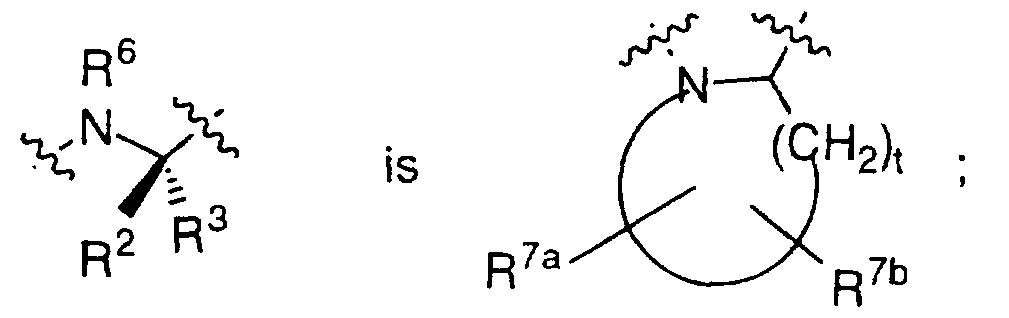TITLE OF THE INVENTION
A METHOD OF TREATING CANCER
BACKGROUND OF THE INVENTION
The present invention relates to a method of treating cancer using a combination of a compound which has Raf antagonist activity and a compound which has famesyl transferase inhibiting activity.
The Raf antagonist compounds used in the present invention demonstrate anti-cancer activity through antagonism of the kinase, Raf . The raf genes code for a family of proteins which can be oncogenically activated through N-terminal fusion, truncation or point mutations. Raf is a member of the MAP Kinase cascade, which also includes MEK's and MAP Kinase (ERK). Raf can be activated and undergoes rapid phosphorylation in response to treatment of cells with PDGF, EGF, insulin, thrombin, endothelin, acidic FGF, CSF1 or TPA, as well as in response to oncoproteins v-fms, v-src, v-sis, Hras and polyoma middle T antigen. Antisense constructs which reduce cellular levels of c-Raf, and hence Raf activity, inhibit the growth of oncogene-transformed rodent fibroblasts in soft agar, while exhibiting little or no general cytotoxicity. Since inhibition of growth in soft agar is highly predictive of tumor responsiveness in whole animals, these studies suggest that the antagonism of Raf is an effective means by which to treat cancers in which Raf plays a role.
Examples of cancers where Raf is implicated through overexpression include cancers of the brain, genitourinary tract, lymphatic system, stomach, larynx and lung. More particularly, such examples include histiocytic lymphoma, lung adenocarcinoma and small cell lung cancers. Additional examples include cancers in which overexpression or activation of Raf-activating oncogenes (e.g., K-ras, erb-B) is observed. More particularly, such cancers include pancreatic and breast carcinoma.
The Ras protein is part of a signalling pathway that links cell surface growth factor receptors to nuclear signals initiating cellular proliferation. Biological and biochemical studies of Ras action
indicate that Ras functions like a G-regulatory protein. In the inactive state, Ras is bound to GDP. Upon growth factor receptor activation, Ras is induced to exchange GDP for GTP and undergoes a conformational change. The GTP -bound form of Ras propagates the growth stimulatory signal until the signal is terminated by the intrinsic GTPase activity of Ras, which returns the protein to its inactive GDP bound form (D.R. Lowy and D.M. Willumsen,
Ann. Rev. Biochem. 62:851-891 (1993)). Activation of Ras leads to activation of multiple intracellular signal transduction pathways, including the MAP Kinase pathway and the Rho/Rac pathway (Joneson et al., Science 271.-810-812).
Mutated ras genes are found in many human cancers, including colorectal carcinoma, exocrine pancreatic carcinoma, and myeloid leukemias. The protein products of these genes are defective in their GTPase activity and constitutively transmit a growth stimulatory signal.
The Ras protein is one of several proteins that are known to undergo post-translational modification. Famesyl-protein transferase utilizes famesyl pyrophosphate to covalently modify the Cys thiol group of the Ras CAAX box with a famesyl group (Reiss et al. , Cell, 62:81 -88 (1990): Schaber et al. , J. Biol. Chem., 265: 14701 - 14704 ( 1990); Schafer et al. , Science, 249: 1 133-1 139 ( 1990); Marine et al., Proc. Natl Acad. Sci USA, 57:7541 -7545 (1990)).
Ras must be localized to the plasma membrane for
both normal and oncogenic functions. At least 3 post-translational modifications are involved with Ras membrane localization, and all 3 modifications occur at the C-terminus of Ras. The Ras C-terminus contains a sequence motif termed a "CAAX" or "Cys-Aaa1-Aaa2-Xaa" box (Cys is cysteine, Aaa is an aliphatic amino acid, the Xaa is any amino acid) (Willumsen et al. , Nature 310:583-586 (1984)). Depending on the specific sequence, this motif serves as a signal sequence for the enzymes farn esyl-protein transferase or geranylgeranyl-protein transferase, which catalyze the alkylation of the cysteine residue of the
CAAX motif with a C15 or C20 isoprenoid, respectively. (S. Clarke., Ann. Rev. Biochem. 61 :355-386 (1992); W.R. Schafer and J. Rine, Ann. Rev. Genetics 30:209-231 (1992)). However, direct inhibition of famesyl-protein transferase would be more specific and attended by fewer side effects than would occur with the required dose of a general inhibitor of isoprene biosynthesis.
Other farnesylated proteins include the Ras-related GTP- binding proteins such as Rho, fungal mating factors, the nuclear lamins, and the gamma subunit of transducin. James, et al., J. Biol Chem. 269, 14182 (1994) have identified a peroxisome associated protein Pxf which is also farnesylated. James, et al., have also suggested that there are farnesylated proteins of unknown structure and function in addition to those listed above.
Inhibitors of famesyl-protein transferase (FPTase) have been described in two general classes. The first class includes analogs of famesyl diphosphate (FPP), while the second is related to protein substrates (e.g., Ras) for the enzyme. The peptide derived inhibitors that have been described are generally cysteine containing molecules that are related to the CAAX motif that is the signal for protein prenylation. (Schaber et al, ibid; Reiss et. al, ibid; Reiss et al., PNAS, 88:132-136 (1991 )). Such inhibitors may inhibit protein prenylation while serving as altemate substrates for the famesyl-protein transferase enzyme, or may be purely competitive inhibitors (U.S.
Patent 5,141 ,851 , University of Texas; N.E. Kohl et al, Science,
260:1934-1931 (1993); Graham, et al., J. Med. Chem., 37, 725 (1994)).
Inhibition of famesyl-protein transferase has been shown to block the growth of ras-transformed cells in soft agar and to modify other aspects of their transformed phenotype. It has also been
demonstrated that certain inhibitors of famesyl-protein transferase selectively block the processing of the Ras oncoprotein intracellularly (N.E. Kohl et al., Science, 260:1934-1937 (1993) and G.L. James et al., Science, 260:1931-1942 (1993). Recently, it has been shown that an inhibitor of famesyl-protein transferase blocks the growth of ras- dependent tumors in nude mice (N.E. Kohl et al., Proc. Natl. Acad. Sci
U.S.A., 91 :9141 -9145 (1994) and induces regression of mammary and salivary carcinomas in ras transgenic mice (N.E. Kohl et al., Nature Medicine, 1 :792-797 (1995).
Indirect inhibition of famesyl-protein transferase in viva has been demonstrated with lovastatin (Merck & Co., Rahway, NJ) and compactin (Hancocket al., ibid; Caseyet al., ibid; Schafer et al., Science 245:319 (1989)). These drugs inhibit HMG-CoA reductase, the rate limiting enzyme for the production of polyisoprenoids including famesyl pyrophosphate. Inhibition of famesyl pyrophosphate
biosynthesis by inhibiting HMG-CoA reductase blocks Ras membrane localization in cultured cells.
A Raf antagonist compound and a famesyl protein transferase inhibitor are used in the present invention to treat cancer, such as in tumor cells which are not particularly Raf or FPTase dependent. The Raf antagonist compound and a famesyl protein transferase inhibiting compound are used in combination.
SUMMARY OF THE INVENTION
A method of treating cancer is disclosed which is comprised of administering to a mammalian patient in need of such treatment an effective amount of a Raf antagonist compound and an effective amount of a famesyl protein transferase inhibiting compound.
DETAILED DESCRIPTION OF THE INVENTION
The present invention relates to a method of treating cancer which is comprised of admininstering to a mammalian patient in need of such treatment an effective amount of a Raf antagonist compound and an effective amount of a famesyl protein transferase inhibiting compound. Any compound which antagonizes Raf and any compound which inhibits famesyl protein transferase can be used.
As used herein the term Raf antagonist is used in the general sense to relate to compounds which antagonize, inhibit or counteract the activity of the raf gene or the protein produced in response thereto.
The term famesyl protein transferase inhibiting compound is likewise used in the general sense and refers to compounds which antagonize, inhibit or counteract the activity of the gene coding famesyl protein transferase or the protein produced in response thereto.
Cancers which are treatable in accordance with the invention described herein include cancers of the brain, genitourinary tract, lymphatic system, stomach, larynx, liver and lung. More particularly, such cancers include histiocytic lymphoma, lung adenocarcinoma and small cell lung cancers. Additional examples include cancers in which overexpression or activation of Raf-activating oncogenes (e.g., K-ras, erb-B) is observed. More particularly, such cancers include pancreatic, mammary and salivary carcinomas, colorectal carcinoma, exocrine pancreatic carcinoma and myeloid leukemias.
Examples of compounds which antagonize Raf are as follows:
(a) a compound represented by formula (I-a):
or a pharmaceutically acceptable salt thereof, wherein:
AR represents an aromatic group containing 6-10 atoms;
X and X' each independently represent -(CH2)m-Y-(CH2)n -, wherein m and n represent integers within the range of from 0 - 4, such that the sum of m and n is from 0 - 6; Y represents a member selected from the group consisting of: a direct bond: O; S(O)y, with y equal to
0, 1 or 2; NRq', with Rq' as defined below; C(O); OC(O); C(O)O; SO
xNRq' with x equal to 1 or 2 and Rq' as defined below; NRq'SO
x; C(O)NRq' and NRq'C(O);
represents a 4 to 10 membered non-aromatic heterocycle containing at least one N atom, and optionally containing 1-2 additional N atoms and 0-1 O or S atom; R
x represents H, C
1-6 alkyl(R
q)
3, OC
1-6 alkyl(Rq)
3 or
C(O)C1-6 alky l(Rq)3; each R and R" independently represents a member selected from the group consisting of: halo; hydroxy; C1-6 alkyl(Rq)3;
OC1-6alkyl(Rq)3; C3-8 cycloalkyl(Rq)3; CN; CONH2; CONHC1-6 alkyl(Rq)3; CON(C1-6alkyl(Rq)3)2; NH2; NHC1-6 alkyl(Rq)3;
N(C1-6alkyl(Rq)3)2; CO2H; CO2C1-6 alkyl(Rq)3; C(O)C1-6
alkyl(Rq)3; aryl(Rq)3; heteroaryl(Rq)3; CF3; SH; NO2; SOyC1-6 alkyl(Rq)3, with y as defined above; SO2NH2; SO2NHC1-6 alkyl(Rq)3; SO2N(C1-6alkyl(Rq)3)2 ; NHSO2C1-6alkyl(Rq)3, NHSO2aryl(Rq)3, NHSO2heteroary(Rq)3, N(Rq')C(O)C1-6alkyl(Rq)3; NRq'C(O)NH (C1-6alkyl(Rq)3);
C2-4 alkenyl(Rq)2-3 and C2-4 alkynyl(Rq)1-3; each R' independently represents a member selected from the group consisting of: CONH2; CONHC1-6 alkyl(Rq)3;
CON(C1-6 alkyl(Rq)3)2; CONHC3-8 cycloalkyl(Rq)3;
CON(C3-8 cycloalkyl(Rq)3)2; CO2H; CO2C1-6aIkyl(Rq)3;
C(O)C1-6 alkyl(Rq)3; CO2C3-8 cycloalkyl(Rq)3;
C(O)C3-8 cycloalkyl(Rq)3; -[C(O)(CH2)j-CR5R6-(CH2)k-NR7]p-R8;
-C(O)C3-8 cycloalkyl(Rq)3; -C(O)heterocyclyl(Rq)3; CON[C1-
6alkyl(Rq)3][C3-8 cycloalkyl(Rq)3]; C(O)aryl(Rq)3,
C(O)heteroaryl(Rq)3;
wherein p represents 1 , 2 or 3;
j and k are integers independently selected from 0 - 3; each R5 and R6 independently represents H, aryl, C1 -6 alkyl(Rq)3, or each CR5R6 taken in combination represents a 3, 4, 5 or 6 membered cycloalkyl or heterocyclyl group, an aryl group or a heteroaryl group, wherein when p equals 1 , at least one of j and k is 1 , 2 or 3; each R7 and R8 independently represents H, C1 -6 alkyl or aryl;
Rq represents a member selected from the group consisting of: Rq'; CN; CO2H; CO2C1 -4 alkyl; C(O)C1 -4 alkyl ; aryl(Ra)3; NH2; NHC1 -6 alkyl(Ra)3; N(C1 -6 alkyl(Ra)3)2; heteroaryl(Ra)3;
CONH2 ; SH ; S(O)y C1-6 alkyl(Ra)3; C(O)NHC1-6 alkyl(Ra )3;
C(O)N(C1-6 aLkyl(Ra)3)2; -heteroalkyl(Ra)3; -NHC(O)NH2;
-NHC(NH)NH2 ;
wherein
independently represent mono or bicyclic ring systems, non-aromatic or partially aromatic, containing from 5-10 ring atoms, 1 -4 of which are N and 0-1 of which are O or S(O)y, with y equal to 0, 1 or 2, optionally containing 1 -2 carbonyl groups; each R
a independently represents a member selected from the group consisting of: H, C
1 -6 alkyl, OC
1 -6 alkyl, aralkyl, substituted aralkyl, heteroaralkyl, substituted heteroaralkyl, aralkoxy, substituted
aralkoxy , halo, hydroxy, CN, CONH
2, CONHC
1 -6 alkyl, CON(C
1 -6 alkyl)
2, CO
2H, CO
2C
1 -6 alkyl, C(O)C
1 -6 alkyl, phenyl, CF
3, SH, NO
2, SO
yC
1 -6 alkyl, with y as defined above; SO
2NH
2, SO
2NHC
1 -6 alkyl, NHSO
2(substituted aryl), NHSO
2(substituted heteroaryl), NHSO
2C
1 -6alkyl, NHSO
2aryl, NHSO
2heteroaryl, NH
2, NHC
1 -6 alkyl, N(C
1 -6 alkyl)
2, NHC(O)C
1 -6 alkyl, NHC(O)NH(C
1 -6 alkyl), C
2-4 alkenyl and C
2-4 alkynyl; and Rq' represents H, OH, C
1 -4 alkyl, -OC
1 -4 alkyl, aryl or C(O)C
1 -4 alkyl;
(b) a compound represented by formula (I-b)
or a pharmaceutically acceptable salt thereof, wherein:
are as defined above with respect to formula (I-a);
each R' independently represents a member selected from the group consisting of: hydroxy; C
1 -6 alkyl(Rq)
3;
C3-8 cycloalkyl(Rq)3; OC1 -6 alkyl(Rq)3; OC3-8 cycloalkyl(Rq)3;
heterocyclyl(Rq)3; CN; NH(Rq"); NHC1 -6 alkyl(Rq)3; N(C1 -6 alkyl(Rq)3)2; NHC3-8 cycloalkyl(Rq)3; N(C3-8 cycloalkyl(Rq)3)2; CF3; SH; NO2; C2-4 alkenyl(Rq)2-3 , aryl(Rq)3 , heteroaryl(Rq)3 ; C2-4 alkynyl(Rq)1 -3 -OC(O) C3-8 cycloalkyI(Rq)3; SO2NH2;
SO2NHC1 -6 alkyl(Rq)3; SO2N(C1 -6 alkyl(Rq)3)2; NHSO2C1 -6 alkyl(Rq)3, NHSO2aryl(Rq)3, NHSO2heteroary(Rq)3,
-OC(O)heterocyclyl(Rq)3; N(Rq')C(O)C1 -6 alkyl(Rq)3;
NRq'C(O)NH(C1 -6 alkyl(Rq)3); -OC(O)C1 -6 alkyl(Rq)3;
-OC(O)aryl(Rq)3, -OC(O)heteroaryl(Rq)3; -C(=NRq')NH2 ;
-C(=Nq')NHC1 -6 alkyl(Rq)3, -C(=Nq')N(C1 -6 alkyl(Rq)3)2;
R5 and R6 are independently H, aryl, C1 -6 alkyl(Rq)3, or CR5R6 in combination represents a 3, 4, 5 or 6 membered cycloalkyl or heterocyclyl group, an aryl group or a heteroaryl group; p represents 1 , 2 or 3, with the proviso that when p represents 1 , CR5R6 represents a 3, 4, 5 or 6 membered cycloalkyl group or a heterocyclyl group, an aryl group or a heteroaryl group, and at least one of j and k is 1 , 2 or 3;
R9 represents H, a negative charge balanced by a positively charged group or a protecting group;
Rq represents a member selected from the group consisting of: Rq'; CN; CO2H; CO2C1 -4 alkyl; C(O)C1 -4 alkyl ; NH(Rq ") ;
aryl(Ra)3; heteroaryl(Ra)3; NHC1 -4 alkyl ; N(C1 -4 alkyl)2 ; CONH2;
SH;S(O)yC1-6alkyl(Ra)3; C(O)NHC1-6 alkyl(Ra)3; C(O)N(C1-6 alkyl(Ra)3)2; NHC(NH)NH2 ; -heteroalkyl(Ra)3; -NHC(O)NH2;
and Rq" represents H, OH or OC
1-4 alkyl, and (c) a compound represented by formula (I-c):
or a pharmaceutically acceptable salt thereof,
wherein:
R1 is 4-pyridyl, pyrimidinyl, quinazolin-4-yl, quinolyl, isoquinolinyl, 1 -imidazolyl or 1-benzimidazolyl which is optionally substituted with one or two substituents each of which is independently selected from C1-4 alkyl, halogen, C1-4 alkoxy, C1-4 alkylthio, NR10R20, or N- heterocyclyl ring which ring has from 5 to 7 members and optionally contains an additional heteroatom selected from oxygen, sulfur or NR22;
R2 is hydrogen, -(CR10R20)n OR12, heterocyclyl, heterocyclyl C1-10 alkyl, C1-10 alkyl, halo-substituted C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, C3-7 cycloalkyl, C3-7 cycloalkyl C1-10 alkyl,
C5-7 cycloalkenyl, aryl, aryl C1-10 alkyl, heteroaryl, heteroaryl C1-10 alkyl, (CR10R20)n'OR13, (CR10R20)n'S(O)mR25,
(CR10R20)n'NHS(O)2R25, (CR10R20)n'NR8R9, (CR10R20)n'NO2, (CR10R20)n'CN, (CR10R20)n'S(O)mNR8R9,
(CR10R20)n'C(Z)R 13, (CR 1 oR20)n'C(Z)OR 13,
(CR10R20)n'NR10C(Z)NR8R9, (CR10R20)n'C(Z)NR13OR12, (CR10R20)n'NR10C(Z)R13, (CR10R20)n'NR10C(Z)NR8R9,
(CR10R20)n'N(OR21)C(Z)NR8R9, (CR10R20)n'N(OR21 )C(Z)R13, (CR10R20)n'C(=NOR21)R1 3, (CR10R20)n'NR10C(=NR27)NR8R9, (CR10R20)n'OC(Z)NR8R9, (CR10R20)n'NR10C(Z)NR8R9,
(CR10R20)n'C(Z)OR 10, 5-(R25)-1,2,4-oxadiazol-3-yl or 4-(R1 2)- 5-(R1 8R19)-4,5-dihydro-1 ,2,4-oxadiazol-3-yl; wherein the aryl,
arylalkyl, heteroaryl, heteroarylalkyl, heterocyclyl or
heterocyclyalkyl moieties may be optionally substituted;
n' is an integer having a value of 1 to 10;
m is 0 or the integer 1 or 2;
R3 is Q-(Y1)t;
Q is an aryl or heteroaryl group;
t is a number having a value of 1 , 2 or 3;
Z is oxygen or sulfur;
n is 0 or an integer from 1 to 10;
Y 1 is independently selected from hydrogen, C1 -5 alkyl, halo- substituted C1 -5 alkyl, halogen, or -(CR10R20)nY2;
Y2 is -OR8, -NO2, -S(O)m'R1 1 , -SR8, -S(O))m'OR8, -S(O)mNR8R9, -NR8R9, -O(CR10R20)nNR8R9, -C(O)R8, -CO2R8,
-CO2(CR10R20)n'CONR8R9, -ZC(O)R8, -CN, -C(Z)NR8R9, NR-NR10C(Z)R8, -C(Z)NR8OR9, -NR10C(Z)NR8R9,
-NR10S(O)mR1 1 , -N(OR21)C(Z)NR8R9, -N(OR21)C(Z)R8, -C(=NOR21)R8, -NR10C(=NR15)SR1 1 , -NR10C(=NR15)NR8R9, -NR10C(=CR14R24)SR1 1 , -NR10C(=CR14R24)NR8R9,
-NR10C(O)C(O)NR8R9, -NR10C(O)C(O)OR10,
-C(=NR13)NR8R9, -C(=NOR13)NR8R9, -C(=NR13)ZR1 1 ,
-OC(Z)NR8R9 , -NR10S(O)mCF3, -NR10C(Z)OR10, 5-(R1 8)- 1 ,2,4-oxadizaoI-3-yl or 4-(R12)-5-(R18R1 9)-4,5-dihydro-1 ,2,4- oxadiazol-3-yl;
m' is a number having a value of 1 or 2;
R4 is phenyl, naphth-1 -yl or naphth-2-yl which is optionally substituted by one or two substituents, each of which is independently selected, and which, for a 4-phenyl, 4-naphth-1 -yl or 5-naphth-1 -yl
substituent, is halo, cyano,-C(Z)NR7R17, -C(Z)OR23,
-(CR10R20)m"'COR36, SR5, -SOR5, OR36, halo-substituted-C 1 -4
alkyl, C1-4 alkyl, -ZC(Z)R36, -NR10C(Z)R23 or
-(CR10R20)m'"NR10R20 and which, for other positions of substitution, is halo, cyano, -C(Z)NR16R26, -C(Z)OR8,
-(CR10R20)m"'COR8, -S(O)mR8, -OR8, halo-substituted-C1-4 alkyl, C1-4 alkyl, -(CR10R20)m"NR10C(Z)R8, -NR10S(O)m'R11,
-NR10S(O)m'NR7R17, -ZC(Z)R8 or -(CR10R20)m'NR16R26; wherein m" is 0 to 5 and m'" is 0 or 1;
R5 is hydrogen, C1-4 alkyl, C2-4 alkenyl, C2-4 alkynyl or NR7R17, excluding the moieties -SR5 being -SNR7R17 and -SOR5 being -SOH;
R6 is C1-4 alkyl, halo-substituted-C1-4 alkyl, C1-4 alkenyl, C2-4
alkynyl or C3-5 cycloalkyl;
R7 and R17 are each independently selected from hydrogen or C1-4 alkyl, or R7 and R17 together with the nitrogen to which they are attached form a heterocyclic ring of 5 to 7 members which ring optionally contains an additional heteroatom selected from oxygen, sulfur or NR22;
R8 is hydrogen, heterocyclyl, heterocyclylalkyl or R11;
R9 is hydrogen, C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, C3-7
cycloalkyl, C5-7 cycloalkenyl, aryl, arylalkyl, heteroaryl or heteroarylalkyl or R8 and R9 may together with the nitrogen to which they are attached form a heterocyclic ring of 5 to 7 members which ring optionally contains an additional heteroatom selected from oxygen, sulfur or NR12;
R10 and R20 are each independently selected from hydrogen and C1-4 alkyl;
R11 is C1-10 alkyl, halo-substituted C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, C3-7 cycloalkyl, C5-7 cycloalkenyl, aryl, arylalkyl, heteroaryl or heteroarylalkyl;
R12 is hydrogen. -C(Z)R13 or optionally substituted C1-4 alkyl,
optionally substituted arylCl .4 alkyl or S(O)2R25;
R13 is hydrogen, C1-10 alkyl, C3-7 cycloalkyl, heterocyclyl,
heterocyclyl C1-10 alkyl, aryl, aryl C1-10 alkyl, heteroaryl or heteroaryl C1-10 alkyl;
R14 and R24 is each independently selected from hydrogen, alkyl, nitro or cyano;
R15 is hydrogen, cyano, C1-4 alkyl, C3-7 cycloalkyl or aryl;
R16 and R26 is each independently selected from hydrogen or
optionally substituted C1-4 alkyl, optionally substituted aryl or optionally substituted aryl-C1-4 alkyl, or together with the nitrogen which they are attached form a heterocyclic ring of 5 to 7 members which ring optionally contains an additional heteroatom selected from oxygen, sulfur or NR12;
R18 and R19 is each independently selected from hydrogen, C1-4 alkyl. substituted alkyl, optionally substituted aryl, optionally substituted arylalkyl or together denote a oxygen or sulfur;
R21 is hydrogen, a pharmaceutically acceptable cation, C1-10 alkyl, C3-7 cycloalkyl, aryl, aryl C1-4 alkyl, heteroaryl, heteroarylalkyl, heterocyclyl, aroyl, or C1-10 alkanoyl;
R22 is R10 or C(Z)-C1-4 alkyl;
R23 is C1-4 alkyl, halo-substituted-C1-4 alkyl or C3-5 cycloalkyl;
R36 is hydrogen or R23;
R25 is C1-10 alkyl, C3-7 cycloalkyl, heterocyclyl, aryl, arylalkyl,
heterocyclyl, heterocyclyl-C1-10 alkyl, heteroaryl or
heteroarylalkyl;
R27 is hydrogen, cyano, C1-4 alkyl, C3-7 cycloalkyl or aryl; or a pharmaceutically acceptable salt thereof.
Examples of famesyl protein transferase inhibiting
compounds include the following:
(a) a compound represented by formula (Il-a) through (II-c):
R
1a and R
1b are independently selected from:
a) hydrogen,
b) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl,
C2-C6 alkynyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-,
c) C1-C6 alkyl unsubstituted or substituted by aryl,
heterocyclyl, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)-NR10-; R2 and R3 are independently selected from: H; unsubstituted or
substituted C1-8 alkyl, unsubstituted or substituted C2-8 alkenyl,
unsubstituted or substituted C
2-8 alkynyl, unsubstituted or substituted aryl, unsubstituted or substituted heterocycle,
wherein the substituted group is substituted with one or more of:
1) aryl or heterocycle, unsubstituted or substituted with: a) C1-4 alkyl,
b) (CH2)pOR6,
c) (CH2)pNR6R7,
d) halogen,
2) C3-6 cycloalkyl,
3) OR6,
R2 and R3 are attached to the same C atom and are combined to form (CH2)u - wherein one of the carbon atoms is optionally replaced by a moiety selected from: O, S(O)m, -NC(O)-, and -N(COR10)-;
R4 and R5 are independently selected from H and CH3; and any two of R2, R3, R4 and R5 are optionally attached to the same carbon atom;
R6, R7 and R7a are independently selected from: H; C1-4 alkyl, C3-6 cycloalkyl, heterocycle, aryl, aroyl, heteroaroyl, arylsulfonyl, heteroarylsulfonyl, unsubstituted or substituted with:
a) C1-4 alkoxy,
b) aryl or heterocycle,
c) halogen,
d) HO, ,
f) - SO2R11 , or g) N(R10)2; or
R6 and R7 may be joined in a ring;
R7 and R7a may be joined in a ring; R8 is independently selected from:
a) hydrogen,
b) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl,
C2-C6 alkynyl, perfluoroalkyl, F, Cl, Br, R10O-,
R11s(O)m-, R10C(O)NR10-, CN, NO2, R10 2N-C(NR10)-,
R10C(O)-, R10OC(O)-, N3, -N(R10)2, or
R11OC(O)NR10-,and
c) C1-C6 alkyl unsubstituted or substituted by aryl,
heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, perfluoroalkyl, F, Cl, Br, R10O-, R11S(O)m-,
R10C(O)NH-, CN, H2N-C(NH)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, or R10OC(O)NH-; R9 is selected from:
a) hydrogen,
b) C2-C6 alkenyl, C2-C6 alkynyl, perfluoroalkyl, F, Cl, Br, R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C-(NR10)-,R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-, and
c) C1-C6 alkyl unsubstituted or substituted by perfluoroalkyl,
F, Cl, Br, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-; R10 is independently selected from hydrogen, C1-C6 alkyl, benzyl and aryl;
R11 is independently selected from C1-C6 alkyl and aryl; A1 and A2 are independently selected from: a bond, -CH=CH-, -CΞC-, -C(O)-, -C(O)NR10-, -NR10C(O)-, O, -N(R10)-, -S(O)2N(R10)-,
-N(R10)S(O)2-,or S(O)m;
V is selected from:
a) hydrogen,
b) heterocycle,
c) aryl,
d) C1-C20 alkyl wherein from 0 to 4 carbon atoms are
replaced with a a heteroatom selected from O, S, and N, and
e) C2-C20 alkenyl,
provided that V is not hydrogen if A1 is S(O)m and V is not hydrogen if A1 is a bond, n is 0 and A2 is S(O)m;
W is a heterocycle;
X is -CH2-, -C(=O)-, or -S(=O)m-;
Y is aryl, heterocycle, unsubstituted or substituted with one or more of:
1 ) C 1 -4 alkyl, unsubstituted or substituted with a) C1 -4 alkoxy,
b) NR6R7,
c) C3-6 cycloalkyl,
d) aryl or heterocycle,
e) HO,
f) -S(O)mR6, or
g) -C(O)NR6R7,
2) aryl or heterocycle,
3) halogen,
4) OR6,
5) NR6R7,
6) CN,
7) NO2,
8) CF3;
9) -S(O)mR6,
10) -C(O)NR6R7, or
11 ) C3-C6 cycloalkyl; m is 0, 1 or 2;
n is 0, 1, 2, 3 or 4;
p is 0, 1 , 2, 3 or 4;
r is 0 to 5, provided that r is 0 when V is hydrogen; s is 0 or 1 ;
t is 0 or 1; and
u is 4 or 5; with respect to formula (Il-b):
or a pharmaceutically acceptable salt thereof,
R1a, R1b, R10, R11, m, R2, R3, R6, R7, p, R7a, u, R8, A1, A2, V, W, X, n, p, r, s, t and u are as defined above with respect to formula (Il-a);
R4 is selected from H and CH3; and any two of R2, R3 and R4 are optionally attached to the same carbon atom; R9 is selected from:
a) hydrogen,
b) alkenyl. alkynyl, perfluoroalkyl, F, Cl, Br, R10O-,
R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C-(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-, and
c) C1-C6 alkyl unsubstituted or substituted by perfluoroalkyl, F, Cl, Br, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-;
G is H2 or O; Z is aryl, heteroaryl, arylmethyl, heteroarylmethyl,
arylsulfonyl, heteroarylsulfonyl, unsubstituted or substituted with one or more of the following:
1) C1-4 alkyl, unsubstituted or substituted with:
a) C1-4 alkoxy,
b) NR6R7,
c) C3-6 cycloalkyl,
d) aryl or heterocycle
e) HO,
f) -S(O)mR6, or
g) -C(O)NR6R7,
2) aryl or heterocycle,
3) halogen,
4) OR6,
5) NR6R7,
6) CN,
7) NO2,
8) CF3;
9) -S(O)mR6,
10) -C(O)NR6R7, or
1 1 ) C3-C6 cycloalkyl; with respect to formula (II-c):
or a pharmaceutically acceptable salt thereof,
R 1 a, R1 b, R10, R11, m, R2, R3, R6, R7, p, u, R7a, R8, A1 , A2, V, W, X, n, r and t are as defined above with respect to formula (Il-a); R4 is selected from H and CH3; and any two of R2, R3 and R4 are optionally attached to the same carbon atom;
G is O;
Z is aryl, , heteroaryl, arylmethyl, heteroarylmethyl, arylsulfonyl, heteroarylsulfonyl, unsubstituted or substituted with one or more of the following:
1 ) C1 -4 alkyl, unsubstituted or substituted with a) C 1 -4 alkoxy,
b) NR6R7,
c) C3-6 cycloalkyl,
d) aryl or heterocycle,
e) HO,
f) -S(O)mR6, or
g) -C(O)NR6R7,
2) aryl or heterocycle,
3) halogen,
4) OR6,
5) NR6R7,
6) CN,
7) NO2,
8) CF3;
9) -S(O)mR6,
10) -C(O)NR6R7 , or
1 1) C
3-C
6 cycloalkyl; and s is 1 ; (b) a compound represented by formula (Il-d) through (Il-g):
or a pharmaceutically acceptable salt thereof,
R11, V, W, m, n, p and r are as defined above with respect to formula (Il-a);
R1a and R1b are independently selected from:
a) hydrogen,
b) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl,
C2-C6 alkynyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN,
NO2,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-,
c) C1-C6 alkyl unsubstituted or substituted by aryl,
heterocyclyl, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)-NR10-;
R2a and R2b are independently selected from:
a) hydrogen,
b) C1-C6 alkyl unsubstituted or substituted by C2-C6
alkenyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, N3, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, -N(R10)2, or R11OC(O)NR10-,
c) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl,
R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R1 1OC(O)NR 10-, and
d) C1 -C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocyclyl and C3-C1 0 cycloalkyl;
R3 and R4 are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone, and
c) substituted or unsubstituted C1 -C20 alkyl, C2-C20 alkenyl, C3-C1 0 cycloalkyl, aryl or heterocyclyl group, wherein the substituent is selected from F, Cl, Br, N(R 10)2, NO2, R10O-, R 1 1 S(O)m-, R 1 0C(O)NR 1 0-,
CN, (R10)2N-C(NR 10)-, R 10C(O)-, R 10OC(O)-, N3, -N(R1 0)2, R11OC(O)NR10- and C1-C20 alkyl, and
d) C1 -C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3- C10 cycloalkyl; or
R3 and R4 are combined to form - (CH2)s - ; R5a and R5b are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone,
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C1 0 cycloalkyl, aryl or heterocycle group,
wherein the substituent is selected from F, Cl, Br. CF3, N(R1 0)2, NO2, R 1 0O-, R1 1 S(O)m-,
R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl,
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3-C10 cycloalkyl; or
R5a and R5b are combined to form - (CH2)s - wherein one of the carbon atoms is optionally replaced by a moiety selected from: O, S(O)m, -NC(O)-, and-N(COR10)-;
X-Y is
R
7a is selected from
a) hydrogen,
b) unsubstituted or substituted aryl,
c) unsubstituted or substituted heterocycle,
d) unsubstituted or substituted C3-C1 0 cycloalkyl, and e) C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C1 0 cycloalkyl;
R7b is selected from
a) hydrogen,
b) unsubstituted or substituted aryl,
c) unsubstituted or substituted heterocycle,
d) unsubstituted or substituted C3-C1 0 cycloalkyl,
e) C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C1 0 cycloalkyl,
f) a carbonyl group which is bonded to an unsubstituted or substituted group selected from aryl, heterocycle, C3-C1 0 cycloalkyl and C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C1 0 cycloalkyl, and
g) a sulfonyl group which is bonded to an unsubstituted or substituted group selected from aryl, heterocycle, C3-C1 0 cycloalkyl and C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C1 0 cycloalkyl; R8 is independently selected from:
a) hydrogen,
b) aryl, heterocycle, C3-C1 0 cycloalkyl, C2-C6 alkenyl,
C2-C6 alkynyl, perfluoroalkyl, F, Cl, Br, R 1 0O-,
R1 1 S(O)m-, R10C(O)NR 10-, CN, NO2, R 10 2N-C(NR 10)-, R 1 0C(O)-, R 1 0OC(O)-, N3, -N(R 1 0)2, or
R 1 1 OC(O)NR 10-, and
c) C1-C6 alkyl unsubstituted or substituted by aryl,
heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, perfluoroalkyl, F, Cl, Br, R10O-,R11S(O)m-, R10C(O)NH-, CN, H2N-C(NH)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, or R10OC(O)NH-; R9 is selected from:
a) hydrogen,
b) C2-C6 alkenyl, C2-C6 alkynyl, perfluoroalkyl, F,
Cl, Br, R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C-(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-, and
c) C1-C6 alkyl unsubstituted or substituted by perfluoroalkyl, F, Cl, Br, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-; R10 is independently selected from H, C1-C6 alkyl, benzyl, substituted aryl and C1-C6 alkyl substituted with substituted aryl;
A1 and A2 are independently selected from: a bond, -CH=CH-, -C≡C-, -C(O)-, -C(O)NR10-, -NR10C(O)-, O, -N(R10)-, -S(O)2N(R10)-, -N(R10)S(O)2-,or S(O)m; Z is independently H2 or O; s is 4 or 5:
t is 3, 4 or 5; and
u is 0 or 1: with respect to formula (Il-e):
or a pharmaceutically acceptable salt thereof,
R11 , W, m, n, p and r are as defined above with respect to formula (II- a);
R1a and R1b are independently selected from:
a) hydrogen,
b) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl,
C2-C6 alkynyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN,
NO2, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, or R11OC(O)NR10-,
c) C1-C6 alkyl unsubstituted or substituted by aryl,
heterocyclyl, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)-NR10-;
R2a and R2b are independently selected from:
a) hydrogen,
b) C1-C6 alkyl unsubstituted or substituted by C2-C6
alkenyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, N3, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, -N(R10)2, or R11OC(O)NR10-,
c) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl, R10O, R11S(O)m-, R10C(O)NR10-, CN, NO2, (R10)2N-C(NR10), R10C(O)-, R10OC(O)-, N3, -N(R10)2, or
R11OC(O)NR10-,and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocyclyl and C3-C10 cycloalkyl; R3 and R4 are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone,
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocyclyl group, wherein the substituent is selected from F, Cl, Br, N(R10)2, NO2, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3-C10 cycloalkyl; or R3 and R4 are combined to form - (CH2)s -;
R5a and R5b are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone,
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocycle group, wherein the substituent is selected from F, Cl, Br, CF3, N(R10)2, NO2, R10O-, R11S(O)m-,
R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-,
R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl; or
R5a and R5b are combined to form - (CH2)s - wherein one of the carbon atoms is optionally replaced by a moiety selected from: O, S(O)m, -NC(O)-, and-N(COR10)-;
R6 is
a) substituted or unsubstituted C1-C8 alkyl, substituted or unsubstituted C5-C8 cycloalkyl, or substituted or unsubstituted cyclic amine, wherein the substituted alkyl, cycloalkyl or cyclic amine is substituted with 1 or 2 substituents independently selected from:
1) C1-C6 alkyl,
2) aryl,
3) heterocycle,
4) -N(R11)2,
5) -OR10, or
R7a is selected from
a) hydrogen,
b) unsubstituted or substituted aryl,
c) unsubstituted or substituted heterocycle,
d) unsubstituted or substituted C3-C1 0 cycloalkyl, and e) C1 -C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and
C3-C10 cycloalkyl;
R7a is selected from
a) hydrogen,
b) unsubstituted or substituted aryl,
c) unsubstituted or substituted heterocycle,
d) unsubstituted or substituted C3-C10 cycloalkyl,
e) C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl,
f) a carbonyl group which is bonded to an unsubstituted or substituted group selected from aryl, heterocycle, C3-C10 cycloalkyl and C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl, and
g) a sulfonyl group which is bonded to an unsubstituted or substituted group selected from aryl, heterocycle, C3-C10 cycloalkyl and C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl; R8 is independently selected from:
a) hydrogen,
b) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl,
C2-C6 alkynyl, perfluoroalkyl F, Cl, Br, R10O-,
R11S(O)m-, R10C(O)NR10-, CN, NO2, R10 2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, or
R11OC(O)NR10-, and
c) C1-C6 alkyl unsubstituted or substituted by aryl,
heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, perfluoroalkyl, F, Cl, Br, R10O-, R11S(O)m-, R10C(O)NH-, CN, H2N-C(NH)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, or R10OC(O)NH-;
R9 is selected from:
a) hydrogen,
b) C2-C6 alkenyl, C2-C6 alkynyl, perfluoroalkyl, F, Cl,
Br, R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C-(NR10)-,R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R1lOC(O)NR10-, and
c) C1-C6 alkyl unsubstituted or substituted by perfluoroalkyl, F, Cl, Br, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-; R10 is independently selected from H, C1-C6 alkyl, benzyl, substituted aryl and C1 -C6 alkyl substituted with substituted aryl;
R12 is hydrogen or C1-C6 alkyl; R13 is C1-C6 alkyl;
A1 and A2 are independently selected from: a bond, -CH=CH-, -C≡C-, -C(O)-, -C(O)NR10-, -NR10C(O)-, O, -N(R10)-, -S(O)2N(R10)-, -N(R10)S(O)2-,or S(O)m; Z is independently H2 or O; s is 4 or 5;
t is 3, 4 or 5; and
u is 0 or 1; with respect to formula (Il-f):
or a pharmaceutically acceptable salt thereof,
R11, V, W, m, n, p and r are as defined above with respect to formula (Il-a);
R1a and R1b are independently selected from:
a) hydrogen,
b) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl,
C2-C6 alkynyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN,
NO2, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2 or R11OC(O)NR10-,
c) C1-C6 alkyl unsubstituted or substituted by aryl,
heterocyclyl, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)-NR10-;
R2a and R2b are independently selected from:
a) hydrogen,
b) C1-C6 alkyl unsubstituted or substituted by C2-C6
alkenyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, N3, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, -N(R10)2, or R11OC(O)NR10-,
c) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6
alkenyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocyclyl and C3-C10 cycloalkyl;
R3 and R4 are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone, and
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocyclyl group,
wherein the substituent is selected from F, Cl, Br, N(R10)2, NO2, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-,R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3-C10 cycloalkyl; or R3 and R4 are combined to form - (CH2)s - ;
X-Y is
R7a is selected from
a) hydrogen,
b) unsubstituted or substituted aryl,
c) unsubstituted or substituted heterocycle,
d) unsubstituted or substituted C3-C1 0 cycloalkyl, and e) C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C1 0 cycloalkyl;
R7a is selected from
a) hydrogen,
b) unsubstituted or substituted aryl,
c) unsubstituted or substituted heterocycle,
d) unsubstituted or substituted C3-C1 0 cycloalkyl,
e) C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C1 0 cycloalkyl,
f) a carbonyl group which is bonded to an unsubstituted or substituted group selected from aryl, heterocycle, C3-C1 0 cycloalkyl and C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C1 0 cycloalkyl, and
g) a sulfonyl group which is bonded to an unsubstituted or substituted group selected from aryl, heterocycle, C3-C1 0 cycloalkyl and C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C1 0 cycloalkyl; R8 is independently selected from:
a) hydrogen,
b) aryl, heterocycle, C3-C1 0 cycloalkyl, C2-C6 alkenyl,
C2-C6 alkynyl, perfluoroalkyl, F, Cl, Br, R 1 0O-,
R 1 1 S(O)m-, R 10C(O)NR 10-, CN. NO2, R 10 2N-C(NR 10)-, R 10C(O)-, R 1 0OC(O)-, N3, -N(R10)2, or
R 1 1OC(O)NR 1 0-, and
c) C1-C6 alkyl unsubstituted or substituted by aryl,
heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, perfluoroalkyl, F, Cl, Br, R10O-, R11S(O)m-, R10C(O)NH-, CN, H2N-C(NH)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, or R10OC(O)NH-;
R9 is selected from:
a) hydrogen,
b) C2-C6 alkenyl, C2-C6 alkynyl, perfluoroalkyl, F, Cl,
Br, R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C-(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-, and
c) C1-C6 alkyl unsubstituted or substituted by perfluoroalkyl, F, Cl, Br, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-; R10 is independently selected from H, C1-C6 alkyl, benzyl, substituted aryl and C1-C6 alkyl substituted with substituted aryl;
R12 is hydrogen or C1-C6 alkyl;
R13 is C1-C6 alkyl; A1 and A2 are independently selected from: a bond, -CH=CH-, -C≡C-, -C(O)-, -C(O)NR10-, -NR10C(O)-, O, -N(R10)-, -S(O)2N(R10)-,
-N(R10)S(O)2-,or S(O)m;
Z is independently H2 or O; q is 0, 1 or 2;
s is 4 or 5;
t is 3, 4 or 5; and
u is 0 or 1;
with respect to formula (Il-g):
or a pharmaceutically acceptable salt thereof,
R11, V, W, m, n, p and r are as previously defined with respect to formula (Il-a);
R1a and R1b are independently selected from:
a) hydrogen,
b) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl,
C2-C6 alkynyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN,
NO2, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, or R11OC(O)NR10-,
c) C1-C6 alkyl unsubstituted or substituted by aryl,
heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)-NR10-;
R2a and R2b are independently selected from:
a) hydrogen,
b) C1-C6 alkyl unsubstituted or substituted by C2-C6
alkenyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, N3, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, -N(R10)2, or R11OC(O)NR10-,
c) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6
alkenyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2 or R11OC(O)NR10-, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocyclyl and C3-C10 cycloalkyl; R3 and R4 are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone,
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocycle group, wherein the substituent is selected from F, Cl, Br, N(R10)2, NO2, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-,R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3-C10 cycloalkyl; or R3 and R4 are combined to form - (CH2)s -;
X-Y is
R7a is selected from
a) hydrogen,
b) unsubstituted or substituted aryl,
c) unsubstituted or substituted heterocycle,
d) unsubstituted or substituted C3-C1 0 cycloalkyl, and e) C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C1 0 cycloalkyl;
R7a is selected from
a) hydrogen,
b) unsubstituted or substituted aryl,
c) unsubstituted or substituted heterocycle,
d) unsubstituted or substituted C3-C10 cycloalkyl,
e) C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl,
f) a carbonyl group which is bonded to an unsubstituted or substituted group selected from aryl, heterocycle, C3-C10 cycloalkyl and C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl, and
g) a sulfonyl group which is bonded to an unsubstituted or substituted group selected from aryl, heterocycle, C3-C10 cycloalkyl and C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl; R8 is independently selected from:
a) hydrogen,
b) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl,
C2-C6 alkynyl, perfluoroalkyl, F, Cl, Br, R10O-,
R11S(O)m-, R10C(O)NR10-, CN, NO2, R10 2N-C(NR10)-,
R10C(O)-, R10OC(O)-, N3, -N(R10)2, or
R11OC(O)NR10-,and
c) C1-C6 alkyl unsubstituted or substituted by aryl,
heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, perfluoroalkyl, F, Cl, Br, R10O-, R11S(O)m-,
R10C(O)NH-, CN, H2N-C(NH)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, or R10OC(O)NH-;
R9 is selected from:
a) hydrogen,
b) C2-C6 alkenyl, C2-C6 alkynyl, perfluoroalkyl, F, Cl,
Br, R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C-(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-, and
c) C1-C6 alkyl unsubstituted or substituted by perfluoroalkyl, F, Cl, Br, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2,or R11OC(O)NR10-; R10 is independently selected from H, C1-C6 alkyl, benzyl, substituted aryl and C1-C6 alkyl substituted with substituted aryl;
R12 is hydrogen or C1-C6 alkyl;
R13 is C1-C6 alkyl;
A1 and A2 are independently selected from: a bond, -CH=CH-, -C=C-, -C(O)-, -C(O)NR10-, -NR10C(O)-, O, -N(R10)-, -S(O)2N(R10)-, -N(R10)S(O)2-,or S(O)m; Z is independently H2 or O; qis 0, 1 or 2;
s is 4 or 5;
tis 3, 4 or 5; and
u is 0 or 1;
(c) a compound represented by formula (II-h) through (Il-k):
R
1a, R
1b, R
8, R
9, R
10, R
11, A
1, A
2, V, W, m, n, p and r are as previously defined with respect to formula (Il-a); R
2 and R
3 are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone, and
c) substituted or unsubstituted C1 -C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocyclyl group, wherein the substituent is selected from F, Cl, Br, N(R10)2, NO2, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2,R10OC(O)NR10-and C1-C20 alkyl, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3-C10 cycloalkyl; or R2 and R3 are combined to form - (CH2)s-; or
R2 or R3 are combined with R6 to form a ring such that
R4a, R4b, R7a and R7a are independently selected from:
a) hydrogen,
b) C1 -C6 alkyl unsubstituted or substituted by alkenyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, N3, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, -N(R10)2, or R11OC(O)NR10-, c) aryl, heterocycle, cycloalkyl, alkenyl, R10O-,
R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, or R11OC(O)NR10-, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocyclyl and C3-C10 cycloalkyl;
R5a and R5b are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone,
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocycle group, wherein the substituent is selected from F, Cl, Br,
N(R10)2, NO2, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3-C10 cycloalkyl; or
R5a and R5b are combined to form - (CH2)s - wherein one of the carbon atoms is optionally replaced by a moiety selected from: O, S(O)m, -NC(O)-, and -N(COR10)- ;
R6 is independently selected from hydrogen or C1-C6 alkyl;
Q is a substituted or unsubstituted nitrogen-containing C4-C9 mono or bicyclic ring system, wherein the non-nitrogen containing ring may be an aromatic ring, a C5-C7 saturated ring or a heterocycle; X, Y and Z are independently H2 or O; s is 4 or 5;
t is 3, 4 or 5; and
u is 0 or 1 ; with respect to formula (Il-i):
or a pharmaceutically acceptable salt thereof,
wherein:
R 1 a, R 1 b, R8, R9, R 10, R 1 1 , A 1 , A2, V, W, m, n, p and r are as previously defined with respect to formula (Il-a); R2 and R3 are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone, and
c) substituted or unsubstituted C1 -C20 alkyl, C2-C20 alkenyl, C3-C1 0 cycloalkyl, aryl or heterocyclyl group,
wherein the substituent is selected from F, Cl, Br, N(R10)2, NO2, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3-C10 cycloalkyl; or R2 and R3 are combined to form - (CH2)s-; or
R2 or R3 are combined with R6 to form a ring such that
R4a, R4b, R7a and R7a are independently selected from:
a) hydrogen,
b) C1-C6 alkyl unsubstituted or substituted by alkenyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, N3, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, -N(R10)2, or R11OC(O)NR10-, c) aryl, heterocycle, cycloalkyl, alkenyl,
R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2 or R11OC(O)NR10-, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocyclyl and
C3-C10 cycloalkyl;
R5a and R5b are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone,
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocycle group, wherein the substituent is selected from F, Cl, Br, N(R10)2, NO2, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3-C10 cycloalkyl; or
R5a and R5b are combined to form - (CH2)s - wherein one of the carbon atoms is optionally replaced by a moiety selected from: O, S(O)m, -NC(O)-, and-N(COR10)-;
R6 is independently selected from hydrogen or C1-C6 alkyl; R12 is
a) substituted or unsubstituted C1-C8 alkyl or substituted or unsubstituted C5-C8 cycloalkyl, wherein the substituent on the alkyl or cycloalkyl is selected from:
1) aryl,
2) heterocycle,
3) -N(R11)2,
R13 is independently selected from hydrogen and C1-C6 alkyl;
R14 is independently selected from C1-C6 alkyl;
Q is a substituted or unsubstituted nitrogen-containing C4-C9 mono or bicyclic ring system, wherein the non-nitrogen containing ring may be an aromatic ring, a C5-C7 saturated ring or a heterocycle;
X, Y and Z are independently H2 or O; s is 4 or 5;
t is 3, 4 or 5; and
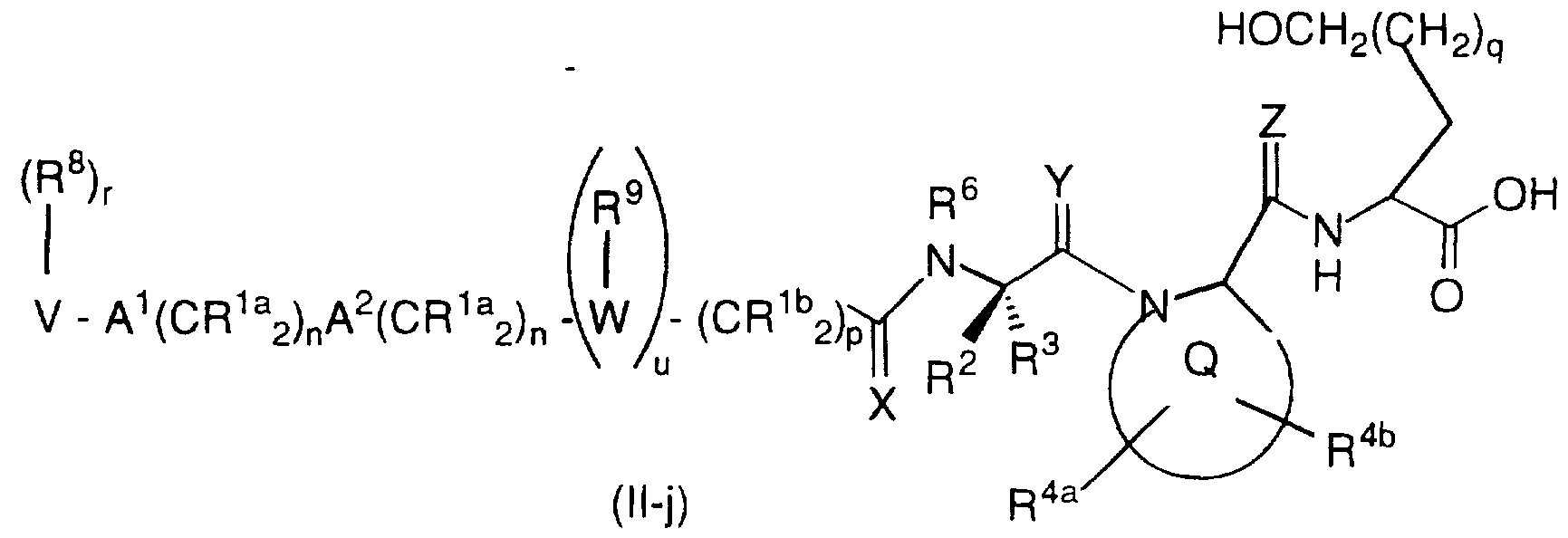
u is 0 or 1 ; with respect to formula (II-j):
or a pharmaceutically acceptable salt thereof,
R 1 a, R1 b, R8, R9, R 10, R1 1 , A 1 , A2, V, W, m, n, p and r are as previously defined with respect to formula (Il-a); R2 and R3 are independently selected from:
a) a side chain of a naturally occurring amino acid, b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone, and
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocyclyl group, wherein the substituent is selected from F, Cl, Br, N(R10)2, NO2, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10-and C1-C20 alkyl, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3-C10 cycloalkyl; or R2 and R3 are combined to form - (CH2)s-; or
R2 or R3 are combined with R6 to form a ring such that
R4a, R4b, R7a and R7a are independently selected from:
a) hydrogen,
b) C1-C6 alkyl unsubstituted or substituted by alkenyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, N3, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, -N(R10)2, orR11OC(O)NR10-, c) aryl, heterocycle, cycloalkyl, alkenyl,
R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2, (R10)2N-C(NR10)-,R10C(O)-, R10OC(O)-, N3,
-N(R10)2or R11OC(O)NR10-, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocyclyl and
C3-C10 cycloalkyl;
R6 is independently selected from hydrogen or C1-C6 alkyl;
Q is a substituted or unsubstituted nitrogen-containing C4-C9 mono or bicyclic ring system, wherein the non-nitrogen containing ring may be an aromatic ring, a C5-C7 saturated ring or a heterocycle;
X, Y and Z are independently H2 or O; q is 0, 1 or 2;
s is 4 or 5;
t is 3, 4 or 5; and
u is 0 or 1; with respect to formula (II-k):
or a pharmaceutically acceptable salt thereof,
R1 a, R 1 b, R8, R9, R 10, R1 1, A1 , A2, V, W, m, n, p, and r are as defined above with respect to formula (Il-a); R2 and R3 are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone, and
c) substituted or unsubstituted C1 -C20 alkyl, C2-C20 alkenyl, C3-C1 0 cycloalkyl, aryl or heterocyclyl group,
wherein the substituent is selected from F, Cl, Br, N(R10)2, NO2, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3- C10 cycloalkyl; or R2 and R3 are combined to form - (CH2)s-; or
R2 or R3 are combined with R6 to form a ring such that
R4a, R4b, R7a and R7a are independently selected from:
a) hydrogen,
b) C1-C6 alkyl unsubstituted or substituted by alkenyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, N3, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, -N(R10)2, or R11OC(O)NR10-, c) aryl, heterocycle, cycloalkyl, alkenyl, R10O-,
R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-,
N3, -N(R10)2 or R11OC(O)NR10-, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocyclyl and
C3-C10 cycloalkyl;
R6 is independently selected from hydrogen or C1-C6 alkyl;
Q is a substituted or unsubstituted nitrogen-containing C4-C9 mono or bicyclic ring system, wherein the non-nitrogen containing ring may be an aromatic ring, a C5-C7 saturated ring or a heterocycle; X, Y and Z are independently H2 or O; q is 0, 1 or 2;
s is 4 or 5;
t is 3, 4 or 5; and
u is 0 or 1 ; and
(d) a compound represented by formula (II-I) through (II-o):
or a pharmaceutically acceptable salt thereof:
R1 a, R 1 b, R8, R9, R 10, R1 1, A1 , A2, V, W, m, n, p and r are as defined above with respect to formula (Il-a); R2 and R3 are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone, and
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocyclyl group, wherein the substituent is selected from F, Cl, Br, N(R10)2, NO2, Rl°0-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3- C10 cycloalkyl; or R2 and R3 are combined to form - (CH2)s-; or
R2 or R3 are combined with R6 to form a ring such that
R4a, R4b, R7a and R7a are independently selected from:
a) hydrogen,
b) C1-C6 alkyl unsubstituted or substituted by alkenyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, N3, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, -N(R10)2, or R11OC(O)NR10-, c) aryl, heterocycle, cycloalkyl, alkenyl, R10O-,
R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-,
N3, -N(R10)2 or R11OC(O)NR10-, and
d) C1 -C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocyclyl and C3-C1 0 cycloalkyl; R5a and R5b are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone,
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C1 0 cycloalkyl, aryl or heterocycle group, wherein the substituent is selected from F, Cl, Br, N(R 10)2, NO2, R10O-, R1 1S(O)m-, R 10C(O)NR 10-, CN, (R1 0)2N-C(NR 10)-, R 1 0C(O)-, R 1 0OC(O)-, N3, -N(R1 0)2, R1 1 OC(O)NR 1 0- and C1 -C20 alkyl, d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3-C1 0 cycloalkyl; or
R5a and R5b are combined to form - (CH2)s - wherein one of the carbon atoms is optionally replaced by a moiety selected from: O, S(O)m, -NC(O)-, and -N(COR 1 0)- ; R6 is independently selected from hydrogen or C1-C6 alkyl;
0 is a substituted or unsubstituted nitrogen-containing C4-C9 mono or bicyclic ring system, wherein the non-nitrogen containing ring may be an aromatic ring, a C5-C7 saturated ring or a heterocycle;
X, Y and Z are independently H2 or O; s is 4 or 5;
t is 3, 4 or 5; and
u is 0 or 1; with respect to formula (Il-m):
or a pharmaceutically acceptable salt thereof,
R1a, R1b, R8, R9, R10, R11, A1, A2, V, W, m, n, p and r are as defined above with respect to formula (Il-a); R2 and R3 are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone, and
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocyclyl group, wherein the substituent is selected from F, Cl, Br, N(R10)2, NO2, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3-C10 cycloalkyl; or R2 and R3 are combined to form - (CH2)s-; or
R2 or R3 are combined with R6 to form a ring such that
R4a, R4b, R7a and R7a are independently selected from:
a) hydrogen,
b) C1 -C6 alkyl unsubstituted or substituted by alkenyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, N3, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, -N(R10)2, orR11OC(O)NR10-, c) aryl, heterocycle, cycloalkyl, alkenyl, R10O-,
R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2 or R11OC(O)NR10-, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocyclyl and C3-C10 cycloalkyl;
R5a and R5b are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone,
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocycle group, wherein the substituent is selected from F, Cl, Br, N(R10)2, NO2, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl,
d) C1-C6 alkyl substituted with an unsubstituted or substituted group selected from aryl, heterocycle and C3-C1 0 cycloalkyl; or
R5a and R5b are combined to form - (CH2)s - wherein one of the carbon atoms is optionally replaced by a moiety selected from: O, S(O)m, -NC(O)-, and -N(COR10)- ;
R6 is independently selected from hydrogen or C 1 -C6 alkyl; R 12 is
a) substituted or unsubstituted C1 -C8 alkyl or substituted or unsubstituted C5-C8 cycloalkyl, wherein the substituent on the alkyl or cycloalkyl is selected from:
1 ) aryl,
2) heterocycle,
3) -N(R1 1)2,
4) -OR 1 0, or
R1 3 is independently selected from hydrogen and C1-C6 alkyl;
R14 is independently selected from C1-C6 alkyl;
Q is a substituted or unsubstituted nitrogen-containing C4-C9 mono or bicyclic ring system, wherein the non-nitrogen containing ring may be an aromatic ring, a C5-C7 saturated ring or a heterocycle; X, Y and Z are independently H2 or O; s is 4 or 5;
t is 3, 4 or 5; and
u is 0 or 1; with respect to formula (Il-n):
or a pharmaceutically acceptable salt thereof:
R1a, R1b, R8, R9, R10, R11, A1, A2, V, W, m, n, p and r are as defined above with respect to formula (Il-a); R2 and R3 are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone, and
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocyclyl group, wherein the substituent is selected from F, Cl, Br,
N(R10)2, NO2, R10O-, R11S(O)m-, R10C(O)NR10- CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl; or
R2 and R3 are combined to form - (CH2)s-; or
R2 or R3 are combined with R6 to form a ring such that
R4a, R4b, R7a and R7a are independently selected from:
a) hydrogen,
b) C1-C6 alkyl unsubstituted or substituted by alkenyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, N3, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, -N(R10)2, or R11OC(O)NR10-, c) aryl, heterocycle, cycloalkyl, alkenyl, R10O-,
R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, or R11OC(O)NR10-, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocyclyl and C3-C10 cycloalkyl; R6 is independently selected from hydrogen or C1-C6 alkyl;
Q is a substituted or unsubstituted nitrogen-containing C4-C9 mono or bicyclic ring system, wherein the non-nitrogen containing ring may be an aromatic ring, a C5-C7 saturated ring or a heterocycle;
X, Y and Z are independently H2 or O; q is 0, 1 or 2;
s is 4 or 5;
t is 3, 4 or 5; and
u is 0 or 1:
and with respect to formula (II-o):
or a pharmaceutically acceptable salt thereof:
R1a, R1b, R8, R9, R10, R11, A1, A2, V, W, m, n, p and r are as defined above with respect to formula (Il-a); R2 and R3 are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone, and
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocyclyl group, wherein the substituent is selected from F, Cl, Br, N(R10)2, NO2, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3-C10 cycloalkyl; or R2 and R3 are combined to form - (CH2)s-; or
R
2 or R
3 are combined with R
6 to form a ring such that
^
R4a, R4b, R7a and R7a are independently selected from:
a) hydrogen,
b) C1-C6 alkyl unsubstituted or substituted by alkenyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, N3, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, -N(R10)2, or R11OC(O)NR10-, c) aryl, heterocycle, cycloalkyl, alkenyl, R10O-,
R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2 or R11OC(O)NR10-, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocyclyl and C3-C10 cycloalkyl;
R6 is independently selected from hydrogen or C1-C6 alkyl;
Q is a substituted or unsubstituted nitrogen-containing C4-C9 mono or bicyclic ring system, wherein the non-nitrogen containing ring may be an aromatic ring, a C5-C7 saturated ring or a heterocycle;
X, Y and Z are independently H2 or O; q is 0, 1 or 2;
s is 4 or 5;
t is 3, 4 or 5; and
u is 0 or 1. Specific compounds which antagonize Raf include the following:
4-[5-(4-fluorophenyl)-4-pyridin-4-yl-1H-imidazol-2-yl]-piperidine-1- carboxylic acid tert-butyl ester;
4-[4-fluorophenyl)-3-pyridin-yl-1H-imidazol-2-yl]-1-acetyl-piperidine:
3-[5-(4-fluorophenyl)-4-pyridin-4-yl-1H-imidazol-2-yl]-piperidine-1- carboxylic acid tert-butyl ester;
3-[4-fluorophenyl)-3-pyridin-yl-1H-imidazol-2-yl]-1-acetyl-piperidine; and
4-benzyl-[4-(4-fluorophenyl)-5-pyridin-4-yI-1H-imidazol-2-yl]- piperidine-1-carboxylic acid tert-butyl ester.
4-[5-(4-fluorophenyl)-4-pyridin-4-yl-1H-imidazol-2-yl]-piperidine;
4-[5-(4-fluorophenyl)-4-pyridin-4-yl-1H-imidazol-2-yl]-1-methyl- piperidine;
4-[5-(4-fluorophenyl)-4-pyridin-4-yl-1H-imidazoI-2-yl]-1-benzyl- piperidine;
4-[5-(4-fluorophenyI)-4-pyridin-4-yl-1H-imidazol-2-yl]-1-ethyl- pipe ridine; 4-[5-(3,4-dichlorophenyl)-4-pyridin-4-yl-1H-imidazol-2-yl]-piperidine;
4-[5-(3,4-dichlorophenyl)-4-pyridin-4-yl-1H-imidazol-2-yl]-1-methyl- pipe ridine; 2-(4-{4-[5-(3,4-dichlorophenyl)-4-pyridin-4-yl-1H-imidazol-2-yl]- piperidin-1-yl}-butyl)-isoindole-1,3-dione;
2-(5-{4-[5-(3,4-dichlorophenyI)-4-pyridin-4-yl-1H-imidazol-2-yl]- piperidin-1-yl}-pentyl)-isoindoIe-1,3-dione;
2-(6-{4-[5-(3,4-dichlorophenyl)-4-pyridin-4-yl-1H-imidazol-2-yl]- piperidin-1-yl}-hexyl)-isoindole-1,3-dione; 4-[5-(3,4-dichlorophenyl)-4-pyridin-4-yl-1H-imidazol-2-yl]-1-benzyl- piperidine;
2-(5-{4-[5-(3,4-dichlorophenyl)-4-pyridin-4-yl-1H-imidazol-2-yl]- piperidin-1-y1}-pentyl)-2,3-dihydro-isoindol-1-oneditrifluoroacetic acid salt;
4-(4-{4-[5-(3,4-dichlorophenyl)-4-pyridin-4-yl-1H-imidazol-2-yI]- piperidin-1-yl}-ethyl)-pyridine; 2-(5-{4-[5-(3,4-dichlorophenyl)-4-pyridin-4-yl-1H-imidazol-2-yl]- piperidin-1-yl}-pentyl)-1,1-dioxobenzo[d]isothiazol-3-one;
2-(4-{4-[5-(3,4-dichlorophenyl)-4-pyridin-4-yl-1H-imidazol-2-yl]- piperidin-1-yl}-butyl)-1,1-dioxobenzo[d]isothiazol-3-one;
2-amino-1-{5-[4-(3,4-dichlorophenyl)-4-pyridin-4-yl-1H-imidazol-2- yl]-piperidin-1-yl}-ethanone dihydrochloride;
4-[5-(3-hydroxyphenyl)-4-pyridin-4-yl-1H-imidazol-2-yl]-1-methyl- piperidine;
3-[5-(4-fluorophenyl)-4-pyridin-4-yl-1H-imidazol-2-yl]-piperidine-1 carboxylic acid tert-butylester; 3-[5-(4-fluorophenyl)-4-pyridin-4-yl-1H-imidazol-2-yl]-piperidine:
3-[5-(4-fluorophenyl)-4-pyridin-4-yl-1H-imidazoI-2-yl]-1-methyl- piperidine;
4-[5-(4-fluorophenyl)-4-pyridin-4-yI-1H-imida2ol-2-yl]- 1 ,4-dimethyl- piperidine;
4-benzyl-[4-(4-fluorophenyl)-5-pyridin-4-yl-1H-imidazol-2-yl]- piperidine-1 -carboxylic acid tert-butyl ester;
4-benzyl-[4-(4-fluorophenyl)-5-pyridin-4-yl-1H-imidazol-2-yl]- piperidine; 4-{5-(3,4-dichlorophenyl)-2-[1 -(2-phenylethyl)-piperidin-4-yl]-1H- imidazol-4-yl}-pyridine;
4-{5-(3,4-dichlorophenyl)-2-{ 1 -(3-phenylpropyI)-piperidin-4-yl]-1H- imidazol-4-yl}-pyridine;
2-(6-{4-[5-(3,4-dichlorophenyI)-4-pyridin-4-yl-1H-imidazol-2-yl]- piperidin-1 -yl}-hexyl)-1 ,1-dioxobenzo[d]isothiazol-3-one;
2-(3-{4-[5-(3,4-dichlorophenyl)-4-pyridin-4-yl-1H-imidazol-2-yl]- piperidin-1 -yl }-propyl)-1 ,1 -dioxobenzo[d]isothiazol-3-one;
4-(5-{4-[5-(3,4-dichlorophenyI)-4-pyridin-4-yl-l H-imidazol-2-yl]- piperidin-1 -yl-methyl } -imidazol-l -yl-methyl)-benzonitrile; 4-[2-[1 -(4-benzyloxybenzyl)-piρeridin-4-yl-5-(3,4-dichlorophenyl)-1 H- imidazoI-4-yl-pyridine;
2-(3-{4-[5-(3,4-dichlorophenyl)-4-pyridin-4-yl-1H-imidazol-2-yl]- piperidin- 1 -yl ) -propyl)-isoindole-1 ,3-dione;
4-[4-(4-fluorophenyl)-5-(4-pyridyl)imidazol-2-yl]benzamidoxime; 4 -(1 -naphthyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)imidazole;
4-(1 -naphthyl)-2-(4-methylthiophenyl)-5-(4-pyridyl)imidazole;
4-(2-naphthyl)-2-(4-methylthiophenyl)-5-(4-pyridyl)imidazole; 4-(2-naphthyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)imidazole;
4-(4-fluorophenyl)-2-(3-thiophenyl)-5-(4-pyridyl)imidazole;
4-(4-fluorophenyl)-2-(2-thiophenyl)-5-(4-pyridyl)imidazole;
4-(4-fluorophenyl)-2-(3-methylthiophenyl)-5-(4-pyridyl)imidazole;
4-(4-fluorophenyl)-2-(3-methylsulfinylphenyl)-5-(4-pyridyl)imidazole; 4-(4-fluorophenyl)-2-(3-methylsulfonylphenyl)-5-(4-pyridyl)imidazole;
4-(4-fluorophenyl)-2-(2-methylthiophenyl)-5-(4-pyridyl)imidazole;
4-(4-fluorophenyl)-2-(2-methylsulfinylphenyl)-5-(4-pyridyl)imidazole;
4-(4-fluorophenyl)-2-(2-methylsulfonylphenyl)-5-(4-pyridyl)imidazole;
4-(4-fluorophenyl)-2-(4-methoxyphenyl)-5-(4-pyridyl)imidazole; 4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)- 1 -methyl-5-(4-pyridyl) imidazole;
4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-1 -(N- morpholinopropyl)-5-(4-pyridyl)imidazole;
4-(4-fluorophenyl)-2-(4-methylthiophenyl)- 1-(N-morpholinopropyl)-5- (4-pyridyl)imidazole;
4-(4-fluorophenyl)-2-(4-methylsulfonylphenyl)- 1 -(N-morpholino-
propyl)-5-(4-pyridyl)imidazole;
4-(4-fluorophenyI)-1-(methylthio-1-proρyl)-2-([4-N- morpholinomethyl]phenyl)-5-(4-pyridyl)imidazole;
4-(4-fluorophenyl)-1-(methylsulfinyl-1-propyl)-2-([4-N- morpholinomethyl]phenyl)-5-(4-pyridyl)imidazole; and
4-(4-fluorophenyl)-1-(methylsulfonyl-1-propyl)-2-([4-N- morpholinomethyl]phenyl)-5-(4-pyridyI)imidazole.
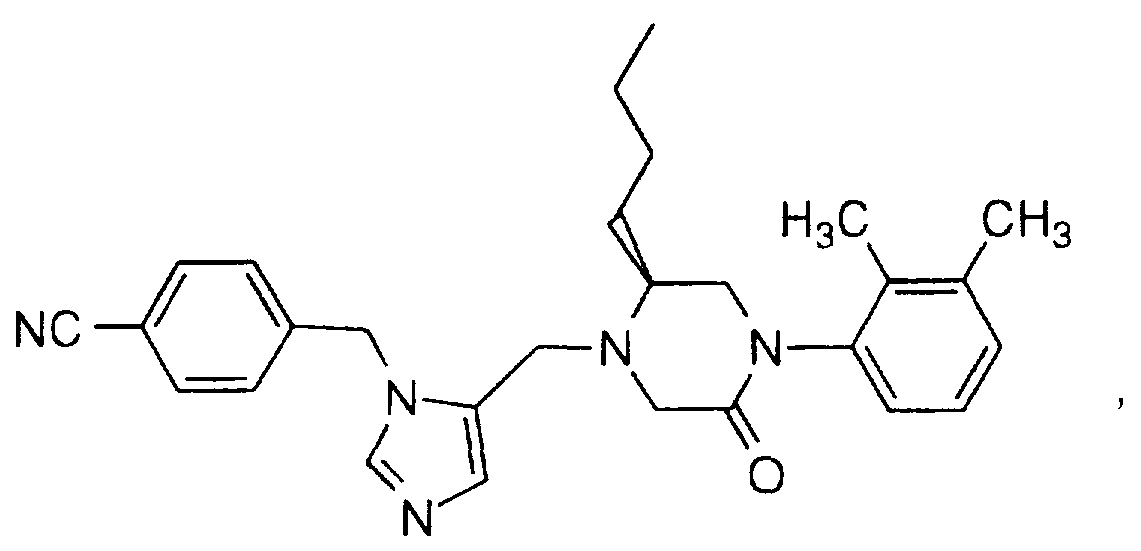
Examples of compounds which antagonize or inhibit famesyl protein transferase include the following:
2(S)-Butyl-1-(2,3-diaminoprop-1-yl)-1-(1-naphthoyl)piperazine;
1-(3-Amino-2-(2-naphthylmethylamino)prop-1-yl)-2(S)-butyl-4-(1- naphthoyl)piperazine; 2(S)-Butyl-1-{5-[1-(2-naphthylmethyl)]-4,5-dihydroimidazol}methyl-4- (1-naphthoyl)piperazine;
1-[5-(1-BenzyIimidazol)methyl]-2(S)-butyl-4-(1-naphthoyl)piperazine; 1-{5-[1-(4-nitrobenzyl)]imidazolylmethyl}-2(S)-butyl-4-(1- naphthoyl)piperazine;
1-(3-Acetamidomethylthio-2(R)-aminoprop-1-yl)-2(S)-butyl-4-(1- naphthoyl)piperazine;
2(S)-Butyl-1-[2-(l-imidazolyl)ethyl]sulfonyI-4-(1-naphthoyl)piperazine; 2(R)-Butyl-1-imidazolyl-4-methyl-4-(1-naphthoyl)piperazine; 2(S)-Butyl-4-(1-naphthoyl)-1-(3-pyridylmethyl)piρerazine;
1-2(S)-butyl-(2(R)-(4-nitrobenzyl)amino-3-hydroxypropyI)-4-(1- naphthoyl)piperazine; 1-(2(R)-Amino-3-hydroxyheptadecyl)-2(S)-butyl-4-(1-naphthoyl)- piperazine;
2(S)-Benzyl-1-imidazolyl-4-methyl-4-(1-naphthoyl)piperazine; 1-(2(R)-Amino-3-(3-benzylthio)propyl)-2(S)-butyl-4-(1- naphthoyl)piperazine;
1-(2(R)-Amino-3-[3-(4-nitrobenzylthio)propyl])-2(S)-butyl-4-(1- naphthoyl)piperazine;
2(S)-Butyl-1-[(4-imidazolyl)ethyl]-4-(1-naphthoyl)piperazine;
2(S)-Butyl-1-[(4-imidazolyl)methyl]-4-(1-naphthoyl)piperazine; 2(S)-Butyl-1-[(1-naphth-2-ylmethyl)-1H-imidazol-5-yl)acetyl]-4-(1- naphthoyl)piperazine;
2(S)-Butyl-1-[(1-naphth-2-ylmethyl)-1H-imidazol-5-yl)ethyl]-4-(1- naphthoyl)piperazine;
1-(2(R)-Amino-3-hydroypropyl)-2(S)-butyI-4-(1-naphthoyl)piperazine;
1-(2(R)-Amino-4-hydroxybutyl)-2(S)-butyl-4-(1-naphthoyl)piperazine; 1-(2-Amino-3-(2-benzyloxyphenyl)propyl)-2(S)-butyl-4-(1- naphthoyl)piperazine;
1-(2-Amino-3-(2-hydroxyphenyl)propyl)-2(S)-butyl-4-(1- naphthoyl)piperazine;
1-[3-(4-imidazolyl)propyl]-2(S)-butyI-4-(1-naphthoyl)-piperazine;
2(S)-n-Butyl-4-(2,3-dimethylphenyl)-1-(4-imidazolylmethyl)- piperazin-5-one;
2(S)-n-Butyl-1-[1-(4-cyanobenzyI)imidazol-5-ylmethyl]-4-(2,3- dimethyIphenyl)piperazin-5-one; 1-[1-(4-Cyanobenzyl)imidazoI-5-ylmethyll-4-(2,3-dimethylphenyl)- 2(S)-(2-methoxyethyl)piρerazin-5-one;
2(S)-n-Butyl-4-(1-naphthoyl)-1-[1-(1-naphthylmethyl)imidazol-5- ylmethyl]-piperazine;
2(S)-n-Butyl-4-(1-naphthoyl)-1-[1-(2-naphthylmethyl)imidazol-5- ylmethy1]-piperazine;
2(S)-n-Butyl-1-[1-(4-cyanobenzyl)imidazol-5-ylmethy1]-4-(1- naphthoyl)piperazine;
2(S)-n-Butyl-1-[1-(4-methoxybenzyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)piperazine; 2(S)-n-Butyl-1-[1-(3-methyl-2-butenyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)piperazine;
2(S)-n-Butyl-1-[1-(4-fluorobenzyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)piperazine;
2(S)-n-Butyl-1-[1-(4-chlorobenzyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)piperazinn;
1-[1-(4-Bromobenzyl)imidazol-5-ylmethyl]-2(S)-n-butyl-4-(1- naphthoyl)piperazine;
2(S)-n-Butyl-4-(1 -naphthoyl)-1-[1-(4-trifluoromethylbenzyl)imidazol-5- ylmethyll-piperazine;
2(S)-n-Butyl-1-[1-(4-methylbenzyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)-piperazine; 2(S)-n-Butyl-1-[1-(3-methylbenzyl)imidazol-5-ylmethyl]-4-(1- naphthoyI)-piperazine;
1-[1-(4-Phenylbenzyl)imidazol-5-ylmethyl]-2(S)-n-butyl-4-(1- naphthoyl)-piperazine;
2(S)-n-Butyl-4-(1-naphthoyl)-1-[1-(2-phenylethyl)imidazol-5-ylmethyl]- piperazine;
2(S)-n-Butyl-4-(1-naphthoyl)-1-[1-(4-trifluoromethoxy)imidazol-5- ylmethyl]piperazine;
1-{[1-(4-cyanobenzyI)-1H-imidazol-5-yllacety1}-2(S)-n-butyl-4-(1- naphthoyl)piperazine;
1-{5-[1-(4-nitrobenzyl)]imidazolylmethyl}-2(S)-butyl-4-(1- naphthoyl)piperazine
1-[5-(1-Benzylimidazol)methyl]-2(S)-butyl-4-(1-naphthoyl)piperazine
1-(2(R)-Amino-3-(3-benzylthio)propyl)-2(S)-butyl-4-(1- naphthoyl)piperazine
1-(2(R)-Amino-3-[3-(4-nitrobenzylthio)propyl])-2(S)-butyl-4-(1- naphthoyl)piperazine
2(S)-n-Butyl-1-[1-(4-cyanobenzyl)imidazol-5-yImethyl]-4-(1- naphthoyl)piperazine
2(S)-n-Butyl-1-[1-(4-cyanobenzyl)imidazol-5-ylmethyl]-4-(2,3- dimethylphenyl)piperazin-5-one
2(S)-n-Butyl-1-[1-(4-chlorobenzyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)piperazine
1-{[1-(4-cyanobenzyl)-1H-imidazol-5-yl]acetyl}-2(S)-n-butyl-4-(1- naphthoyl)piperazine
1-[1-(4-Cyanobenzyl)imidazol-5-ylmethyl]-4-(2,3-dimethylphenyl)- 2(S)-(2-methoxyethyl)piperazin-5-one
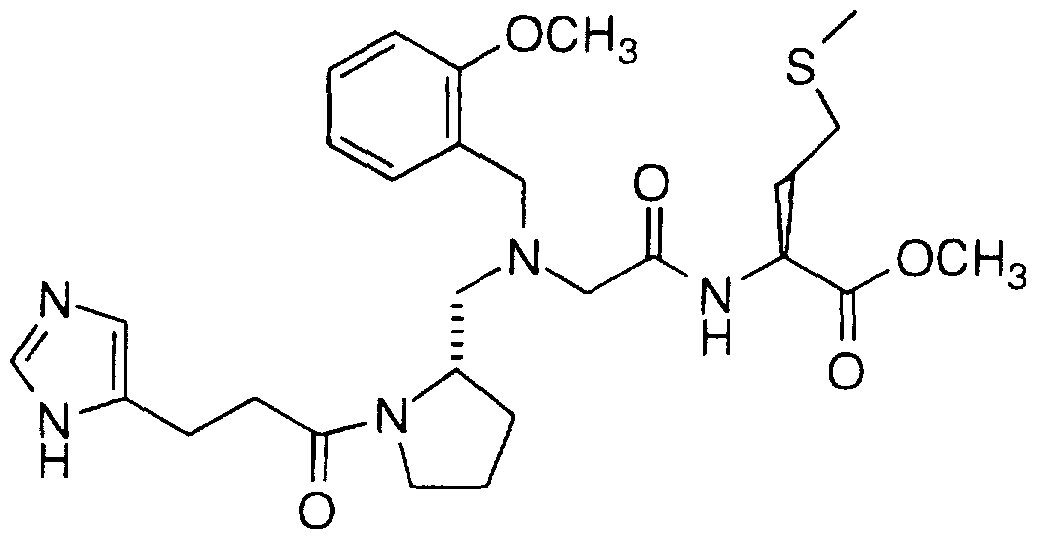
N-[1-(4-Imidazoleacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycylmethionine
N-[1-(4-Imidazoleacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthyl- methyl)glycyl-methionine methyl ester; N-[1-(2(S),3-Diaminopropionyl)pyrrolidin-2(S)-yImethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(2(S),3-Diaminopropionyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(3-Aminopropionyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(3-Aminopropionyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(2(S)-Amino-3-benzyloxycarbonylaminopropionyl)pyrrolidin- 2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl-methionine; N-[1-(2(S)-Amino-3-benzyloxycarbonylaminopropionyl)pyrrolidin- 2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(3-Amino-2(S)-benzyloxycarbonylaminopropionyl)pyrrolidin- 2(S)-ylmethyll-N-(1-naphthylmethyl)glycyl-methionine;
N-[1-(3-Amino-2(S)-benzyloxycarbonylaminopropionyl)pyrrolidin- 2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(L-Glutaminyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine; N-[1-(L-Glutaminyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(L-Histidyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(L-Histidyl)pyrrolidin-2(S)-yImethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(D-Histidyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(D-Histidyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester; N-[1-(L-Pyroglutamyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(L-Pyroglutamyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester ;
2(S)-[1-(2(S)-Pyroglutamyl)pyrrolidin-2(S)-ylmethyloxy]-3- phenylpropionyl-methionine;
2(S)-[1-(2(S)-Pyroglutamyl)pyrrolidin-2(S)-ylmethyloxy]-3- phenylpropionyl-methionine methyl ester;
2(S)-[1-(2(S)-Pyroglutamyl)pyrrolidin-2(S)-ylmethyloxy]-3- phenylpropionyl-methionine isopropyl ester;
2(S)-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyloxy]-3- phenylpropionyl-methionine;
2(S)-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyloxy]-3- phenylpropionyl-methionine methyl ester;
2(S)-[1-(2(S)-Pyroglutamyl)pyrrolidin-2(S)-ylmethyloxy]-3- phenylpropionyl-methionine sulfone; 2(S)-[1-(2(S)-Pyroglutamyl)pyrrolidin-2(S)-ylmethyloxyl-3- phenylpropionyl-methionine sulfone methyl ester;
2(S)-[1-(Pyrid-3-ylcarboxy)pyrrolidin-2(S)-ylmethyloxy]-3- phenylpropionyl-methionine;
2(S)-[1-(Pyrid-3-ylcarboxy)pyrrolidin-2(S)-ylmethyloxyJ-3- phenylpropionyl-methionine methyl ester;
2(R)-{2-[1-(Naphth-2-yl)-1H-imidazol-5-ylacetyl]pyrrolidin-2(S)- ylmethoxy}-3-phenylpropionyl-methionine;
2(R)-{2-[1-(Naphth-2-yl)-1H-imidazol-5-ylacetyl]pyrrolidin-2(S)- ylmethoxy}-3-phenylpropionyl-methionine methyl ester; 2(S)-[1-(Pyrid-3-ylmethyl)pyrrolidin-2(S)-ylmethyloxy]-3- phenylpropionyl-methionine;
2(S)-[1-(Pyrid-3-ylmethyl)pyrrolidin-2(S)-ylmethyloxy]-3- phenylpropionyl-methionine methyl ester;
N-[1-(lH-imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine isopropyl ester;
N-[1-(1H-imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine sulfone isopropyl ester;
N-[1-(1H-imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethy1]-N-(1- naphthylmethyl)glycyl-methionine sulfone;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine methyl ester; N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine isopropyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine sulfone methyl ester ;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine sulfone;
N-[1-(Sarcosyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine methyl ester;
N-[1-(Sarcosy1)pyrroIidin-2(S)-ylmethylJ-N-(1-naphthylmethyl)glycyl- methionine;
N-[1-(N,N-Dimethylglycyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester ;
N-[1-(N,N-Dimethylglycyl)pyrrolidin-2(S)-ylmethyl]-N-(1 - naphthylmethyl)glycyl-methionine;
N-[1-(1H-imidazol-4-ylacetyl)pyrroIidin-3(S)-ethyl-2(S)-ylmethyl]-N- (1-naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(1H-imidazol-4-ylacetyl)pyrrolidin-3(S)-ethyl-2(S)-ylmethyl]-N- (1-naphthylmethyl)glycyl-methionine; N-[1-(Glycyl)pyrrolidin-3(S)-ethyl-2(S)-ylmethyll-N-(1- naphthylmethyl)glycyl-methionine methyl ester; N-[1-(Glycyl)pyrrolidin-3(S)-ethyl-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(4-Cyanobenzyl)-1H-imidazol-5-ylacetyl)pyrrolidin-2(S)- ylmethyl]-N-(1-naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(4-Cyanobenzyl)-1H-imidazol-5-ylacetyl)pyrrolidin-2(S)- ylmethyl]-N-(1-naphthylmethyl)glycyl-methionine;
N-[1-(2-Acetylamino-3(S)- benzyloxycarbonylaminopropionyl)pyrrolidin-2(S)-ylmethy1]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(2-Acetylamino-3(S)-aminopropionyI)pyrrolidin-2(S)-ylmethyl]- N-(1-naphthylmethyl)glycyl-methionine;
N-[1-(2-Amino-3(S)-acetylaminopropionyl)pyrrolidin-2(S)-ylmethyl]- N-(1-naphthylmethyl)glycyl-methionine;
2(S)-[1-(1H-Imidazol-4-ylacetyI)pyrrolidin-3(S)-ethyl-2(S)- ylmethyloxy]-3-phenylpropionyl-methionine methyl ester;
2(S)-[1-(1H-imidazol-4-ylacetyl)pyrrolidin-3(S)-ethyl-2(S)- ylmethyloxy]-3-phenylpropionyl-methionine;
2(R)-{2-[1-(4-Cyanobenzyl)-1H-imidazol-5-ylacetyl]pyrrolidin-2(S)- ylmethoxy}-3-phenyl propionyl-methionine methyl ester ;
2(R)-{2-[ 1 -(4-CyanobenzyI)-1H-imidazol-5-ylacetyl]ρyrrolidin-2(S)- ylmethoxy}-3-phenyl propionyl-methionine;
2(R)-{ 2-[ 1-(4-Nitrobenzyl)-1H-imidazol-5-ylacetyl]pyrrolidin-2(S)- ylmethoxy}-3-phenyl propionyl-methionine methyl ester; 2(R)-{ 2-[ 1-(4-Nitrobenzyl)- 1 H-imidazol-5-ylacetyl]pyrrolidin-2(S)- ylmethoxy}-3-phenyl propionyl-methionine;
2(R)-{2-[1 -(4-Methoxybenzyl)-1 H-imidazol-5-ylacetyl]pyrrolidin-2(S)- ylmethoxy}-3-phenyl propionyl-methionine methyl ester;
2(R)-{ 2-f 1 -(4-MethoxybenzyI)-1 H-imidazol-5-ylacetyl]pyrrolidin-2(S)- ylmethoxy}-3-phenyl propionyl-methionine;
2(R)-{2-[1 -(4-Cyanobenzyl)-1H-imidazol-5-ylacetyI]pyrrolidin-3(S)- ethyl-2(S)-ylmethoxy} -3-pheny] propionyl-methionine methyl ester;
2(R)-{2-f 1 -(4-Cyanobenzyl)-1H-imidazol-5-ylacetyl]pyrrolidin-3(S)- ethyl-2(S)-ylmethoxy}-3-phenyl propionyl-methionine; N-[1-(1H-imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]- N-(1 - naphthylmethyl)glycyl-(β-acetylamino)alanine methyl ester;
N-[1 -(1H-imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]- N-(1 - naphthylmethyl)glycyl-(β-acetylamino)alanine;
N-[ 1 -(Glycyl) pyrrolidin-2(S)-ylmethyl]-N-( 1 -naphthylmethyl)glycyl- (β-acetylamino)alanine methyl ester;
N-[1-(Glycyl) pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- (β-acetylamino)alanine;
N-[1-(Seryl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine methyl ester;
N-[1-(D-Alanyl) pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine methyl ester; N-[1-(1H-imidazol-4-carbonyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(Isoasparagyl) pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(1H-Imidazol-4-propionyl) pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(3-Pyridylacetyl) pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(2-Pyridylacetyl) pyrrolidin-2(S)-ylmethyl)-N-(1- naphthylmethyl)glycyl-methionine methyl ester; N-[1-(4-Pyridylglycyl) pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(Seryl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine;
N-[1-(D-Alanyl)pyrrolidin-2(S)-yImethyl]-N-(1-naphthylmethyl)glycyl- methionine;
N-[1-(1H-Imidazol-4-carbonyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(Isoasparagyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(1H-Imidazol-4-propionyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(3-Pyridylacetyl) pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine; N-[1-(2-Pyridylacetyl) pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(4-Pyridylglycyl) pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(1H-Imidazol-4-ylmethyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(2-Aminoethyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- (2-thienyl)alanine; N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(trifluoromethyl)alanine;
N-[1-(1H-Imidazol-4-yIacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(2(S)-amino-4-acetylamino)butyric acid ;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(N,N-dimethyl)glutamine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyI]-N- (benzyl)glycyl-methionine;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(benzyl)glycyl-methionine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(4- methoxybenzyl)glycyl-methionine;
N-[1-(Glycyl)pyrrolidin-3(S)-ethyl-2(S)-ylmethyl]-N-(benzyl)glycyl- methionine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-3(S)-ethyl-2(S)-ylmethyl]-N- (benzyl)glycyl-methionine; N-((4-Imidazolyl)methyl-(2S)-pyrrolidinylmethyl)-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- (2-thienyl)alanine methyl ester;
N-[1-(1H-imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(N,N-dimethyl)glutamine methyl ester ;
N-[1-(1H-imidazol-4-ylacetyl)pyrrolidin-2(S)-yImethyl]-N-(1- naphthylmethyl)glycyl-(trifluoromethyl)aIanine methyl ester;
N-[1-(1H-imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(2(S)-amino-4-acetylamino)butyric acid methyl ester;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N- (benzyl)glycyl-methionine methyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(benzyl)glycyl-methionine methyl ester;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyI]-N-(4- methoxybenzyl)glycyl-methionine methyl ester;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-3(S)-ethyl-2(S)-ylmethyl]-N- (benzyl)glycyl-methionine methyl ester;
N-[1-(Glycyl)pyrrolidin-3(S)-ethyl-2(S)-ylmethyl]-N-(benzyl)glycyl- methionine methyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine isopropyl ester;
N-[1-(Glycyl) pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine cyclohexyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine benzyl ester;
N-[1-(Glycyl) pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine ethyl ester;
N-[1-(Sarcosyl) pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine isopropyl ester;
N-[1-(N,N-Dimethylglycyl) pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine isopropyl ester;
N-[1-(Glycyl) pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine (2-pyridylmethyl) ester;
N-[1-(Glycyl) pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine (1-glyceryl) ester;
N-[1-L-Prolylpyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(L-Prolyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(1-Morpholinoacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester; N-[1-(1-Morpholinoacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(4-Piperidinecarbonyl)pyrrolidin-2(S)-yImethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(4-Piperidinecarbonyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(3-Piperidinecarbonyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(3-Piperidinecarbonyl)pyrrolidin-2(S)-ylmethyI]-N-(1- naphthylmethyl)glycyl-methionine; N-[1-(2-Pyridylglycyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(2-Pyridylglycyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(4-Pyridylglycyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(4-Pyridylglycyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(4-Pyridyl(N-methyl)glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(4-Pyridyl(N-methyl)glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine; N-[1-(1H-Imidazol-4-ylpropionyl)ρyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(β-acetylamino)alanine;
N-[1-(1H-Imidazol-4-ylpropionyl) pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(β-acetylamino)alanine methyl ester;
N-[1-(4-Pyridylglycyl) pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(β-acetylamino)alanine;
N-[1-(4-Pyridylglycyl) pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(β-acetylamino)alanine methyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- (β-acetylamino)alanine cyclohexyl ester; N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyI)glycyl-(N-methyl)glutamine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(N-methyl)glutamine methyl ester ;
N-[1-(1H-Imidazol-4-yIacetyl) pyrrolidin-2(S)-ylmethy1]-N-(1- naphthylmethyl)glycyl-(β-methylcarbonylamino)alanine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(β-methylcarbonylamino)alanine methyl ester;
N-[1-(1H-Imidazol-4-ylacetyl) pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(β-methylsulfonylamino)alanine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(β-methylsulfonylamino)alanine methyl ester; N-[1-(1H-Imidazol-4-ylacetyl) pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(β-propionylamino)alanine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(β-propionylamino)alanine methyl ester;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-yImethyl]-N-(1- naphthylmethyl)glycyl-(β-pyrrolidinon-1-ylamino)alanine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyI)glycyl-(β-pyrrolidinon-l-ylamino)alanine methyl ester;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(3- methoxybenzyl)glycyl-methionine; N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(3- methoxybenzyl)glycyl-methionine methyl ester;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(2- methoxybenzyl)glycyl-methionine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(2- methoxybenzyl)glycyl-methionine methyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(3-methoxybenzyl)glycyl- methionine;
N-[1-(Glycyl)ρyrrolidin-2(S)-ylmethyl]-N-(3-methoxybenzyl)glycyl- methionine methyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(2-methoxybenzyl)glycyl- methionine; N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(2-methoxybenzyl)glycyl- methionine methyl ester;
N-[1-(1H-Imidazol-4-ylpropionyl)pyrrolidin-2(S)-ylmethyl]-N-(2- methoxybenzyl)glycyl-methionine;
N-[1-(1H-Imidazol-4-ylpropionyl)pyrrolidin-2(S)-yImethyl]-N-(2- methoxybenzyl)glycyl-methionine methyl ester;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyI]-N-(3- cyanobenzyl)glycyl-methionine;
N-[1-(1H-Imidazol-4-ylacetyI)pyrrolidin-2(S)-ylmethy1]-N-(3-cyanobenzyl)glycyl-methioninemethylester; N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(4- cyanobenzyl)glycyl-methionine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(2- cyanobenzyl)glycyl-methionine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-yImethyl]-N-(2- cyanobenzyl)glycyl-methionine methyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(2-cyanobenzyl)glycyl- methionine;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(2-cyanobenzyl)glycyI- methionine methyl ester;
N-[1-(1H-Imidazol-4-ylpropionyl)pyrrolidin-2(S)-ylmethyl]-N-(2- cyanobenzyl)glycyl-methionine; N-[1-(1H-Imidazol-4-ylρropionyl)pyrrolidin-2(S)-ylmethyl]-N-(2- cyanobenzyl)glycyl-methionine methyl ester;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-yImethyl]-N-(2- methylbenzyl)glycyl-methionine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrroIidin-2(S)-yImethyl]-N-(2- methyIbenzyl)glycyl-methionine methyl ester;
N-[ 1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyI]-N-(2- trifluoromethyIbenzyl)glycyl-methionine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(2- trifluoromethylbenzyl)glycyl-methionine methyl ester; N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylsulfonyl)glycyl-methionine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylsulfonyl)glycyl-methionine methyl ester;
N-[1-(Glycyl) pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine 4-N-methylpiperidinyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine tert-butyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyI)glycyl- methionine 3-pentyl ester;
N-[1-(4-Pyridylglycyl) pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine isopropyl ester; N-[ 1-(1H-Imidazol-4-ylpropionyl)pyrrolidin-2(S)-yImethyl]-N-(11- naphthylmethyl)glycyl-methionine isopropyl ester;
N-[1-(4-Imidazoleacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester
N-[1-(4-Imidazoleacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine isopropyl ester
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl-methioninemethylester
N-[1-(Glycyl)pyrroIidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine isopropyl ester
N-[1-(L-Pyroglutamyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine
N-[1-(L-Pyroglutamyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester
2(S)-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-3(S)-ethyl-2(S)- ylmethyloxy]-3-phenylpropionyl-methionine methyl ester
2(S)-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-3(S)-ethyl-2(S)- ylmethyloxy]-3-phenylpropionyl-methionine
N-[1-(Sarcosyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine
N-[1-(Sarcosyl)pyrrolidin-2(S)-yImethyl]-N-(1-naphthylmethyl)glycyl- methionine methyl ester o
N-[1-(N,N-Dimethylglycyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthyImethyl)glycyl-methionine
N-[1-(N,N-Dimethylglycyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine mtthyl ester
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(β-acetylamino)aIanine methyl ester
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethy1]-N-(1- naphthylmethyl)glycyl-(β-acetylamino)alanine
N-[1-(Glycyl) pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- (β-acetylamino)alanine cyclohexyl ester
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- (β-acetylamino)alanine
N-[1-(4-PyridyIglycyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine isopropyl ester
N-[1-(4-Pyridylglycyl)pyrroIidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-yimethyl]-N-(2- methoxybenzyl)glycyl-methionine
N-[1-(1H-Imidazol-4-ylacetyI)pyrrolidin-2(S)-ylmethyl]-N-(2- methoxybenzyl)glycyl-methionine methyl ester
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(2-methoxybenzyl)glycyl- methionine methyl ester
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(2-methoxybenzyl)glycyl- methionine
N-[1-(1H-Imidazol-4-ylpropionyl)pyrrolidin-2(S)-ylmethyl]-N-(2- methoxybenzyl)glycyl-methionine methyl ester
N-[1-(1H-Imidazol-4-ylpropionyl)pyrrolidin-2(S)-ylmethyl]-N-(2- methoxybenzyl)glycyl-methionine
N-[1-(1H-Imidazol-4-yIacetyl)pyrrolidin-2(S)-ylmethyl]-N-(2- cyanobenzyl)glycyl-methionine
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(2- cyanobenzyl)glycyl-methionine methyl ester
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine 4-N-methylpiperidinyI ester
N-[1-(1H-Imidazol-4-ylpropionyl)pyrrolidin-2(S)-ylmethy1]-N-(1- naphthylmethyl)glycyl-methionine isopropyl ester
N-[(1H-imidazol-4-ylacetyl-2(S)-amino)-3(S)-methylpentyl]-1,2,3,4- tetrahydro-3(S)-isoquinolinecarbonyl-methionine methyl ester;
N-[(1H-imidazol-4-ylacetyl-2(S)-amino)-3(S)-methylpentyl]-1,2,3,4- tetrahydro-3(S)-isoquinolinecarbonyl-methionine;
N-[1 -(1H-imidazol-4-ylacetyl)-3(S)-ethylpyrrolidin-2(S)-ylmethyl ]- prolyl-methionine methyl ester;
N-[ 1 -(1H-imidazol-4-ylacetyl)-3(S)-ethylρyrrolidin-2(S)-ylmethyl]- prolyl-methionine;
N-[ 1 -Glycylpyrrolidin-2(S)-ylmethyl]-3(S)-ethylprolyl-methionine methyl ester;
N-[ 1 -Glycylpyrrolidin-2(S)-ylmethyI]-3(S)-ethylprolyl-methionine;
N-[L-Pyroglutamyl-2(S)-amino-3(S)-methylpentyl]-1 ,2,3,4-tetrahydro- 3(S)-isoquinolinecarbonyl-methionine
N-[L-Pyroglutamyl-2(S)-amino-3(S)-methylpentyl]-1 ,2,3,4-tetrahydro- 3(S)-isoquinolinecarbonyl-methionine methyl ester
N-[1-(1H-imidazol-4-ylacetyl)-pyrrolidin-2(S)-yImethyl]-3(S)- ethylprolyl-methionine
N-[ 1 -(1H-imidazol-4-ylacetyl)-pyrrolidin-2(S-)ylmethyl ]-3(S)- ethylprolyl-methionine methyl ester
N-[(1H-imidazol-4-ylacetyl-2(S)-amino)-3(S)-methylpentyl]-prolyl- methionine methyl ester
N-[(1H-imidazol-4-ylacetyl-2(S)-amino)-3(S)-methylpentyl]-prolyl- methionine
N-[( 1 H-imidazol-4-ylacetyl-2(S)-amino)-3(S)-methylpentyl]-1 ,2,3,4- tetrahydro-3(S)-isoquinolinecarbonyl-methionine methyl ester N-[(1H-imidazol-4-ylacetyl-2(S)-amino)-3(S)-methylρentyl]-1 ,2,3,4- tetrahydro-3(S)-isoquinolinecarbonyl-methionine
N-[L-Pyroglutamyl-2(S)-amino-3(S)-methylpentyl]-1 ,2,3,4-tetrahydro- 3(S)-isoquinolinecarbonyI-methionine methyl ester
N-[L-Pyroglutamyl-2(S)-amino-3(S)-methylpentyl]-1 ,2,3,4-tetrahydro- 3(S)-isoquinolinecarbonyl-methionine
N-[(1H-imidazol-4-ylacetyl-2(S)-amino)-3(S)-methylpentyl]-prolyl- methionine methyl ester
N-[(1H-imidazol-4-ylacetyl-2(S)-amino)-3(S)-methylpentyl]-prolyl- methionine N-[ 1-(1H-imidazol-4-ylacetyl)-3(S)-ethylpyrrolidin-2(S)-ylmethyl]- prolyl-methionine methyl ester
N-[1 -(1H-imidazol-4-ylacetyl)-3(S)-ethylpyrrolidin-2(S)-ylmethyl]- prolyl-methionine
N-[ 1 -(1 H-imidazol-4-ylacetyl)-pyrroIidin-2(S)-ylmethyl]-3(S)- ethylprolyl-methionine methyl ester
N-[1-(1H-imidazol-4-ylacetyl)-pyrrolidin-2(S)-ylmethyl]-3(S)- ethylprolyl-methionine N-[1-Glycylpyrrolidin-2(S)-ylmethyl]-3(S)-ethylprolyl-methionine methyl ester
N-[1-Glycylpyrrolidin-2(S)-ylmethyl]-3(S)-ethylprolyl-methionine 2(S)-Butyl-1-(2,3-diaminoprop-1-yl)-1-(1-naphthoyl)piperazine
1-(3-Amino-2-(2-naphthylmethylamino)prop-1-yl)-2(S)-butyl-4-(1- naphthoyl)piperazine
2(S)-Butyl-1-{5-[1-(2-naphthylmethyl)]-4,5-dihydroimidazol}methy1-4- (1-naphthoyl)piperazine
1-[5-(1-BenzyIimidazol)methyl]-2(S)-butyl-4-(1-naphthoyl)piperazine
1-{5-[1-(4-nitrobenzyI)]imidazolylmethyl}-2(S)-butyl-4-(1- naphthoyl)piperazine
1-(3-Acetamidomethylthio-2(R)-aminoprop-1-yl)-2(S)-butyl-4-(1- naphthoyl)piperazine 2(S)-Butyl-1-[2-(1-imidazolyl)ethyl]sulfonyI-4-(1-naphthoyl)piperazine
2(R)-Butyl-1-imidazoly1-4-methyl-4-(1-naphthoyl)piperazine
2(S)-Butyl-4-(1-naphthoyl)-1-(3-pyridylmethyl)piperazine
1-2(S)-butyl-(2(R)-(4-nitrobenzyl)amino-3-hydroxypropyl)-4-(1- naphthoyl)piperazine
1-(2(R)-Amino-3-hydroxyheptadecyl)-2(S)-butyl-4-(1-naphthoyl)- piperazine
2(S)-Benzyl-1-imidazolyl-4-methyl-4-(1-naphthoyl)piperazine
1-(2(R)-Amino-3-(3-benzylthio)propyl)-2(S)-butyl-4-(1- naphthoyl)piperazine
1-(2(R)-Amino-3-f3-(4-nitrobenzylthio)propyl])-2(S)-butyl-4-(1- naphthoyl)piperazine
2(S)-Buty1-1-[(4-imidazolyl)ethyl]-4-(1-naphthoyl)piperazine
2(S)-Butyl-1-[(4-imidazolyl)methyl]-4-(1-naphthoyl)piperazine
2(S)-Butyl-1-[(1-naphth-2-ylmethyl)-1H-imidazol-5-yl)acetyl]-4-(1- naphthoyl)piperazine
2(S)-Butyl-1-[(1-naphth-2-ylmethyl)-1H-imidazol-5-yl)ethyl]-4-(1- naphthoyl)piperazine
1-(2(R)-Amino-3-hydroypropyl)-2(S)-butyl-4-(1-naphthoyl)piperazine
1-(2(R)-Amino-4-hydroxybutyl)-2(S)-butyl-4-(1-naphthoyl)piperazine
1-(2-Amino-3-(2-benzyloxyphenyl)propyl)-2(S)-butyl-4-(1- naphthoyl)piperazine
1-(2-Amino-3-(2-hydroxyphenyl)propyl)-2(S)-butyl-4-(1- naphthoyl)piperazine
1-[3-(4-imidazolyl)propyl]-2(S)-butyl-4-(1-naphthoyl)-piperazine
2(S)-n-Butyl-4-(2,3-dimethylphenyl)-1-(4-imidazolylmethyl)- piperazin-5-one
2(S)-n-Butyl-1-[1-(4-cyanobenzyl)imidazol-5-ylmethyl]-4-(2,3- dimethylphenyl)piperazin-5-one
1-[1-(4-Cyanobenzyl)imidazol-5-ylmethyl]-4-(2,3-dimethylphenyl)- 2(S)-(2-methoxyethyl)piperazin-5-one 2(S)-n-Butyl-4-(1-naphthoyl)-1-[1-(1-naphthylmethyl)imidazol-5- ylmethyl]-piperazine
2(S)-n-Butyl-4-(1-naphthoyl)-1-[1-(2-naphthylmethyl)imidazol-5- ylmethyl]-piperazine
2(S)-n-Butyl-1-[1-(4-cyanobenzyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)piperazine
2(S)-n-Butyl-1-f1-(4-methoxybenzyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)piperazine
2(S)-n-Butyl-1-|1-(3-methyl-2-butenyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)piperazine 2(S)-n-Butyl-1-[1-(4-fluorobenzyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)piperazine
2(S)-n- Butyl-1-[1-(4-chlorobenzyl)imidazol-5-yImethyl]-4-(1- naphthoyl)piperazine
1-[1-(4-Bromobenzyl)imidazol-5-ylmethyl]-2(S)-n-butyl-4-(1- naphthoyl)piperazine
2(S)-n-Butyl-4-(1-naphthoyl)-1-[1-(4-trifluoromethylbenzyl)imidazol-5- ylmethyl]-piperazine
2(S)-n-Butyl-1-[1-(4-methylbenzyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)-piperazine
2(S)-n-Butyl-1-[1-(3-methylbenzyl)imidazoI-5-ylmethyl]-4-(1- naphthoyl)-piperazine 1-[1-(4-Phenylbenzyl)imidazol-5-ylmethyl]-2(S)-n-butyl-4-(1- naphthoyl)-piperazine
2(S)-n-Butyl-4-(1-naphthoyl)-1-[1-(2-phenylethyl)imidazol-5-ylmethyl]- piperazine
2(S)-n-Butyl-4-(1-naphthoyl)-1-[1-(4-trifluoromethoxy)imidazol-5- ylmethyl]piperazine
1-{[1-(4-cyanobenzy1)-1H-imidazol-5-yl]acety1}-2(S)-n-butyl-4-(1- naphthoyl)piperazine
(N-[1-Cyanobenzyl)-1H-imidazol-5-yl)acetyl]pyrrolidin-2(S)-ylmethyl]- 3(S)-ethyl-prolyl methionine
(N-[1-Cyanobenzyl)-1H-imidazol-5-yl)acetyl]pyrrolidin-2(S)-ylmethyl]- 3(S)-ethyl-proIyl methionine methyl ester
(N-[1-Cyanobenzyl)-1H-imidazol-5-yl)acetyl]pyrrolidin-2(S)-ylmethyl]- 3(S)-ethyl-prolyl methionine isopropyl ester
N-[1-(1H-Imidazol-4-propionyl)ρyrrolidin-2(S)-ylmethyl]-N-(2- methoxybenzyl)glycyl-methionine isopropyl ester
Compounds which are useful in the present invention, and methods of synthesis thereof, can be found in the following patents, pending applications and publications:
USSN 60/005,059 filed on October 6, 1995;
USSN 60/005,063 filed on October 6, 1995
USSN 60/005,521 filed on October 13, 1995
WO 95/32987 published on 7 December 1995.
U. S. Pat. No. 5,420,245;
European Pat. Publ. 0 618 221 ;
WO 94/26723;
WO 95/08542 ;
WO 95/1 1917;
WO 95/12612.
U. S. Pat. No. 5,238,922 granted on August 24, 1993; ;
U. S. Pat. No. 5,340,828 granted on August 23, 1994; ;
U. S. Pat. No. 5,352,705 granted on October 4, 1994; U. S. Pat. No. 5,326,773 granted on July 5, 1994; USSN 07/968,022 filed on October 29, 1992 ;
USSN 08/968,025 filed on October 29, 1992 and USSN 08/143,943 filed on October 27, 1993 ; USSN 08/080,028 filed on June 18, 1993 and USSN 08/237,586 filed on May 1 1, 1994 ;
USSN 08/314,987 filed on September 29, 1994 USSN 08/315,171 filed on September 29, 1994
USSN 08/315,046 filed on September 29, 1994 ;
USSN 08/315,161 filed on September 29, 1994; USSN 08/399,282 filed on March 6, 1995; USSN 472,077 filed on June 6, 1995 and USSN 08/527,972 filed on September 14, 1995
USSN 08/315,151 filed on September 29, 1994 ;
USSN 08/314,974 filed on September 29, 1994
USSN 08/412,621 filed on March 29, 1995 and USSN 08/448,865 filed on May 24, 1995 ;
USSN 08/413,137 filed on March 29, 1995; ;
USSN 08/412,828 filed on March 29, 1995;
USSN 08/412,829 filed on March 29, 1995; and USSN 08/470,690 filed on June 6, 1995;
USSN 08/412,830 filed on March 29, 1995;
USSN 08/449,038 filed on May 24, 1995; ; USSN 08/468, 160 filed on June 6, 1995; ;
All patents, publications and pending patent applications identified are hereby incorporated by reference.
The Raf antagonists are described herein using the terms defined below unless otherwise specified.
The term "alkyl" refers to a monovalent alkane (hydrocarbon) derived radical containing from 1 to 15 carbon atoms unless otherwise defined. It may be straight, branched or cyclic.
Preferred straight or branched alkyl groups include methyl, ethyl, propyl, isopropyl, butyl and t-butyl. Preferred cycloalkyl groups include cyclopentyl and cyclohexyl.
Alkyl also includes a straight or branched alkyl group which contains or is interrupted by a cycloalkylene portion.
Examples include the following:
wherein: x and y = from 0-10; and w and z = from 0-9.
The alkylene and monovalent alkyl portion(s) of the alkyl group can be attached at any available point of attachment to the cycloalkylene portion.
When substituted alkyl is present, this refers to a straight, branched or cyclic alkyl group as defined above, substituted with 1-3 groups as defined with respect to each variable.
Heteroalkyl refers to an alkyl group having from 2-15 carbon atoms, and interrupted by from 1 -4 heteroatoms selected from O, S and N.
The term "alkenyl" refers to a hydrocarbon radical straight, branched or cyclic containing from 2 to 15 carbon atoms and at least one carbon to carbon double bond. Preferably one carbon to carbon double bond is present, and up to four non- aromatic (non-resonating) carbon-carbon double bonds may be present. Examples of alkenyl groups include vinyl, allyl, isopropenyl, pentenyl, hexenyl, heptenyl, cyclopropenyl,
cyclobutenyl, cyclopentenyl, cyclohexenyl, 1 -propenyl, 2-butenyl, 2-methyl-2-butenyl, isoprenyl, famesyl, geranyl, geranylgeranyl and the like. Preferred alkenyl groups include ethenyl, propenyl, butenyl and cyclohexenyl. As described above with respect to alkyl, the straight, branched or cyclic portion of the alkenyl group may contain double bonds and may be substituted when a substituted alkenyl group is provided.
The term "alkynyl" refers to a hydrocarbon radical straight, branched or cyclic, containing from 2 to 15 carbon atoms and at least one carbon to carbon triple bond. Up to three carbon- carbon triple bonds may be present. Preferred alkynyl groups include ethynyl, propynyl and butynyl. As described above with respect to alkyl, the straight, branched or cyclic portion of the alkynyl group may contain triple bonds and may be substituted
when a substituted alkynyl group is provided.
Aryl refers to aromatic rings e.g., phenyl, substituted phenyl and like groups as well as rings which are fused, e.g., naphthyl and the like. Aryl thus contains at least one ring having at least 6 atoms, with up to two such rings being present, containing up to 10 atoms therein, with alternating (resonating) double bonds between adjacent carbon atoms. The preferred aryl groups are phenyl and naphthyl.
Aryl groups may likewise be substituted as defined below. Preferred substituted aryls include phenyl and naphthyl substituted with one or two groups. With regard to the famesyl transferase inhibitors, "aryl" is intended to include any stable monocyclic, bicyclic or tricyclic carbon ring(s) of up to 7 members in each ring, wherein at least one ring is aromatic. Examples of aryl groups include phenyl, naphthyl, anthracenyl, biphenyl, tetrahydronaphthyl, indanyl, phenanthrenyl and the like.
The term "heteroaryl" refers to a monocyclic aromatic hydrocarbon group having 5 or 6 ring atoms, or a bicyclic aromatic group having 8 to 10 atoms, containing at least one heteroatom,
O, S or N, in which a carbon or nitrogen atom is the point of
attachment, and in which one additional carbon atom is optionally replaced by a heteroatom selected from O or S, and in which from
1 to 3 additional carbon atoms are optionally replaced by nitrogen heteroatoms. The heteroaryl group is optionally substituted with up to three groups.
Heteroaryl thus includes aromatic and partially aromatic groups which contain one or more heteroatoms. Examples of this type are thiophene, purine, imidazopyridine, pyridine, oxazole, thiazole, oxazine, pyrazole, tetrazole, imidazole, pyridine, pyrimidine, pyrazine and triazine. Examples of partially aromatic groups are tetrahydro- imidazof4,5-c]pyridine, phthalidyl and saccharinyl, as defined below.
With regard to the famesyl transferase inhibitors, the term heterocycle or heterocyclic, as used herein, represents a stable 5- to 7- membered monocyclic or stable 8- to 1 1 -membered bicyclic or stable 1 1 -15 membered tricyclic heterocycle ring which is either saturated or
unsaturated, and which consists of carbon atoms and from one to four heteroatoms selected from the group consisting of N, O, and S, and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring. The heterocyclic ring may be attached at any heteroatom or carbon atom which results in the creation of a stable structure. Examples of such heterocyclic elements include, but are not limited to, azepinyl, benzimidazolyl, benzisoxazolyl, benzofurazanyl, benzopyranyl, benzothiopyranyl, benzofuryl,
benzothiazolyl, benzothienyl, benzoxazolyl, chromanyl, cinnolinyl, dihydrobenzofuryl, dihydro-benzothienyl. dihydrobenzothiopyranyl, dihydrobenzothio-pyranyl sulfone, furyl, imidazolidinyl, imidazolinyl, imidazolyl, indolinyl, indolyl, isochromanyl, isoindolinyl, isoquinolinyl, isothiazolidinyl, isothiazolyl, isothiazolidinyl, morpholinyl,
naphthyridinyl, oxadiazolyl, 2-oxoazepinyl, 2-oxopiperazinyl, 2- oxopiperidinyl, 2-oxopyrrolidinyl, piperidyl, piperazinyl, pyridyl, pyridyl N-oxide, pyridonyl, pyrazinyl, pyrazolidinyl, pyrazolyl, pyrimidinyl, pyrrohdinyl, pyrrolyl, quinazolinyl, quinolinyl, quinolinyl N-oxide, quinoxalinyl, tetrahydrofuryl, tetrahydroisoquinolinyl, tetrahydro-quinolinyl, thiamorpholinyl, thiamorpholinyl sulfoxide, thiazolyl, thiazolinyl, thienofuryl, thienothienyl, and thienyl.
Preferably, heterocycle is selected from imidazolyl, 2-oxopyrrolidinyl, piperidyl, pyridyl and pyrrolidinyl.
Substituted alkyl, aryl and heteroaryl, and the substituted portions of aralkyl, aralkoxy, heteroaralkyl, heteroaralkoxy and like groups are substituted with from 1 -3 groups selected from the group consisting of: halo, hydroxy, cyano, acyl, acylamino, aralkoxy, alkylsulfonyl, arylsulfonyl, alkylsulfonylamino, arylsulfonylamino, alkylaminocarbonyl, alkyl, alkoxy, aryl, aryloxy, aralkoxy, amino, alkylamino, dialkylamino, and sulfonylamino.
With regard to the famesyl transferase inhibitors, the terms
"substituted aryl", "substituted heterocycle" and "substituted cycloalkyl" are intended to include the cyclic group which is substituted with 1 or
2 substitutents selected from the group which includes but is not limited to F, Cl, Br, CF3, NH2, N(C1-C6 alkyl)2, NO2, CN, (C1 -C6 alkyl)O-, -OH, (C1-C6 alkyl)S(O)m-, (C1 -C6 alkyl)C(O)NH-, H2N-C(NH)-, (C1-C6 alkyl)C(O)-, (C1 -C6 alkyl)OC(O)-, N3,(C1 -C6
alkyl)OC(O)NH- and C1-C20 alkyl.
The terms "heterocycloalkyl" and "heterocyclyl" refer to a cycloalkyl group (nonaromatic) in which one of the carbon atoms in the ring is replaced by a heteroatom selected from O, S(O)y or N, and in which up to three additional carbon atoms may be replaced by said heteroatoms. When three heteroatoms are present in the heterocycle, they are not all linked together.
Examples of heterocyclyls are piperidinyl, morpholinyl, pyrrohdinyl, tetrahydrofuranyl, imidazolinyl, piperazinyl, pyrolidine- 2-one, piperidine-2-one and the like.
Acyl as used herein refers to -C(O)C1 -6 alkyl and -C(O)- aryl.
Acylamino refers to the group -NHC(O)C1 -6 alkyl and -NHC(O)aryl.
Aralkoxy refers to the group -OC1 -6 alkylaryl. Alkylsulfonyl refers to the group -SO2C1-6 alkyl.
Alkylsulfonylamino refers to the group -NHSO2C1 -6alkyl.
Arylsulfonylamino refers to the group -NHSO2aryl.
Alkylaminocarbonyl refers to the group -C(O)NHC1 -6 alkyl.
Aryloxy refers to the group -O-aryl.
Aralkoxy refers to the group -O-C1 -6 alkylaryl.
Sulfonylamino refers to the group -NHSO3H.
Halo means Cl, F, Br and I selected on an independent basis.
Within -[C(O)(CH2)j-CR5R6-(CH2)k-NR7]p-R8, there may be from 1 to 3 groups -[C(O)(CH2)j-CR5R6-(CH2)k-NR7]-.
Thus, -[C(O)(CH2)j-CR5R6-(CH2)k-NR7]p-R8 with p equal to 1 , 2 or 3 means the following:
-C(O)(CH2)j-CR5R6-(CH2)k-NR7-RX; -C(O)(CH2)j-CR5R6-(CH2)k-NR7-C(O)(CH2)j-CR5R6-(CH2)k-NR7R8; and
C(O)(CH2)jCR5R6(CH2)kNR7C(0)(CH2)jCR5R6(CH2)kNR7C(0)(CH2)jCR5R6(CH2)kNR7R8.
Within these groups, the variables are determined independently. For example, when more than one j is present, they may be the same or different. When CR5R6 is taken in combination, it represents a 3, 4, 5 or 6 membered cycloalkyl or heterocyclyl group, an aryl group or a heteroaryl group. Examples of suitable cycloalkylene attachment are as follows:
In each of the pattems of attachment noted above, the ring may also be heterocyclic as defined above.
are optional substituents linked to the HETCy group.
independently represent mono or bicyclic ring systems, non-aromatic or partially aromatic, containing from 5-10 ring atoms, 1 -4 of which are N and 0- 1 of which are O or S(O)
y, with y equal to 0, 1 or 2, and when partially aromatic, the non-aromatic portion thereof optionally containing 1-2 carbonyl groups. Hence, these ring systems can be heteroaryl or heterocyclic as defined above.
is linked to HETCy through a nitrogen atom contained in the ring system, either directly or through a linking group which is part of R'. Examples include phthalidyl and saccharinyl, as further defined below.
is likewise linked to HETCy, but through a carbon atom contained in the ring system.
The term phthalidyl refers to the heteroaryl group:
The term saccharinyl refers to the heteroaryl group:
In the present method, amino acids which are disclosed are identified both by conventional 3 letter and single letter abbreviations as indicated below:
The compounds used in the present method may have asymmetric centers and occur as racemates, racemic mixtures, and as individual diastereomers, with all possible isomers, including optical isomers, being included in the present invention. Unless otherwise
specified, named amino acids are understood to have the natural "L" stereoconfiguration
The following structure:
represents a cyclic amine moiety having 5 or 6 members in the ring, such a cyclic amine which may be optionally fused to a phenyl or cyclohexyl ring. Examples of such a cyclic amine moiety include, but are not limited to, the following specific structures:
It is also understood that substitution on the cyclic amine moiety by R
2a and R2b
may be on different carbon atoms or on the same carbon atom.
When R
3 and R
4 are combined to form - (CH
2)
s -, cyclic moieties are formed. Examples of such cyclic moieties include, but are not limited to:
When R
5a and R
5b are combined to form - (CH
2)
s -, cyclic moieties as described hereinabove for R
3 and R
4 are formed. In addition, such cyclic moieties may optionally include a heteroatom(s). Examples of such heteroatom-containing cyclic moieties include, but are not limited to:
The pharmaceutically acceptable salts of the compounds of this invention include the conventional non-toxic salts of the compounds of this invention as formed, e.g., from non-toxic inorganic or organic acids. For example, such conventional non-toxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like: and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenyl-acetic, glutamic, benzoic, salicylic, sulfanihc, 2-acetoxy-benzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, isethionic, trifluoroacetic and the like.
It is intended that the definition of any substituent or variable (e.g., Rl O, Z, n, etc.) at a particular location in a molecule be independent of its definitions elsewhere in that molecule. Thus, -N(R 10)2 represents -NHH, -NHCH3, -NHC2H5, etc. It is understood that substituents and substitution pattems on the compounds of the instant invention can be selected by one of ordinary skill in the art to provide compounds that are chemically stable and that can be readily synthesized by techniques known in the art as well as those methods set forth below.
The pharmaceutically acceptable salts of the compounds of this invention can be synthesized from the compounds of this invention which contain a basic moiety by conventional chemical methods. Generally, the salts are prepared by reacting the free base with stoichiometric amounts or with an excess of the desired salt-
forming inorganic or organic acid in a suitable solvent or various combinations of solvents.
The compounds of formulas (ITa) through (Il-k) can be synthesized from their constituent amino acids by conventional peptide synthesis techniques, and the additional methods described below. Standard methods of peptide synthesis are disclosed, for example, in the following works: Schroeder et al, "The Peptides", Vol. I, Academic Press 1965, or Bodanszky et al, "Peptide Synthesis", Interscience Publishers, 1966, or McOmie (ed.) "Protective Groups in Organic Chemistry", Plenum Press, 1973, or Barany et al, "The Peptides: Analysis, Synthesis, Biology" 2, Chapter 1 , Academic Press, 1980, or Stewartet al., "Solid Phase Peptide Synthesis", Second Edition, Pierce Chemical Company, 1984. Also useful in exemplifying syntheses of specific unnatural amino acid residues are European Pat. Appl. No. 0 350 163 A2 (particularly page 51 -52) and J. E. Baldwin et al.
Tetrahedron, 50:5049-5066 (1994). With regards to the synthesis of instant compounds containing a (β-acetylamino)alanine residue at the C-terminus, use of the commercially available Nα-Z-L-2,3- diaminopropionic acid (Fluka) as a starting material is preferred.
Abbreviations used in the description of the chemistry and in the Examples that follow are:
Ac2O Acetic anhydride;
Boc t-Butoxycarbonyl;
DBU 1 ,8-diazabicyclo[5.4.0]undec-7-ene;
DMAP 4-Dimethylaminopyridine;
DME 1 ,2-Dimethoxyethane;
DMF Dimethylformamide;
EDC 1 -(3-dimethylaminopropyl)-3-ethyl-carbodiimide- hydrochloride;
HOBT 1-Hydroxybenzotriazole hydrate;
Et3N Triethylamine;
EtOAc Ethyl acetate;
FAB Fast atom bombardment;
HOOBT 3-Hydroxy- 1 ,2,2-benzotriazin-4(3H)-one;
HPLC High-performance liquid chromatography;
MCPBA m-Chloroperoxybenzoic acid;
MsCl Methanesulfonyl chloride;
NaHMDS Sodium bis(trimethylsilyl)amide;
Py Pyridine;
TFA Trifluoroacetic acid;
THF Tetrahydrofuran. The compounds of formula (I-a) and (I-b) are prepared in accordance with U. S. Application No. 60/005,059 filed on October 6, 1995 and 60/005,063 filed on October 6, 1995. Two general methods for preparation of the imidazole nucleus are outlined. In the first, a suitably protected picolyl alcohol is deprotonated with a strong base such as n-butyl lithium or lithium diisopropyl amide and the resulting anion is reacted with an appropriate N,O-dimethylhydrox- amide to give a protected alpha hydroxy ketone. The protected alpha hydroxy ketone is then condensed with a suitably functionalized and protected aminoaldehyde in the presence of ammonium acetate, acetic acid and copper acetate.
The aldehydes typically used contain a suitably protected nitrogen atom. After the imidazole nucleus has been formed, the nitrogen is deprotected and then reacted with an appropriate
electrophilic reagent to provide the final compounds.
In the second method, a suitably protected picolyl alcohol is deprotonated with a strong base such as n-butyl lithium or lithium diisopropyl amide and the resulting anion is reacted with an appropriate aryl or alkyl aldehyde to give a mono-protected diol. The protecting group is removed and the resulting diol is oxidized (by the method of Swem or Moffat) to a dione. The dione is then condensed with a suitably functionalized and protected aminoaldehyde in the presence of ammonium acetate in acetic acid to give the imidazole.
In this same manner, the nitrogen is deprotected and then reacted with an appropriate electrophilic reagent to provide the compounds of formula I.
TBDMSO refers to t-butyldimethylsilyloxy, TFAA refers to trifluoroacetic anhydride, TBDMS refers to t-butyldimethylsilyl, TBAF refers to tetrabutyl ammonium fluoride, Cbz refers to carboxyl- benzyl, Ac refers to acetyl, and LDA refers to lithium diisopropyl amide.
E represents an electrophile attached to the heterocyclic ring nitrogen atom. Examples of suitable electrophiles include alkyl halides, alkyl triflates, alkyl mesylates, benzyl halides, vinyl pyridine and the like. Hence, E represents alkyl, benzyl, vinyl and the like.
The compounds are useful in various pharmaceutically acceptable salt forms. The term "pharmaceutically acceptable salt" refers to those salt forms which would be apparent to the pharmaceutical chemist, i.e., those which are substantially non-toxic and which provide the desired pharmacokinetic properties, palatability, absorption,
distribution, metabolism or excretion. Other factors, more practical in nature, which are also important in the selection, are cost of the raw materials, ease of crystallization, yield, stability, hygroscopicity and flowability of the resulting bulk drug. Conveniently, pharmaceutical compositions may be prepared from the active ingredients in
combination with pharmaceutically acceptable carriers.
Pharmaceutically acceptable salts include conventional non-toxic salts or quarternary ammonium salts formed, e.g., from non-toxic inorganic or organic acids. Non-toxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like; and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, sulfanihc, 2-acetoxybenzoic, fumaric, toluenesulfonic, methane- sulfonic, ethane disulfonic, oxalic, isethionic, trifluoroacetic and the like.
The pharmaceutically acceptable salts of the present invention can be synthesized by conventional chemical methods.
Generally, the salts are prepared by reacting the free base or acid with stoichiometric amounts or with an excess of the desired salt- forming inorganic or organic acid or base, in a suitable solvent or solvent combination.
Compounds of formula (I-c)
may be prepared using procedures described in PCT/US94/08297 published on 2 February 1995 and in U.S. Application No. 60/005,521 filed on October 13, 1995. Suitable procedures are also described in U.S. Patent Nos. 3,707,475 and 3,940,486.
The Raf antagonists described herein are useful in various pharmaceutically acceptable salt forms. The term "pharmaceutically acceptable salt" refers to those salt forms which would be apparent to the pharmaceutical chemist, i.e., those which are substantially non-toxic and which provide the desired pharmacokinetic properties, palatabihty, absorption, distribution, metabolism or excretion. Other factors, more practical in nature, which are also important in the selection, are cost of the raw materials, ease of crystallization, yield, stability, hygroscopicity and flowability of the resulting bulk drug. Conveniently,
pharmaceutical compositions may be prepared from the active
ingredients in combination with pharmaceutically acceptable carriers.
Pharmaceutically acceptable salts include conventional non-toxic salts or quartemary ammonium salts formed, e.g., from non-toxic inorganic or organic acids. Non-toxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like; and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, sulfanihc, 2-acetoxybenzoic, fumaric, toluenesulfonic, methane- sulfonic, ethane disulfonic, oxalic, isethionic, trifluoroacetic and the like.
The pharmaceutically acceptable salts can be synthesized by conventional chemical methods. Generally, the salts are prepared by reacting the free base or acid with stoichiometric amounts or with an excess of the desired salt-forming inorganic or organic acid or base, in a suitable solvent or solvent combination.
The famesyl transferase inhibitors of formula (Il-a) through (II-c) can be synthesized in accordance with reaction schemes 1-16, in addition to other standard manipulations such as ester
hydrolysis, cleavage of protecting groups, etc., as may be known in the literature or exemplified in the experimental procedures. Substituents Ra and RD, as shown in the Schemes, represent the substituents R2, R3, R4, and R5; however their point of attachment to the ring is illustrative only and is not meant to be limiting.
These reactions may be employed in a linear sequence to provide the compounds of the invention or they may be used to synthesize fragments which are subsequently joined by the alkylation reactions described in the Reaction Schemes.
Synopsis of reaction Schemes 1 -16:
The requisite intermediates are in some cases commercially available, or can be prepared according to literature procedures, for the most part. In Scheme 1 , for example, the synthesis of 2-aIkyl substituted piperazines is outlined, and is essentially that described by J. S. Kiely and S. R. Priebe in Organic Preparations and Proceedings Int., 1990, 22, 761-768. Boc -protected amino acids I, available commercially or by procedures known to those skilled in the art, can be coupled to N-benzyl amino acid esters using a variety of dehydrating agents such as DCC (dicyclohexycarbodiimide) or EDC·HCl (1-ethyl- 3-(3-dimethylaminopropyl)carbodiimide hydrochloride) in a solvent such as methylene chloride , chloroform, dichloroethane, or in dimethylformamide. The product II is then deprotected with acid, for example hydrogen chloride in chloroform or ethyl acetate, or trifluoroacetic acid in methylene chloride, and cyclized under weakly basic conditions to give the diketopiperazine III. Reduction of III with lithium aluminum hydride in refluxing ether gives the piperazine IV, which is protected as the Boc derivative V. The N-benzyl group can be cleaved under standard conditions of hydrogenation, e.g., 10% palladium on carbon at 60 psi hydrogen on a Parr apparatus for
24-48 h. The product VI can be treated with an acid chloride, or a carboxylic acid under standard dehydrating conditions to furnish the carboxamides VII. A final acid deprotection step gives the intermediate VIII (Scheme 2). Intermediate VIII can be reductively alkylated with a variety of aldehydes, such as IX, prepared by standard procedures, such as that described by O. P. Goel, U. Krolls, M. Stier and S. Kesten in Organic Syntheses. 1988, 67, 69-75, from the appropriate amino acid (Scheme 3). The reductive alkylation can be accomplished at pH 5-7 with a variety of reducing agents, such as sodium triacetoxyborohydride
or sodium cyanoborohydride, in a solvent such as dichloroethane, methanol or dimethylformamide. The product X can be deprotected to give the final compounds XI with trifluoroacetic acid in methylene chloride. The final product XI is isolated in the salt form, for example, as a trifluoroacetate, hydrochloride or acetate salt, among others. The product diamine XI can further be selectively protected to obtain XII, which can subsequently be reductively alkylated with a second aldehyde to obtain XIII. Removal of the protecting group, and conversion to the cyclized product such as the dihydroimidazole XV, can be accomplished by literature procedures.
Alternatively, the protected piperazine intermediate VII can be reductively alkylated with other aldehydes such as 1 -trityl-4- carboxaldehyde or l-trityl-4-imidazolylacetaldehyde, to give products such as XVI (Scheme IV) (Tr = trityl). The trityl protecting group can be removed from XVI to give XVII, or alternatively, XVI can first be treated with an alkyl halide then subsequently deprotected to give the alkylated imidazole XVIII. Alternatively, the intermediate VIII can be acylated or sulfonylated by standard techniques. The imidazole acetic acid XIX can be converted to the acetate XXI by standard procedures, and XXI can be first reacted with an alkyl halide, then treated with refluxing methanol to provide the regiospecifically alkylated imidazole acetic acid ester XXII. Hydrolysis and reaction with piperazine VIJJ in the presence of condensing reagents such as 1 -(3-dimethylaminopropyl)- 3-ethylcarbodiimide (EDC) leads to acylated products such as XXIV.
If the piperazine VIII is reductively alkylated with an aldehyde which also has a protected hydroxyl group, such as XXV in Scheme 6, the protecting groups can be subsequently removed to unmask the hydroxyl group (Schemes 6, 7). The alcohol can be oxidized under standard conditions to e.g. an aldehyde, which can then be reacted with a variety of organometalhc reagents such as Grignard reagents, to obtain secondary alcohols such as XXIX. In addition, the fully deprotected amino alcohol XXX can be reductively alkylated
(under conditions described previously) with a variety of aldehydes to obtain secondary amines, such as XXXI (Scheme 7), or tertiary amines.
The protected amino alcohol XXVII can also be utilized to synthesize 2-aziridinylmethylpiperazines such as XXXII (Scheme 8). Treating XXVII with 1 ,1 '-sulfonyldiimidazole and sodium hydride in a solvent such as dimethylformamide leads to the formation of aziridine XXXII. The aziridine reacts in the presence of a nucleophile, such as a thiol, in the presence of base to yield the ring-opened product XXXIII.
Piperazine VIII can be reacted with an aldehyde derived from an amino acid, such as an O-alkylated tyrosine, to obtain compounds such as XXXIX. When R' is an aryl group, XXXIX can first be hydrogenated to unmask the phenol, and the amine group deprotected with acid to produce XL. Alternatively, the amine protecting group in XXXIX can be removed, and O-alkylated phenolic amines such as XLI produced.
Depending on the identity of the amino acid I, various side chains can be incorporated onto the piperazine. For example, when I is a protected β-benzyl ester of aspartic acid, the intermediate diketopiperazine XLII (where n=1 and R=benzyl) is obtained, as shown in Scheme 10. Subsequent reduction reduces the ester to the alcohol XLIII, which can then be reacted with a variety of alkylating agents such as an alkyl iodide, under basic conditions, for example, sodium hydride in dimethylformamide or tetrahydrofuran. The resulting ether XLIV can then be carried on to final products as described in Schemes 3-9.
N-Aryl piperazines can be prepared as described in Scheme 1 1. An aryl amine XLV is reacted with bis -chloroethyl amine hydrochloride (XLVI) in refluxing n -butanol to furnish compounds XLVII. The resulting piperazines XLVII can then be carried on to final products as described in Schemes 3-9.
Piperazin-5-ones can be prepared as shown in Scheme 12. Reductive amination of protected amino aldehydes XLIX (prepared from I as described previously) gives rise to compound L. This is then reacted with bromoacetyl bromide under Schotten-Baumann conditions. Ring closure is effected with a base, such as sodium hydride, in a polar
aprotic solvent, such as dimethylformamide, to give LI. The carbamate protecting group is removed under acidic conditions, such as trifluoroacetic acid in methylene chloride or hydrogen chloride gas in methanol or ethyl acetate, and the resulting piperazine can then be carried on to final products as described in Schemes 3-9.
The isomeric piperazin-3-ones can be prepared as described in Scheme 13. The imine formed from arylcarboxamides LII and 2- aminoglycinal diethyl acetal (LIII) can be reduced under a variety of conditions, including sodium triacetoxyborohydride in dichloroethane, to give the amine LIV. Amino acids I can be coupled to amines LIV under standard conditions, and the resulting amide LV when treated with aqueous acid in tetrahydrofuran can cyclize to the unsaturated LVI. Catalytic hydrogenation under standard conditions gives the requisite intermediate LVll, which is elaborated to final products as described in Schemes 3-9.
Access to alternatively substituted piperazines is described in Scheme 14. Following deprotection, e.g., with trifluoroacetic acid, the N-benzyl piperazine V can be acylated with an aryl carboxylic acid. The resulting N-benzyl aryl carboxamide LIX can be hydrogenated in the presence of a catalyst to give the piperazine carboxamide LX which can then be carried on to final products as described in Schemes 3-9.
Reaction Scheme 15 provides an example of the synthesis of compounds wherein the substituents R2 and R3 are combined to form - (CH2)u -. For example, 1-aminocyclohexane-1 -carboxylic acid LXI can be converted to the spiropiperazine LXVI essentially according to the procedures outlined in Schemes 1 and 2. The piperazine intermediate LXIX can be deprotected as before, and carried on to final products as described in Schemes 3-9. It is understood that reagents utilized to provide the substituent Y which is 2-(naphthyl) and the imidazolylalkyl substituent may be readily replaced by other reagents well known in the art and readily available to provide other
N-substituents on the piperazine.
The aldehyde XLIX from Scheme 12 can also be reductively alkylated with an aniline as shown in Scheme 16. The
product LXXl can be converted to a piperazinone by acylation with chloroacetyl chloride to give LXXII, followed by base-induced cyclization to LXXIII. Deprotection, followed by reductive alkylation with a protected imidazole carboxaldehyde leads to LXXV, which can be alkylation with an arylmethylhalide to give the imidazolium salt LXXVI. Final removal of protecting groups by either solvolysis with a lower alkyl alcohol, such as methanol, or treatment with triethylsilane in methylene chloride in the presence of trifluoroacetic acid gives the final product LXXVII.
The famesyl transferase inhibitors can be synthesized in accordance with the general reaction schemes in addition to other standard manipulations such as ester hydrolysis, cleavage of protecting groups, etc., as may be known in the literature or exemplified in the experimental procedures. Some key bond-forming and peptide modifying reactions are:
Reaction A Amide bond formation and protecting group cleavage using standard solution or solid phase methodologies.
Reaction B Preparation of a reduced peptide subunit by reductive
alkylation of an amine by an aldehyde using sodium cyanoborohydride or other reducing agents.
Reaction C Alkylation of a reduced peptide subunit with an alkyl or aralkyl halide or, alternatively, reductive alkylation of a reduced peptide subunit with an aldehyde using sodium cyanoborohydride or other reducing agents.
Reaction D Peptide bond formation and protecting group cleavage
using standard solution or solid phase methodologies.
Reaction E Preparation of a reduced subunit by borane reduction of the amide moiety.
Reaction Schemes A-E illustrate bond-forming and peptide modifying reactions incorporating acyclic peptide units. Such reactions are equally useful when the - NHC(RA) - moiety of the reagents and compounds illustrated is replaced with the following moiety:
which can be substituted with R
4a, R
4D, R
7a and R
7 b in accordance with structures (Il-d) through (II-o). These reactions may be employed in a linear sequence to provide the compounds of the invention or they may be used to synthesize fragments which are subsequently joined by the alkylation reactions described in the Reaction Schemes.
REACTION SCHEME A Reaction A. Coupling of residues to form an amide bond
ALTERNATIVE REACTION SCHEME A FOR
COMPOUNDS (ll-h) THROUGH (ll-o)
Coupling of residues to form an amide bond
Preparation of reduced peptide subunits by reductive alkylation
ALTERNATIVE REACTION SCHEME B FOR COMPOUNDS
(Il-h) THROUGH (II-o)
Preparation of reduced peptide subunits by reductive alkylation
Alkylation/reductive alkylation of reduced peptide subunits e
where R
A and R
B are R
3, R
4, R
5a or R
5b as previously defined; R
C is R
6 as previously defined or a carboxylic acid protectiwg group; X
L is a leaving group, e.g., Br, I- or MsO-; and Ry is defined such that R
7a is generated by the reductive alkylation process.
ALTERNATIVE REACTION SCHEME C for COMPOUNDS
(ll-h) THROUGH (ll-o)
Deprotection of reduced peptide subunits
REACTION SCHEME D
Coupling of residues to form an amide bond
ALTERNATIVE REACTION SCHEME D FOR COMPOUNDS
(Il-h) THROUGH (II-o)
Coupling of residues to form an amide bond
REACTION SCHEME E
Preparation of reduced dipeptides from peptides
ALTERNATIVE REACTION SCHEME E FOR COMPOUNDS
(Il-h) THROUGH (II-o)
Preparation of reduced dipeptides from peptides
All variables are as defined above.
Certain compounds wherein X-Y is an ethenylene or ethylene unit are prepared by employing the reaction sequences shown in Reaction Schemes F and G. Scheme F outlines the preparation of the alkene isosteres utilizing standard manipulations such as Weinreb amide formation, Grignard reaction, acetylation, ozonolysis, Wittig reaction,
ester hydrolysis, peptide coupling reaction, mesylation, cleavage of peptide protecting groups, reductive alkylation, etc., as may be known in the literature or exemplified in the Experimental Procedure. For simplicity, substituents R2a and R2b on the cyclic amine moiety are not shown. It is, however, understood that the reactions illustrated are also applicable to appropriately substituted cyclic amine compounds. The key reactions are: stereoselective reduction of the Boc-aminoenone to the corresponding syn aminoalcohol (Scheme F, Step B, Part 1), and stereospecific boron triflouride or zinc chloride activated organo- magnesio, organo-lithio, or organo-zinc copper (1) cyanide SN2' displacement reaction (Scheme F, Step G). Through the use of optically pure N-Boc amino acids as starting material and these two key reactions, the stereochemistry of the final products is well defined. In Step H of Scheme F, the amino terminus sidechain, designated Rx is incorporated using coupling reaction A and RxCOOH; the alkylation reaction C using RxCHO and a reducing agent; or alkylation reaction C using RxCH2XL. Such reactions as described in Step H are described in more detail in Reaction Schemes J-X hereinbelow.
The alkane analogs are prepared in a similar manner by including an additional catalytic hydrogenation step as outlined in
Reaction Scheme G.
The oxa isostere compounds of this invention are prepared according to the route outlined in Scheme H. An aminoalcohol 1 is acylated with alpha-chloroacetyl chloride in the presence of trialkyl- amines to yield amide 2. Subsequent reaction of 2 with a deprotonation reagent (e.g., sodium hydride or potassium t-butoxide) in an ethereal solvent such as THF provides morpholinone 3. Alkylation of 3 with R3XL, where XL is a leaving group such as Br, I- or Cl- in THF/DME (1 ,2-dimethoxyethane) in the presence of a suitable base, preferably NaHMDS [sodium bis(trimethylsilyl)amide], affords 4, which is retreated with NaHMDS followed by either protonation or the addition of an alkyl halide R4X to give 5a or 5b, respectively, as a enantiomeric mixture. Alternatively, 5a can be prepared from 3, via an aldol
condensation approach. Namely, deprotonation of 3 with NaHMDS followed by the addition of a carbonyl compound RyRzCO gives the adduct 6. Dehydration of 6 can be effected by mesylation and subsequent elimination catalyzed by DBU (1 ,8-diazabicyclo[5.4.0] undec-7-ene) or the direct treatment of 6 with phosphorus oxychloride in pyridine to give olefin 7. Then, catalytic hydrogenation of 7 yields 5a (wherein -CHRyRz constitutes R3). Direct hydrolysis of 5 with lithium hydrogen peroxide in aqueous THF, or aqueous HCl, produces acid 8a. Compound 8a is then derivatized with BOC-ON or BOC anhydride to give 8b . The peptide coupling of acid 8b with either an alpha-aminolactone (e.g., homoserine lactone, etc.) or the ester of an amino acid is carried out under the conditions exemplified in the previously described references to yield derivative 9. Treatment of 2 with gaseous hydrogen chloride gives 10, which undergoes further elaboration as described in Reaction Schemes J- hereinbelow.
An alternative method for the preparation of the prolyl oxa isostere (compounds 23 and 24 ) is shown in Scheme H-1.
Referring to Scheme H-1 , the aminoalcohol 1 is protected with trifluoroacetic anhydride and the blocked compound 15 treated with diphenyl disulfide in the presence of tributylphosphine to provide the thioether 16. Chlorination of compound 16 provides compound 17 which can be reacted with the appropriate carboxylic acid alcohol in the presence of silver perchlorate and tin (II) chloride, to afford the mixed acetal 18 . Removal of the phenylmercapto moiety with Raney nickel provides compound 19. Compound 19 is doubly deprotected, then selectively BOC protected to provide the acid 20, which undergoes the steps previously described for incorporating terminal amino acid. Still another alternative method for the preparation of the prolyl oxa isostere (compounds 23 and 24 ) is described in the literature [Ruth E.
TenBrink, J. Org. Chem., 52, 418-422 (1987)].
The thia, oxothia and dioxothia isostere compounds of this invention are prepared in accordance to the route depicted in Scheme I. Aminoalcohol 1 is derivatized with BOC2O to give 25. Mesylation of 25 followed by reaction with methyl alpha-mercaptoacetate in the presence of cesium carbonate gives sulfide 26. Removal of the BOC group in 26 with TFA followed by neutralization with di-isopropyl- ethylamine leads to lactam 27. Sequential alkylation of 27 with the alkyl halides R3X and R4X in THF/DME using NaHDMS as the deprotonation reagent produces 28. Hydrolysis of 28 in hydro-chloride to yield 29a, which is derivatized with Boc anhydride to yield 29b. The coupling of 29b with an alpha-aminolactone (e.g., homoserine lactone, etc.) or the ester of an amino acid is carried out under conventional conditions as
exemplified in the previously described references to afford 30. Sulfide 30 is readily oxidized to sulfone 31 by the use of MCPBA (m-chloro- peroxybenzoic acid). The N-BOC group of either 30 or 31 is readily removed by treatment with gaseous hydrogen chloride.
Reaction Schemes J - R illustrate reactions wherein the non- sulfhydryl-containing moiety at the N-terminus of the compounds of the instant invention is attached to the fully elaborated cyclic amino peptide unit, prepared as described in Reaction Schemes A-I. It is understood
that the reactions illustrated may also be performed on a simple cyclic amino acid, which may then be further elaborated utilizing reactions described in Reaction Schemes A- l to provide the instant compounds.
The intermediates whose synthesis are illustrated in
Reaction Schemes A-I can be reductively alkylated with a variety of aldehydes, such as V, as shown in Reaction Scheme J. The aldehydes can be prepared by standard procedures, such as that described by O. P. Goel, U. Krolls, M. Stier and S. Kesten in Organic Syntheses.
1988, 67, 69-75, from the appropriate amino acid (Reaction Scheme F). The reductive alkylation can be accomplished at pH 5-7 with a variety of reducing agents, such as sodium triacetoxyborohydride or sodium cyanoborohydride in a solvent such as dichloroethane, methanol or dimethylformamide. The product VI can be deprotected with
trifluoroacetic acid in methylene chloride to give the final compounds VII. The final product VII is isolated in the salt form, for example, as a trifluoroacetate, hydrochloride or acetate salt, among others. The product diamine VII can further be selectively protected to obtain VIII, which can subsequently be reductively alkylated with a second aldehyde to obtain IX. Removal of the protecting group, and
conversion to cyclized products such as the dihydroimidazole XI can be accomplished by literature procedures.
Alternatively, the protected cyclic aminopeptidyl intermediate can be reductively alkylated with other aldehydes such as 1 -trityl-4-carboxaldehyde or l-trityI-4-imidazolylacetaIdehyde, to give products such as XII (Reaction Scheme K). The trityl protecting group can be removed from XII to give XIII, or alternatively, XII can first be treated with an alkyl halide then subsequently deprotected to give the alkylated imidazole XIV. Alternatively, the dipeptidyl analog intermediate can be acylated or sulfonylated by standard techniques.
The imidazole acetic acid XV can be converted to the protected acetate XVII by standard procedures, and XVII can be first reacted with an alkyl halide, then treated with refluxing methanol to provide the regiospecif ically alkylated imidazole acetic acid ester XVIII. Hydrolysis and reaction with the protected dipeptidyl analog
intermediate in the presence of condensing reagents such as l-(3- dime thy laminopropyl)-3 -ethyl carbodiimide (EDC) leads to acylated products such as XIX.
If the protected dipeptidyl analog intermediate is
reductively alkylated with an aldehyde which also has a protected hydroxyl group, such as XX in Reaction Scheme N, the protecting groups can be subsequently removed to unmask the hydroxyl group (Reaction Schemes N, P). The alcohol can be oxidized under standard conditions to e.g. an aldehyde, which can then be reacted with a variety of organometallic reagents such as Grignard reagents, to obtain secondary alcohols such as XXIV. In addition, the fully deprotected amino alcohol XXV can be reductively alkylated (under conditions described previously) with a variety of aldehydes to obtain secondary amines, such as XXVI (Reaction Scheme P), or tertiary amines.
The Boc protected amino alcohol XXII can also be utilized to synthesize 2-aziridinylmethylpiperazines such as XXVII (Reaction Scheme Q). Treating XXII with l j '-sulfonyldiimidazole and sodium hydride in a solvent such as dimethylformamide led to the formation of aziridine XXVII . The aziridine may be reacted in the presence of a nucleophile, such as a thiol, in the presence of base to yield the ring- opened product XXVIII .
In addition, the protected dipeptidyl analog intermediate can be reacted with aldehydes derived from amino acids such as
O-alkylated tyrosines, according to standard procedures, to obtain compounds such as XXXIV, as shown in Reaction Scheme R. When R' is an aryl group, XXXIV can first be hydrogenated to unmask the phenol, and the amine group deprotected with acid to produce XXXV. Alternatively, the amine protecting group in XXXIV can be removed, and O-alkylated phenolic amines such as XXXVI produced.
The intermediates whose synthesis are illustrated in Reaction Schemes A and C can be reductively alkylated with a variety
of aldehydes, such as I, as shown in Reaction Scheme F. The aldehydes can be prepared by standard procedures, such as that described by O. P. Goel, U. Krolls, M. Stier and S. Kesten in Organic Syntheses, 1988, 67, 69-75, from the appropriate amino acid (Reaction Scheme F). The reductive alkylation can be accomplished at pH 5-7 with a variety of reducing agents, such as sodium triacetoxyborohydride or sodium cyanoborohydride in a solvent such as dichloroethane, methanol or dimethylformamide. The product II can be deprotected to give the final compounds III with trifluoroacetic acid in methylene chloride. The final product III is isolated in the salt form, for example, as a trifluoroacetate, hydrochloride or acetate salt, among others. The product diamine III can further be selectively protected to obtain IV, which can subsequently be reductively alkylated with a second aldehyde to obtain V. Removal of the protecting group, and conversion to cyclized products such as the dihydroimidazole VII can be accomplished by literature procedures.
Alternatively, the protected dipeptidyl analog intermediate can be reductively alkylated with other aldehydes such as 1 -trityl-4- carboxaldehyde or 1-trityl-4-imidazolylacetaldehyde, to give products such as VIII (Alternative Reaction Scheme G). The trityl protecting group can be removed from VIII to give IX, or alternatively, VIII can first be treated with an alkyl halide then subsequently deprotected to give the alkylated imidazole X. Alternatively, the dipeptidyl analog intermediate can be acylated or sulfonylated by standard techniques.
The imidazole acetic acid XI can be converted to the acetate
XIII by standard procedures, and XIII can be first reacted with an alkyl halide, then treated with refluxing methanol to provide the regiospecifically alkylated imidazole acetic acid ester XIV (Alternative Reaction Scheme H). Hydrolysis and reaction with the protected dipeptidyl analog intermediate in the presence of condensing reagents such as 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC) leads to acylated products such as XV.
If the protected dipeptidyl analog intermediate is reductively alkylated with an aldehyde which also has a protected
hydroxyl group, such as XVI in Reaction Scheme I, the protecting groups can be subsequently removed to unmask the hydroxyl group (Reaction Schemes I, J). The alcohol can be oxidized under standard conditions to e.g. an aldehyde, which can then be reacted with a variety of organometallic reagents such as Grignard reagents, to obtain secondary alcohols such as XX. In addition, the fully deprotected amino alcohol XXI can be reductively alkylated (under conditions described previously) with a variety of aldehydes to obtain secondary amines, such as XXII (Reaction Scheme K), or tertiary amines.
The Boc protected amino alcohol XVIII can also be utilized to synthesize 2-aziridinylmethylpiperazines such as XXIII (Reaction Scheme L). Treating XVIII with 1 ,1 '-sulfonyldiimidazole and sodium hydride in a solvent such as dimethylformamide led to the formation of aziridine XXIII . The aziridine reacted in the presence of a nucleophile, such as a thiol, in the presence of base to yield the ring-opened product XXIV .
In addition, the protected dipeptidyl analog intermediate can be reacted with aldehydes derived from amino acids such as
O-alkylated tyrosines, according to standard procedures, to obtain compounds such as XXX, as shown in Reaction Scheme M. When R' is an aryl group, XXX can first be hydrogenated to unmask the phenol, and the amine group deprotected with acid to produce XXXI. Alternatively, the amine protecting group in XXX can be removed, and O-alkylated phenolic amines such as XXXII produced.
Similar procedures as are illustrated in Reaction Schemes
F-M may be employed using other peptidyl analog intermediates such as those whose synthesis is illustrated in Reaction Schemes B - E.
Certain compounds used in the invention are described below.
EXAMPLE 1
(S)-1 -(3-chlorophenyl)-4-[ 1 -(4-cyanobenzyl)-imidazolylmethyl]-5-[2- (methanesulfonyl)ethyl]-2-piperazinone dihydrochloride
Step A: 1 -triphenylmethyl-4-(hydroxymethyl)-imidazole
To a solution of 4-(hydroxymethyl)imidazole hydrochloride (35.0 g, 260 mmol) in 250 mL of dry DMF at room temperature was added triethylamine (90.6 mL, 650 mmol). A white solid precipitated from the solution. Chlorotriphenylmethane (76.1 g, 273 mmol) in 500 mL of DMF was added dropwise. The reaction mixture was stirred for 20 hours, poured over ice, filtered, and washed with ice water. The resulting product was slurried with cold dioxane, filtered, and dried in vacuo to provide the titled product as a white solid which was sufficiently pure for use in the next step. Step B: 1 -triphenylmethyl-4-(acetoxymethyl)-imidazole
Alcohol from Step A (260 mmol, prepared above) was suspended in 500 mL of pyridine. Acetic anhydride (74 mL, 780 mmol) was added dropwise, and the reaction was stirred for 48 hours during which it became homogeneous. The solution was poured into 2 L of EtOAc, washed with water (3 x 1 L), 5% aq. HCl soln. (2 x 1 L), sat. aq. NaHCO3, and brine, then dried (Na2Sθ4), filtered, and concentrated in vacua to provide the crude product. The acetate was isolated as a white powder which was sufficiently pure for use in the next reaction.
Step C: 1 -(4-cyanobenzyl)-5-(acetoxymethyl)-imidazoIe
hydrobromide
A solution of the product from Step B (85.8 g, 225 mmol) and α-bromo-p -tolunitrile (50.1 g, 232 mmol) in 500 mL of EtOAc was stirred at 60°C for 20 hours, during which a pale yellow precipitate
formed. The reaction was cooled to room temperature and filtered to provide the solid imidazolium bromide salt. The filtrate was concentrated in vacua to a volume 200 mL, reheated at 60°C for two hours, cooled to room temperature, and filtered again. The filtrate was concentrated in vacuo to a volume 100 mL, reheated at 60°C for another two hours, cooled to room temperature, and concentrated in vacuo to provide a pale yellow solid. All of the solid material was combined, dissolved in 500 mL of methanol, and warmed to 60°C. After two hours, the solution was reconcentrated in vacua to provide a white solid which was triturated with hexane to remove soluble materials. Removal of residual solvents in vacuo provided the titled product hydrobromide as a white solid which was used in the next step without further purification. Step D: 1 -(4-cyanobenzyl)-5-(hydroxymethyl)-imidazole
To a solution of the acetate from Step C (50.4 g, 150 mmol) in 1.5 L of 3: 1 THF/water at 0°C was added lithium hydroxide monohydrate ( 18.9 g, 450 mmol). After one hour, the reaction was concentrated in vacua, diluted with EtOAc (3 L), and washed with water, sat. aq. NaHCO3 and brine. The solution was then dried
(Na2SO4), filtered, and concentrated in vacuo to provide the crude product as a pale yellow fluffy solid which was sufficiently pure for use in the next step without further purification. Step E: 1-(4-cyanobenzyl)-5-imidazolecarboxaldehyde
To a solution of the alcohol from Step D (21.5 g, 101 mmol) in 500 mL of DMSO at room temperature was added triethylamine (56 mL, 402 mmol), then SO3-pyridine complex (40.5 g, 254 mmol). After 45 minutes, the reaction was poured into 2.5 L of EtOAc, washed with water (4 x 1 L) and brine, dried (Na2Sθ4), filtered, and concentrated in vacuo to provide the aldehyde as a white powder which was sufficiently pure for use in the next step without further
purification.
Step F: (S)-2-(t ert-butoxycarbonylamino)-N -methoxy-N-methyl-4-
(methylthio)butanamide
L-N-Boc-methionine (30.0 g, 0.120 mol), N,O-dimethyl- hydroxylamine hydrochloride (14.1 g, 0.144 mol), EDC hydrochloride (27.7 g, 0.144 mol) and HOBT (19.5 g, 0.144 mol) were stirred in dry DMF (300 mL) at 20°C under nitrogen. More N,O -dimethylhydroxyl- amine hydrochloride (2.3 g, 23 mmol) was added to obtain pH 7-8. The reaction was stirred ovemight, the DMF distilled to half the original volume under high vacuum, and the residue partitioned between ethyl acetate and sat. NaHCO3 soln. The organic phase was washed with saturated sodium bicarbonate, water, 10% citric acid, and brine, and dried with sodium sulfate. The solvent was removed in vacuo to give the title compound. Step G: (S)-2-(tert-butoxycarbonylamino)-4-(methylthio)butanal
A suspension of lithium aluminum hydride (5.02 g, 0.132 mol) in ether (500 mL) was stirred at room temperature for one hour. The solution was cooled to -50°C under nitrogen, and a solution of the product from Step F (39.8 g, ca. 0.120 mol) in ether (200 mL) was added over 30 min, maintaining the temperature below -40°C. When the addition was complete, the reaction was warmed to 5°C, then recooled to -45°C. Analysis by tic revealed incomplete reaction. The solution was rewarmed to 5°C, stirred for 30 minutes, then cooled to -50°C. A solution of potassium hydrogen sulfate (72 g, 0.529 mol) in 200 mL water was slowly added, maintaining the temperature below -20°C. The mixture was wasmed to 5°C, filtered through Celite, and concentrated in vacuo to provide the title aldehyde.
Step H: (S)-2-(tert-butoxycarbonylamino)-N-(3-chlorophenyl)-4- (methylthio)butanamine
To a solution of 3-chloroaniline (10.3 mL, 97.4 mmol), the product from Step G (23.9 g, 97.4 mmol), and acetic acid (27.8 mL, 487 mmol) in dichloroethane (250 mL) under nitrogen was added sodium triacetoxyborohydride (41.3 g, 195 mmol). The reaction was
stirred overnight, then quenched with saturated sodium bicarbonate solution. The solution was diluted with CHCl3, and the organic phase was washed with water, 10% citric acid and brine. The solution was dried over sodium sulfate and concentrated in vacuo to provide the crude product (34.8 g) which was chromatographed on silica gel with 20% ethyl acetate in hexane to obtain the title compound .
Step I: (S)-4-(tert-butoxycarbonyl)-1-(3-chlorophenyl)-5-[2-
(methylthio)ethyl]piperazin-2-one
A solution of the product from Step H (22.0 g, 63.8 mmol) in ethyl acetate (150 mL) was vigorously stirred at 0°C with saturated sodium bicarbonate (150 mL). Chloroacetyl chloride (5.6 mL, 70.2 mmol) was added dropwise, and the reaction stirred at 0°C for 2h.
The layers were separated, and the ethyl acetate phase was washed with 10% citric acid and saturated brine, and dried over sodium sulfate. After concentration in vacuo, the resulting crude product (27.6 g) was dissolved in DMF (300 mL) and cooled to 0°C under argon. Cesium carbonate (63.9 g, 196 mmol) was added, and the reaction was stirred for two days, allowing it to warm to room temperature. Another portion of cesium carbonate (10 g, 30 mmol) was added, and the reaction was stirred for 16 hours. The DMF was distilled in vacuo, and the residue partitioned between ethyl acetate and water. The organic phase was washed with saturated brine, and dried over sodium sulfate. The crude product was chromatographed on silica gel with 20-25% ethyl acetate in hexane to obtain the title compound.
Step J: (S)-4-(tert-butoxycarbonyl)-1 -(3-chlorophenyl)-5-[ 2-
(methanesulfonyl)ethyl]piperazin-2-one
A solution of the product from Step I ( 14.2 g, 37 mmol) in methanol (300 mL) was cooled to 0 °C, and a solution of magnesium monoperoxyphthalate (54.9 g, 1 1 1 mmol) in 210 mL MeOH was added over 20 minutes. The ice bath was removed, and the solution was allowed to warm to room temperature. After 45 minutes, the reaction was concentrated in vacua to half the original volume, then quenched by
the addition of 2N Na2S2O3 soln. The solution was poured into EtOAc and sat NaHCO3 solution, and the organic layer was washed with brine, dried (Na2SO4), filtered, and concentrated in vacuo to provide the crude sulfone. This material was chromatographed on silica gel with 60-100% ethyl acetate in hexane to obtain the titled compound.
Step K: (S)- 1 -(3-chlorophenyl)-5-[2-
(methanesulfonyl)ethyllpiperazin-2-one
Through a solution of Boc-protected piperazinone from Step J (1.39 g, 3.33 mmol) in 30 mL of EtOAc at 0 °C was bubbled anhydrous HCl gas. The saturated solution was stirred for 35 minutes, then concentrated in vacua to provide the hydrochloride salt as a white powder. This material was suspended in EtOAc and treated with dilute aqueous NaHC03 solution. The aqueous phase was extracted with EtOAc, and the combined organic mixture was washed with brine, dried (Na2S04), filtered, and concentrated in vacuo. The resulting amine was reconcentrated from toluene to provide the titled material suitable for use in the next step. Step L: (S)- 1 -(3-chlorophenyl)-4-[ 1 -(4- cyanobenzyl)imidazolylmethyl]-5-[2-(methanesulfonyl)- ethyl]-2-piperazinone dihydrochloride
To a solution of the amine from Step K (898 mg, 2.83 mmol) and imidazole carboxaldehyde from Step E (897 mg, 4.25 mmol) in 15 mL of 1 ,2-dichloroethane was added sodium triacetoxyboro- hydride (1.21 g, 5.7 mmol). The reaction was stirred for 23 hours, then quenched at 0 °C with sat. NaHCO3 solution. The solution was poured into CHCl3, and the aqueous layer was back-extracted with CHCl3. The combined organics were washed with brine, dried
(Na2Sθ4), filtered, and concentrated in vacua. The resulting product was purified by silica gel chromatography (95:5:0.5-90: 10:0
EtOAc:MeOH:NH4Cl), and the resultant product was taken up in
EtOAc/methanol and treated with 2.1 equivalents of 1 M HCl/ether
solution. After concentrated in vacua, the product dihydrochloride was isolated as a white powder.
EXAMPLE 2
1-(3-chlorophenyl)-4-[ 1 -(4-cvanobenzyl)imidazolyl-methyl]-2- piperazinone dihydrochloride
Step A: N-(3-chlorophenyl)ethylenediamine hydrochloride
To a solution of 3-chloroaniline (30.0 mL, 284 mmol) in 500 mL of dichloromethane at 0°C was added dropwise a solution of 4 N HCl in 1 ,4-dioxane (80 mL, 320 mmol HCl). The solution was warmed to room temperature, then concentrated to dryness in vacua to provide a white powder. A mixture of this powder with 2-oxazolidinone (24.6 g, 282 mmol) was heated under nitrogen atmosphere at 160°C for 10 hours, during which the solids melted, and gas evolution was observed. The reaction was allowed to cool, forming the crude diamine hydrochloride salt as a pale brown solid. Step B: N-(tert-butoxycarbonyl)-N'-(3- chlorophenyl)ethylenediamine
The amine hydrochloride from Step A (ca. 282 mmol, crude material prepared above) was taken up in 500 mL of THF and 500 mL of sat. aq. NaHCO3 soln., cooled to 0°C, and di-tert- butylpyrocarbonate (61.6 g, 282 mmol) was added. After 30 h, the reaction was poured into EtOAc, washed with water and brine, dried (Na2Sθ4), filtered, and concentrated in vacuo to provide the titled carbamate as a brown oil which was used in the next step without further purification.
Step C: N-[2-(ter t-butoxycarbamoyl)ethyl]-N-(3-chlorophenyl)- 2-chloroacetamide
A solution of the product from Step B (77 g, ca. 282 mmol) and triethylamine (67 mL, 480 mmol) in 500 mL of CH2CI2 was cooled to 0°C. Chloroacetyl chloride (25.5 mL, 320 mmol) was added dropwise, and the reaction was maintained at 0°C with stirring. After 3 h, another portion of chloroacetyl chloride (3.0 mL) was added dropwise. After 30 min, the reaction was poured into EtOAc (2 L) and washed with water, sat. aq. NH4CI soln, sat. aq. NaHCO3 soln., and brine.
The solution was dried (Na2SO4), filtered, and concentrated in vacuo to provide the chloroacetamide as a brown oil which was used in the next step without further purification.
Step D: 4-(tert-butoxycarbonyl)-1 -(3-chlorophenyl)-2- piperazinone
To a solution of the chloroacetamide from Step C (ca. 282 mmol) in 700 mL of dry DMF was added K2CO3 (88 g, 0.64 mol).
The solution was heated in an oil bath at 70-75°C for 20 hours, cooled to room temperature, and concentrated in vacua to remove ca. 500 mL of DMF. The remaining material was poured into 33% EtOAc/hexane, washed with water and brine, dried (Na2SO4), filtered, and concen- trated in vacuo to provide the product as a brown oil. This material was purified by silica gel chromatography (25-50% EtOAc/hexane) to yield pure product, along with a sample of product (ca. 65% pure by HPLC) containing a less polar impurity. Step E: 1-(3-chlorophenyl)-2-piperazinone
Through a solution of Boc-protected piperazinone from Step D (17.19 g, 55.4 mmol) in 500 mL of EtOAc at -78°C was bubbled anhydrous HCl gas. The saturated solution was warmed to 0°C, and stirred for 12 hours. Nitrogen gas was bubbled through the reaction to remove excess HCl, and the mixture was warmed to room temperature. The solution was concentrated in vacuo to provide the hydrochloride as a white powder. This material was taken up in 300 mL of CH2CI2 and treated with dilute aqueous NaHCO3 solution. The aqueous phase was extracted with CH2CI2 (8 x 300 mL) until tic analysis indicated
complete extraction. The combined organic mixture was dried
(Na2Sθ4), filtered, and concentrated in vacua to provide the titled free amine as a pale brown oil. Step F: 1-(3-chlorophenyl)-4-[ 1 -(4-cyanobenzyl)imidazolylmethyl]-
2-piperazinone dihydrochloride
To a solution of the amine from Step E (55.4 mmol, prepared above) in 200 mL of 1 ,2-dichloroethane at 0°C was added 4Å powdered molecular sieves (10 g), followed by sodium triacetoxyboro- hydride (17.7 g, 83.3 mmol). The imidazole carboxaldehyde from Step E of Example 1 (1 1.9 g, 56.4 mmol) was added, and the reaction was stirred at 0°C. After 26 hours, the reaction was poured into EtOAc, washed with dilute aq. NaHCO3, and the aqueous layer was back- extracted with EtOAc. The combined organics were washed with brine, dried (Na2SO4), filtered, and concentrated in vacuo. The resulting product was taken up in 500 mL of 5:1 benzene:CH2Cl2, and propyl- amine (20 mL) was added. The mixture was stirred for 12 hours, then concentrated in vacuo to afford a pale yellow foam. This material was purified by silica gel chromatography (2-7% MeOH/CH2Cl2), and the resultant white foam was taken up in CH2CI2 and treated with 2.1 equivalents of 1 M HCl/ether solution. After concentrated in vacua, the product dihydrochloride was isolated as a white powder.
N-[ 1 -(1H-Imidazol-4-propionyl) pyrrolidin-2(S)-ylmethyl]-N-(2- methoxybenzyl)glycyl-methionine isopropyl ester
Step A: 2-Methoxybenzylglycine methyl ester
2-MethoxybenzyI alcohol (53.5 g, 0.39 mol) was dissolved in CH2CI2 (200 mL), treated with diisopropylethylamine (81 mL, 0.74 mol), cooled to 0°C. with stirring in an ice-CH3OH bath under Ar, and treated dropwise with methanesulfonyl chloride (33 mL, 0.43 mol). After 0.5 hr, the reaction mixture was left to warm to ambient temperature and stirred for 4 hr. This solution and diisopropylethylamine (202.5 mL, 1.16 mol) were added alternately portionwise with to a slurry of glycine methyl ester hydrochloride ( 146.5 g, 1.17 mol) in DMF (250 mL) with stirring at 0°C. The reaction mixture was left to stir and warm to room temperature overnight. The DMF was removed under reduced pressure, and the residue was partitioned between EtOAc (1 L) and satd NaHCO3 solution ( 1 L). The aqueous layer was washed with EtOAc (2 x 600 mL), the organics combined, washed with brine and dried (MgSO4). Filtration and concentration to dryness gave the title compound after chromatography (SiO2, 1 -5% CH3OH/CH2CI2).
Step B: N-[(2S)-(t-Butoxycarbonylpyrrolidinyl-methyl)-N-(2- methoxybenzyl)glycine methyl ester
2-Methoxybenzylglycine methyl ester (27.4 g, 0.131 mol) was dissolved in 1 ,2-dichloroethane (500 ml), 3Å molecular sieves (20 g) were added, and the pH of the reaction mixture adjusted to pH 5 with acetic acid (7.5 mL, 0.131 mol). N-(t-Butoxycarbonyl)-L-prolinal (26.1 g, 0.131 mol) (J. Org. Chem. (1994) 59, [21 ], 6287-95) was added followed by sodium triacetoxyborohydride (33.2 g, 0.157 mol). The mixture was stirred at ambient temperature for 18 h, filtered through celite and concentrated. The residue was partitioned between EtOAc and sat. NaHCO3 (500 ml/100 ml). The aqueous layer was washed with EtOAc (3x100 ml). The organic layers were combined, dried with Na2SO4, filtered, and concentrated to give the title
compound.
Step C: N-[(2S)-(t-Butoxycarbonylpyrrolidinyl-methyl)-N-(2- methoxybenzyl)glycine
N-[(2S)-(t-ButoxycarbonyIpyrrolidinylmethyI)-N-(2- methoxybenzyl)glycine methyl ester (7.0 g, 0.018 mol) was dissolved in CH3OH (150 ml) and 2N NaOH (71 ml, 0.071 mol) was added with cooling in an ice-water bath. The mixture was stirred at ambient temperature for 2 hr, neutralized with 1 N HCl (71 ml, 0.071 mol), concentrated to remove the CH3OH, and the residue extracted with EtOAc (3x200 mL). The organic layers were combined, dried with Mg2SO4, filtered, and concentrated to give the title compound as a foam.
Step D: Methionine isopropyl ester hydrochloride
N-(t-Butoxycarbonyl)methionine (25 g, 0.1 mol), isopropyl alcohol (1 1.8 mL, 0.15 mol), EDC (21.1 g, 0.1 1 mol), and 4-dimethyl- aminopyridine (DMAP) (1.22 g, 0.01 mol) were dissolved in CH2CI2 (400 mL) with stirring in an ice-water bath. After 2 h the bath was removed, and the solution was left to stir o.n. at RT. The reaction mixture was concentrated to dryness, then partitioned between EtOAc and H2O, the aqueous layer washed with EtOAc (2 x 50 mL), the organics combined, washed with NaHCO3 soln, brine, and dried
(Na2SO4). Filtration and concentration to dryness gave a yellow oil after chromatography (flash silica gel column, hexane: EtOAc, 6: 1 to 5: 1 ).
N-(t-Butoxycarbonyl)methionine isopropyl ester (20.5 g,
0.07 mol) was dissolved in EtOAc (200 mL) with stirring and cooling to -20°C in a dry ice- acetone bath. HCl gas was bubbled into the solution for 10 min, the flask was stoppered and stirred for 1 h. Tic (EtOAc: hexane, 1 :3) indicates loss of starting material. Argon was bubbled through the soln for 5 min, then it was concentrated to dryness to give the title compound as a white solid.
Step E: N-[(2S)-(t-Butoxycarbonylpyrrolidinyl-methyl)-N-
(2-methoxybenzyl)glycyl-methionine isopropyl ester
N-[(2S)-(t-Butoxycarbonylpyrrolidinylmethyl)-N- (2-methoxybenzyl)glycine (from step C) (5.98 g, 0.0158 mol), dissolved in CH2Cl2 (100 mL), was treated with HOBT (2.57 g, 0.019 mol), EDC (4.54 g, 0.024 mol), and methionine isopropyl ester hydrochloride (4.33 g, 0.019 mol). The pH was adjusted to 7.5 with Et3N (8.81 mL, 0.063 mol) and the mixture was stirred at ambient temperature for 18 h. The mixture was diluted with EtOAc ( 150 mL) and washed sequentially with 10% citric acid soln, saturated NaHC03 solution, brine, and dried (MgSθ4). Filtration and concentration to dryness gave the title compound as a thick oil. This was used without further purification.
Step F: N-((2S)-Pyrrolidinylmethyl)-N-(2-methoxybenzyl)- glycyl-methionine isopropyl ester bis hydrochloride
N-[(2S)-(t-Butoxycarbonylpyrrolidinylmethyl)-N-(2- methoxybenzyl)glycyl-methionine isopropyl ester (0.85 g, 1.54 mmol) was dissolved in EtOAc (25 mL) and cooled to 0°C. HCl was bubbled through the mixture until the soln was saturated, and it was stoppered and stirred for 3 hr. Argon was bubbled through the mixture to remove excess HCl and the mixture was then concentrated to give the title compound.
Step G: N-[1 -(1H-Imidazol-4-propionyl) pyrrolidin-2(S)- yImethyl]-N-(2-methoxybenzyl)glycy1-methionine
isopropyl ester
N-((2S)-Pyrrolidinylmethyl)-N-(2-methoxybenzyl)glycyl methionine isopropyl ester bis hydrochloride (0.800 g, 1.53 mmol), dissolved in DMF (30 mL), was treated with 1H-imidazol-4-propionic acid (0.43 g, 3.05 mmol) (prepared by catalytic hydrogenation of urocanic acid in 20% acetic acid with Pd on carbon), and BOP reagent (1.35 g, 3.05 mmol). The pH was adjusted to 7.5 with N-methyl- morpholine (0.756 mL, 6.89 mmol), and the mixture was stirred
at ambient temperature for 18 h. The mixture was concentrated to dryness, diluted with EtOAc (100 mL), washed with 5% Na2CO3 solution, brine, and dried (MgSO4). Filtration and concentration to dryness gave an oil which was purified by chromatography (silica gel, 95:5 CH2Cl2/MeOH) to give the title compound as a foam.
1 H NMR (CD3OD); δ 7.58 (d, 1H, J=l Hz), 7.25-7.31 (m, 2H), 6.89- 7.00 (m, 2H), 6.81 (s, 1H), 5.00-5.06 (m, 1H), 4.49-4.56 (m, 1H), 4.23- 4.30 (m, 1H), 3.91 (d, 1H, J=13 Hz), 3.86 (s, 3H), 3.54 (d, 1H, J=13Hz), 3.30-3.41 (m, 2H), 3.36 (d, I H, J= 17 Hz), 3.15 (d, 1H, J=17 Hz), 2.85- 2.92 (m, 2H), 2.56-2.77 (m, 3H), 2.30-2.45 (m, 3H), 2.05-2.17 (m, 1H), 2.04 (s, 3H), 1.69- 1.86 (m, 5H), 1.24 (d, 6H, 5=6 Hz).
Anal, calculated for C29H43N5O5S• 0.6 H2O:
C, 59.59; H, 7.62; N, 1 1.98;
Found: C, 59.58; H, 7.43; N, 12.02.
EXAMPLE 4
(N-[1-Cyanobenzyl)-1 H-imidazol-5-yl)acetyl]pyrrolidin-2(S)-ylmethyl]- 3(S)-ethyl-prolyl methionine isopropyl ester
Step A: Diethyl 1-Acetyl-5-hydroxy-3-ethylpyrroIidine-2,2- dicarboxylate
Sodium (4.02 g, 0.175 mol) was dissolved in a stirred solution of diethyl acetamidomalonate (235.4 g, 1.19 mol) in abs EtOH
(1.4 L) at ambient temperature under argon. The reaction mixture was cooled to 0°C, and trans-2-pentenal (100 g, 1.08 mol) was added dropwise maintaining the reaction temperature at <5°C. After the addition, the reaction was allowed to warm to room temperature, stirred for 4 h, then quenched with acetic acid (28 mL). The solution was concentrated in vacua, and the residue dissolved in EtOAc (1.5 L), washed with 10% NaHC03 solution (2 x 300 mL), brine, and dried (MgSO4). The solution was filtered and concentrated to 700 mL, then heated to reflux and treated with hexane ( 1 L). On cooling, the title compound
precipitated and was collected, mp 106 - 109°C. 1 H NMR (CD3OD) δ 5.65 (d, 1H, J= 5 Hz), 4.1 - 4.25 (m, 4H), 2.7-2.8 (m, 1H), 2.21 (s, 3H), 2.10 (dd, 1H, J = 6, 13, Hz),1.86- 1.97 (m, 2H), 1.27 (t, 3H, J= 7 Hz), 1.23 (t, 3H, J= 7 Hz), 1.1- 1.25 (m, 1H), 0.97 (t, 3H, J= 7 Hz). Step B: Diethyl 1 -Acetyl-3-ethylpyrrolidine-2,2-dicarboxylate
To a solution of diethyl 1 -acetyl-5-hydroxy-3-ethyl- pyrrolidine-2,2-dicarboxylate (287 g, 0.95 mol) and triethylsilane (228 mL, 1.43 mol) in CH2CI2 (3 L) under argon was added trifluoroacetic acid (735 mL, 9.53 mol) dropwise with stirring while maintaining the internal temperature at 25 °C by means of an ice bath. After stirring for 3 h at 23°C, the solution was concentrated in vacua. , the residue diluted with CH2CI2 (1.5 L), then treated with H2O (1 L) and solid Na2CO3 with vigorous stirring until the solution was basic. The organic layer was separated, dried (Na2SO4), filtered, then concentrated to give the title compound as a yellow oil which was used without further purification.
Step C: 3-Ethylproline hydrochloride (Cis:Trans Mixture)
Diethyl 1-acetyl-3-ethylpyrrolidine-2,2-dicarboxylate (373 g, 0.95 mol) was suspended in 6N HCl (2 L) and HOAc (500 mL) and heated at reflux for 20 h. The reaction mixture was cooled, washed with EtOAc (1 L). then concentrated in vacuo to give an oil which crystallized upon trituration with ether to give the title compound.
1 H NMR (D2O) δ 4.23 (d, 1H, J= 8 Hz), 3.84 (d, 1 H, J= 8 Hz), 3.15- 3.4 (m, 4H), 2.33- 2.44 (m, 1H), 2.19-2.4 (m, 1H), 2.02- 2.15 (m, 2H), 1.53- 1.72 (m, 3H), 1.23- 1.43 (m, 2H), 1.0- 1.15 (m, 1H), 0.75 - 0.83 (m, 6H).
Step D: N-[(tert-Butyloxy)carbonyl]-cis:transs-3-ethylprolme methyl ester
3-Ethylproline hydrochloride (Cis.Trans Mixture)
(20 g, 0.1 1 mol) was dissolved in CH3OH (200 mL), and the solution was saturated with HCl gas, then stirred at 23°C for 24 h. Argon was bubbled through the solution to remove excess HCl. The solution was treated with NaHCO3 (>84 g) to a pH of 8, then di-tert-butyl
dicarbonate (25.1 g, 0.1 15 mol) dissolved in CH3OH (20 mL) was added slowly. After stirring for 18 h at 23°C, the mixture was filtered, the filtrate concentrated, and the residue triturated with EtOAc, filtered again, and concentrated to give the title compound as an oil.
Step E: N-[(tert-Butyloxy)carbonyI]-trans-3-ethylproline and N- [(tert-Butyloxy)carbonyl]-cis-3-ethylproline methyl ester
N-[(tert-Butyloxy)carbonyl]-c is,tran s-3-ethylproline methyl ester (29.1 g, 0.1 13 mol) was dissolved in CH3OH (1 14 mL) with cooling to 0°C, then treated with 1 N NaOH (1 14 mL). After stirring for 20 h at 23°C, the solution was concentrated to remove the CH3OH and then extracted with EtOAc (3 x). The organic layers were
combined, dried (MgSO4), filtered, and concentrated to give 12.8 g of N-[(ter t-Butyloxy)carbonyl]-cis -3-ethylproline methyl ester as an oil. The aqueous layer was acidified with solid citric acid and extracted with EtOAc (2 x), the organic layers combined, dried (MgSO4), filtered, and concentrated to give N-[(tert-Butyloxy)carbonyl]-trans-3-ethylproline as an oil. 1H NMR (CD3OD) δ 3.86 and 3.78 (2 d, 1H, J = 6 Hz), 3.33 - 3.58 (m, 2H), 2.01 - 2.22 (m, 2H), 1.5 - 1.74 (m, 2H), 1.33 - 1.5 (m, I H), 1.45 and 1.42 (2 s, 9H), 0.98 (t, 3H, J= 8 Hz).
Step F: 3(S)-Ethyl-2(S)-proline hydrochloride
N-[(tert-Butyloxy)carbonyl]-trans-3-ethylproline (15.5 g, 0.064 mol), S-α-methylbenzylamine (9.03 mL, 0.070 mol), HOBT (10.73 g, 0.70 mol), and N-methylmorpholine (8 mL, 0.076 mol) were dissolved in CH2CI2 (150 mL) with sitrring in an ice-H2O bath, treated with EDC (13.4 g, 0.070 mol) stirred at 23°C for 48 h. The reaction mixture was partitioned between EtOAc and 10% citric acid solution, the organic layer washed with satd NaHCθ3 solution, brine and dried (MgSO4), filtered, and concentrated to give an oil. This oil was dissolved in a minimum amount of ether ( 10 mL) to crystallize the desired S,S,S diastereomer (4.2 g), mp 1 18-121 °C. A solution of this product in 8N HCl (87 mL) and glacial acetic acid (22 mL) was heated at reflux overnight. The solution was concentrated on a rotary
evaporator, and the residue taken up in H2O and extracted with ether. The aqueous layer was concentrated to dryness to give a 1 : 1 mixture of 3(S)-ethyl-2(S)-proline hydrochloride and α-methylbenzylamine.
3(S)-Ethyl-2(S)-proline containing α-methylbenzylamine (2.0 g, 0.0128 mol) was dissolved in dioxane (10 mL) and H2O (10 mL) with stirring and cooling to 0°C. N,N-diisopropylethylamine (2.2 mL, 0.0128 mol) and di-ter t-butyl-dicarbonate (2.79 g, 0.0128 mol) were added and stirring was continued at 23°C for 48 h. The reaction mixture was partitioned between EtOAc (60 mL) and H2O (30 mL), the organic layer washed with 0.5N NaOH (2 x 40 mL), the aqueous layers combined and washed with EtOAc ( 30 mL) and this layer back-extracted with 0.5 N NaOH (30 mL). The aqueous layers were combined and carefully acidified at 0°C with 1N HCl to pH 3. This mixture was extracted with EtOAc (3 x 40 mL), the organics combined, dried (MgSO4), filtered and concentrated to give N-[(tert-Butyloxy) carbonyl-3(S)-ethyl-2(S)-proline as a colorless oil. N-[(tert-Butyloxy) carbonyl-3(S)-ethyl-2(S)-proline was dissolved in EtOAc (50 mL) and the solution was saturated with HCl gas with cooling in an ice-H2θ bath. The solution was stoppered and stirred at 0°C. for 3 hr. Argon was
bubbled through the solution to remove excess HCl, and the solution was concentrated to dryness to give 3(S)-ethyl-2(S)-proline hydrochloride.
Step G: N-(t-Butyloxycarbonyl)-pyrrolidin-2(S)-ylmethyl]-3(S)- ethyl-pro line
3(S)-Ethyl-2(S)-proIine hydrochloride (2.33 g, 0.013 mol) was dissolved in CH3OH (20 mL), treated with 3A molecular sieves (2 g) and KOAc ( 1.27 g, 0.013 mol) to adjust the pH of the reaction mixture to 4.5-5, then N-[(ter t-Butyloxy)carbonyl-prolinal (Pettit et al., J. Org. Chem. (1994) 59, [21 ] 6287-95) (3.36 g, 0.017 mol) was added, and the mixture was stirred for 16 hrs at room temperature. The reaction mixture was filtered, quenched with aq satd NaHCO3 (5 mL) and concentrated to dryness. The residue was extracted with CHCl3. The extract was dried (MgSO4), filtered, and concentrated to give the title compound and inorganic salts.
Step H: N-(t-Butyloxycarbonyl)-pyrrolidin-2(S)-ylmethyl]-3(S)- ethyl-prolyl methionine isopropyl ester N-(t-Butyloxycarbonyl)~pyrrolidin-2(S)-ylmethylJ-
3(S)-ethyl-proline (2.4 g, 0.008 mol), methionine isopropyl ester hydrochloride (2.21 g, 0.0097 mol), HOBT (1.49 g, 0.0097 mol) and EDC (1.86 g, 0.0097 mol) were dissolved in DMF (15 mL) at room temperature and treated with N-methylmorphoIine (3 mL, 0.024 mol). The reaction mixture was stirred overnight at room temperature, then concentrated and partitioned between EtOAc and H2O. The organic layer was washed with aq satd NaHCO3 solution, brine, and dried
(MgSO4). The crude product was chromatographed on a flash silica gel column eluting with hexane: EtOAc, 7:3 to give N-(t-butyloxy- carbonyl)-pyrrolidin-2(S)-ylmethyl]-3(S)-ethyl-prolyl methionine isopropyl ester.
isopropyl ester hydrochloride
N-(t-butyloxycarbonyl)-pyrrolidin-2(S)-ylmethyl]-3(S)- ethyl-prolyl methionine isopropyl ester (1.38 g, 0.0028 mol) was dissolved in EtOAc (40 mL), cooled to -20°C, saturated with HCl gas, and stirred at 0°C. for 1.25 hr, and room temperature for 0.25 hr. Concentration to dryness gave pyrrolidin-2(S)-ylmethyl]-3(S)-ethyl- prolyl methionine isopropyl ester hydrochloride.
Step J: Preparation of 1H-Imidazole-4- acetic acid methyl ester hydrochloride
A solution of 1 H-imidazole-4-acetic acid hydrochloride (4.00g, 24.6 mmol) in methanol (100 ml) was saturated with gaseous hydrogen chloride. The resulting solution was allowed to stand at room temperature (RT) for 18hr. The solvent was evaporated in vacuo to afford the title compound as a white solid.
1H NMR(CDCl3, 400 MHz) δ 8.85(1H, s),7.45( 1H, s), 3.89(2H, s) and 3.75(3H, s) ppm.
Step K: Preparation of 1 -(Triphenylmethyl)-1 H-imidazol-4- ylacetic acid methyl ester
To a solution of 1 H-Imidazole-4- acetic acid methyl ester hydrochloride (24.85g, 0.141 mol) in dimethyl formamide (DMF) (1 15ml) was added triethylamine (57.2 ml, 0.412mol) and triphenyl- methyl bromide(55.3g, 0.17 lmol) and the suspension was stirred for 24hr. After this time, the reaction mixture was diluted with ethyl acetate (EtOAc) (1 1) and water (350 ml). The organic phase was washed with sat. aq. NaHCO3 (350 ml), dried (Na2SO4) and evaporated in vacuo. The residue was purified by flash chromatography (SiO2, 0-100% ethyl acetate in hexanes; gradient elution) to provide the title compound as a white solid.
1 H NMR (CDCl3, 400 MHz) δ 7.35(1H, s), 7.31 (9H, m), 7.22(6H, m), 6.76(1H, s), 3.68(3H, s) and 3.60(2H, s) ppm.
Step L: Preparation of [ 1 -(4-Cyanobenzyl)-1H-imidazol-5-yl]acetic acid methyl ester
To a solution of 1 -(Triphenylmethyl)-1H-imidazol-4- ylacetic acid methyl ester (8.00g, 20.9mmol) in acetonitrile (70 ml) was added bromo-p-toluonitrile (4.10g, 20.92 mmol) and heated at 55°C for 3 hr. After this time, the reaction was cooled to room temperature and the resulting imidazolium salt (white precipitate) was collected by filtration. The filtrate was heated at 55°C for 18hr. The reaction mixture was cooled to room temperature and evaporated in vacuo. To the residue was added EtOAc (70 ml) and the resulting white precipitate collected by filtration. The precipitated imidazolium salts were combined, suspended in methanol ( 100 ml) and heated to reflux for 30min. After this time, the solvent was removed in vacuo, the resulting residue was suspended in EtOAc (75ml) and the solid isolated by filtration and washed (EtOAc). The solid was treated with sat aq NaHCO3 (300ml) and CH2CI2 (300ml) and stirred at room temperature for 2 hr. The organic layer was separated, dried (MgSO4) and evaporated in vacuo to afford the title compound as a white solid :
1HNMR(CDCl3, 400 MHz) δ 7.65(1H, d, J=8Hz), 7.53(1H, s), 7.15(1H, d, J=8Hz), 7.04(1H, s), 5.24(2H, s), 3.62(3H, s) and 3.45(2H, s) ppm.
Step M: Preparation of [1 -(4-cyanobenzyl)-1H-imidazol-5-yl]acetic acid
A solution of [1-(4-cyanobenzyl)-1H-imidazol-5-yI] acetic acid methyl ester (4.44g, 17.4mmol ) in THF (100ml) and 1 M lithium hydroxide (17.4 ml, 17.4 mmol) was stirred at RT for 18 hr. 1 M HCl (17.4 ml) was added and the THF was removed by evaporation in vacuo. The aqueous solution was lyophilized to afford the title compound containing lithium chloride as a white solid.
1H NMR(CD3OD, 400 MHz) δ 8.22(1H, s), 7.74(1H, d, J=8.4Hz), 7.36(1 H, d, J=8.4Hz), 7.15(1H, s), 5.43(2H, s) and 3.49(2H, s) ppp.
Step N: Preparation of N-[(1 -(4-Cyanobenzyl)-1H-imidazol-5- yl)acetyl]pyrrolidin-2(S)-ylmethyl]-3(S)-ethyl-prolyl methionine isopropyl ester
[1 -(4-Cyanobenzyl)-1H-imidazol-5-yl]acetic acid• LiCl (0.416 g, 1.47 mmol), pyrroIidin-2(S)-ylmethyl]-3(S)-ethyl-prolyl methionine isopropyl ester hydrochloride (Step I) ( 0.63 g, 1.33 mmol), HOOBT (0.239 g, 1.47 mmol), and EDC (0.281 g, 1.47 mmol) were dissolved in degassed DMF (20 mL) with stirring at room temperature, N-methylmorpholine (0.8 mL, 5.32 mmol) was added to achieve a pH of 7, and stirring was continued overnight. The reaction mixture was concentrated to remove most of the DMF, and the residue was
partitioned between EtOAc and aq satd NaHCO3 solution. The aq layer was washed with EtOAc, the organics combined, washed with brine and dried (MgSO4). Filtration and concentration to dryness gave the title compound after chromatography on silica gel eluting with
CH2Cl2:CH3OH, 95:5.
Anal, calcd for C33H46N6O4S• 0.7 H2O:
C, 62.38; H, 7.52; N, 13.23;
Found: C, 62.40; H, 7.17; N, 13.1 1.
FAB MS 623 (M+1)
EXAMPLE 5
2(S)-n -Butyl- 1 -[1 -(4-cyanobenzyl)imidazol-5-ylmethyl]-4-(2,3- dimethylphenyI)piperazin-5-one
1 -[ 1 -(4-Cyanobenzyl)imidazol-5-ylmethyl]-4-(2,3-dimethylphenyl)- 2(S)-(2-methoxyethyl)piperazin-5-one ditrifluoroacetic acid salt
Step A: N-Methoxy-N-methyl 4-benzyloxy-2(S)-(tert- butoxycarbonylamino)butanamide
4-Benzyloxy-2(S)-(tert-butoxycarbonylamino)butanoic acid (1.00 g, 3.23 mmol) was converted to the title compound following the procedure described in Example 24, Step A, using EDC• HCl (0.680 g, 3.55 mmol). HOBT (0.436 g, 3.23 mmol) and N,O-dimethylhydroxyl- amine hydrochloride (0.473 g, 4.85 mmol) in DMF (50 mL) at pH 7. After workup. the title compound was obtained as a clear gum.
Step B: 4-(1 -Benzyloxyethyl)-2(S)-(tm-butoxycarbonylamino) butanal
The title compound was obtained by lithium aluminum hydride reduction of the product of Step A using the procedure described in Example 24, Step B.
Step C: N-(2,3-Dimethylphenyl)-4-(2-benzyloxyethyl)-2-(S)-(tert- butoxycarbonylamino)butanamine
The title compound was prepared from the product of Step C according to the procedure described in Example 24, Step C, using 2,3-dimethylaniline (0.505 mL, 4.14 mmol), sodium triacetoxyborohydride (1.20 g, 5.65 mmol) and crushed molecular sieves (1 g) at pH 5 in dichloroethane (20 mL). The title compound was obtained after purification on silica gel, eluting with 15% ethyl acetate in hexane.
Step D: 2(S)-(2-Benzyloxyethyl)-1 -tert-butoxycarbonyI-4-(2,3- dimethylphenyl)piperazin-5-one
The title compound was prepared from the product of Step
C according to the procedure described in Example 24, Step D, using chloroacetyl chloride (0.21 mL, 2.57 mmol) in 60 mL 1 :1 ethyl acetate: saturated sodium bicarbonate, followed by reaction of the crude product with sodium hydride (0.373 g, 60% dispersion in oil, 9.32 mmol) in
DMF (30 mL). After workup, the crude product was chromatographed on silica gel with 30% ethyl acetate in hexane to obtain the title compound. Step E: 1 -ter t-Butoxycarbonyl-4-(2,3-dimethylphenyl)-2(S)-(2- hydroxyethyl)piperazin-5-one
The product from Step D was dissolved in methanol (40 mL) and 10% Pd/C was added (0.160 g). The reaction was shaken under 60 psi hydrogen overnight. The catalyst was removed by filtration, and the solvent evaporated to give the title compound.
Step F: 1 -tert-Butoxycarbonyl-4-(2,3-dimethylphenyl)-2(S)-(2- methoxyethyl)piperazin-5-one
The product from Step E (0.241 g, 0.688 mmol) was dissolved in DMF (10 mL) containing methyl iodide (0.21 mL, 3.44 mmol) and the stirred solution cooled to 0°C under nitrogen. Sodium hydride (0.070 g, 60% dispersion in oil, 1.72 mmol) was added and the reaction stirred for lh. The reaction was quenched with water, and the DMF removed under vacuum. The residue was partitioned between ethyl acetate and water, and the organic phase washed with saturated brine and dried over magnesium sulfate. The crude product was chromatographed on silica gel with 40% ethyl acetate in hexane to give the title compound. Step G: 4-(2,3-Dimethylphenyl)-2(S)-(2-methoxyethyl)- 1 -[4-(1 - triphenylmethylimidazolyl)methyllpiperazin-5-one
The product from Step F (0.1 13 g, 0.312 mmol) was converted to the title compound according to the procedure described in Example 24, Step E, except using 30% trifluoroacetic acid in dichloromethane (10 mL) for 1 h for the initial deprotection. The volatiles were removed in vacuo, and the residue dissolved in
dichloroethane. Triethylamine was added to obtain pH 5. Sodium triacetoxyborohydride (0.100 g, 0.468 mmol) and 1 -triphenylmethyl-
4-imidazolylcarboxaldehyde (0.1 164 g, 0.343 mmol) was added. The reaction was stirred overnight at 20°C then poured into saturated sodium bicarbonate solution. The organic phase was washed with saturated brine and dried over magnesium sulfate. Silica gel
chromatography using 5% methanol in chloroform as eluant yielded the title compound.
Step H: 1-[1 -(4-Cyanobenzyl)imidazol-5-ylmethyl]-4-(2,3- dimethylphenyl)-2(S)-(2-methoxyethyl)piperazin-5-one ditrifluoroacetic acid salt
The product from Step G (0.182 g, 0.312 mmol) was converted to the title compound according to the procedure described in Example 25, using 4-cyanobenzylbromide (0.061 g, 0.312 mmol) in acetonitrile (10 mL), followed by reaction of the crude imidazolium salt with triethylsilane (0.13 mL) and trifluoroacetic acid (2 mL) in dichloromethane (6 mL). Purification was accomplished by reverse phase preparative HPLC with a mixed gradient of 0%-70%
acetonitrile/0.1 % TFA; 100%-30% 0.1 % aqueous TFA over 60 min. The title compound was isolated after lyophilization from water. FAB ms (m+1 ) 458.
Anal. Calc. for C27H3 1N5O2• 0.35 H2O• 2.0 TFA:
C, 53.81 ; H, 4.91 ; N, 10.21.
Found: C, 53.83; H, 4.95; N, 10.29. EXAMPLE 6
N-[2(S)-N'-(1 -(4-Cyanophenyl-methyl)- 1 H-imidazol-5-ylacetyl)amino- 3(S)-methylpentyl]-N-1 -naphthylmethyl-glycyl-methionine methyl ester
Preparation of N-[2(S)-N'-(1 -(4-Cyanophenylmethyl)-1H-imidazol-5- ylacetyl)amino-3(S)-methylpentyl]-N- 1 -naphthylmethyl-glycyl- methionine bis trifluoroacetate
Step A: Preparation of 1 -(TriphenyImethyl)-1 H-imidazol-4-ylacetic acid methyl ester (23)
To a suspension of 1H-imidazole-4-acetic acid methyl ester hydrochloride (1, 7.48, 42.4 mmol) in methylene chloride (200 ml) was added triethylamine (17.7 ml, 127 mmol) and triphenylmethyl bromide ( 16.4 g, 50.8 mmol) and stirred for 72 h. After this time, reaction mixture was washed with sat. aq. sodium bicarbonate (100 ml) and water (100 ml). The organic layer was evaporated in vacua and purified by flash chromatography (30- 100% ethyl acetate/hexanes gradient elution) to provide 23 as a white solid.
1 H NMR (CDCI3, 400 MHz) δ 7.35 (1H, s), 7.31 (9H, m), 7.22 (6H, m), 6.76 (1H, s), 3.68 (3H, s) and 3.60 (2H, s) ppm.
Step B: Preparation of 1-(4-NitrophenyImethyl)-1H-imidazoI-5- ylacetic acid methyl ester (16)
To a solution of 1 -(triphenylmethyl)- 1H-imidazol-4-ylacetic acid methyl ester (23, 274 mg, 0.736 mmol) in acetonitrile ( 10 ml) was added 4-nitrobenzylbromide (159 mg, 0.736 mmol) and heated to 55°C for 16 h. After this time, the reaction was cooled to room temperature, treated with ethyl acetate (20 ml) and the resulting precipitate was filtered. The filtrate was concentrated to dryness in vacuo and the residue was redissolved in acetonitrile (4 ml) and heated to 65°C for
3 h. After this time, the reaction mixture was evaporated to dryness and combined with initial precipitate. This residue was dissolved in methanol (5 ml ) and heated to reflux for 30 min. The resulting solution was evaporated in vacuo and the residue was purified by flash chromatography (2-5% methanol/methylene chloride gradient elution ) to provide 16.
1 H NMR (CDCl3, 400 MHz) δ 8.20 (2H, d, J=8.8 Hz), 7.53 ( 1H, s), 7.19 (2H, d, J=8.8 Hz), 7.03 (1H, s), 5.28 (2H, s), 3.61 (3H, s) and 3.44 (2H, s) ppm.
Step C: 1 -(4-Nitrophenylmethyl)- 1 H-imidazol-5-ylacetic acid
hydrochloride
1 -(4-Nitropheny Imethyl)- 1H-imidazol-5-ylacetic acid methyl ester (0.1 15 g, 0.42 mmol ) was dissolved in 1.0N hydrochloric acid (10 ml ) and heated at-55°C for 3 h. The solution was evaporated in vacuo to give the compound as a white solid.
1H NMR (CD3OD, 400 MHz) δ 9.06 (1H, s), 8.27 (2H, d, J=8.8 Hz), 7.61 (1H, s), 7.55 (2H, d, J=8.8 Hz), 5.63 (2H, s) and 3.81 (2H, s) ppm. Step D: N-[2(S)-N'-(1 -(4-Nitrophenylmethyl)-1H-imidazol-5- ylacetyI)amino-3(S)-methylpentyl]-N-1-naphthylmethyl- glycyl-methionine methyl ester bis trifluoroacetate
To a solution of 1 -(4-nitrophenylmethyl)-1H-imidazol- 5-ylacetic acid hydrochloride, N-[2(S)-amino-3(S)-methylpentyl]-N- naphthylmethyl-glycyl-methionine methyl ester bis hydrochloride (209 mg, 0.392 mmol) and 3-hydroxy-1 ,2,3-benzotriazin-4(3H)-one (HOOBT, 64 mg, 0.39 mmol) in methylene chloride (10 ml) was added 1 -(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, 75.2 mg, 0.392 mmol) and triethylamine (219 μl, 1.57 mmol) and the mixture stirred overnight at room temperature. After this time, satd. aq. sodium bicarbonate (10 ml) was added and the mixture was extracted with methylene chloride. The combined extracts were washed with satd. aq. sodium bicarbonate (10 ml) and the solvent evaporated in vacuo.
EXAMPLE 7
2(S)-[2(S)-[2(R)-Amino-3-mercapto]propylamino-3(S)-methyl]- pentyloxv-3-phenylpropionyl-methionine sulfone isopropyl ester
The title compound is prepared in accordance with WO 94/10138 published on May 1 1 , 1994, incorporated by reference.
BIOLOGICAL ASSAYS.
The ability of compounds of the present invention to inhibit cancer can be demonstrated using the following assays.
Raf kinase assay
Raf kinase activity in vitro is measured by the phosphorylation of its physiological substrate MEK (Map/ERK kinase).
Phosphorylated MEK is subsequently trapped on a filter membrane and incorporation of radio-labeled phosphate is quantitated by scintillation counting.
MATERlALS
Activated Raf
Produced in Sf9 insect cells coinfected with three
different baculoviruses expressing epitope-tagged Raf, and the upstream activators Val ^-H-Ras, and Lck. The epitope sequence Glu-Tyr-Met- Pro-Met-Glu ("Glu-Glu") was fused to the carboxy-terminus of full- length c-Raf. MEK
Catalytically inactive MEK is produced in Sf9 cells infected with baculovirus expressing epitope-tagged MEK with a lysine97 to alanine mutation (K97A). The epitope sequence Glu-Tyr-Met-Pro-Met- Glu ("Glu-Glu") was fused to the amino-terminus of full-length MEK 1.
Anti "Glu-Glu" antibody
A hybridoma cell line expressing an antibody specific for the "Glu-Glu" epitope was obtained from Gernot Walter, UCSD. Cells were grown and antibodies were purified as described (Grussenmeyer et al., Proc. Natl. Acad. Sci. U.S.A., 82, pp. 7952-7954, 1985).
Column buffer
20 mM Tris, pH 8, 100 mM NaCl, 1 mM EDTA, 2.5 mM EGTA, 10 mM MgCl2, 2 mM DTT, 0.4 mM AEBSF, 0.1 % n-octyl glucopyranoside, 1 nM okadeic acid, and 10 μg/ml each of benzamidine, leupeptin, pepstatin, and aprotinin (all SIGMA).
5x reaction buffer
125 mM HEPES pH=8.0, 25 mM MgCl2, 5 mM EDTA, 5 mM Na3VO4, 100 μg/ml BS A
Enzyme dilution buffer
25 mM HEPES pH=8.0, 1 mM EDTA, 1 mM Na3VO4, 400 μg/ml BSA.
Stop solution
100 mM EDTA, 80 mM sodium pyrophosphate.
Filter plates
Millipore Multiscreen #SE3M078E3, Immobilon-P (PVDF).
METHOD
A. Protein purification
1. Sf9 insect cells were infected with baculovirus and grown as described (Williams et al., Proc. Natl. Acad. Sci. U.S.A., 89, pp. 2922-2926, 1992).
2. All subsequent steps were performed on ice or at 4°C.
Cells were pelleted and lysed by sonication in column buffer. Lysates were spun at 17,000x g for 20 min, followed by 0.22 μm filtration.
3. Epitope-tagged proteins were purified by chromatography over a GammaBind Plus (Pharmacia) affinity column to which "Glu-Glu" antibody had been coupled. Proteins were loaded on the column, followed by washes with two column volumes of column buffer, and eluted with 50 μg/ml of peptide antigen (Glu-Tyr-Met-Pro-Met-Glu) in column buffer.
B. Raf kinase assay
1. Add 10 μl of inhibitor or control in 10% DMSO to assay plate.
2. Add 30 μl of reaction mix containing 10 μl 5x reaction
buffer and 0.5 μl ImM "P-γ-ATP (20 μCi/ml), 0.5 μl MEK (2.5 mg/ml), 1 μl 50 mM β-mercaptoethanol.
3. Start reaction by addition of 10 μl enzyme dilution buffer containing 1 mM DTT and an empirically determined amount of activated Raf that produces linear incorporation kinetics over the reaction time course.
4. Mix and incubate at room temperature for 90 min.
5. Stop reaction by addition of 50 μl stop solution.
6. Prewet filter plate with 70% ethanol and rinse with water.
7. Transfer 90 μl aliquots of stopped reaction to filter plate.
8. Aspirate and wash four times with 200 μl H20.
9. Add 50 μl scintillation cocktail, seal plate, and count in Packard TopCount scintillation counter.
Map Kinase Phosnhorylation assay
Inhibition of Raf kinase activity in intact cells is measured by determining the phosphorylation state of Map Kinase in TPA- stimulated C-33a human epithelial cells. Phosphorylated Map Kinase is detected by "Western" blot using an anti-phospho-Map Kinase antibody.
Materials
C33a Human Epithelial Cells
The C33a cell line is obtained from the ATCC repository, catalog # H TB31 , and is maintained in DMEM (Mediatech) + 10% fetal bovine serum +1 % penicillin/streptomycin (Gibco) according to the instructions provided.
Anti-phospho-MAP Kinase antibody
The rabbit polyclonal anti-phospho-MAP kinase antibody is obtained from New England Biolabs (Beverly, MA)
Secondary antibody
The anti-rabbit antibody-alkaline phosphatase conjugate is obtained from New England Biolabs
Acrylamide Gel
Ten percent bi s-acrylamide electrophoresis gels were obtained from Novex.
Blocking Buffer
1 x Phosphate-buffered saline, 0.1 % Tween-20, 5% nonfat dry milk.
Antibody dilution buffer
1x phosphate-buffered saline, 0.05% Tween-20, 5% bovine serum albumin Alkaline phosphatase substrate
The chemiluminescent alkaline phosphatase substrate, CDP-Star™, is obtained from New England Biolabs.
Assay Buffer
0.1 M diethanolamine, 1 mM MgCl2.
Method
1. C33a cells are grown to confluency in 24 well plates, then starved for 24 hr in DMEM + 0.5 % charcoal -stripped serum.
2. Compound to be tested, dissolved in DMSO at 1000x concentration, is added to each well. 3. One hour later, TPA (dissolved in DMSO at 1000x concentratrion) is added at a final concentration of 100 ng/ml.
4. Twenty minutes later, the media is removed from all wells, and 100 μl of boiling hot reducing Laemmli sample buffer is added to each well. The plate is agitated, and the cell lysate is transferred to a 1.5 ml plastic microcentrifuge tube. Each lysate is then sonicated for 10 s, and placed in a boiling water bath for 5-10 minutes. Fifteen microliters of each sample is then loaded on a 10 % Laemmli polyacrylamide gel (Novex), and the gel electrophoresed according to the manufacturer's
instructions.
5. Proteins in the gel are electroblotted to a PVDF membrane, which is then rinsed in PBS and blocked with Blocking Buffer for approximately 1 hr at room temperature.
6. The PVDF membrane is rinsed in PBS. The anti-phospho-MapK antibody, diluted approximately 1 :500 in antibody dilution buffer, is incubated with the PVDF membrane with gentle agitation overnight at 4 °C.
7. The PVDF membrane is rinsed 3 times for 5 minutes with Blocking Buffer, then incubated with the secondary antibody, diluted
approximately 1 : 1000 in antibody dilution buffer, for 1 hr with gentle agitation at room temperature.
8. The PVDF membrane is rinsed 5 times for 5 minutes with Blocking Buffer, then incubated with the chemi luminescent alkaline phosphatase substrate dissolved in Assay Buffer for approximately 5 minutes. The membrane is then rinsed, wrapped in plastic, and exposed to x-ray film to detect blotted proteins.
In the Raf kinase inhibition assay, the IC50 ranges from about 0.001 μM to about 1.5 μM.
In vitro inhibition of ras famesyl transferase
Assays of famesyl-protein transferase.
Partially purified bovine FPTase and Ras peptides
(Ras-CVLS, Ras-CVIM and Ras-CAIL) were prepared as described by Schaber et al., J. Biol. Chem. 265: 14701 - 14704 (1990), Pompliano, et al., Biochemistry 31 :3800 (1992) and Gibbs et al., PNAS U.S.A.
86:6630-6634 (1989), respectively. Bovine FPTase was assayed in a volume of 100 μl containing 100 mM N-(2-hydroxy ethyl) piperazine- N'-(2-ethane sulfonic acid) (HEPES), pH 7.4, 5 mM MgCl2, 5 mM dithiothreitol (DTT), 100 mM [3H]-farnesyl diphosphate ([3H]-FPP; 740 CBq/mmol, New England Nuclear), 650 nM Ras-CVLS and 10 μg/ml FPTase at 31°C for 60 min. Reactions were initiated with FPTase and stopped with 1 ml of 1.0 M HCL in ethanol. Precipitates were collected
onto filter-mats using a TomTec Mach II cell harvestor, washed with 100% ethanol, dried and counted in an LKB β-plate counter. The assay was linear with respect to both substrates, FPTase levels and time; less than 10% of the l3H]-FPP was utilized during the reaction period.
Purified compounds were dissolved in 100% dimethyl sulfoxide
(DMSO) and were diluted 20-fold into the assay. Percentage inhibition is measured by the amount of incorporation of radioactivity in the presence of the test compound when compared to the amount of incorporation in the absence of the test compound.
Human FPTase was prepared as described by Omer
et al., Biochemistry 32:5167-5176 ( 1993). Human FPTase activity was assayed as described above with the exception that 0.1 % (w/v) polyethylene glycol 20,000, 10 μM ZnCl2 and 100 nM Ras-CVIM were added to the reaction mixture. Reactions were performed for 30 min., stopped with 100 μl of 30% (v/v) trichloroacetic acid (TCA) in ethanol and processed as described above for the bovine enzyme.
The famesyl protein transferase inhibiting compounds are tested for inhibitory activity against human FPTase by the assay described above and the compounds can generally be found to have IC50 of approximately 50 μM.
In viva ras famesylation assay
The cell line used in this assay is a v-ras line derived from either Ratl or N1H3T3 cells, which expressed viral Ha-ras p21. The assay is performed essentially as described in DeClue, J.E. et al., Cancer Research 51 :712-717, (1991). Cells in 10 cm dishes at 50-75%
confluency are treated with the test compound (final concentration of solvent, methanol or dimethyl sulfoxide, is 0.1 %). After 4 hours at 37°C, the cells are labelled in 3 ml methionine -free DMEM supple- meted with 10% regular DMEM, 2% fetal bovine serum and 400 mCi[35S]methionine ( 1000 Ci/mmol). After an additional 20 hours, the cells are lysed in 1 ml lysis buffer (1 % NP40/20 mM HEPES, pH 7.5/5 mM MgCl2/lmM DTT/10 mg/ml aprotinen/2 mg/ml leupeptin/2 mg/ml antipain/0.5 mM PMSF) and the lysates cleared by centrifugation at
100,000 x g for 45 min. Aliquots of lysates containing equal numbers of acid-precipitable counts are bought to 1 ml with IP buffer (lysis buffer lacking DTT) and immunoprecipitated with the ras-specific monoclonal antibody Y 13-259 (Furth, M.E. et al. , J. Virol. 43:294-304, ( 1982)). Following a 2 hour antibody incubation at 4°C, 200 ml of a 25% suspension of protein A-Sepharose coated with rabbit anti rat IgG is added for 45 min. The immunoprecipitates are washed four times with IP buffer (20 nM HEPES, pH 7.5/1 mM EDTA/1 % Triton X- 100.0.5% deoxycholate/0.1 %/SDS/0.1 M NaCl) boiled in SDS-PAGE sample buffer and loaded on 13% acrylamide gels. When the dye front reached the bottom, the gel is fixed, soaked in Enlightening, dried and autoradiographed. The intensities of the bands corresponding to farnesylated and nonfarnesylated ras proteins are compared to
determine the percent inhibition of famesyl transfer to protein.
In viva growth inhibition assay
To determine the biological consequences of FPTase inhibition, the effect of the compounds of the instant invention on the anchorage-independent growth of Ratl cells transformed with either a v-ras, v-raf, or v-mos oncogene is tested. Cells transformed by v-Raf and v-Mos maybe included in the analysis to evaluate the specificity of instant compounds for Ras-induced cell transformation.
Rat 1 cells transformed with either v-ras, v-raf, or v-mos are seeded at a density of 1 x 104 cells per plate (35 mm in diameter) in a 0.3% top agarose layer in medium A (Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum) over a bottom agarose layer (0.6%). Both layers contain 0.1 % methanol or an appropriate concentration of the instant compound (dissolved in
methanol at 1000 times the final concentration used in the assay).
The cells are fed twice weekly with 0.5 ml of medium A containing 0.1 % methanol or the concentration of the instant compound.
Photomicrographs are taken 16 days after the cultures are seeded and comparisons are made.