WO1999015539A2 - Pentopyranosyl-nucleosid, seine herstellung und verwendung - Google Patents
Pentopyranosyl-nucleosid, seine herstellung und verwendung Download PDFInfo
- Publication number
- WO1999015539A2 WO1999015539A2 PCT/EP1998/005997 EP9805997W WO9915539A2 WO 1999015539 A2 WO1999015539 A2 WO 1999015539A2 EP 9805997 W EP9805997 W EP 9805997W WO 9915539 A2 WO9915539 A2 WO 9915539A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- nucleoside
- pentopyranosyl
- benzoyl
- ribopyranosyl
- group
- Prior art date
Links
- 0 C**(OC1C=CC([N+]([O-])=O)=CC1)=O Chemical compound C**(OC1C=CC([N+]([O-])=O)=CC1)=O 0.000 description 1
- CEWDTFLISAWJHG-YFKPBYRVSA-N NCCCC[C@@H](C(O)=O)O Chemical compound NCCCC[C@@H](C(O)=O)O CEWDTFLISAWJHG-YFKPBYRVSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/549—Sugars, nucleosides, nucleotides or nucleic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/001—Preparation for luminescence or biological staining
- A61K49/0013—Luminescence
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/02—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with ribosyl as saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/04—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with deoxyribosyl as saccharide radical
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/531—Production of immunochemical test materials
- G01N33/532—Production of labelled immunochemicals
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S977/00—Nanotechnology
- Y10S977/70—Nanostructure
- Y10S977/724—Devices having flexible or movable element
- Y10S977/727—Devices having flexible or movable element formed from biological material
- Y10S977/728—Nucleic acids, e.g. DNA or RNA
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S977/00—Nanotechnology
- Y10S977/70—Nanostructure
- Y10S977/788—Of specified organic or carbon-based composition
- Y10S977/797—Lipid particle
- Y10S977/798—Lipid particle having internalized material
- Y10S977/799—Containing biological material
- Y10S977/80—Nucleic acid, e.g. DNA or RNA
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S977/00—Nanotechnology
- Y10S977/70—Nanostructure
- Y10S977/832—Nanostructure having specified property, e.g. lattice-constant, thermal expansion coefficient
- Y10S977/835—Chemical or nuclear reactivity/stability of composition or compound forming nanomaterial
- Y10S977/836—Chemical or nuclear reactivity/stability of composition or compound forming nanomaterial having biological reactive capability
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S977/00—Nanotechnology
- Y10S977/84—Manufacture, treatment, or detection of nanostructure
- Y10S977/894—Manufacture, treatment, or detection of nanostructure having step or means utilizing biological growth
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S977/00—Nanotechnology
- Y10S977/84—Manufacture, treatment, or detection of nanostructure
- Y10S977/895—Manufacture, treatment, or detection of nanostructure having step or means utilizing chemical property
- Y10S977/896—Chemical synthesis, e.g. chemical bonding or breaking
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S977/00—Nanotechnology
- Y10S977/902—Specified use of nanostructure
- Y10S977/904—Specified use of nanostructure for medical, immunological, body treatment, or diagnosis
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S977/00—Nanotechnology
- Y10S977/902—Specified use of nanostructure
- Y10S977/904—Specified use of nanostructure for medical, immunological, body treatment, or diagnosis
- Y10S977/914—Protein engineering
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S977/00—Nanotechnology
- Y10S977/902—Specified use of nanostructure
- Y10S977/904—Specified use of nanostructure for medical, immunological, body treatment, or diagnosis
- Y10S977/915—Therapeutic or pharmaceutical composition
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S977/00—Nanotechnology
- Y10S977/902—Specified use of nanostructure
- Y10S977/904—Specified use of nanostructure for medical, immunological, body treatment, or diagnosis
- Y10S977/924—Specified use of nanostructure for medical, immunological, body treatment, or diagnosis using nanostructure as support of dna analysis
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S977/00—Nanotechnology
- Y10S977/902—Specified use of nanostructure
- Y10S977/904—Specified use of nanostructure for medical, immunological, body treatment, or diagnosis
- Y10S977/925—Bioelectrical
Definitions
- Pentopyranosyl nucleoside its production and use
- the present invention relates to a pentopyranosyl nucleoside of the formula (I) or of the formula (II)
- p-NA's Pyranosyl nucleic acids
- p-NA's are generally structural isomers isomeric to natural RNA, in which the pentose units are in the pyranose form and are repetitively linked by phosphodiester groups between positions C-2 'and C-4' (Fig.
- nucleobase is understood to mean the canonical nucleobases A, T, U, C, G, but also the pairs isoguanine / isocytosine and 2,6-diamondopu ⁇ n / xanthine and in the sense of the present invention also other purines and pyrimidines p-NAs, and The p-RNAs derived from the ribose were first described by Eschenmoser et al (see Pitsch, S et al Helv Chim Acta 1993, 76, 2161, Pitsch, S et al.
- This specificity which is valuable for the construction of supramolecular units, is related to the relatively low flexibility of the ribopyranosephosphate backbone as well as the strong inclination of the base plane to the strand axis and the consequent tendency to mtercatenary base stacking in the resulting duplex and can ultimately be attributed to the participation of a 2 ', Recycle 4'-cis-disubstituted ribopyranose rings on the structure of the backbone.
- These significantly better pairing properties make p-NAs preferred pairing systems compared to DNA and RNA for the application of the structure of supramolecular units. They form a pairing system orthogonal to natural nucleic acids, ie they do not pair with in the naturally occurring DNA's and RNA's, which is particularly important in the diagnostic field
- a suitable protected nucleobase was reacted with the anomeric gene of tetrabenzoyl ribopyranose by the action of bis (tmethylsilyl) acetamid and a Lewis acid such as, for example, tmethylsilyltrifluoromethanesulfonate (analogously to H Vorbruggen, K Kroluaiewicz, Ber, 1981, Bennol.
- the carrier-bound component was repeatedly de-acidified in 4'-position, a phosphoramidite coupled under the action of a coupling reagent, for example a tetrazole derivative, acetylated free 4'-oxygen atoms and the phosphorus atom oxidized so as to form the oligomeric product
- a coupling reagent for example a tetrazole derivative, acetylated free 4'-oxygen atoms and the phosphorus atom oxidized so as to form the oligomeric product
- the remaining protective groups were then split off, the product was purified by HPLC and desalted.
- 5- (4-nitrophenyl) -lH-tetrazole is used as a coupling reagent in automated p-RNA synthesis.
- concentration of this reagent in the solution of tetrazole in acetonitrile is so high that the 5- (4-N ⁇ trophenyl) -lH-tetrazole crystallized in the thin tubes of the synthesizer and the synthesis thus came to an early end. It was also observed that the oligomers were contaminated with 5- (4-nitrophenyl) -lH-tetrazole
- the object of the present invention was therefore to provide new pentopyranosyl nucleosides and a process for their preparation in which the production of the pentopyranosyl nucleosides is to be made possible on a larger scale than by known processes and the disadvantages described above are avoided.
- the present invention therefore relates to a pentopyranosyl nucleoside of the formula (I)
- R 1 is equal to H. OH. Shark with shark equal to Br or Cl, or a rest selected from
- n H 2n + ⁇ with n is an integer from 1-12, preferably 1-8, in particular 1-4, a ß-eliminable group, preferably a group of the formula -OCH 2 CH R 18 where R 18 is a cyano or p-nitrophenyl radical or a fluorenylmethyloxycarbonyl (Fmoc) radical, or (C n H 2n ) NR 10 R n with R , 0 R n equal to H, or R 10 R ⁇ connected via a radical of the formula
- wo ⁇ n R 1 is H, OH, shark with shark is Br or Cl or a group selected from
- the pentopyranosyl nucleoside according to the invention is generally a ribo-, arabino-, lyxo- and / or xylopyranosyl nucleoside, preferably a ribopyranosyl nucleoside, whereby the pentopyranosyl part can be D-configured but also L-configured
- the pentopyranosvl nucleoside according to the invention is a pentopyranosyl-pu ⁇ n, -2,6-d ⁇ am ⁇ nopu ⁇ n, -6-pu ⁇ nth ⁇ ol, -py ⁇ din, -py ⁇ midin, -adenosine, - guanosine, -isoguanosine, -6-th ⁇ oguanos , -hypoxanthine, -thymidine, -cytosine, -isocytosine, -mdol, -tryptamm, -N-phthaloyltryptamine, -uracil, -caffeine, -theobromine, - theophyln, -benzot ⁇ azol or -ac ⁇ din, especially around a pentopyranosyl-pu ⁇ n - py ⁇ midin, -adenosin, -guanosin, -thy
- the compounds according to the invention also include pentopyranosyl nucleosides which can be used as linkers, ie as compounds with functional groups which occur covalently on biomolecules, such as, for example, in their natural form modified nucleic acids such as DNA, RNA but also p-NA's, preferably pRNA's, can bind. This is surprising since no linkers are known for p-NA's
- this includes pentopyranosyl nucleosides in which R 2 , R 3 , R 4 , R 2 , R 3 and / or R 4 is a 2-phthalimidoethyl or allyloxy radical.
- Uracil-based linkers are preferred according to the present invention, for example, in which the 5-position of the uracil has preferably been modified, for example N-phthaloylaminoethyluracil, but also indole-based linkers, preferably tryptamine pvate, such as N-phthaloyltryptamine
- the present invention also provides easier-to-handle pentopyranosyl-N, N-dacyl nucleosides, preferably confine, in particular adenosine, guanosine or 6-thioguanosine, whose nucleobase can be completely deprotected in a simple manner.
- the invention also includes pentopyranosyl nucleosides in which R 2 , R 3 , R 4 , R 2 , R J and / or R 4 is a radical of the formula -N [C (O) R 9 ] 2 , in particular N 6 , N 6 -dibenzoyl-9- (ß-D-ribopyranosyl) -adenos ⁇ n
- the present invention provides pentopyranosyl nucleosides which carry a protective group, preferably a base- or metal-catalyzed protective group, in particular an acyl group, particularly preferably a benzoyl group, exclusively on the 3 'oxygen atom of the pentopyranoside part.
- a protective group preferably a base- or metal-catalyzed protective group, in particular an acyl group, particularly preferably a benzoyl group, exclusively on the 3 'oxygen atom of the pentopyranoside part.
- a further protective group preferably an acid- or base-labile protective group, in particular a trityl group, particularly preferably a dimethoxytyl group, to the 4'-oxygen atom of the pentopyranoside part without additional steps which reduce the yield, such as additional steps Cleaning steps
- the present invention provides pentopyranosyl nucleosides which carry a protective group, preferably an acid- or base-labile protective group, in particular a trityl group, particularly preferably a dimethoxy-methyl group, exclusively on the 4 ′ oxygen atom of the pentopyranoside part.
- a protective group preferably an acid- or base-labile protective group, in particular a trityl group, particularly preferably a dimethoxy-methyl group, exclusively on the 4 ′ oxygen atom of the pentopyranoside part.
- a further protective group preferably a base- or metal-catalyzed removable protective group, in particular an acyl group, particularly preferably a benzoyl group, for. B. on the 2'-oxygen atom of the pentopyranoside part, without additional, the yield-reducing steps such. B. additional cleaning steps.
- the pentopyranoside nucleosides according to the invention can be reacted in a so-called one-pot reaction, which increases the yields and is therefore particularly advantageous.
- ⁇ -ribopyranosyl nucleosides in particular a ⁇ -ribopyranosyl adenine, guanine, cytosine, thymidine or uracil, xanthine or hypoxanthine, and an N-benzoyl, N-isobutyroyl, O 6 - (2 -Cyanoethyl) - or O 6 - (2- (4-nitrophenyl) ethyl) -N 2 -isobutylroyl-ß-ribopyranosyl nucleoside.
- 4'-DMT-pentopyranosyl nucleosides preferably a 4'-DMT-ribopyranosyl nucleoside, in particular a 4'-DMT-ribopyranosyl adenine, guanine, cytosine, thymidine, uracil, xanthine, or - hypoxanthine, and an N-benzoyl-4'-DMT-ribopyranosyl nucleoside, in particular an N-benzoyl-4'-DMT-ribopyranosyl-adenine, guanine or cytosine, and an N-isobutyroyl-4'-DMT-ribopyranosyl Nucleoside, especially an N-isobutyroyl-4'- DMT-ribopyranosyl adenine, guanine or cytosine and an O 6 - (2-cyanoethyl) -N 2 - isobutyroyroy
- Suitable precursors for oligonucleotide synthesis are, for example, 4'-DMT-pentopyranosyl-nucleosides-2'-phosphitamide / -H-phosphonate, preferably a 4'DMT-ribopyranosyl-nucleoside-2'-phosphitamide / -H-phosphonate, in particular a 4 '-DMT- ribopyranosyl-adenine, guanine, cytosine, thymidine, xanthine, or hypoxanthine, or uracil-2'-phosphitamide / -H-phosphonate and an N-benzoyl-4' -DMT-ribopyranosyl-adenine, -guanine or -cytosine-2'-phosphitamide / -H-phosphonate and an N-isobutylroyl-4'-DMT-ribopyranosyl-a
- guanine or cytosine 2'-succinate O- (2-cyanoethyl) -4'-DMT-ribopyranosyl-guanine, xanthine or hypoxanthine-2'-succinate and an O 6 - (2- ( 4-nitrophenyl) ethyl) -N 2 -isobutyroyl-4'-DMT-ribopyranosyl-guanine-2'-succinate.
- Another object of the present invention is a method for producing a pentopyranosyl nucleoside of the formula (I) or (II) according to the invention, in which starting from the unprotected pentopyranoside
- the 2'-, 3'- or 4'-position of the pentopyranoside is first protected, and preferably (b) in a second step the other position at the 2'-, 3'- or 4'-position.
- the method according to the invention is not limited to the nucleobases described in the cited literature, but can surprisingly be carried out successfully with a large number of natural and synthetic nucleobases.
- it is particularly surprising that the process according to the invention can be carried out in large yields and with an average time saving of 60% compared to the process known from the literature, which is particularly advantageous for industrial use.
- the cleaning steps required in the method described in the literature for example chromatographic intermediate purifications, are not necessary and the reactions can in part be carried out as a so-called one-pot reaction, which significantly increases the space / time yields
- the protective group in the case of a 2 'protected position, is rearranged from the 2' position to the 3 'position, which is generally carried out in the presence of a base, in particular in the presence of N-ethyldiisopropylamine and / or triethylamine According to the present invention, this reaction can be carried out particularly advantageously in the same reaction container as a one-pot reaction
- the pyranosyl nucleoside is protected by an acid-labile, base-labile or metal-catalyzed protective group S c ⁇ , S c2 , S c r or S c2 -, with the protective groups S c! and S cr are different from the protective groups S o2 and S c2 -.
- the protective groups mentioned are an acyl group, preferably an acetyl, benzoyl, nitrobenzoyl and / or methoxybenzoyl group, trityl groups, preferably a 4, 4'-dimethoxytrityl (DMT) group or a ß-eliminable group, preferably a group of the formula -OCH 2 CH 2 R 18 where R 18 is a cyano or p-nitrophenyl radical or a fluorenylmethyloxycarbonyl (Fmoc) group.
- acyl group preferably an acetyl, benzoyl, nitrobenzoyl and / or methoxybenzoyl group
- trityl groups preferably a 4, 4'-dimethoxytrityl (DMT) group or a ß-eliminable group, preferably a group of the formula -OCH 2 CH 2 R 18 where R 18 is a cyano or p-nitrophenyl radical
- the 2 'or 3' position is protected by a base-labile or metal-catalyzed protective group, preferably by an acyl group, in particular by an acetyl, benzoyl, nitrobenzoyl and / or methoxybenzoyl group, and / or that 4 'position is protected by an acid- or base-labile protective group, preferably by a trityl and / or Fmoc group, in particular by a DMT group.
- a base-labile or metal-catalyzed protective group preferably by an acyl group, in particular by an acetyl, benzoyl, nitrobenzoyl and / or methoxybenzoyl group, and / or that 4 'position is protected by an acid- or base-labile protective group, preferably by a trityl and / or Fmoc group, in particular by a DMT group.
- the process according to the invention does not require any acetal protecting groups, such as acetals or ketals, which avoids additional chromatographic intermediate purifications and consequently allows the reactions to be carried out as one-pot reactions with surprisingly high space / time yields
- the protective groups mentioned are preferably introduced at low temperatures, since surprisingly they can thereby be introduced selectively.
- a benzoyl group is introduced by reaction with benzoyl chloride in pyridine or in a pyridine / methylene chloride mixture at low temperatures.
- a DMT group can be introduced, for example, by reaction with DMTCl in the presence of a base, for example N-ethyldiisopropylamine (Hunig- Base), and for example pyridine, methylene chloride or a pyridine / methylene chloride mixture at room temperature
- reaction products are purified by chromatography after the acylation and / or after the possible rearrangement from the 2 'to the 3' position. Purification after tritylation is not necessary in accordance with the process of the invention, which is particularly advantageous .
- the end product can be further purified by crystallization.
- Another object of the present invention is a method for producing an R bopyranosyl nucleoside, in which (a) a protected nucleobase is reacted with a protected ribopyranose,
- step (b) the protective groups are removed from the ribopyranosyl part of the product from step (a), and
- step (c) the product from step (b) is reacted according to the method described in more detail above.
- pentopyranoses such as, for. B tetrabenzoyl-pentopyranoses, preferably ⁇ -tetrabenzoyl-ribopyranoses (R. Jeanloz, J Am. Chem. Soc 1948. 70, 4052).
- a linker of the formula (II) in which R 4 is (C n H 2n ) NR 10, R u ' and R ltr R ⁇ r is linked via a radical of the formula (III) to the meaning already described , advantageously produced by the following process:
- reaction product from (b) is reacted with a corresponding phthalimide, for example N-ethoxycarbonylphthalimide, and
- indole derivatives as linkers have the advantage of fluorescence capability and are therefore the smallest for nanotechnology applications, where detection may be required
- indole-1-ribosides were found in N.N. Suvorov et al., Biol. Aktivn. Soedin., Akad. Nauk SSSR 1965, 60 and Tetrahedron 1967, 23, 4653 already described.
- indole-1-ribosides are prepared by forming an amine of the unprotected sugar component and an indoline, which is then converted into the indole-1-riboside by oxidation.
- B. indole-1-glucoside and -1-arabinoside (YV Dobriynin et al, Khim.-Farm. Zh. 1978, 12, 33), whose 3 -substituted derivatives were mostly prepared via the Dahlsmeier reaction.
- this route of introducing aminoethyl units in the 3-position of the indole is too complex for industrial use.
- this method can be used not only for ribopyranoses, but also for ribo relationsanoses and 2'-deoxyribo disorders or 2'-deoxyribopyranoses, which is particularly advantageous.
- Tryptamine in particular N-acyl derivatives of tryptamine, especially N-phthaloyltryptamine, is preferably used as the nucleosidation partner of the sugars.
- the 4'-protected, preferably the 3 ', 4'-protected pentopyranosyl nucleosides are phosphitylated in a further step or bound to a solid phase.
- the phosphitylation is carried out, for example, by phosphorous acid monoallyl ester diisopropyl amide chloride in the presence of a base, for.
- the product is a phosphoramidite and in the second case an H-phosphonate.
- the compounds obtained serve e.g. B. for the production of pentopyranosyl nucleic acids.
- Another object of the present invention is therefore a method for producing a pentopyranosyl nucleic acid, with the following steps: (a) in a first step, a protected pentopyranosyl nucleoside is bound to a solid phase, as already described above, and
- step (b) in a second step, the 3'-, 4'-protected pentopyranosyl nucleoside bound to a solid phase according to step (a) is extended by a phosphitylated 3'-, 4 '-protected pentopyranosyl nucleoside and then z.
- B. is oxidized by an aqueous iodine solution, and
- step (c) Repeat step (b) with the same or different phosphitylated 3'-, 4'-protected pentopyranosyl nucleosides until the desired pentopyranosyl nucleic acid is present.
- Acidic activators such as pyridinium hydrochloride are particularly suitable as coupling reagents when using phosphoramidites, especially benzimidazolium triflate, preferably after recrystallization in acetonitrile and after dissolving in acetonitrile, since in contrast to 5- (4-nitrophenyl) -lH-tetrazole as coupling reagent there is no blockage of the coupling reagent. Pipes and contamination of the product takes place.
- Arylsulfonyl chlorides, diphenyl chlorophosphate, pivaloyl chloride or adamantoyl chloride are particularly suitable as coupling reagents when using H-phosphonates. Furthermore, it is advantageous to protect against ring formation which would destroy the oligonucleotide by adding a salt, such as sodium chloride, for the hydrazinolysis of oligonucleotides, in particular p-NAs, preferably p-RNAs, preferably p-RNAs, especially uracil and thymine, which would destroy the oligonucleotide Allyloxy groups can preferably be split off by palladium [Pd (0)] complexes, for example before the hydrazinolysis
- pentofuranosyl nucleosides for example the naturally occurring adenosine, guanosine
- step (a) and / or step (b) can also be used in step (a) and / or step (b).
- Cytidine, thymidine and / or uracil. be installed, which leads, for example, to a mixed p-NA-DNA or p-NA-RNA
- an allyloxy linker of the formula can be used in a further step
- S L4 and S 7 independently of one another, identical or different, each have a protective group selected in particular from Fmoc and / or DMT,
- a particularly preferred allyloxy linker is (2- (S) -N-Fmoc-O 1 -DMT-O - allyloxyd ⁇ isopropylaminophosphinyl-6-amino-1,2-hexanediol)
- amino-terminal linkers can be built up in a few reaction steps, which are both an activatable phosphorus compound and a acid-labile protecting group, such as DMT, and can therefore be easily used in automatable oligonucleotide synthesis (see, for example, PS Nelson et al., Nucleic Acid Res. 1989, 17, 7179, LJ Arnold et al., WO 8902439).
- Another object of the present invention is therefore also a pentopyranosyl nucleic acid.
- p-NA's and especially the p-RNA's form stable duplexes with each other and generally do not pair with the DNA's and RNA's occurring in their natural form. This property makes p-NA's the preferred pairing system.
- Such pairing systems are supramolecular systems of non-covalent interaction, which are characterized by selectivity, stability and reversibility, and whose properties are preferably influenced thermodynamically, ie by temperature, pH and concentration.
- Such pairing systems can also be used, for example, as a “molecular adhesive” due to their selective properties 'can be used for the merging of different metal clusters into cluster assemblies with potentially new properties [see, for example, BRL Letsinger, et al, Nature 1996, 382, 607-9, PG Schultz et al, Nature 1996, 382, 609-11]
- the p-NAs are also suitable for use in the field of nanotechnology, for example for the production of new materials, diagnostics and therapeutics as well as microelectronic, photonic or optoelectronic components and for the controlled merging of molecular species into supramolecular units, such as e.g.
- Another object of the invention is therefore the use of a pentopyranosyl nucleoside of the formula (I) or (II) according to the invention or a pentopyranosyl nucleic acid according to the invention for the production of a medicament, such as a therapeutic, a diagnostic and / or an electronic component
- a biomolecule e.g. DNA or RNA
- DNA or RA can be used for powerful covalent linking (linking) with another biomolecule.
- DNA or RA are used if both biomolecules contain sections which can bind to one another due to complementary sequences of nucleobases by forming hydrogen bonds.
- Such biomolecules are used, for example, in analytical systems for signal amplification, where a DNA molecule to be analyzed in its sequence via such a non-covalent DNA linker on the one hand immobilized on a solid support and on the other hand to be bound to a signal-enhancing branchedDNA molecule (bDNA) (see, for example, BS Urdea, Bio / Technol 1994, 12, 926 or US patent No.
- bDNA signal-enhancing branchedDNA molecule
- a major disadvantage of the systems described last is that they are so far inferior to the methods for nucleic acid diagnosis by polymerase chain reaction (PCR) (K Muliis. Methods Enzymol 1987, 155, 335) in terms of sensitivity among others attributable to the fact that the non-covalent binding of the solid support to the DNA molecule to be analyzed as well as the non-covalent binding of the DNA molecule to be analyzed is not always specific, which leads to a mixing of the functions "sequence recognition" and "not -covalent binding "comes
- p-NA's as an orthogonal pairing system, which does not interfere with the DNA or RNA pairing process, solves this problem in an advantageous manner, whereby the sensitivity of the analytical methods described can be increased significantly.
- the present invention therefore furthermore relates to the use of a pentopyranosyl nucleoside of the formula (I) or (II) according to the invention or a pentopyranosyl nucleic acid according to the invention for the preparation of a conjugate containing a pentopyranosyl nucleoside of the formula (I) or (II) according to the invention or a pentopyranosyl nucleic acid according to the invention and a biomolecule
- conjugates are covalently bound hybrids composed of p-NAs and other biomolecules, preferably a peptide, protein or a nucleic acid, for example an antibody or a functional part thereof or a DNA and / or RNA functional part of parts occurring in their natural form
- Antibodies are, for example, Fv fragments (Skerra & Pluckthun (1988) Science 240, 1038), single-chain Fv fragments (scFv, Bird et al (1988), Science 242, 423, Huston et al (1988) Proc Natl Acad Sei USA, 85, 5879) or Fab fragments (Better et al (1988) Science 240, 1041)
- biomolecule is understood to mean a naturally occurring substance or a substance derived from a naturally occurring substance
- these are p-RNA / DNA or p-RNA / RNA conjugates
- Conjugates are preferably used when the functions “sequence recognition and non-covalent binding must be implemented in one molecule, since the conjugates according to the invention contain two mutually orthogonal pairing systems
- a DNA oligonucleotide is further synthesized.
- This process can also be carried out in reverse order In a convergent process, e.g. B. p-RNA oligomers with amino terminal linkers and z. B. DNA oligomers with z. B. synthesized thiol linkers in separate processes. This is preferably followed by iodoacetylation of the p-RNA oligomer and the coupling of the two units according to protocols known from the literature (T. Zhu et al, Bioco ⁇ jug. Chem. 1994, 5, 312).
- conjugate in the sense of the present invention also means so-called arrays.
- arrays are arrays of immobilized recognition species that play an important role in the simultaneous determination of analytes, especially in analysis and diagnostics. Examples are peptide arrays (Fodor et al., Nature 1993, 364, 555) and nucleic acid arrays (Southern et al. Genomics 1992, 13, 1008; Heller, U.S. Patent No. 5,632,957).
- a higher flexibility of these arrays can be achieved by binding the recognition species to coding oligonucleotides and the associated, complementary strands at certain positions on a solid support.
- the recognition species are non-covalently bound at the desired positions.
- different types of recognition species such as DNA segments, antibodies, can only be
- hybridization conditions can be arranged simultaneously on a solid support (see FIG. 3), but this requires extremely strong, selective - in order to keep the coding sections as short as possible - and codons and anticodons which do not interfere with natural nucleic acid p-RNAs are particularly suitable for this in a particularly advantageous manner.
- the term “carrier” means material, in particular chip material, which is in solid or gel-like form.
- suitable carrier materials are ceramic, metal, in particular noble metal, glasses, plastics, crystalline materials or thin layers of Carrier, in particular the materials mentioned or thin layers of the carrier, in particular the materials mentioned, or (bio) molecular filaments such as cellulose, framework proteins.
- the present invention therefore also relates to the use of the pentopyranosyl nucleic acids according to the invention, preferably ribopyranosyl nucleic acids for coding recognition species, preferably natural DNA or RNA strands or proteins, in particular antibodies or functional parts of antibodies. These can then be shown in FIG.
- Codons can be hybridized on a fixed carrier
- a fixed carrier which is equipped with codons in the form of an array
- Analyte for example a biological sample such as serum or the like
- the species to be detected are bound in a certain pattern on the array, which is then indirect (for example by fluorescent labeling of the recognition species) or directly (for example by impedance measurement) ng at the point of attachment of the codons)
- the hybridization is canceled by a suitable condition (temperature, salts, solvents, electrophoretic processes) so that again only the carrier remains with the codons.
- Another object of the present invention therefore relates in particular to a diagnostic agent containing a pentopyranosyl nucleoside described above or a conjugate according to the invention, as already described in more detail above
- FIGURES 1 shows a section of the structure of RNA in its naturally occurring form (left) and in the form of a p-NA (right).
- FIG. 3 schematically shows an arrangement of immobilized recognition structures (arrays) on a solid support.
- the reaction mixture was mixed with 2.46 g (20.5 mmol, 0.1 eq) 4-dimethylaminopy ⁇ din (DMAP), cooled to -6 ° C and between -6 and -1 ° C within 15 min 27.9 ml (0.24 mol, 1.2 eq) benzoylchloride (BzCl) in 30 ml Py ⁇ dm was added dropwise and the mixture was stirred for 10 min. To complete the reaction, 2.8 ml (24 mmol, 0.12 eq) BzCl were then added at intervals of 25 mm added with cooling and finally stirred for 20 min
- Phase was dried with sodium sulfate, filtered and concentrated (220 g residue) It was first filtered through silica gel 60 (20 ⁇ 10 cm) with step gradient heptane / ethyl acetate, 1 1 to 0 1) for pre-cleaning, then it was chromatographed on silica gel 60 (30 ⁇ 10 cm; step gradient dichloromethane / ethyl acetate, 1 0 to 1.1)
- N ⁇ 4-benzoyl-1- ( ⁇ -D-ribopyranosyl) cytosine 1 were dried in 830 ml of dimethylformamide (DMF) and 1.5 l of pyridine (both solution m. And dried over molecular sieve 3 ⁇ ) dissolved while warming to 124 ° C.
- the G-triol A (393 mg, 1.0 mmol) was dissolved in 4 ml of dry dichloromethane. Trimethylorthoester (0.52 ml, 3.0 mmol) and camphorsulfonic acid were added
- the diol B (101 mg, 0 2 mmol) was suspended in 3 2 ml of dry dichloromethane. 171 ⁇ l (1 0 mmol) of N-ethyldiisopropylamine, 320 ⁇ l (3 96 mmol) of pyridine and 102 mg (0 3 mmol) of DMTCl were added and stirred at room temperature After 24 h, a further 102 mg (0.3 mmol) of DMTCl were added and the mixture was stirred again for 24 h. Then, the mixture was diluted with 30 ml of dichloromethane.
- Hydroxyethyluracil 28 can be prepared on a large scale by known methods (JD Fissekis, A. Myles, GB Brown, J. Org. Chem. 1964, 29, 2670).
- G-Butyrolactone 25 was formylated with methyl formate, the sodium salt 26 to the urea derivative 27 implemented and this cyclized to hydroxyethyl uracil 28 (Scheme 4).
- Hydroxyethyluracil 28 was mesylated with methanesulfonic acid chloride in pyridine to 29 (J.D. Fissekis, F. Sweet, J. Org. Chem. 1973, 38, 264).
- reaction mixture was then heated to 50.degree. After stirring for 2 5 h at 50 ° C. (DC control), the mixture was cooled to RT, thoroughly stirred on an ice-cold mixture of 250 ml of AcOEt and 190 ml of saturated NaHCO 3 solution and for 10 min. The mixture was washed again with 100 ml of NaHCO solution and extracted the aqueous phases again with 100 ml AcOEt Die ver org. Phases were dried with MgSO 4 and the solvents i V i RV removed.
- Indole-based linker N-phthaloyltryptamine is obtained from phthalic anhydride and tryptamm as described (Kuehne et al J Org. Chem 43, 13, 1978, 2733-2735). This is reduced to indoline with borane-THF (analogous to A Giannis, et al, Angew Chem 1989, 101, 220)
- the 3-substituted indoline is first reacted with ribose to form the nucleoside triol and then with acetic anhydride to form the triacetate.
- Oxidized with 2,3-dichloro-5,6-dicyanoparachinone and the acetates are cleaved with sodium methoxide, selectively benzoylated in the 2'-position, DM-t ⁇ ylated selectively in 4'-position, and carries out the migration reaction to the 3'-benzoate.
- the formation of the phosphoramidite takes place as usual. This can be used for automated oligonucleotide synthesis without changing the synthesis protocols
- 6-Amino-2 (S) -hydroxyhexanoic acid (1) was prepared in a manner known from the literature by diazotization and subsequent hydrolysis from L-lysine (K -I Aketa. Chem Pharm Bull 1976, 24, 621)
- indollinker phosphoramidite and 244 mg A phosphoramidite are weighed into Svnthesizer glasses and left for 3 h in a desiccator over KOH together with the pill filled with 28.1 mg CPG carrier, loaded with A building block, at the HV.
- the phosphoramidites are in Dissolve 1 ml (Indollinker) or 2.5 ml (A-phosphoramidite) acetonitrile and add a few beads from the molecular sieve and leave them closed in the desiccator over KOH. 200 mg of iodine are dissolved in 50 ml of acetonitrile with vigorous stirring.
- the carrier is slurried with aqueous 0.1 molar sodium diethyldithiocarbamate solution and 45 min at RT. leave. You suck off, wash with water, acetone,
- Triethylammonium hydrogen carbonate buffer (TEAB buffer) diluted to 7 ml. Over a
- Triethylammonium acetate buffer Triethylammonium acetate buffer.
- the oligonucleotide provides standard desalination (Waters Sep-Pak Cartridge). Yield: 17.6 OD.
- a p-RNA oligomer of sequence A 8 is produced on the Eppendorf Ecosyn D 300+ and then the following reagents are exchanged: 6% dichloroacetic acid against 2% trichloroacetic acid, Iodine in collidine against iodine in pyridine, benzimidazolium triflate solution against tetrazole solution.
- a DNA oligomer of the sequence GATTC is further synthesized according to known methods (MJ Gait, Oligonucleotide Synthesis, IRL Press, Oxford, UK 1984). The deallylation, hydrazinolysis, HPL chromatography and desalting are carried out as described for the p-RNA oligomer (see above) and provide the desired conjugate.
- a p-RNA oligomer with the sequence 4'-indollinker-Ag-2 ' is prepared, purified, and iodoacetylated.
- a DNA oligomer of the sequence GATTC thiol linker is synthesized and purified by known methods (MJ Gait, Oligonucleotide Synthesis, ERL Press, Oxford, UK 1984) (3'-thiol linker from Glen Research: No. 20-2933) .
- the two fragments T. Zhu et al., Bioconjug. Chem. 1994, 5, 312
- the conjugate is formed, which is then purified by HPLC.
- Zuert was analogous to that described in Example 6, a p-RNA oligomer of the sequence TAGGCAAT, which at the 4 'end using the 5'-amino modifier 5 from Eurogentec (2- (2- (4-monomethoxytrityl) aminoethoxy) ethyl- (2 -cyanoethyl) - (N, N-diisopropyl) phosphorus amidite) is provided with an amino group, synthesized and worked up.
- the oligonucleotide (17.4 OD, 0.15 ⁇ mol) was taken up in 0.5 ml of basic buffer, 1.14 mg (2.5 ⁇ mol) of biotin-N-hydroxysuccinimide ester was dissolved in 1 14 ⁇ l of DMF (abs.) And lh left at RT.
- the resulting conjugate was purified by preparative HPLC and the pure product was desalted with a Sepak.
- Example 9 Production of cyanine or biotin-labeled p-RNA online
- a synthesis cycle consists of the following steps
- the last DMT (dimethoxytrityl) or MMT (monomethoxytrityl) protective group was not split off from biotin or cyanine monomers.
- the last coupling with the modified phosphoramidites is detected after synthesis with 1% of the resin by means of trityl cation absorption in UV (503 nm).
- allyl ether protecting groups were cleaved off with a solution of tetrakis (triphenylphosphine) palladium (272mg), triphenylphosphine (272 mg) and
- the “Trityl ON” oligonucleotide was activated using an (acetonitrile, 20 ml) water Sep-Pak Cartridge freed of the hydrazine
- the hydrazine was washed with TEAB 0.1M (30ml)
- the oligonucleotide was then eluted with acetonitrile / TEAB 0.1M (10ml)
- the mixture was purified by HPLC (to separate termination sequences) and the DMT- Deprotection (30 ml of 80% aqueous formic acid) by final desalination (via Sep-Pak Kartuche, with TEAB buffer 0, 1 M / acetonitrile 1/1) provided the pure cyanine or biotin-labeled oligomers.
- the oligos were freeze-dried for storage
- the p-RNA was dissolved in a 0.1 molar sodium bicarbonate solution (pH 8.4) (1 ml per 350 nmol) and a solution of N- (iodoacetyloxy) succinimide in DMSO (40 ⁇ l per mg) was darkened Approach with aluminum foil and leave it at room temperature for 30-90 minutes. The progress of the reaction was monitored by means of analytical HPLC
- Buffer A 0.1 molar t ⁇ ethylammonium acetate buffer in water
- Buffer B 0.1 molar t ⁇ ethylammonium acetate buffer in water
- a Waters Sep-Pak cartridge RP-18 (from 15 OD 2 g filling) was activated with 2 ⁇ 10 ml acetonitrile and 2 ⁇ 10 ml water, the oligo was applied , let it sink in, washed the reaction vessel with 2 x 10 ml water, washed with 3 x 10 ml water to remove salt and reagent, and eluted first with 5 x 1 ml 50 1 water acetonitrile and then with 1 1 the product eluted in the 1 1 fractions in very good purity. The fractions were concentrated in the cold and in the dark, combined, and concentrated again
- the yields were determined by means of UV absorption spectrometry at 260 nm. They reached 70-95% of the Theo ⁇ e.
- Buffer system Borax / HCl buffer from Riedel-de Haen, pH 8.0 was mixed in a ratio of 1 l with a 10 millimolar solution of EDTA-Dinat ⁇ umsalz in water and adjusted to pH 6.6 with HC1. which contains 5 mM Na 2 EDTA
- the standard conditions of analytical HPLC are: Buffer A 0.1 molar triethylammonium acetate buffer in water Buffer B 0.1 molar triethylammonium acetate buffer in water.
- the mixture was diluted to four times the volume with water.
- a Waters Sep-Pak cartridge RP-18 (from 15 OD 2g filling) was activated with 3 x 10 ml acetoniril and 3 x 10 ml water, the oligo was applied, let it sink in, the reaction vessel was washed with 2 x 10 ml water After, the cartridge was washed with 3 ⁇ 10 ml of water to remove salt and excess peptide, and eluted with 1: 1 water: acetonitrile until no more product eluted by UV spectroscopy. The frankings were concentrated in the cold and in the dark, combined and then concentrated again.
Abstract
Description





























Claims

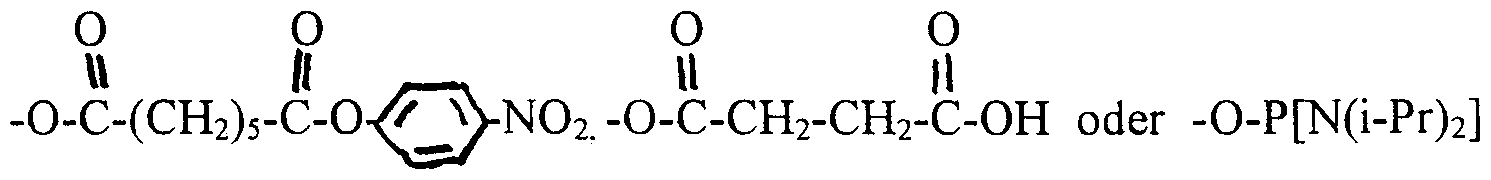



Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU10245/99A AU757983B2 (en) | 1997-09-22 | 1998-09-21 | Pentopyranosyl nucleoside, and production and use of the same |
| DE59811296T DE59811296D1 (de) | 1997-09-22 | 1998-09-21 | Pentopyranosyl-nucleosid, seine herstellung und verwendung |
| US09/509,058 US6608186B1 (en) | 1997-09-22 | 1998-09-21 | Pyranosyl nucleic acid conjugates |
| AT98952610T ATE265462T1 (de) | 1997-09-22 | 1998-09-21 | Pentopyranosyl-nucleosid, seine herstellung und verwendung |
| KR1020007003010A KR100571299B1 (ko) | 1997-09-22 | 1998-09-21 | 펜토피라노실 뉴클레오시드 및 이의 제조방법 |
| BR9812381-5A BR9812381A (pt) | 1997-09-22 | 1998-09-21 | Peptopiranosilnucleosìdeo, seu preparo e uso |
| CA002303229A CA2303229C (en) | 1997-09-22 | 1998-09-21 | Pentopyranosylnucleoside, its preparation and use |
| JP2000512844A JP4674964B2 (ja) | 1997-09-22 | 1998-09-21 | ペントピラノシルヌクレオシド、その製造および使用 |
| EP98952610A EP1017703B1 (de) | 1997-09-22 | 1998-09-21 | Pentopyranosyl-nucleosid, seine herstellung und verwendung |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE19741715.9 | 1997-09-22 | ||
| DE19741715A DE19741715A1 (de) | 1997-09-22 | 1997-09-22 | Pentopyranosyl-Nucleosid, seine Herstellung und Verwendung |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO1999015539A2 true WO1999015539A2 (de) | 1999-04-01 |
| WO1999015539A3 WO1999015539A3 (de) | 1999-06-17 |
Family
ID=7843183
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP1998/005998 WO1999015540A2 (de) | 1997-09-22 | 1998-09-21 | Verfahren zur herstellung eines pentopyranosyl-nucleosids |
| PCT/EP1998/005997 WO1999015539A2 (de) | 1997-09-22 | 1998-09-21 | Pentopyranosyl-nucleosid, seine herstellung und verwendung |
| PCT/EP1998/005999 WO1999015541A2 (de) | 1997-09-22 | 1998-09-21 | Verwendung eines pentopyranosyl-nucleosids zur herstellung eines elektronischen bauteils sowie konjugate davon |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP1998/005998 WO1999015540A2 (de) | 1997-09-22 | 1998-09-21 | Verfahren zur herstellung eines pentopyranosyl-nucleosids |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP1998/005999 WO1999015541A2 (de) | 1997-09-22 | 1998-09-21 | Verwendung eines pentopyranosyl-nucleosids zur herstellung eines elektronischen bauteils sowie konjugate davon |
Country Status (10)
| Country | Link |
|---|---|
| US (8) | US6613894B1 (de) |
| EP (3) | EP1019423B1 (de) |
| JP (3) | JP4674964B2 (de) |
| KR (3) | KR20010030656A (de) |
| AT (3) | ATE233272T1 (de) |
| AU (3) | AU1024699A (de) |
| BR (3) | BR9812381A (de) |
| CA (3) | CA2302414A1 (de) |
| DE (4) | DE19741715A1 (de) |
| WO (3) | WO1999015540A2 (de) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6608186B1 (en) * | 1997-09-22 | 2003-08-19 | Nanogen Recognomics Gmbh | Pyranosyl nucleic acid conjugates |
| US6893822B2 (en) | 2001-07-19 | 2005-05-17 | Nanogen Recognomics Gmbh | Enzymatic modification of a nucleic acid-synthetic binding unit conjugate |
| US7700761B2 (en) | 1998-08-18 | 2010-04-20 | Nanogen Recognomics Gmbh | 3-deoxypentopyranosyl nucleic acid, its production and its use |
Families Citing this family (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE19741739B4 (de) * | 1997-09-22 | 2006-04-27 | Nanogen Recognomics Gmbh | Supramolekulares Paarungssystem, dessen Herstellung und Verwendung |
| DE19741738A1 (de) * | 1997-09-22 | 1999-03-25 | Hoechst Ag | Linker-Nucleosid, seine Herstellung und Verwendung |
| DE19815901A1 (de) * | 1998-04-08 | 1999-10-14 | Aventis Res & Tech Gmbh & Co | Verfahren zur Herstellung von Pentopyranosyl-Nucleosiden |
| US20010049111A1 (en) * | 1999-08-13 | 2001-12-06 | Norbert Windhab | Methods, procedures, and formats for using microelectronic array devices to perform multiplex immunoassay analyses |
| DE10010118A1 (de) * | 2000-03-03 | 2001-09-20 | Henkel Kgaa | Beschichtungssystem auf der Basis von Pyranosyl-Nukleinsäuren |
| DE10111681A1 (de) * | 2001-03-09 | 2002-10-02 | Nanogen Recognomics Gmbh | Verbessertes Verfahren zur Herstellung von Pentopyranosyl-Nucleosiden |
| AU2003219576A1 (en) * | 2003-04-03 | 2004-10-25 | Korea Advanced Institute Of Science And Technology | Conjugate for gene transfer comprising oligonucleotide and hydrophilic polymer, polyelectrolyte complex micelles formed from the conjugate, and methods for preparation thereof |
| CA2463719A1 (en) * | 2003-04-05 | 2004-10-05 | F. Hoffmann-La Roche Ag | Nucleotide analogs with six membered rings |
| US20070286863A1 (en) * | 2006-05-17 | 2007-12-13 | Christopher Sinal | CMKLR regulation of adipogenesis and adipocyte metabolic function |
| JP5126706B2 (ja) * | 2007-02-16 | 2013-01-23 | 独立行政法人農業・食品産業技術総合研究機構 | 2以上の水酸基を有する化合物のライブラリー合成方法 |
| SG171914A1 (en) | 2008-12-02 | 2011-07-28 | Chiralgen Ltd | Method for the synthesis of phosphorus atom modified nucleic acids |
| KR101885383B1 (ko) | 2009-07-06 | 2018-08-03 | 웨이브 라이프 사이언시스 리미티드 | 신규한 핵산 프로드러그 및 그의 사용 방법 |
| EP2593892A1 (de) * | 2010-07-16 | 2013-05-22 | Elitech Holding B.V. | Orthogonale nukleinsäureaffinitätspaare |
| JP5868324B2 (ja) | 2010-09-24 | 2016-02-24 | 株式会社Wave Life Sciences Japan | 不斉補助基 |
| DK2734208T3 (en) | 2011-07-19 | 2017-06-19 | Wave Life Sciences Ltd | PROCEDURES FOR SYNTHESIS OF FUNCTIONALIZED NUCLEIC ACIDS |
| CN104684923B (zh) | 2012-07-13 | 2018-09-28 | 株式会社新日本科学 | 手性核酸佐剂 |
| KR101850319B1 (ko) | 2012-07-13 | 2018-04-20 | 웨이브 라이프 사이언시스 리미티드 | 비대칭 보조 그룹 |
| CN104661664B (zh) | 2012-07-13 | 2020-07-03 | 波涛生命科学有限公司 | 手性控制 |
| WO2014175974A2 (en) | 2013-03-13 | 2014-10-30 | Elitech Holding B.V. | Artificial nucleic acids |
| EP3095460A4 (de) | 2014-01-15 | 2017-08-23 | Shin Nippon Biomedical Laboratories, Ltd. | Chirales nukleinsäureadjuvans mit antiallergischer wirkung und antiallergikum |
| JPWO2015108048A1 (ja) | 2014-01-15 | 2017-03-23 | 株式会社新日本科学 | 抗腫瘍作用を有するキラル核酸アジュバンド及び抗腫瘍剤 |
| JPWO2015108047A1 (ja) | 2014-01-15 | 2017-03-23 | 株式会社新日本科学 | 免疫誘導活性を有するキラル核酸アジュバンド及び免疫誘導活性剤 |
| SG10201912897UA (en) | 2014-01-16 | 2020-02-27 | Wave Life Sciences Ltd | Chiral design |
| US10870845B2 (en) | 2014-07-01 | 2020-12-22 | Global Life Sciences Solutions Operations UK Ltd | Methods for capturing nucleic acids |
| US10472620B2 (en) | 2014-07-01 | 2019-11-12 | General Electric Company | Method, substrate and device for separating nucleic acids |
| US9593368B2 (en) | 2014-07-01 | 2017-03-14 | General Electric Company | Methods for amplifying nucleic acids on substrates |
| CN107556355B (zh) * | 2016-06-30 | 2021-10-22 | 上海兆维科技发展有限公司 | 一种核苷双亚磷酰胺及其制备方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998025943A1 (de) * | 1996-12-11 | 1998-06-18 | Aventis Research & Technologies Gmbh & Co Kg | Nicht-helikale supramolekulare nanosysteme |
Family Cites Families (113)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2644817A (en) * | 1949-10-28 | 1953-07-07 | Merck & Co Inc | 1-glycosido-5, 6 dimethyl benzimidazoles and process therefor |
| GB690119A (en) * | 1950-07-13 | 1953-04-15 | British Drug Houses Ltd | Improvements in or relating to the manufacture of heterocyclic compounds |
| US2719843A (en) * | 1951-10-24 | 1955-10-04 | Davoll John | Method of synthesizing nucleosides and analogous compounds and compounds prepared thereby |
| GB866815A (en) * | 1957-06-14 | 1961-05-03 | Pfizer & Co C | Piperazine salts |
| US2993039A (en) * | 1959-11-06 | 1961-07-18 | Upjohn Co | Preparing nu-glycosides of aldose and ketose sugars |
| US3658788A (en) * | 1969-06-06 | 1972-04-25 | Salk Inst For Biological Studi | Aminooxazolines and products thereof and processes for synthesizing same |
| US3752804A (en) | 1969-06-14 | 1973-08-14 | Takeda Chemical Industries Ltd | 2-allyloxyinosine-5'-phosphate and physiologically acceptable salts thereof |
| JPS5036087A (de) * | 1973-07-13 | 1975-04-04 | ||
| JPS5821005B2 (ja) * | 1974-08-26 | 1983-04-26 | 株式会社クボタ | 球状黒鉛鋳鉄製造における黒鉛球状化剤 |
| US3995190A (en) * | 1974-09-19 | 1976-11-30 | Butler, Binion, Rice, Cook & Knapp | Mobile ion film memory |
| US4283773A (en) * | 1977-08-30 | 1981-08-11 | Xerox Corporation | Programmable master controller communicating with plural controllers |
| US4225410A (en) * | 1978-12-04 | 1980-09-30 | Technicon Instruments Corporation | Integrated array of electrochemical sensors |
| JPS55153796A (en) * | 1979-05-21 | 1980-11-29 | Meiji Seika Kaisha Ltd | 5-fluorouracil nucleoside and its preparation |
| US4352795A (en) | 1981-01-29 | 1982-10-05 | Warner-Lambert Company | 7-β-D-Arabinofuranosyl-7H-pyrrolo[2,3-d]pyrimidine compounds and methods for their production |
| JPS57146796A (en) * | 1981-03-09 | 1982-09-10 | Haruo Ogura | Glycoside derivative |
| FI63596C (fi) * | 1981-10-16 | 1983-07-11 | Orion Yhtymae Oy | Mikrobdiagnostiskt foerfarande som grundar sig pao skiktshybridisering av nukleinsyror och vid foerfarandet anvaenda kombinationer av reagenser |
| US4476301A (en) * | 1982-04-29 | 1984-10-09 | Centre National De La Recherche Scientifique | Oligonucleotides, a process for preparing the same and their application as mediators of the action of interferon |
| CA1223831A (en) * | 1982-06-23 | 1987-07-07 | Dean Engelhardt | Modified nucleotides, methods of preparing and utilizing and compositions containing the same |
| US4580895A (en) * | 1983-10-28 | 1986-04-08 | Dynatech Laboratories, Incorporated | Sample-scanning photometer |
| EP0143623A3 (de) * | 1983-11-25 | 1987-09-23 | Mars Incorporated | Automatisches Prüfgerät |
| US4661451A (en) * | 1984-02-06 | 1987-04-28 | Ortho Diagnostic Systems, Inc. | Methods for immobilizing and translocating biological cells |
| FI71768C (fi) | 1984-02-17 | 1987-02-09 | Orion Yhtymae Oy | Foerbaettrade nykleinsyrareagenser och foerfarande foer deras framstaellning. |
| DE3434039A1 (de) * | 1984-09-17 | 1986-03-27 | Behringwerke Ag, 3550 Marburg | Glycopeptide, verfahren zu ihrer herstellung und ihre verwendung |
| US4828979A (en) * | 1984-11-08 | 1989-05-09 | Life Technologies, Inc. | Nucleotide analogs for nucleic acid labeling and detection |
| US4584075A (en) * | 1984-11-26 | 1986-04-22 | Ionics Incorporated | Process and apparatus for electrically desorbing components selectively sorbed on an electrolytically conducting barrier |
| GB8432118D0 (en) | 1984-12-19 | 1985-01-30 | Malcolm A D B | Sandwich hybridisation technique |
| US4594135A (en) * | 1985-02-20 | 1986-06-10 | Ionics Incorporated | Process and apparatus for electrically desorbing components selectively sorbed on granules |
| US5096807A (en) * | 1985-03-06 | 1992-03-17 | Murex Corporation | Imaging immunoassay detection system with background compensation and its use |
| JPH0337770Y2 (de) | 1985-03-30 | 1991-08-09 | ||
| DE3513168A1 (de) | 1985-04-12 | 1986-10-16 | Thomas 8000 München Dandekar | Biosensor bestehend aus einem halbleiter auf silizium oder kohlenstoffbasis (elektronischer teil) und nukleinbasen (od. anderen biol. monomeren) |
| US4751177A (en) * | 1985-06-13 | 1988-06-14 | Amgen | Methods and kits for performing nucleic acid hybridization assays |
| US4816418A (en) * | 1985-07-22 | 1989-03-28 | Sequoia-Turner Corporation | Method and apparatus for performing automated, multi-sequential immunoassays |
| EP0213825A3 (de) * | 1985-08-22 | 1989-04-26 | Molecular Devices Corporation | Chemisch-modulierte Mehrfachkapazitanz |
| US4822566A (en) * | 1985-11-19 | 1989-04-18 | The Johns Hopkins University | Optimized capacitive sensor for chemical analysis and measurement |
| EP0228075B1 (en) | 1986-01-03 | 1991-04-03 | Molecular Diagnostics, Inc. | Eucaryotic genomic dna dot-blot hybridization method |
| US5242797A (en) * | 1986-03-21 | 1993-09-07 | Myron J. Block | Nucleic acid assay method |
| US5125748A (en) * | 1986-03-26 | 1992-06-30 | Beckman Instruments, Inc. | Optical detection module for use in an automated laboratory work station |
| FR2604438B1 (fr) * | 1986-09-26 | 1988-12-23 | Centre Nat Rech Scient | Nouveaux conjugues de couplage entre des sequences d'arn ou d'adn et une proteine, leur procede de preparation et leur application biologique |
| US4918056A (en) * | 1986-10-14 | 1990-04-17 | Health Research, Inc. (Roswell Park Division) | 2-substituted arabinopyranosyl nucleosides and nucleotides |
| ATE111083T1 (de) * | 1986-10-22 | 1994-09-15 | Abbott Lab | Chemilumineszierende acridinium- und phenantridiniumsalze. |
| US4859538A (en) * | 1986-11-20 | 1989-08-22 | Ribi Hans O | Novel lipid-protein compositions and articles and methods for their preparation |
| US4885211A (en) * | 1987-02-11 | 1989-12-05 | Eastman Kodak Company | Electroluminescent device with improved cathode |
| YU57087A (en) | 1987-04-01 | 1990-08-31 | Centar Za Genticko Inzenjerstv | Process for obtaining genome by hebridization and oligonucleotidic tests |
| US5202231A (en) * | 1987-04-01 | 1993-04-13 | Drmanac Radoje T | Method of sequencing of genomes by hybridization of oligonucleotide probes |
| US5114674A (en) | 1987-05-01 | 1992-05-19 | Biotronic Systems Corporation | Added array of molecular chains for interfering with electrical fields |
| US4787963A (en) * | 1987-05-04 | 1988-11-29 | Syntro Corporation | Method and means for annealing complementary nucleic acid molecules at an accelerated rate |
| US5074977A (en) * | 1987-05-05 | 1991-12-24 | The Washington Technology Center | Digital biosensors and method of using same |
| JP2682859B2 (ja) | 1987-07-27 | 1997-11-26 | コモンウェルス サイエンティフィック アンド インダストリアル リサーチ オーガニゼーション | レセプター膜 |
| JP2603480B2 (ja) * | 1987-08-05 | 1997-04-23 | 住友製薬株式会社 | 安定化されたアンスラサイクリン系製剤 |
| KR970005898B1 (ko) | 1987-09-21 | 1997-04-21 | 젠- 프로우브 인코퍼레이티드 | 뉴클레오티드 프로브에 대한 비-뉴클레오티드 연결시약 |
| US5359100A (en) | 1987-10-15 | 1994-10-25 | Chiron Corporation | Bifunctional blocked phosphoramidites useful in making nucleic acid mutimers |
| GB8810400D0 (en) | 1988-05-03 | 1988-06-08 | Southern E | Analysing polynucleotide sequences |
| US4908112A (en) * | 1988-06-16 | 1990-03-13 | E. I. Du Pont De Nemours & Co. | Silicon semiconductor wafer for analyzing micronic biological samples |
| US5075077A (en) * | 1988-08-02 | 1991-12-24 | Abbott Laboratories | Test card for performing assays |
| US5188963A (en) * | 1989-11-17 | 1993-02-23 | Gene Tec Corporation | Device for processing biological specimens for analysis of nucleic acids |
| WO1990001564A1 (en) | 1988-08-09 | 1990-02-22 | Microprobe Corporation | Methods for multiple target analyses through nucleic acid hybridization |
| WO1990002327A1 (en) * | 1988-08-18 | 1990-03-08 | AUSTRALIAN MEMBRANE AND BIOTECHNOLOGY RESEARCH INSTITUTE LTD., Commonwealth Scientific and Industrial Research Organization | Improvements in sensitivity and selectivity of ion channel membrane biosensors |
| US5096669A (en) * | 1988-09-15 | 1992-03-17 | I-Stat Corporation | Disposable sensing device for real time fluid analysis |
| US5849482A (en) * | 1988-09-28 | 1998-12-15 | Epoch Pharmaceuticals, Inc. | Crosslinking oligonucleotides |
| US5063081A (en) * | 1988-11-14 | 1991-11-05 | I-Stat Corporation | Method of manufacturing a plurality of uniform microfabricated sensing devices having an immobilized ligand receptor |
| US5200051A (en) * | 1988-11-14 | 1993-04-06 | I-Stat Corporation | Wholly microfabricated biosensors and process for the manufacture and use thereof |
| US5391723A (en) * | 1989-05-31 | 1995-02-21 | Neorx Corporation | Oligonucleotide conjugates |
| US5219726A (en) * | 1989-06-02 | 1993-06-15 | The Salk Institute For Biological Studies | Physical mapping of complex genomes |
| US5143854A (en) * | 1989-06-07 | 1992-09-01 | Affymax Technologies N.V. | Large scale photolithographic solid phase synthesis of polypeptides and receptor binding screening thereof |
| DE3924454A1 (de) * | 1989-07-24 | 1991-02-07 | Cornelis P Prof Dr Hollenberg | Die anwendung von dna und dna-technologie fuer die konstruktion von netzwerken zur verwendung in der chip-konstruktion und chip-produktion (dna chips) |
| US5681941A (en) * | 1990-01-11 | 1997-10-28 | Isis Pharmaceuticals, Inc. | Substituted purines and oligonucleotide cross-linking |
| US5126022A (en) * | 1990-02-28 | 1992-06-30 | Soane Tecnologies, Inc. | Method and device for moving molecules by the application of a plurality of electrical fields |
| US5750015A (en) * | 1990-02-28 | 1998-05-12 | Soane Biosciences | Method and device for moving molecules by the application of a plurality of electrical fields |
| CA2079005A1 (en) * | 1990-04-11 | 1991-10-12 | Geoffrey Stephen Begg | Methods and apparatus allowing sequential chemical reactions |
| US5166063A (en) * | 1990-06-29 | 1992-11-24 | Eli Lilly And Company | Immobolization of biomolecules by enhanced electrophoretic precipitation |
| US5527670A (en) * | 1990-09-12 | 1996-06-18 | Scientific Generics Limited | Electrochemical denaturation of double-stranded nucleic acid |
| GB2247889A (en) | 1990-09-12 | 1992-03-18 | Scient Generics Ltd | DNA denaturation by an electric potential |
| WO1992004470A1 (en) | 1990-09-12 | 1992-03-19 | Scientific Generics Limited | Electrochemical denaturation of double-stranded nucleic acid |
| US5227265A (en) * | 1990-11-30 | 1993-07-13 | Eastman Kodak Company | Migration imaging system |
| US5679757A (en) | 1990-12-12 | 1997-10-21 | The Regents Of The University Of California | Highly organic solvent soluble, water insoluble electroluminescent polyphenylene vinylenes having pendant steroid groups and products and uses thereof |
| WO1992019960A1 (en) * | 1991-05-09 | 1992-11-12 | Nanophore, Inc. | Methods for the electrophoretic separation of nucleic acids and other linear macromolecules in gel media with restrictive pore diameters |
| US5605662A (en) * | 1993-11-01 | 1997-02-25 | Nanogen, Inc. | Active programmable electronic devices for molecular biological analysis and diagnostics |
| US6048690A (en) | 1991-11-07 | 2000-04-11 | Nanogen, Inc. | Methods for electronic fluorescent perturbation for analysis and electronic perturbation catalysis for synthesis |
| US6017696A (en) | 1993-11-01 | 2000-01-25 | Nanogen, Inc. | Methods for electronic stringency control for molecular biological analysis and diagnostics |
| US5849486A (en) * | 1993-11-01 | 1998-12-15 | Nanogen, Inc. | Methods for hybridization analysis utilizing electrically controlled hybridization |
| US5632957A (en) | 1993-11-01 | 1997-05-27 | Nanogen | Molecular biological diagnostic systems including electrodes |
| US5846708A (en) * | 1991-11-19 | 1998-12-08 | Massachusetts Institiute Of Technology | Optical and electrical methods and apparatus for molecule detection |
| US5349203A (en) * | 1991-12-09 | 1994-09-20 | Mitsubishi Denki Kabushiki Kaisha | Organic electric-field switching device |
| JPH05236997A (ja) * | 1992-02-28 | 1993-09-17 | Hitachi Ltd | ポリヌクレオチド捕捉用チップ |
| US5573905A (en) | 1992-03-30 | 1996-11-12 | The Scripps Research Institute | Encoded combinatorial chemical libraries |
| GB9207086D0 (en) * | 1992-03-31 | 1992-05-13 | Sharp Kk | Improvements relating to information technology |
| US5304487A (en) * | 1992-05-01 | 1994-04-19 | Trustees Of The University Of Pennsylvania | Fluid handling in mesoscale analytical devices |
| ATE256143T1 (de) * | 1992-07-23 | 2003-12-15 | Isis Pharmaceuticals Inc | 2'-0-alkyl-nukleoside und-phosphoramidite, verfahren zu ihrer herstellung und ihre verwendungen |
| US5312527A (en) * | 1992-10-06 | 1994-05-17 | Concordia University | Voltammetric sequence-selective sensor for target polynucleotide sequences |
| US5433819A (en) * | 1993-05-26 | 1995-07-18 | Pressac, Inc. | Method of making circuit boards |
| CA2170264A1 (en) | 1993-09-10 | 1995-03-16 | Michael W. Konrad | Optical detection of position of oligonucleotides on large dna molecules |
| US5965452A (en) | 1996-07-09 | 1999-10-12 | Nanogen, Inc. | Multiplexed active biologic array |
| US5445525A (en) * | 1994-05-12 | 1995-08-29 | Intel Corporation | Interconnection scheme for integrated circuit card with auxiliary contacts |
| JPH10507763A (ja) * | 1994-10-24 | 1998-07-28 | ジェネンコア インターナショナル インコーポレーテッド | L−ピラノシルヌクレオシド |
| US5718915A (en) * | 1994-10-31 | 1998-02-17 | Burstein Laboratories, Inc. | Antiviral liposome having coupled target-binding moiety and hydrolytic enzyme |
| US5585069A (en) * | 1994-11-10 | 1996-12-17 | David Sarnoff Research Center, Inc. | Partitioned microelectronic and fluidic device array for clinical diagnostics and chemical synthesis |
| US5464517A (en) * | 1995-01-30 | 1995-11-07 | Bio-Rad Laboratories | Electrophoresis in low conductivity buffers |
| US5559222A (en) * | 1995-02-03 | 1996-09-24 | Eli Lilly And Company | Preparation of 1-(2'-deoxy-2',2'-difluoro-D-ribo-pentofuranosyl)-cytosine from 2-deoxy-2,2-difluoro-β-D-ribo-pentopyranose |
| US5602262A (en) * | 1995-02-03 | 1997-02-11 | Eli Lilly And Company | Process for the preparation of 2-deoxy-2,2-difluoro-β-D-ribo-pentopyranose |
| PT739898E (pt) * | 1995-03-13 | 2002-03-28 | Aventis Pharma Gmbh | Mono-esteres de acidos fosfonucleicos processo para a sua preparacao e sua utilizacao |
| WO1996039414A1 (en) * | 1995-06-01 | 1996-12-12 | Hybridon, Inc. | Novel base protecting groups for oligonucleotide synthesis |
| KR19990028344A (ko) * | 1995-06-23 | 1999-04-15 | 로버터 에이.커리 | 치료 화합물 |
| AU6514096A (en) | 1995-07-27 | 1997-02-26 | Carsten Behrens | A new achiral linker reagent for the incorporation of multiple amino groups into oligonucleotides |
| US5912340A (en) | 1995-10-04 | 1999-06-15 | Epoch Pharmaceuticals, Inc. | Selective binding complementary oligonucleotides |
| AU7286696A (en) * | 1995-10-13 | 1997-05-07 | F. Hoffmann-La Roche Ag | Antisense oligomers |
| JP2002515738A (ja) * | 1996-01-23 | 2002-05-28 | アフィメトリックス,インコーポレイティド | 核酸分析法 |
| GB9602028D0 (en) | 1996-02-01 | 1996-04-03 | Amersham Int Plc | Nucleoside analogues |
| US5660701A (en) * | 1996-02-29 | 1997-08-26 | Bio-Rad Laboratories, Inc. | Protein separations by capillary electrophoresis using amino acid-containing buffers |
| JPH10306907A (ja) * | 1997-05-06 | 1998-11-17 | Kobe Steel Ltd | 流動層熱分解方法及び熱分解炉並びに被燃焼物処理装置 |
| ID24054A (id) * | 1997-06-10 | 2000-07-06 | Glaxo Group Ltd Glaxo | Turunan turunan benzimidazol |
| DE19741715A1 (de) * | 1997-09-22 | 1999-03-25 | Hoechst Ag | Pentopyranosyl-Nucleosid, seine Herstellung und Verwendung |
| DE19741738A1 (de) * | 1997-09-22 | 1999-03-25 | Hoechst Ag | Linker-Nucleosid, seine Herstellung und Verwendung |
| CA2381750A1 (en) * | 1999-08-13 | 2001-02-22 | Nanogen, Inc. | Microelectronic molecular descriptor array devices, methods, procedures, and formats for combinatorial selection of intermolecular ligand binding structures and for drug screening |
-
1997
- 1997-09-22 DE DE19741715A patent/DE19741715A1/de not_active Withdrawn
-
1998
- 1998-09-21 JP JP2000512844A patent/JP4674964B2/ja not_active Expired - Fee Related
- 1998-09-21 DE DE59810856T patent/DE59810856D1/de not_active Expired - Lifetime
- 1998-09-21 CA CA002302414A patent/CA2302414A1/en not_active Abandoned
- 1998-09-21 KR KR1020007003006A patent/KR20010030656A/ko not_active Application Discontinuation
- 1998-09-21 AT AT98950062T patent/ATE233272T1/de active
- 1998-09-21 WO PCT/EP1998/005998 patent/WO1999015540A2/de not_active Application Discontinuation
- 1998-09-21 KR KR1020007003010A patent/KR100571299B1/ko not_active IP Right Cessation
- 1998-09-21 BR BR9812381-5A patent/BR9812381A/pt not_active IP Right Cessation
- 1998-09-21 CA CA002303229A patent/CA2303229C/en not_active Expired - Fee Related
- 1998-09-21 AT AT98952610T patent/ATE265462T1/de active
- 1998-09-21 WO PCT/EP1998/005997 patent/WO1999015539A2/de active IP Right Grant
- 1998-09-21 JP JP2000512846A patent/JP4674965B2/ja not_active Expired - Fee Related
- 1998-09-21 BR BR9812501-0A patent/BR9812501A/pt not_active IP Right Cessation
- 1998-09-21 DE DE59807330T patent/DE59807330D1/de not_active Expired - Lifetime
- 1998-09-21 DE DE59811296T patent/DE59811296D1/de not_active Expired - Lifetime
- 1998-09-21 AT AT98952611T patent/ATE260290T1/de active
- 1998-09-21 US US09/509,010 patent/US6613894B1/en not_active Expired - Lifetime
- 1998-09-21 AU AU10246/99A patent/AU1024699A/en not_active Abandoned
- 1998-09-21 BR BR9812376-9A patent/BR9812376A/pt not_active IP Right Cessation
- 1998-09-21 EP EP98950062A patent/EP1019423B1/de not_active Expired - Lifetime
- 1998-09-21 AU AU96271/98A patent/AU751058B2/en not_active Ceased
- 1998-09-21 JP JP2000512845A patent/JP4601817B2/ja not_active Expired - Fee Related
- 1998-09-21 KR KR1020007002982A patent/KR20010024202A/ko not_active Application Discontinuation
- 1998-09-21 AU AU10245/99A patent/AU757983B2/en not_active Ceased
- 1998-09-21 US US09/509,058 patent/US6608186B1/en not_active Expired - Lifetime
- 1998-09-21 US US09/509,039 patent/US6506896B1/en not_active Expired - Lifetime
- 1998-09-21 WO PCT/EP1998/005999 patent/WO1999015541A2/de active IP Right Grant
- 1998-09-21 EP EP98952610A patent/EP1017703B1/de not_active Expired - Lifetime
- 1998-09-21 EP EP98952611A patent/EP1017704B1/de not_active Expired - Lifetime
- 1998-09-21 CA CA002303784A patent/CA2303784A1/en not_active Abandoned
-
2002
- 2002-05-16 US US10/150,402 patent/US7153955B2/en not_active Expired - Fee Related
-
2003
- 2003-08-19 US US10/644,592 patent/US20040198966A1/en active Pending
- 2003-09-02 US US10/654,274 patent/US20050053945A1/en not_active Abandoned
-
2006
- 2006-08-03 US US11/499,543 patent/US7501506B2/en not_active Expired - Fee Related
-
2009
- 2009-02-20 US US12/389,789 patent/US7777024B2/en not_active Expired - Fee Related
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998025943A1 (de) * | 1996-12-11 | 1998-06-18 | Aventis Research & Technologies Gmbh & Co Kg | Nicht-helikale supramolekulare nanosysteme |
Non-Patent Citations (9)
| Title |
|---|
| B.DOBOSZEWSKI ET AL.: "Synthesis of 3'-Deoxy-3'-C-Hydroxymethyl-aldopentopyran osyl Nucleosides and their Incorporation into Oligonucleotides. Part II." TETRAHEDRON, Bd. 51, Nr. 45, 1995, Seiten 12319-12336, XP002096831 * |
| CHEMICAL ABSTRACTS, vol. 98, no. 13, 28. M{rz 1983 Columbus, Ohio, US; abstract no. 107682z, H.OGURA : "Glucoside Derivatives." Seite 636; Spalte 2; XP002097208 & JP 57 146796 A10. September 1982 * |
| I.SCHL\NVOGT ET AL.: "188. Pyranosyl-RNA ('p-RNA'): NMR and Molecular Dynamics Study of the DUplex Formed by Self-Pairing of Ribopyranosyl-(C-G-A-A-T-T-C-G)" HELVETICA CHIMICA ACTA., Bd. 79, Nr. 8, 1996, Seiten 2316-2345, XP002096829 BASEL CH * |
| J.A.KELLEY ET AL.: "Furanose-pyranose Isomerization of Reduced Pyrimidine and Cyclic Urea Ribosides." JOURNAL OF MEDICINAL CHEMISTRY., Bd. 29, Nr. 11, November 1986, Seiten 2351-2358, XP002097206 WASHINGTON US * |
| M.B\HRINGER ET AL.: "110. Warum Pentose- und Nicht Hexose-Nucleins{uren ? Oligonucleotide aus 2',3'-Dideoxy-B-D-glucopyranosyl-Bausteine n ('Homo-DNS'): Hersellung." HELVETICA CHIMICA ACTA., Bd. 75, Nr. 5, 13. August 1992, Seiten 1416-1477, XP002096856 BASEL CH * |
| M.BOLLI ET AL.: "131. Pyranosyl-RNA : Further Observations on Replication." HELVETICA CHIMICA ACTA., Bd. 80, Nr. 6, 22. September 1997, Seiten 1901-1951, XP002096832 BASEL CH * |
| M.MELGUIZO ET AL.: "A New One-Step Synthesis of 8-Aminopurines Nucleoside Analogs from 6-(glycosylamino)-5-nitrosopyrimidines." JOURNAL OF ORGANIC CHEMISTRY., Bd. 57, Nr. 2, 1992, Seiten 559-565, XP002097207 EASTON US * |
| S.PITSCH ET AL.: "122. Pyranosyl-RNA ('p-RNA'): Base-Pairing Selectivity and Potential to Replicate." HELVETICA CHIMICA ACTA., Bd. 78, Nr. 7, 1995, Seiten 1621-1635, XP002096830 BASEL CH in der Anmeldung erw{hnt * |
| S.PITSCH ET AL.: "147. Why Pentose- and Not Hexose-Nucleic Acids ? Pyranosyl-RNA ('p-RNA')" HELVETICA CHIMICA ACTA., Bd. 76, Nr. 6, 1993, Seiten 2161-2183, XP002094190 BASEL CH in der Anmeldung erw{hnt * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6608186B1 (en) * | 1997-09-22 | 2003-08-19 | Nanogen Recognomics Gmbh | Pyranosyl nucleic acid conjugates |
| US6613894B1 (en) * | 1997-09-22 | 2003-09-02 | Nanogen Recognomics Gmbh | Method for producing a pyranosyl nucleic acid conjugate |
| US7700761B2 (en) | 1998-08-18 | 2010-04-20 | Nanogen Recognomics Gmbh | 3-deoxypentopyranosyl nucleic acid, its production and its use |
| US6893822B2 (en) | 2001-07-19 | 2005-05-17 | Nanogen Recognomics Gmbh | Enzymatic modification of a nucleic acid-synthetic binding unit conjugate |
| EP2218726A1 (de) | 2001-07-19 | 2010-08-18 | Nanogen Recognomics GmbH | Sortier- und Immobilisierungssystem für Nukleinsäuren unter Verwendung synthetischer Bindungssysteme |
| EP2298929A1 (de) | 2001-07-19 | 2011-03-23 | Nanogen Recognomics GmbH | Sortierungs und Immobilisationssystem für Nukleinsäuren mittels synthetischer Bindungssysteme |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1017704B1 (de) | Verwendung eines konjugates enthaltend ein pentopyranosyl-nucleotid zur herstellung eines arrays | |
| EP1070079B1 (de) | Verfahren zur herstellung von pentopyranosyl-nucleosiden | |
| EP1034180B1 (de) | Linker-nucleosid, seine herstellung und verwendung | |
| EP1105403B1 (de) | 3'-desoxypentopyranosyl-nucleinsäure und ihre herstellung |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AU BR CA JP KR US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AU BR CA JP KR US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 2303229 Country of ref document: CA Ref country code: CA Ref document number: 2303229 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10245/99 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020007003010 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1998952610 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1998952610 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09509058 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020007003010 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 10245/99 Country of ref document: AU |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1998952610 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1020007003010 Country of ref document: KR |