989-07-26 | 1991-11-05 | Abbott Laboratories | Peptidyl aminodiol renin inhibitors |
| WO1991008221A1 (en) * | 1989-12-04 | 1991-06-13 | Wisconsin Alumni Research Foundation | Peptide inhibitors of hiv protease |
| GB8927915D0 (en) * | 1989-12-11 | 1990-02-14 | Hoffmann La Roche | Novel alcohols |
| GB8927913D0 (en) * | 1989-12-11 | 1990-02-14 | Hoffmann La Roche | Amino acid derivatives |
| CA2032259A1 (en) * | 1989-12-18 | 1991-06-19 | Wayne J. Thompson | Hiv protease inhibitors useful for the treatment of aids |
| CA2056911C (en) * | 1990-12-11 | 1998-09-22 | Yuuichi Nagano | Hiv protease inhibitors |
| CA2060844A1 (en) * | 1991-02-08 | 1992-08-09 | Yabe Yuichiro | Beta-amino-alpha-hydroxycarboxylic acids and their use |
| US5235039A (en) * | 1991-06-10 | 1993-08-10 | Eli Lilly And Company | Substrates for hiv protease |
| US5508407A (en) * | 1991-07-10 | 1996-04-16 | Eli Lilly And Company | Retroviral protease inhibitors |
| CN1071930A (en) * | 1991-07-10 | 1993-05-12 | 伊莱利利公司 | Inhibitor as the human immunodeficiency virus protease who treats acquired immune deficiency syndrome (AIDS) |
| US5220796A (en) | 1991-07-15 | 1993-06-22 | The Boc Group, Inc. | Adsorption condensation solvent recovery system |
| DE4126482A1 (en) * | 1991-08-10 | 1993-02-11 | Bayer Ag | (ALPHA) -TRIFLUORMETHYL-SUBSTITUTED, SATURATED-BICYCLIC AMINES AND METHOD FOR THE PRODUCTION THEREOF |
| US5516784A (en) * | 1991-08-13 | 1996-05-14 | Schering Corporation | Anti-HIV (AIDS) agents |
| EP0534511A1 (en) * | 1991-08-16 | 1993-03-31 | Merck & Co. Inc. | HIV protease inhibitors useful for the treatment of aids |
| US5256783A (en) * | 1991-09-18 | 1993-10-26 | Hoffmann-La Roche Inc. | Method for producing 2-isoquinoline compounds |
| WO1993013066A1 (en) * | 1991-12-20 | 1993-07-08 | Syntex (U.S.A.) Inc. | Cyclic amides of 3-amino-2-hydroxy-carboxylic acids as hiv-protease inhibitors |
| EP1148050A1 (en) * | 1992-05-21 | 2001-10-24 | Monsanto Company | Retroviral protease inhibitors |
| ATE172717T1 (en) * | 1992-08-25 | 1998-11-15 | Searle & Co | HYDROXYETHYLAMINOSULFONAMIDES USABLE AS INHIBITORS OF RETROVIRAL PROTEASES |
| IS2334B (en) * | 1992-09-08 | 2008-02-15 | Vertex Pharmaceuticals Inc., (A Massachusetts Corporation) | Aspartyl protease inhibitor of a new class of sulfonamides |
| US5484926A (en) * | 1993-10-07 | 1996-01-16 | Agouron Pharmaceuticals, Inc. | HIV protease inhibitors |
| EP0604182B1 (en) | 1992-12-22 | 2000-10-11 | Eli Lilly And Company | Inhibitors of HIV protease useful for the treatment of Aids |
| US5554653A (en) * | 1992-12-22 | 1996-09-10 | Eli Lilly And Company | Inhibitors of HIV protease useful for the treatment of AIDS |
| US5475136A (en) * | 1992-12-22 | 1995-12-12 | Eli Lilly And Company | Inhibitors of HIV protease useful for the treatment of AIDS |
| US5846993A (en) * | 1992-12-22 | 1998-12-08 | Agouron Pharmaceuticals, Inc. | HIV protease inhibitors |
| US5733906A (en) * | 1993-10-12 | 1998-03-31 | Eli Lilly And Company | Inhibitors of HIV Protease useful for the treatment of Aids |
| MX9308016A (en) | 1992-12-22 | 1994-08-31 | Lilly Co Eli | HUMAN IMMUNODEFICIENCY VIRUS PROTEASE INHIBITING COMPOUNDS, PROCEDURE FOR THEIR PREPARATION AND PHARMACEUTICAL FORMULATION CONTAINING THEM. |
| MX9308025A (en) * | 1992-12-22 | 1994-08-31 | Lilly Co Eli | HUMAN IMMUNODEFICIENCY VIRUS PROTEASE INHIBITING COMPOUNDS, PROCEDURE FOR SUPREPARATION AND PHARMACEUTICAL FORMULATION CONTAINING THEM. |
| US5434265A (en) * | 1992-12-22 | 1995-07-18 | Eli Lilly And Company | Inhibitors of HIV protease |
| US5491166A (en) * | 1992-12-22 | 1996-02-13 | Eli Lilly And Company | Inhibitors of HIV protease useful for the treatment of AIDS |
| US5480887A (en) * | 1994-02-02 | 1996-01-02 | Eli Lilly And Company | Protease inhibitors |
| US5527829A (en) * | 1994-05-23 | 1996-06-18 | Agouron Pharmaceuticals, Inc. | HIV protease inhibitors |
-
1994
- 1994-02-02 US US08/190,764 patent/US5484926A/en not_active Expired - Lifetime
- 1994-07-10 UA UA96041689A patent/UA56984C2/en unknown
- 1994-10-07 NZ NZ329626A patent/NZ329626A/en not_active IP Right Cessation
- 1994-10-07 AT AT98113006T patent/ATE286025T1/en active
- 1994-10-07 ES ES03011749T patent/ES2287387T3/en not_active Expired - Lifetime
- 1994-10-07 HU HU0900219A patent/HU227885B1/en unknown
- 1994-10-07 AT AT94930609T patent/ATE222240T1/en active
- 1994-10-07 DE DE69434977T patent/DE69434977T2/en not_active Expired - Lifetime
- 1994-10-07 EP EP03011749A patent/EP1340744B1/en not_active Expired - Lifetime
- 1994-10-07 DE DE69431193T patent/DE69431193T2/en not_active Expired - Lifetime
- 1994-10-07 EP EP94930609A patent/EP0722439B1/en not_active Expired - Lifetime
- 1994-10-07 RU RU96109378A patent/RU2139280C1/en active
- 1994-10-07 MY MYPI94002664A patent/MY138860A/en unknown
- 1994-10-07 CZ CZ19961004A patent/CZ290417B6/en not_active IP Right Cessation
- 1994-10-07 SI SI9430421T patent/SI0722439T1/en unknown
- 1994-10-07 ES ES94930609T patent/ES2181725T3/en not_active Expired - Lifetime
- 1994-10-07 AP APAP/P/1996/000844A patent/AP600A/en active
- 1994-10-07 UY UY23840A patent/UY23840A1/en not_active IP Right Cessation
- 1994-10-07 SK SK493-2002A patent/SK284116B6/en not_active IP Right Cessation
- 1994-10-07 RO RO96-00738A patent/RO119363B1/en unknown
- 1994-10-07 DK DK94930609T patent/DK0722439T3/en active
- 1994-10-07 AT AT03011749T patent/ATE362918T1/en not_active IP Right Cessation
- 1994-10-07 WO PCT/US1994/011307 patent/WO1995009843A1/en active IP Right Grant
- 1994-10-07 AU AU79674/94A patent/AU694746B2/en not_active Expired
- 1994-10-07 DK DK98113006T patent/DK0889036T3/en active
- 1994-10-07 CN CNB991051645A patent/CN1195737C/en not_active Expired - Lifetime
- 1994-10-07 BR BR9407782A patent/BR9407782A/en not_active Application Discontinuation
- 1994-10-07 ES ES98113006T patent/ES2236849T3/en not_active Expired - Lifetime
- 1994-10-07 EP EP98113006A patent/EP0889036B1/en not_active Expired - Lifetime
- 1994-10-07 DE DE69434214T patent/DE69434214T2/en not_active Expired - Lifetime
- 1994-10-07 EE EE9600091A patent/EE05399B1/en unknown
- 1994-10-07 JP JP7511006A patent/JP2951724B2/en not_active Expired - Lifetime
- 1994-10-07 PT PT94930609T patent/PT722439E/en unknown
- 1994-10-07 CN CN94193534A patent/CN1046269C/en not_active Expired - Lifetime
- 1994-10-07 PL PL94313871A patent/PL185647B1/en unknown
- 1994-10-07 SI SI9430471T patent/SI0889036T1/en unknown
- 1994-10-07 SK SK439-96A patent/SK284115B6/en not_active IP Right Cessation
- 1994-10-07 TJ TJ96000281A patent/TJ339B/en unknown
- 1994-10-07 CA CA002268709A patent/CA2268709C/en not_active Expired - Lifetime
- 1994-10-07 PT PT98113006T patent/PT889036E/en unknown
- 1994-10-07 GE GEAP19943170A patent/GEP20002209B/en unknown
- 1994-10-07 CA CA002173328A patent/CA2173328C/en not_active Expired - Lifetime
- 1994-10-07 HU HU9600908A patent/HU226814B1/en unknown
- 1994-10-07 NZ NZ275633A patent/NZ275633A/en not_active IP Right Cessation
- 1994-10-07 MD MD96-0141A patent/MD1507G2/en unknown
- 1994-11-19 TW TW083110782A patent/TW432049B/en not_active IP Right Cessation
-
1995
- 1995-06-07 US US08/484,706 patent/US5852043A/en not_active Expired - Lifetime
- 1995-06-07 US US08/478,020 patent/US5827858A/en not_active Expired - Lifetime
- 1995-06-07 US US08/473,363 patent/US5824688A/en not_active Expired - Lifetime
- 1995-06-07 US US08/482,504 patent/US5834467A/en not_active Expired - Lifetime
- 1995-06-07 US US08/481,831 patent/US5952343A/en not_active Expired - Lifetime
- 1995-06-07 US US08/474,138 patent/US5859002A/en not_active Expired - Lifetime
- 1995-06-07 US US08/479,765 patent/US5837710A/en not_active Expired - Lifetime
- 1995-06-07 US US08/478,934 patent/US5827859A/en not_active Expired - Lifetime
- 1995-06-07 US US08/478,600 patent/US6271235B1/en not_active Expired - Lifetime
- 1995-06-07 US US08/478,599 patent/US5827891A/en not_active Expired - Lifetime
-
1996
- 1996-02-23 KR KR1019960700897A patent/KR100190517B1/en not_active IP Right Cessation
- 1996-03-27 BG BG100455A patent/BG62567B1/en unknown
- 1996-03-29 FI FI961449A patent/FI114794B/en not_active IP Right Cessation
- 1996-04-03 OA OA60810A patent/OA10718A/en unknown
- 1996-04-03 NO NO961382A patent/NO307050B1/en not_active IP Right Cessation
-
1998
- 1998-12-23 HK HK98114956A patent/HK1013650A1/en not_active IP Right Cessation
-
1999
- 1999-01-14 HK HK99100152A patent/HK1014950A1/en not_active IP Right Cessation
- 1999-02-04 JP JP06723199A patent/JP3703647B2/en not_active Expired - Lifetime
- 1999-04-01 US US09/283,152 patent/US6162812A/en not_active Expired - Lifetime
-
2001
- 2001-06-21 US US09/885,056 patent/US6525215B2/en not_active Expired - Fee Related
-
2002
- 2002-11-21 US US10/300,638 patent/US6693199B2/en not_active Expired - Fee Related
-
2003
- 2003-11-20 HK HK03108464A patent/HK1056172A1/en not_active IP Right Cessation
-
2008
- 2008-01-08 GE GEAP200810469A patent/GEP20084497B/en unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0346847A2 (en) * | 1988-06-13 | 1989-12-20 | F. Hoffmann-La Roche Ag | Amino acid derivatives |
| EP0539192A1 (en) * | 1991-10-23 | 1993-04-28 | Merck & Co. Inc. | HIV Protease inhibitors |
| EP0560268A1 (en) * | 1992-03-13 | 1993-09-15 | Bio-Mega/Boehringer Ingelheim Research Inc. | Substituted pipecolinic acid derivatives as HIV protease inhibitors |
Cited By (101)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| USRE43596E1 (en) | 1992-08-25 | 2012-08-21 | G.D. Searle Llc | α- and β-amino acid hydroxyethylamino sulfonamides useful as retroviral protease inhibitors |
| USRE42889E1 (en) | 1992-08-25 | 2011-11-01 | G.D. Searle Llc | α- and β- amino acid hydroxyethylamino sulfonamides useful as retroviral protease inhibitors |
| EP0741719A1 (en) * | 1994-02-02 | 1996-11-13 | Eli Lilly And Company | Intermediate and process for making |
| EP0744948A1 (en) * | 1994-02-02 | 1996-12-04 | Eli Lilly And Company | Protease inhibitors |
| EP0746320A1 (en) * | 1994-02-02 | 1996-12-11 | Eli Lilly And Company | Hiv protease inhibitors and intermediates |
| EP0741719A4 (en) * | 1994-02-02 | 1997-04-02 | Lilly Co Eli | Intermediate and process for making |
| EP0744948A4 (en) * | 1994-02-02 | 1997-06-11 | Lilly Co Eli | Protease inhibitors |
| EP0746320A4 (en) * | 1994-02-02 | 1997-06-11 | Lilly Co Eli | Hiv protease inhibitors and intermediates |
| US6703403B2 (en) | 1995-06-29 | 2004-03-09 | Abbott Laboratories | Method for improving pharmacokinetics |
| EP2295052A1 (en) | 1995-06-29 | 2011-03-16 | Abbott Laboratories | Use of ritonavir for improving the pharmacokinetics of drugs metabolized by cytochrome P450 |
| EP2130534A1 (en) | 1995-06-29 | 2009-12-09 | Abbott Laboratories | Use of ritonavir for improving the pharmacokinetics of drugs metabolized by cytochrome P450 |
| US6476232B2 (en) | 1995-09-26 | 2002-11-05 | Japan Tobacco Inc. | 4-(2-amino-1-hydroxyethyl)oxazoline derivative and method for producing same |
| WO1997011937A1 (en) * | 1995-09-26 | 1997-04-03 | Japan Tobacco Inc. | Process for producing amide derivatives and intermediates |
| EP0983999A1 (en) * | 1995-09-26 | 2000-03-08 | Japan Tobacco Inc. | Process for producing amide derivatives and intermediates |
| US6252085B1 (en) | 1995-09-26 | 2001-06-26 | Japan Tobacco Inc. | Production of amide derivatives and intermediates therefor |
| WO1997011938A1 (en) * | 1995-09-26 | 1997-04-03 | Japan Tobacco Inc. | Process for producing amide derivatives and intermediate compounds |
| US6596903B2 (en) | 1995-09-26 | 2003-07-22 | Japan Tobacco Inc. | Methods for producing amide derivative |
| EP0983999A4 (en) * | 1995-09-26 | 2002-09-11 | Japan Tobacco Inc | Process for producing amide derivatives and intermediates |
| US6121468A (en) * | 1995-09-26 | 2000-09-19 | Japan Tobacco Inc. | Process for producing amide derivatives and intermediate compounds |
| US6133461A (en) * | 1995-09-26 | 2000-10-17 | Japan Tobacco Inc. | Process for producing amide derivatives and intermediates therefor |
| US6333416B2 (en) | 1995-09-26 | 2001-12-25 | Japan Tobacco Inc. | (Oxazolin-4-yl) oxirane derivative |
| BG64457B1 (en) * | 1996-03-22 | 2005-03-31 | Glaxo Group Limited | Hiv protease inhibitor-containing pharmaceutical form for oral administration |
| US6605721B2 (en) | 1996-09-05 | 2003-08-12 | Agouron Pharmaceuticals, Inc. | Intermediates for making HIV-protease inhibitors and methods for making HIV-protease inhibitors |
| US6303786B1 (en) | 1996-09-05 | 2001-10-16 | Agouron Pharmaceuticals, Inc. | Processes for making nelfinavir mesylate |
| US6316625B1 (en) | 1996-09-05 | 2001-11-13 | Agouron Pharmaceuticals, Inc. | Methods of making HIV-protease inhibitors and intermediates for making HIV-protease inhibitors |
| US6512135B2 (en) | 1996-09-05 | 2003-01-28 | Agouron Pharmaceuticals, Inc. | Intermediates for making HIV-protease inhibitors and methods for making HIV-protease inhibitors |
| US6117999A (en) * | 1996-09-05 | 2000-09-12 | Agouron Phramaceuticals, Inc. | Methods of making HIV-protease inhibitors and intermediates for making HIV-protease inhibitors |
| US6392067B1 (en) | 1996-09-05 | 2002-05-21 | Agouron Pharmaceuticals, Inc. | Methods of making HIV-protease inhibitors and intermediates for making HIV-protease inhibitors |
| US6407285B1 (en) | 1996-09-05 | 2002-06-18 | Agouron Pharmaceuticals, Inc. | Intermediates for making HIV-protease inhibitors and methods for making HIV-protease inhibitors |
| US6465661B1 (en) | 1996-09-05 | 2002-10-15 | Agouron Pharmaceuticals, Inc. | Methods of making HIV-protease inhibitors and intermediates for making HIV-protease inhibitors |
| US5962725A (en) * | 1996-09-05 | 1999-10-05 | Agouron Pharmaceuticals, Inc. | Intermediate compounds useful for making HIV protease inhibitors such as nelfinavir |
| US6232333B1 (en) | 1996-11-21 | 2001-05-15 | Abbott Laboratories | Pharmaceutical composition |
| US6521651B1 (en) | 1996-11-21 | 2003-02-18 | Abbott Laboratories | Pharmaceutical composition |
| US6458818B1 (en) | 1996-11-21 | 2002-10-01 | Abbott Laboratories | Pharmaceutical composition |
| US6001851A (en) * | 1997-03-13 | 1999-12-14 | Agouron Pharmaceuticals, Inc. | HIV protease inhibitors |
| KR100511089B1 (en) * | 1997-03-13 | 2005-08-31 | 아구론 파마슈티컬스, 인크. | HIV protease inhibitors |
| EA002378B1 (en) * | 1997-03-13 | 2002-04-25 | Агурон Фармасьютикалз, Инк. | Hiv protease inhibitors |
| WO1998040357A2 (en) * | 1997-03-13 | 1998-09-17 | Agouron Pharmaceuticals, Inc. | Hiv protease inhibitors |
| WO1998040357A3 (en) * | 1997-03-13 | 1998-11-26 | Agouron Pharma | Hiv protease inhibitors |
| AP1358A (en) * | 1997-03-13 | 2004-12-03 | Agouron Pharma | 1-Aryl-2-Hydroxy-3-Isoquinoline Carboxamide HIV Protease Inhibitors, Their Preparation and Use. |
| DE19730602A1 (en) * | 1997-07-17 | 1999-01-21 | Clariant Gmbh | 3-Acetoxy-2-methylbenzoic acid chloride and a process for its preparation |
| US6051732A (en) * | 1997-07-17 | 2000-04-18 | Clariant Gmbh | Process for the preparation of 3-acetoxy-2-methylbenzoyl chloride |
| US6084107A (en) * | 1997-09-05 | 2000-07-04 | Agouron Pharmaceuticals, Inc. | Intermediates for making HIV-protease inhibitors |
| US7592368B2 (en) | 1997-12-24 | 2009-09-22 | Vertex Pharmaceuticals Incorporated | Sulphonamide derivatives as prodrugs of aspartyl protease inhibitors |
| US6436989B1 (en) | 1997-12-24 | 2002-08-20 | Vertex Pharmaceuticals, Incorporated | Prodrugs of aspartyl protease inhibitors |
| US6559137B1 (en) | 1997-12-24 | 2003-05-06 | Vertex Pharmaceuticals Incorporated | Sulphonamide derivatives as prodrugs of aspartyl protease inhibitors |
| US6838474B2 (en) | 1997-12-24 | 2005-01-04 | Vertex Pharmaceuticals, Incorporated | Sulphonamide derivatives as prodrugs of aspartyl protease inhibitors |
| US6197966B1 (en) | 1998-01-16 | 2001-03-06 | Japan Tobacco Inc. | Production method of optically active amino alcohols |
| WO1999054297A1 (en) * | 1998-04-16 | 1999-10-28 | Nagase & Company, Ltd. | Process for preparing chloro alcohol derivatives and intermediates |
| US7419967B2 (en) | 1998-06-19 | 2008-09-02 | Vertex Pharmaceuticals Incorporated | Sulfonamide inhibitors of aspartyl protease |
| US6613743B2 (en) | 1998-06-19 | 2003-09-02 | Vertex Pharmaceuticals Incorporated | Sulfonamide inhibitors of aspartyl protease |
| US6107511A (en) * | 1998-07-29 | 2000-08-22 | Kaneka Corporation | Process for the purification or isolation of (2S,3R)-1-halo-2-hydroxy-3-(protected amino)4-phenylthiobutanes or optical antipodes thereof |
| US6617350B2 (en) | 1999-02-12 | 2003-09-09 | Vertex Pharmaceuticals Incorporated | Inhibitors of aspartyl protease |
| US7919523B2 (en) | 1999-02-12 | 2011-04-05 | Vertex Pharmaceuticals, Inc. | Inhibitors of aspartyl protease |
| US6319946B1 (en) | 1999-02-12 | 2001-11-20 | Vertex Pharmaceuticals Incorporated | Inhibitors of aspartyl protease |
| US7442718B2 (en) | 1999-02-12 | 2008-10-28 | Vertex Pharmaceuticals Incorporated | Inhibitors of aspartyl protease |
| US6878728B1 (en) | 1999-06-11 | 2005-04-12 | Vertex Pharmaceutical Incorporated | Inhibitors of aspartyl protease |
| US8455497B2 (en) | 1999-06-11 | 2013-06-04 | Vertex Pharmaceuticals Incorporated | Inhibitors of aspartyl protease |
| EP2269591A2 (en) | 2000-01-19 | 2011-01-05 | Abbott Laboratories | Improved pharmaceutical formulations |
| EP1917958A2 (en) | 2000-01-19 | 2008-05-07 | Abbott Laboratories | Improved HIV protease inhibitors pharmaceutical formulations |
| US6846813B2 (en) * | 2000-06-30 | 2005-01-25 | Pharmacia & Upjohn Company | Compounds to treat alzheimer's disease |
| WO2002064553A1 (en) * | 2001-02-14 | 2002-08-22 | Kureha Chemical Industry Company, Limited | Process for preparation of halogenoalcohol derivatives |
| US6953858B2 (en) | 2001-06-11 | 2005-10-11 | Agouron Pharmaceuticals, Inc. | HIV protease inhibitors, compositions containing the same, their pharmaceutical uses and materials for their synthesis |
| US7179918B2 (en) | 2001-06-11 | 2007-02-20 | Agouron Pharmaceuticals, Inc. | HIV protease inhibitors, compositions containing the same, their pharmaceutical uses and materials for their synthesis |
| US8114901B2 (en) | 2002-07-12 | 2012-02-14 | Eli Lilly And Company | Crystalline 2,5-dione-3-(1-methyl-1H-indol-3-yl)-4-[1-(pyridin-2-ylmethyl)piperidin-4-yl]-1H-indol-3-yl]-1H-pyrrole mono-hydrochloride |
| EP2266946A2 (en) | 2003-02-13 | 2010-12-29 | Wellstat Therapeutics Corporation | Compound For The Treatment Of Metabolic Disorders |
| WO2006127133A2 (en) | 2005-04-01 | 2006-11-30 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| US8076514B2 (en) | 2005-07-20 | 2011-12-13 | Eli Lilly And Company | Phenyl compounds and their use in the treatment of Type II diabetes |
| US8106090B2 (en) | 2005-07-20 | 2012-01-31 | Eli Lilly And Company | 1-amino linked compounds |
| WO2007087504A2 (en) | 2006-01-25 | 2007-08-02 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| WO2007087505A2 (en) | 2006-01-25 | 2007-08-02 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| WO2007092729A2 (en) | 2006-02-02 | 2007-08-16 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| US8779144B2 (en) | 2006-03-16 | 2014-07-15 | Evotec (Us) Inc. | Bicycloheteroaryl compounds as P2X7 modulators and uses thereof |
| US8598364B2 (en) | 2007-03-12 | 2013-12-03 | Nektar Therapeutics | Oligomer-protease inhibitor conjugates |
| US9107956B2 (en) | 2007-03-12 | 2015-08-18 | Nektar Therapeutics | Oligomer-protease inhibitor conjugates |
| EP2522367A1 (en) | 2007-03-12 | 2012-11-14 | Nektar Therapeutics | Oligomer-protease inhibitor conjugates |
| US9095620B2 (en) | 2008-03-12 | 2015-08-04 | Nektar Therapeutics | Reagents |
| WO2009151695A1 (en) | 2008-03-13 | 2009-12-17 | Wellstat Therapeutics Corporation | Compounds and method for reducing uric acid |
| US8716263B2 (en) | 2008-12-23 | 2014-05-06 | Gilead Pharmasset Llc | Synthesis of purine nucleosides |
| US8716262B2 (en) | 2008-12-23 | 2014-05-06 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8957045B2 (en) | 2008-12-23 | 2015-02-17 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US9045520B2 (en) | 2008-12-23 | 2015-06-02 | Gilead Pharmasset Llc | Synthesis of purine nucleosides |
| US8551973B2 (en) | 2008-12-23 | 2013-10-08 | Gilead Pharmasset Llc | Nucleoside analogs |
| WO2010144869A2 (en) | 2009-06-12 | 2010-12-16 | Nektar Therapeutics | Protease inhibitors |
| US11034647B2 (en) | 2009-12-10 | 2021-06-15 | The Trustees Of Columbia University In The City Of New York | Histone acetyltransferase activators and uses thereof |
| US10640457B2 (en) | 2009-12-10 | 2020-05-05 | The Trustees Of Columbia University In The City Of New York | Histone acetyltransferase activators and uses thereof |
| USRE46757E1 (en) | 2010-03-19 | 2018-03-20 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of CF |
| US9365552B2 (en) | 2010-03-19 | 2016-06-14 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of CF |
| US8247436B2 (en) | 2010-03-19 | 2012-08-21 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of CF |
| US10117858B2 (en) | 2010-03-19 | 2018-11-06 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of CF |
| US8476269B2 (en) | 2010-03-19 | 2013-07-02 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of CF |
| US11911371B2 (en) | 2010-03-19 | 2024-02-27 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of chronic bronchitis |
| US8859756B2 (en) | 2010-03-31 | 2014-10-14 | Gilead Pharmasset Llc | Stereoselective synthesis of phosphorus containing actives |
| US9227990B2 (en) | 2012-10-29 | 2016-01-05 | Cipla Limited | Antiviral phosphonate analogues and process for preparation thereof |
| US11833156B2 (en) | 2017-09-22 | 2023-12-05 | Jubilant Epipad LLC | Heterocyclic compounds as pad inhibitors |
| US11426412B2 (en) | 2017-10-18 | 2022-08-30 | Jubilant Epipad LLC | Imidazo-pyridine compounds as PAD inhibitors |
| US11629135B2 (en) | 2017-11-06 | 2023-04-18 | Jubilant Prodell Llc | Pyrimidine derivatives as inhibitors of PD1/PD-L1 activation |
| US11459338B2 (en) | 2017-11-24 | 2022-10-04 | Jubilant Episcribe Llc | Heterocyclic compounds as PRMT5 inhibitors |
| US11529341B2 (en) | 2018-03-13 | 2022-12-20 | Jubilant Prodel LLC | Bicyclic compounds as inhibitors of PD1/PD-L1 interaction/activation |
| WO2021176369A1 (en) | 2020-03-06 | 2021-09-10 | Pfizer Inc. | Methods of inhibiting sars-cov-2 replication and treating coronavirus disease 2019 |
| WO2021209563A1 (en) | 2020-04-16 | 2021-10-21 | Som Innovation Biotech, S.A. | Compounds for use in the treatment of viral infections by respiratory syndrome-related coronavirus |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU694746B2 (en) | HIV protease inhibitors | |
| US5554653A (en) | Inhibitors of HIV protease useful for the treatment of AIDS | |
| US5461154A (en) | Intermediate and process for making | |
| JPH06256277A (en) | Hiv-protease inhibitor being useful for treatment of aids | |
| US5846993A (en) | HIV protease inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 94193534.5 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AM AT AU BB BG BR BY CA CH CN CZ DE DK EE ES FI GB GE HU JP KE KG KP KR KZ LK LR LT LU LV MD MG MN MW NL NO NZ PL PT RO RU SD SE SI SK TJ TT UA UZ VN |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): KE MW SD SZ AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 275633 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1994930609 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 96-0141 Country of ref document: MD |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 961449 Country of ref document: FI |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2173328 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 43996 Country of ref document: SK |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV1996-1004 Country of ref document: CZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 96-00738 Country of ref document: RO |
|
| EX32 | Extension under rule 32 effected after completion of technical preparation for international publication | ||
| LE32 | Later election for international application filed prior to expiration of 19th month from priority date or according to rule 32.2 (b) | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1994930609 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: PV1996-1004 Country of ref document: CZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4932002 Country of ref document: SK |
|
| WWG | Wipo information: grant in national office |
Ref document number: PV1996-1004 Country of ref document: CZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1994930609 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 961449 Country of ref document: FI |