WO2000064484A2 - Conjugate having a cleavable linkage for use in a liposome - Google Patents
Conjugate having a cleavable linkage for use in a liposome Download PDFInfo
- Publication number
- WO2000064484A2 WO2000064484A2 PCT/US2000/010922 US0010922W WO0064484A2 WO 2000064484 A2 WO2000064484 A2 WO 2000064484A2 US 0010922 W US0010922 W US 0010922W WO 0064484 A2 WO0064484 A2 WO 0064484A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- conjugate
- therapeutic drug
- mitomycin
- drug
- dithiobenzyl
- Prior art date
Links
- 239000002502 liposome Substances 0.000 title claims abstract description 161
- 239000003814 drug Substances 0.000 claims abstract description 109
- 150000002632 lipids Chemical class 0.000 claims abstract description 69
- 125000004396 dithiobenzyl group Chemical group 0.000 claims abstract description 36
- 230000002209 hydrophobic effect Effects 0.000 claims abstract description 23
- 238000001727 in vivo Methods 0.000 claims abstract description 15
- 230000004044 response Effects 0.000 claims abstract description 8
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 claims description 195
- 229960004857 mitomycin Drugs 0.000 claims description 137
- 229940079593 drug Drugs 0.000 claims description 100
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 claims description 76
- 239000000203 mixture Substances 0.000 claims description 59
- 229940126585 therapeutic drug Drugs 0.000 claims description 58
- 235000012000 cholesterol Nutrition 0.000 claims description 38
- 238000000034 method Methods 0.000 claims description 29
- LOUPRKONTZGTKE-LHHVKLHASA-N quinidine Chemical compound C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@H]2[C@@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-LHHVKLHASA-N 0.000 claims description 20
- 239000000232 Lipid Bilayer Substances 0.000 claims description 18
- 239000003640 drug residue Substances 0.000 claims description 18
- 229960002949 fluorouracil Drugs 0.000 claims description 18
- -1 thio-carbonate Chemical compound 0.000 claims description 17
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 16
- 229960004630 chlorambucil Drugs 0.000 claims description 15
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 claims description 14
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 claims description 14
- 150000002148 esters Chemical class 0.000 claims description 14
- 150000002430 hydrocarbons Chemical class 0.000 claims description 14
- 150000001412 amines Chemical class 0.000 claims description 13
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 claims description 13
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 claims description 12
- 125000004432 carbon atom Chemical group C* 0.000 claims description 12
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 claims description 11
- 150000001982 diacylglycerols Chemical class 0.000 claims description 11
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 11
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 claims description 10
- 229930003347 Atropine Natural products 0.000 claims description 10
- 108010006654 Bleomycin Proteins 0.000 claims description 10
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 claims description 10
- RKUNBYITZUJHSG-UHFFFAOYSA-N Hyosciamin-hydrochlorid Natural products CN1C(C2)CCC1CC2OC(=O)C(CO)C1=CC=CC=C1 RKUNBYITZUJHSG-UHFFFAOYSA-N 0.000 claims description 10
- XQFRJNBWHJMXHO-RRKCRQDMSA-N IDUR Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(I)=C1 XQFRJNBWHJMXHO-RRKCRQDMSA-N 0.000 claims description 10
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 claims description 10
- HYFMSAFINFJTFH-UHFFFAOYSA-N Mitomycin-A Natural products O=C1C(OC)=C(C)C(=O)C2=C1C(COC(N)=O)C1(OC)N2CC2NC21 HYFMSAFINFJTFH-UHFFFAOYSA-N 0.000 claims description 10
- OIRDTQYFTABQOQ-UHTZMRCNSA-N Vidarabine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1O OIRDTQYFTABQOQ-UHTZMRCNSA-N 0.000 claims description 10
- 229960004150 aciclovir Drugs 0.000 claims description 10
- RKUNBYITZUJHSG-SPUOUPEWSA-N atropine Chemical compound O([C@H]1C[C@H]2CC[C@@H](C1)N2C)C(=O)C(CO)C1=CC=CC=C1 RKUNBYITZUJHSG-SPUOUPEWSA-N 0.000 claims description 10
- 229960000396 atropine Drugs 0.000 claims description 10
- WZPBZJONDBGPKJ-VEHQQRBSSA-N aztreonam Chemical compound O=C1N(S([O-])(=O)=O)[C@@H](C)[C@@H]1NC(=O)C(=N/OC(C)(C)C(O)=O)\C1=CSC([NH3+])=N1 WZPBZJONDBGPKJ-VEHQQRBSSA-N 0.000 claims description 10
- 229960001561 bleomycin Drugs 0.000 claims description 10
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 claims description 10
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 10
- LOUPRKONTZGTKE-UHFFFAOYSA-N cinchonine Natural products C1C(C(C2)C=C)CCN2C1C(O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-UHFFFAOYSA-N 0.000 claims description 10
- 229960005420 etoposide Drugs 0.000 claims description 10
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 claims description 10
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 claims description 10
- 229960004716 idoxuridine Drugs 0.000 claims description 10
- 229960000485 methotrexate Drugs 0.000 claims description 10
- HYFMSAFINFJTFH-NGSRAFSJSA-N mitomycin A Chemical compound O=C1C(OC)=C(C)C(=O)C2=C1[C@@H](COC(N)=O)[C@]1(OC)N2C[C@@H]2N[C@@H]21 HYFMSAFINFJTFH-NGSRAFSJSA-N 0.000 claims description 10
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 claims description 10
- 229960001156 mitoxantrone Drugs 0.000 claims description 10
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 claims description 10
- 229960002340 pentostatin Drugs 0.000 claims description 10
- 150000003904 phospholipids Chemical class 0.000 claims description 10
- 229960001404 quinidine Drugs 0.000 claims description 10
- 229960003636 vidarabine Drugs 0.000 claims description 10
- 229960002555 zidovudine Drugs 0.000 claims description 10
- 229920001184 polypeptide Polymers 0.000 claims description 9
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 9
- 229930195733 hydrocarbon Natural products 0.000 claims description 8
- 150000001408 amides Chemical class 0.000 claims description 7
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 7
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 claims description 6
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 claims description 6
- 229960000975 daunorubicin Drugs 0.000 claims description 6
- 229960004679 doxorubicin Drugs 0.000 claims description 6
- 150000003335 secondary amines Chemical group 0.000 claims description 5
- YIBHDAKUJGXPLH-UNHHQYQHSA-N [(2s)-2-(adamantane-1-carbonyloxy)-3-hydroxypropyl] adamantane-1-carboxylate Chemical compound C1C(C2)CC(C3)CC2CC13C(=O)O[C@@H](CO)COC(=O)C1(C2)CC(C3)CC2CC3C1 YIBHDAKUJGXPLH-UNHHQYQHSA-N 0.000 claims description 2
- 238000012377 drug delivery Methods 0.000 claims description 2
- 239000003981 vehicle Substances 0.000 claims description 2
- MKUXAQIIEYXACX-UHFFFAOYSA-N aciclovir Chemical compound N1C(N)=NC(=O)C2=C1N(COCCO)C=N2 MKUXAQIIEYXACX-UHFFFAOYSA-N 0.000 claims 6
- 238000003776 cleavage reaction Methods 0.000 abstract description 15
- 230000007017 scission Effects 0.000 abstract description 15
- 239000003638 chemical reducing agent Substances 0.000 abstract description 13
- 229940124597 therapeutic agent Drugs 0.000 abstract description 9
- 238000007079 thiolysis reaction Methods 0.000 abstract 1
- 238000009472 formulation Methods 0.000 description 45
- 210000004027 cell Anatomy 0.000 description 44
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 43
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 41
- 235000018417 cysteine Nutrition 0.000 description 41
- 150000001875 compounds Chemical class 0.000 description 35
- 238000006243 chemical reaction Methods 0.000 description 34
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 30
- 239000000243 solution Substances 0.000 description 26
- 230000012010 growth Effects 0.000 description 25
- 238000002360 preparation method Methods 0.000 description 22
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 21
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 18
- 229920001223 polyethylene glycol Polymers 0.000 description 16
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 16
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 15
- 229920000642 polymer Polymers 0.000 description 14
- 239000000047 product Substances 0.000 description 13
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 12
- 238000004128 high performance liquid chromatography Methods 0.000 description 12
- FKLJPTJMIBLJAV-UHFFFAOYSA-N Compound IV Chemical compound O1N=C(C)C=C1CCCCCCCOC1=CC=C(C=2OCCN=2)C=C1 FKLJPTJMIBLJAV-UHFFFAOYSA-N 0.000 description 11
- 210000004369 blood Anatomy 0.000 description 11
- 239000008280 blood Substances 0.000 description 11
- 231100000135 cytotoxicity Toxicity 0.000 description 11
- 230000003013 cytotoxicity Effects 0.000 description 11
- 239000010432 diamond Substances 0.000 description 11
- 238000000338 in vitro Methods 0.000 description 10
- 230000017531 blood circulation Effects 0.000 description 9
- 238000011534 incubation Methods 0.000 description 9
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 8
- 230000015572 biosynthetic process Effects 0.000 description 8
- 238000000354 decomposition reaction Methods 0.000 description 8
- 239000000725 suspension Substances 0.000 description 8
- 241000700159 Rattus Species 0.000 description 7
- 230000000694 effects Effects 0.000 description 7
- 229940093499 ethyl acetate Drugs 0.000 description 7
- 235000019439 ethyl acetate Nutrition 0.000 description 7
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 7
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 7
- 238000003908 quality control method Methods 0.000 description 7
- 238000012360 testing method Methods 0.000 description 7
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 6
- 206010028980 Neoplasm Diseases 0.000 description 6
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical compound ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 description 6
- 239000011248 coating agent Substances 0.000 description 6
- 238000000576 coating method Methods 0.000 description 6
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 6
- 229920001477 hydrophilic polymer Polymers 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 238000010253 intravenous injection Methods 0.000 description 6
- 230000008569 process Effects 0.000 description 6
- 239000011780 sodium chloride Substances 0.000 description 6
- 239000007787 solid Substances 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- 239000002202 Polyethylene glycol Substances 0.000 description 5
- 238000003556 assay Methods 0.000 description 5
- 238000001125 extrusion Methods 0.000 description 5
- 238000010348 incorporation Methods 0.000 description 5
- 230000014759 maintenance of location Effects 0.000 description 5
- 239000002609 medium Substances 0.000 description 5
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 5
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 5
- LVNGJLRDBYCPGB-UHFFFAOYSA-N 1,2-distearoylphosphatidylethanolamine Chemical group CCCCCCCCCCCCCCCCCC(=O)OCC(COP([O-])(=O)OCC[NH3+])OC(=O)CCCCCCCCCCCCCCCCC LVNGJLRDBYCPGB-UHFFFAOYSA-N 0.000 description 4
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 4
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 4
- 229930182558 Sterol Natural products 0.000 description 4
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 4
- 238000013459 approach Methods 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 239000012528 membrane Substances 0.000 description 4
- RMLUKZWYIKEASN-UHFFFAOYSA-M sodium;2-amino-9-(2-hydroxyethoxymethyl)purin-6-olate Chemical compound [Na+].O=C1[N-]C(N)=NC2=C1N=CN2COCCO RMLUKZWYIKEASN-UHFFFAOYSA-M 0.000 description 4
- 150000003432 sterols Chemical class 0.000 description 4
- 235000003702 sterols Nutrition 0.000 description 4
- RBTBFTRPCNLSDE-UHFFFAOYSA-N 3,7-bis(dimethylamino)phenothiazin-5-ium Chemical compound C1=CC(N(C)C)=CC2=[S+]C3=CC(N(C)C)=CC=C3N=C21 RBTBFTRPCNLSDE-UHFFFAOYSA-N 0.000 description 3
- KEPSVHAYVNZDDW-UHFFFAOYSA-N 3-(2,3-dihydroxypropyldisulfanyl)propane-1,2-diol Chemical compound OCC(O)CSSCC(O)CO KEPSVHAYVNZDDW-UHFFFAOYSA-N 0.000 description 3
- 239000004254 Ammonium phosphate Substances 0.000 description 3
- 201000009030 Carcinoma Diseases 0.000 description 3
- 108010024636 Glutathione Proteins 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- DBTDEFJAFBUGPP-UHFFFAOYSA-N Methanethial Chemical compound S=C DBTDEFJAFBUGPP-UHFFFAOYSA-N 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- 235000021355 Stearic acid Nutrition 0.000 description 3
- 238000002835 absorbance Methods 0.000 description 3
- 229910000148 ammonium phosphate Inorganic materials 0.000 description 3
- 235000019289 ammonium phosphates Nutrition 0.000 description 3
- 125000003118 aryl group Chemical group 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 230000010261 cell growth Effects 0.000 description 3
- 230000004087 circulation Effects 0.000 description 3
- 231100000433 cytotoxic Toxicity 0.000 description 3
- 230000001472 cytotoxic effect Effects 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 239000008367 deionised water Substances 0.000 description 3
- 229910021641 deionized water Inorganic materials 0.000 description 3
- 238000011026 diafiltration Methods 0.000 description 3
- MNNHAPBLZZVQHP-UHFFFAOYSA-N diammonium hydrogen phosphate Chemical compound [NH4+].[NH4+].OP([O-])([O-])=O MNNHAPBLZZVQHP-UHFFFAOYSA-N 0.000 description 3
- 235000014113 dietary fatty acids Nutrition 0.000 description 3
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 description 3
- 239000000194 fatty acid Substances 0.000 description 3
- 229930195729 fatty acid Natural products 0.000 description 3
- 150000004665 fatty acids Chemical class 0.000 description 3
- 229960003180 glutathione Drugs 0.000 description 3
- 230000036571 hydration Effects 0.000 description 3
- 238000006703 hydration reaction Methods 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- 125000005647 linker group Chemical group 0.000 description 3
- 229920001427 mPEG Polymers 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 229960000907 methylthioninium chloride Drugs 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 3
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical group CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 3
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- 229940002612 prodrug Drugs 0.000 description 3
- 239000000651 prodrug Substances 0.000 description 3
- 230000008929 regeneration Effects 0.000 description 3
- 238000011069 regeneration method Methods 0.000 description 3
- 238000002390 rotary evaporation Methods 0.000 description 3
- 239000012265 solid product Substances 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 239000008117 stearic acid Substances 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- FMCAFXHLMUOIGG-IWFBPKFRSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2r)-2-formamido-3-sulfanylpropanoyl]amino]-3-methylbutanoyl]amino]-3-(4-hydroxy-2,5-dimethylphenyl)propanoyl]amino]-4-methylsulfanylbutanoic acid Chemical compound O=CN[C@@H](CS)C(=O)N[C@@H](C(C)C)C(=O)N[C@H](C(=O)N[C@@H](CCSC)C(O)=O)CC1=CC(C)=C(O)C=C1C FMCAFXHLMUOIGG-IWFBPKFRSA-N 0.000 description 2
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 2
- PORPENFLTBBHSG-MGBGTMOVSA-N 1,2-dihexadecanoyl-sn-glycerol-3-phosphate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(O)=O)OC(=O)CCCCCCCCCCCCCCC PORPENFLTBBHSG-MGBGTMOVSA-N 0.000 description 2
- FCSKOFQQCWLGMV-UHFFFAOYSA-N 5-{5-[2-chloro-4-(4,5-dihydro-1,3-oxazol-2-yl)phenoxy]pentyl}-3-methylisoxazole Chemical compound O1N=C(C)C=C1CCCCCOC1=CC=C(C=2OCCN=2)C=C1Cl FCSKOFQQCWLGMV-UHFFFAOYSA-N 0.000 description 2
- 108010017384 Blood Proteins Proteins 0.000 description 2
- 102000004506 Blood Proteins Human genes 0.000 description 2
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 2
- DRSHXJFUUPIBHX-UHFFFAOYSA-N COc1ccc(cc1)N1N=CC2C=NC(Nc3cc(OC)c(OC)c(OCCCN4CCN(C)CC4)c3)=NC12 Chemical compound COc1ccc(cc1)N1N=CC2C=NC(Nc3cc(OC)c(OC)c(OCCCN4CCN(C)CC4)c3)=NC12 DRSHXJFUUPIBHX-UHFFFAOYSA-N 0.000 description 2
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 2
- JZNWSCPGTDBMEW-UHFFFAOYSA-N Glycerophosphorylethanolamin Natural products NCCOP(O)(=O)OCC(O)CO JZNWSCPGTDBMEW-UHFFFAOYSA-N 0.000 description 2
- 229930192392 Mitomycin Natural products 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 150000001263 acyl chlorides Chemical class 0.000 description 2
- 235000001014 amino acid Nutrition 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 239000012062 aqueous buffer Substances 0.000 description 2
- 230000004888 barrier function Effects 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 229920001429 chelating resin Polymers 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- MWRBNPKJOOWZPW-CLFAGFIQSA-N dioleoyl phosphatidylethanolamine Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC(COP(O)(=O)OCCN)OC(=O)CCCCCCC\C=C/CCCCCCCC MWRBNPKJOOWZPW-CLFAGFIQSA-N 0.000 description 2
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 229920000578 graft copolymer Polymers 0.000 description 2
- 230000009036 growth inhibition Effects 0.000 description 2
- 229960002163 hydrogen peroxide Drugs 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- PGLTVOMIXTUURA-UHFFFAOYSA-N iodoacetamide Chemical compound NC(=O)CI PGLTVOMIXTUURA-UHFFFAOYSA-N 0.000 description 2
- 239000003456 ion exchange resin Substances 0.000 description 2
- 229920003303 ion-exchange polymer Polymers 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 210000002540 macrophage Anatomy 0.000 description 2
- AGJSNMGHAVDLRQ-IWFBPKFRSA-N methyl (2s)-2-[[(2s)-2-[[(2s)-2-[[(2r)-2-amino-3-sulfanylpropanoyl]amino]-3-methylbutanoyl]amino]-3-(4-hydroxy-2,3-dimethylphenyl)propanoyl]amino]-4-methylsulfanylbutanoate Chemical compound SC[C@H](N)C(=O)N[C@@H](C(C)C)C(=O)N[C@H](C(=O)N[C@@H](CCSC)C(=O)OC)CC1=CC=C(O)C(C)=C1C AGJSNMGHAVDLRQ-IWFBPKFRSA-N 0.000 description 2
- 210000000865 mononuclear phagocyte system Anatomy 0.000 description 2
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 150000008104 phosphatidylethanolamines Chemical class 0.000 description 2
- 150000003905 phosphatidylinositols Chemical class 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 235000018102 proteins Nutrition 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- 230000000717 retained effect Effects 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 239000012086 standard solution Substances 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 238000007910 systemic administration Methods 0.000 description 2
- 230000007704 transition Effects 0.000 description 2
- 150000003626 triacylglycerols Chemical class 0.000 description 2
- 210000004881 tumor cell Anatomy 0.000 description 2
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 1
- GNSFRPWPOGYVLO-UHFFFAOYSA-N 3-hydroxypropyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCCCO GNSFRPWPOGYVLO-UHFFFAOYSA-N 0.000 description 1
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical group C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- ACFIXJIJDZMPPO-NNYOXOHSSA-N NADPH Chemical compound C1=CCC(C(=O)N)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@@H](OP(O)(O)=O)[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)O1 ACFIXJIJDZMPPO-NNYOXOHSSA-N 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- 108010038807 Oligopeptides Proteins 0.000 description 1
- 102000015636 Oligopeptides Human genes 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 1
- 239000004695 Polyether sulfone Substances 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 241000906446 Theraps Species 0.000 description 1
- QNEPTKZEXBPDLF-JDTILAPWSA-N [(3s,8s,9s,10r,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1h-cyclopenta[a]phenanthren-3-yl] carbonochloridate Chemical compound C1C=C2C[C@@H](OC(Cl)=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 QNEPTKZEXBPDLF-JDTILAPWSA-N 0.000 description 1
- FBPDURWKLAOHMN-UHFFFAOYSA-N [O-][NH+](C=C(C(N1)=O)F)C1=O Chemical compound [O-][NH+](C=C(C(N1)=O)F)C1=O FBPDURWKLAOHMN-UHFFFAOYSA-N 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 230000021736 acetylation Effects 0.000 description 1
- 238000006640 acetylation reaction Methods 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- 229930013930 alkaloid Natural products 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 229940126575 aminoglycoside Drugs 0.000 description 1
- 238000004873 anchoring Methods 0.000 description 1
- 229920006187 aquazol Polymers 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- PUJDIJCNWFYVJX-UHFFFAOYSA-N benzyl carbamate Chemical compound NC(=O)OCC1=CC=CC=C1 PUJDIJCNWFYVJX-UHFFFAOYSA-N 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 239000003012 bilayer membrane Substances 0.000 description 1
- 229920000249 biocompatible polymer Polymers 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 229920001400 block copolymer Polymers 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical group 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 238000012602 chemosensitivity assay Methods 0.000 description 1
- QOPVNWQGBQYBBP-UHFFFAOYSA-N chloroethyl chloroformate Chemical compound CC(Cl)OC(Cl)=O QOPVNWQGBQYBBP-UHFFFAOYSA-N 0.000 description 1
- 125000002603 chloroethyl group Chemical group [H]C([*])([H])C([H])([H])Cl 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 230000000875 corresponding effect Effects 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000001212 derivatisation Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 1
- 150000002009 diols Chemical group 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 238000002296 dynamic light scattering Methods 0.000 description 1
- 238000000921 elemental analysis Methods 0.000 description 1
- NLFBCYMMUAKCPC-KQQUZDAGSA-N ethyl (e)-3-[3-amino-2-cyano-1-[(e)-3-ethoxy-3-oxoprop-1-enyl]sulfanyl-3-oxoprop-1-enyl]sulfanylprop-2-enoate Chemical compound CCOC(=O)\C=C\SC(=C(C#N)C(N)=O)S\C=C\C(=O)OCC NLFBCYMMUAKCPC-KQQUZDAGSA-N 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 239000012091 fetal bovine serum Substances 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- 239000012510 hollow fiber Substances 0.000 description 1
- 229920001519 homopolymer Polymers 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical compound O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 description 1
- 229920003063 hydroxymethyl cellulose Polymers 0.000 description 1
- 229940031574 hydroxymethyl cellulose Drugs 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 229910052816 inorganic phosphate Inorganic materials 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 201000005296 lung carcinoma Diseases 0.000 description 1
- 238000001906 matrix-assisted laser desorption--ionisation mass spectrometry Methods 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- AGJSNMGHAVDLRQ-HUUJSLGLSA-N methyl (2s)-2-[[(2r)-2-[[(2s)-2-[[(2r)-2-amino-3-sulfanylpropanoyl]amino]-3-methylbutanoyl]amino]-3-(4-hydroxy-2,3-dimethylphenyl)propanoyl]amino]-4-methylsulfanylbutanoate Chemical compound SC[C@H](N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(=O)N[C@@H](CCSC)C(=O)OC)CC1=CC=C(O)C(C)=C1C AGJSNMGHAVDLRQ-HUUJSLGLSA-N 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- BOPGDPNILDQYTO-NNYOXOHSSA-N nicotinamide-adenine dinucleotide Chemical compound C1=CCC(C(=O)N)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@@H](O)[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)O1 BOPGDPNILDQYTO-NNYOXOHSSA-N 0.000 description 1
- 231100000065 noncytotoxic Toxicity 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- UQDHUTAFUQLDFX-UHFFFAOYSA-N phenyl(sulfanyl)methanol Chemical compound OC(S)C1=CC=CC=C1 UQDHUTAFUQLDFX-UHFFFAOYSA-N 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 238000007747 plating Methods 0.000 description 1
- 229920002492 poly(sulfone) Polymers 0.000 description 1
- 229920002432 poly(vinyl methyl ether) polymer Polymers 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 229920006393 polyether sulfone Polymers 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 239000012264 purified product Substances 0.000 description 1
- ZDYVRSLAEXCVBX-UHFFFAOYSA-N pyridinium p-toluenesulfonate Chemical compound C1=CC=[NH+]C=C1.CC1=CC=C(S([O-])(=O)=O)C=C1 ZDYVRSLAEXCVBX-UHFFFAOYSA-N 0.000 description 1
- 229920005604 random copolymer Polymers 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000006748 scratching Methods 0.000 description 1
- 230000002393 scratching effect Effects 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 238000002943 spectrophotometric absorbance Methods 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 238000007447 staining method Methods 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 238000011146 sterile filtration Methods 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 210000005166 vasculature Anatomy 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/127—Liposomes
- A61K9/1271—Non-conventional liposomes, e.g. PEGylated liposomes, liposomes coated with polymers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/195—Carboxylic acids, e.g. valproic acid having an amino group
- A61K31/196—Carboxylic acids, e.g. valproic acid having an amino group the amino group being directly attached to a ring, e.g. anthranilic acid, mefenamic acid, diclofenac, chlorambucil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/407—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with other heterocyclic ring systems, e.g. ketorolac, physostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/14—Peptides containing saccharide radicals; Derivatives thereof, e.g. bleomycin, phleomycin, muramylpeptides or vancomycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/542—Carboxylic acids, e.g. a fatty acid or an amino acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/543—Lipids, e.g. triglycerides; Polyamines, e.g. spermine or spermidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/543—Lipids, e.g. triglycerides; Polyamines, e.g. spermine or spermidine
- A61K47/544—Phospholipids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/554—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound the modifying agent being a steroid plant sterol, glycyrrhetic acid, enoxolone or bile acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
- A61K47/60—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes the organic macromolecular compound being a polyoxyalkylene oligomer, polymer or dendrimer, e.g. PEG, PPG, PEO or polyglycerol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/69—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit
- A61K47/6905—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a colloid or an emulsion
- A61K47/6911—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a colloid or an emulsion the form being a liposome
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/10—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C323/18—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and singly-bound oxygen atoms bound to the same carbon skeleton having the sulfur atom of at least one of the thio groups bound to a carbon atom of a six-membered aromatic ring of the carbon skeleton
- C07C323/19—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and singly-bound oxygen atoms bound to the same carbon skeleton having the sulfur atom of at least one of the thio groups bound to a carbon atom of a six-membered aromatic ring of the carbon skeleton with singly-bound oxygen atoms bound to acyclic carbon atoms of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/52—Two oxygen atoms
- C07D239/54—Two oxygen atoms as doubly bound oxygen atoms or as unsubstituted hydroxy radicals
- C07D239/545—Two oxygen atoms as doubly bound oxygen atoms or as unsubstituted hydroxy radicals with other hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/553—Two oxygen atoms as doubly bound oxygen atoms or as unsubstituted hydroxy radicals with other hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms with halogen atoms or nitro radicals directly attached to ring carbon atoms, e.g. fluorouracil
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains three hetero rings
- C07D487/14—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/04—Heterocyclic radicals containing only oxygen as ring hetero atoms
- C07H17/08—Hetero rings containing eight or more ring members, e.g. erythromycins
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/12—Triazine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/29—Coated or structually defined flake, particle, cell, strand, strand portion, rod, filament, macroscopic fiber or mass thereof
- Y10T428/2982—Particulate matter [e.g., sphere, flake, etc.]
- Y10T428/2984—Microcapsule with fluid core [includes liposome]
Definitions
- the present invention relates to a conjugate comprised of a hydrophobic moiety, a cleavable linkage, and a therapeutic agent. More particularly, the present invention relates to conjugates comprised of a lipid, a cleavable linkage and a drug inco ⁇ orated into a liposomal formulation. The conjugates are cleavable under mild thiolytic conditions in vivo for release of the drug in an unmodified state.
- Liposomes are closed lipid vesicles used for a variety of therapeutic purposes, a d in particular, for carrying therapeutic agents to a target region or cell by systemic administration of liposomes.
- Liposomes having a surface grafted with chains of water- soluble, biocompatible polymer, in particular polyethylene glycol, have become important drug carries. These liposomes offer an extended blood circulation lifetime over liposomes lacking the polymer coating. The grafted polymer chains shield or mask the liposome, thus minimizing nonspecific interaction by plasma proteins. This in turn slows the rate at which the liposomes are cleared or eliminated in vivo since the liposome circulate unrecognized by macrophages and other cells of the reticuloendothelial system. Furthermore, due to the so-called enhanced permeability and retention effect, the liposomes tend to accumulate in sites of damaged or expanded vasculature, e.g. , tumors, sites of inflammation.
- An extended blood circulation time is often desired to allow systemically administered liposomes to reach a target region, cell or site.
- a blood circulation lifetime of greater than about 12 hours is preferred for liposomal-therapy to a tumor region, as the liposomes must systemically distribute and then extravasate into the tumor region.
- liposome-based therapy One problem associated with liposome-based therapy is retention of drug within the liposome for a time sufficient for systemic distribution. This problem is of particular concern when long-circulating liposomes, i.e., liposomes with grafted polymer chains, are administered. Relatively few drugs can be efficiently loaded and retained for a long duration and subsequently released.
- lipid bilayer components that render the bilayer less permeable to entrapped drug.
- the lipid bilayer should be sufficiently fluidic such that the drug is released, for example by transport across the lipid bilayer or by lipid vesicle breakdown, at the desired time, e.g. , after localization at a target site or sufficient biodistribution.
- Another approach to improving drug retention is to covalently attach the drug to a lipid in the liposomal lipid bilayer (Waalkes, et al., Selective Cancer Therap., 6: 15-22 (1990); Asai, et al, Biol. Pharm. Bull , 21:766-771 (1998)). It would be desirable to formulate a liposome composition having a long blood circulation lifetime and capable of retaining an entrapped drug for a desired time, yet able to release the drug on demand.
- a liposome from a non-vesicle-forming lipid, such as dioleoylphosphatidylethanolamine (DOPE), and a lipid bilayer stabilizing lipid, such as methoxy-polyethylene glycol-distearoyl phosphatidylethanolamine (mPEG-DSPE) (Kirpotin, D, et al, FEBS Lett. 388:115-118 (1996)).
- DOPE dioleoylphosphatidylethanolamine
- mPEG-DSPE methoxy-polyethylene glycol-distearoyl phosphatidylethanolamine
- the mPEG is attached to the DSPE via a cleavable linkage. Cleavage of the linkage destabilizes the liposome for a quick release of the liposome contents.
- Labile bonds for linking PEG polymer chains to liposomes has been described (U.S. Patent Nos. 5,013,556, 5,891,468; WO 98/16201).
- the labile bond in these liposome compositions releases the PEG polymer chains from the liposomes, for example, to expose a surface attached targeting ligand or to trigger fusion of the liposome with a target cell.
- Senter describes a drug-antibody prodrug, where the antibody is linked to a drug using a disulfide benzyl carbamate or carbonate linker, and reduction of the disulfide bond effects release of the drug.
- Senter' s teaching is specific to cleavage of a drug-ligand prodrug molecule, under the action of reducing agents such as 1,4- dithiothreitol, glutathione, NADH and NADPH.
- an extended blood circulation time is a desirable feature of PEG- coated liposomes, with blood circulation lifetimes of greater than about 12 hours being preferred for liposomal-therapy to a tumor region.
- the disclosure of Senter provides no guidance as to the release kinetics of a conjugate incorporated into a liposome under endogenous reducing conditions, such as during blood circulation of the liposome.
- the invention includes a conjugate for use in a liposomal drug- delivery vehicle, the conjugate having the general structural formula:
- L is a hydrophobic moiety suitable for incorporation into a liposomal lipid bilayer
- R 1 represents a therapeutic drug covalently attached to the dithiobenzyl moiety, and where orientation of the CH 2 R' group is selected from the ortho position and the para position.
- the therapeutic drug is covalently attached by a linkage selected from the group consisting of urethane, amine, amide, carbonate, thio- carbonate, ether and ester.
- L is selected from the group consisting of cholesterol, a diacylglycerol, a phospholipid and derivatives thereof.
- L is a diacylglycerol derivative to yield a conjugate having the general structural formula:
- R 2 and R 3 are hydrocarbons having between about 8 to about 24 carbon atoms, or in another embodiment, from about 12 to about 22 carbon atoms. In still another embodiment, R 2 and R 3 are hydrocarbon chains of the same length.
- the drug is selected from the group consisting of mitomycin C, mitomycin A, bleomycin, doxorubicin, daunorubicin, fluorodeoxyuridine, iododeoxyuridine, etoposide, AZT, acyclovir, vidarabine, arabinosyl cytosine, pentostatin, quinidine, atropine, chlorambucil, methotrexate, mitoxantrone and 5-fluorouracil.
- the therapeutic drug is covalently linked to the dithiobenzyl moiety to form a conjugate having the structure:
- R 4 represents a residue of the therapeutic drug.
- R 4 in one embodiment is a therapeutic drug residue containing a primary or a secondary amine moiety thereby forming a urethane linkage between the dithiobenzyl and the therapeutic drug.
- the therapeutic drug can be, for example, mitomycin A, mitomycin C, bleomycin or a polypeptide.
- R 4 is a residue of a carboxyl-containing therapeutic drug, which forms an ester linkage between the dithiobenzyl and the therapeutic drug.
- Examplary drugs in this embodiment include chlorambucil or methotrexate.
- R 4 is a therapeutic drug residue containing a hydroxyl moiety thereby to form a carbonate linkage between the dithiobenzyl and the therapeutic drug.
- exemplary drugs in this embodiment include fluorodeoxyuridine, iododeoxyuridine, etoposide, AZT, acyclovir, vidarabine, arabinosyl cytosine, pentostatin, quinidine, mitoxantrone and atropine.
- the invention includes a liposome composition, comprising liposomes composed of vesicle-forming lipids including from about 1 to about 30 mole percent of a conjugate having the general structural described above.
- the therapeutic drug is released from the conjugate in vivo in response to a physiologic condition or an artificially induced condition.
- the invention includes a method for retaining a drug in a liposome, comprising preparing liposomes comprised of a vesicle-forming lipid and of between about 1 to about 30 mole percent of a conjugate described above.
- the liposomes effectively retain the drug in the liposomes until release from the conjugate in response to a physiologic condition or an artificially induced condition.
- Fig. 1 shows a synthetic reaction scheme for preparation of /r ⁇ ra-diacyldiglycerol- dithiobenzylalcohol for further reaction with amine-, hydroxy- or carboxyl-containing drugs;
- Fig. 2 A shows a general reaction scheme for attachment of an amino-containing drug to a reactive diacyldiglycerol-dithiobenzylcarbonate
- Fig. 2B shows the products after thiolytic cleavage of the conjugate in Fig. 2A;
- Fig. 3 A shows a synthetic reaction scheme for preparation of a diacyldiglycerol- dithiobenzyl-5-fluorouracil conjugate ;
- Fig. 3B shows the products after thiolytic cleavage of the conjugate in Fig. 3 A;
- Fig. 4 shows an alternative synthetic reaction scheme for preparation of a diacyldiglycerol-dithiobenzyl-5-fluorouracil conjugate and the products after thiolytic cleavage of the conjugate;
- Fig. 5 shows a synthetic reaction scheme for preparation of a diacyldiglycerol- dithiobenzyl-chlorambucil conjugate and the products after thiolytic cleavage of the conjugate;
- Fig. 6 A shows a synthetic reaction scheme for preparation of a diacyldiglycerol- difhiobenzy 1-mitomycin-C conjugate ;
- Fig. 6B shows the products after thiolytic cleavage of the conjugate in Fig. 6A
- Fig. 7 shows a synthetic reaction scheme for preparation of a cholesterol- dithiobenzyl-mitomycin-C conjugate ;
- Fig. 8 shows another synthetic reaction scheme for preparation of a cholesterol- dithiobenzy 1-mitomycin-C conjugate
- Figs. 9A-9C show the structures of three lipid-dithiobenzy 1-mitomycin-C conjugates, ⁇ ra-distearoyl-DTB-mitomycin-C (Fig. 9A), /? ⁇ r ⁇ -dipalmitoyl-DTB- mitomycin-C (Fig. 9B) and ⁇ rtb ⁇ -dipalmitoyl-DTB- mitomycin-C (Fig. 9C);
- Figs. 10A-10B are HPLC chromatograms for liposomes comprised of HSPC/mPEG-DSPE/lipid-DTB-mitomycin C (Fig. 10A) and HSPC/cholesterol/mPEG- DSPE/lipid-DTB-mitomycin C (Fig. 10B), where each figure shows a series of chromatograms as a function of time of incubation of the liposomes in the presence of cysteine;
- Fig. 11 is a plot showing the percent of mitomycin C released from liposomes comprised of HSPC/mPEG-DSPE/lipid-DTB-mitomycin C (closed diamonds) and HSPC/cholesterol/mPEG-DSPE/lipid-DTB-mitomycin C (closed circles) as a function of time of incubation in the presence of cysteine;
- Figs. 12A-12B are plots showing the percent of mitomycin C released from liposomes comprised of HSPC/mPEG-DSPE/lipid-DTB-mitomycin C (Fig. 12 A) and HSPC/cholesterol/mPEG-DSPE/lipid-DTB-mitomycin C (Fig. 12B) as a function of time of incubation in the presence of cysteine at concentrations of 150 ⁇ M (closed symbols) and at 1.5 mM (open symbols);
- Fig. 13 is a plot of growth rate of Ml 09 cells, expressed as a percentage based on growth of M 109 cells in the absence of drug and cysteine, as a function of mitomycin C amount, in nM, for free mitomycin c (open triangles), liposomes comprised of HSPC/mPEG-DSPE/lipid-DTB-mitomycin C (closed squares), and liposomes comprised of HSPC/cholesterol/mPEG-DSPE/lipid-DTB-mitomycin C (open circles); Fig.
- 14A is a plot of growth rate of Ml 09 cells, expressed as a percentage based on growth of M109 cells in the absence of drug or cysteine, as a function of mitomycin C concentration in nM. Shown are cells treated mitomycin C in free form (open triangles) and with mitomycin C in free form plus 1000 ⁇ M cystein (closed triangles). Also shown are cells treated with the liposome formulation comprised of HSPC/PEG- DSPE/lipid-DTB-mitomycin C (open circles) and with the liposome formulation with additional cysteine added at concentrations of 150 ⁇ M (open diamonds), 500 ⁇ M (closed circles) and 1000 ⁇ M (open squares);
- Fig. 14B is a plot of growth rate of Ml 09 cells, expressed as a percentage based on growth of M109 cells in the absence of drug or cysteine, as a function of mitomycin C concentration in nM. Shown are cells treated mitomycin C in free form (open triangles) and with mitomycin C in free form plus 1000 ⁇ M cystein (closed triangles). Also shown are cells treated with the liposome formulation comprised of HSPC/cholesterol/mPEG-DSPE/lipid-DTB-mitomycin C (open circles) and with the liposome formulation with additional cysteine added at concentrations of 150 ⁇ M (open diamonds), 500 ⁇ M (closed circles) and 1000 ⁇ M (open squares); Fig. 15 is a plot showing the percent increase in cytotoxicity (as determined by
- Fig. 16A is a plot showing the concentration of mitomycin C in the blood of rats as a function of time in hours following intravenous injection of free mitomycin C (open squares), liposomes comprised of HSPC/cholesterol/mPEG-DSPE/lipid-DTB- mitomycin C (closed diamonds), and liposomes comprised of HSPC/mPEG-DSPE/lipid- DTB-mitomycin C (closed circles); and
- Fig. 16B is a plot showing the percent of injected dose remaining in the blood of rats as a function of time in hours following intravenous injection of free mitomycin C (open squares), liposomes comprised of HSPC/cholesterol/mPEG-DSPE/lipid-DTB- mitomycin C (closed diamonds), and liposomes comprised of HSPC/mPEG-DSPE/lipid- DTB-mitomycin C (closed circles).
- hydrophobic moiety suitable for incorporation into a liposomal lipid bilayer intends any material comprising a hydrophobic portion capable of being integrated with the hydrophobic bilayer region of a liposomal lipid bilayer.
- Such hydrophobic moieties are typically lipids, including amphipathic lipids having a hydrophobic lipid tail and a hydrophilic polar head, such as phospholipids and diacylglycerols.
- Triglycerides, sterols, derivatives of phospholipids, diacylglyerols, sterols and triglycerides and other lipids derived from a natural source or synthetically prepared are also contemplated.
- the term “residue” as in "therapeutic drug residue” intends a drug molecule that has been reacted to form an linkage with another molecule where at least one atom of the drug molecule is replaced or has been sacrificed to from the linkage.
- Polypeptide refers to a polymer of amino acids and does not refer to a specific length of a polymer of amino acids. Thus, for example, the terms peptide, oligopeptide, protein, and enzyme are included within the definition of polypeptide. This term also includes post-expression modifications of the polypeptide, for example, glycosylations, acetylations, phosphorylations, and the like.
- PEG poly(ethylene glycol); mPEG, methoxy-PEG; DTB, dithiobenzyl; DSPE, distearoyl phosphatidylethanolamine; HSPC, hydrogenated soy phosphatidylcholine; MMC, mitomycin C.
- the invention includes a conjugate of the form:
- L is a hydrophobic moiety suitable for incorporation into a liposomal lipid bilayer
- R 1 represents a therapeutic drug residue covalently attached to the dithiobenzyl moiety, and where orientation of the CH 2 R' group is selected from the ortho position and the para position.
- the hydrophobic moiety, L is typically a lipid such as a diacylglycerol, a sterol, a phospholipid, derivatives of these lipids, other naturally-occurring lipids and their synthetic analogs.
- a therapeutic drug is attached to the dithiobenzyl moiety by a covalent linkage, thereby forming a drug residue, represented by R 1 in the structure.
- the linkage will vary according to the drug and the reaction chemistry, as will be appreciated by those of skill in the art.
- the therapeutic drug is covalently attached to the diithiobenzyl moiety by a linkage selected from the group consisting of urethane, amine, amide, carbonate, thio-carbonate, ether and ester.
- a drug containing a primary or secondary amine such as mitomycin C, mitomycin A, bleomycin and therapeutic polypeptides to name a few, is reacted to from a urethane linkage with the amine moiety in the drug.
- Exemplary drugs having such a moiety for reaction with dithiobenzyl alcohol to form a carbonate linkage include fluorodeoxyuridine, iododeoxyuridine, etoposide, AZT, acyclovir, vidarabine, arabinosyl cytosine, pentostatin, quinidine, mitoxantrone and atropine.
- the linkage derives from reaction with a carboxylic acid moiety in the therapeutic drug, and an example of a conjugate having an ester linkage between chlorambucil and dithiobenzyl is described below.
- Methotrexate is another example of a drug capable of forming an ester linkage with the dithiobenzyl moiety of the conjugate.
- Conjugates having a urethane, carbonate or ester linkage attaching the drug to the dithiobenzyl moiety can generally be represented by the following structure:
- R 4 represents a residue of the therapeutic drug.
- the conjugate includes an ether linkage, which takes the form of O-R 4 , where R 4 represents the therapeutic drug residue.
- the linkage typically derives from reaction with an alcohol functionality on the drug.
- a conjugate with the drug 5-fluorouracil where an amine linkage is formed will be described below.
- An amide linkage can also be formed with a peptide as the therapeutic agent, where the free carboxyl of an amino acid residue, such as an aspartic acid or glutamic acid, is condensed with dithiobenzylamine.
- Fig. 1 shows a synthetic reaction scheme for preparation of exemplary conjugates in accord with the invention.
- synthesis of an intermediate compound, ⁇ r ⁇ -diacyldiglyceroldithiobenzalcohol (Compound IV) is prepared for further reaction with a selected therapeutic drug.
- Compound IV is prepared, as described in Example 1 , by reacting 3-mercapto-l ,2-propanediol (Compound I) with hydrogen peroxide to form r ⁇ c-3,3'-dithiobis(l ,2-propanediol) (Compound II).
- Rac- 3,3'-dithiobis(l ,2-propanediol) is acylated with a hydrophobic moiety R.
- R can be a fatty acid having from about 8 to about 24 carbon atoms.
- Example 1 details the reaction procedure where R is stearic acid.
- R is a fatty acid having from about 12 to about 22 carbon atoms.
- Acylation of Compound II yields Rac- 3,3'-dithiobis(l ,2-propanedistearoyl) (Compound III), which is reacted with sulfuryl chloride and 4-mercaptobenzalcohol to form the desired intermediate product, para- diacyldiglycerol-dithiobenzalcohol (Compound IV).
- Compound IV is readily reacted with a drug containing a reactive carboxyl moiety (R'C0 2 H) to form a lipid- dithiobenzyl (DTB)-drug conjugate where the drug is joined to the DTB via an ester linkage (Compound V).
- Compound IV is also readily reacted with a drug containing a reactive amine moiety (R'-NH 2 ) to yield a lipid-DTB-drug conjugate where the drug is joined to the DTB by a urethane linkage (Compound VI).
- Compound IV is also readily reacted with a drug containing a reactive hydroxyl moiety (R'OH) to form a lipid-DTB- drug conjugate where the drug is joined to the DTB by a carbonate linkage (Compound VII).
- drugs are contemplated for use in the conjugate of the invention.
- the invention contemplates drugs having an amine (NH or NH 2 ), carboxyl, sulfhydryl or hydroxyl moiety suitable for reaction.
- "suitable for reaction” implies that the drug has one of the recited moieties capable of reacting with the dithiobenzyl moiety, in the form of, for example, dithiobenzyl alcohol.
- Exemplary drugs include 5-fluorouracil, which has an NH group suitable for reaction, chlorambucil, which has a reactive carboxyl and mitomycin C, which has a reactive amine (aziridine group).
- exemplary drugs contemplated for use include mitomycin C, mitomycin A, bleomycin, doxorubicin, daunorubicin, fluorodeoxyuridine, iododeoxyuridine, etoposide, AZT, acyclovir, vidarabine, arabinosyl cytosine, pentostatin, quinidine, atropine, chlorambucil, methotrexate, mitoxantrone and 5-fluorouracil.
- polypeptides, aminoglycosides, alkaloids are all also suitable for use in the invention.
- Example 1 also details the reaction conditions for preparation of ortho- diacyldiglyceroldithiobenzalcohol, which can serve as a intermediary compound to form the conjugate.
- Figs. 2A-2B show preparation of a lipid-DTB-drug conjugate (Fig. 2A), and thiolytic cleavage of the conjugate in the presence of a reducing agent (Fig. 2B).
- Fig. 2A Compound VII of Fig.
- hydrophobic moiety R is derived from a fatty acid R"(CO)OH, such as stearic acid (CH 3 (CH 2 ) 16 CO 2 H), is reacted with an amine-containing drug, H 2 N-drug, in the presence of phosgene (COCl 2 ).
- This reaction yields the lipid-DTB-drug conjugate illustrated in Fig. 2A.
- the conjugate upon exposure to reducing conditions, i.e., a reducing agent such as cysteine or glutathione, decomposes to yield the products shown in Fig. 2B.
- thiolytic cleavage of the conjugate results in regeneration of the drug in an unmodified, natural state.
- the drug in conjugate can be readily incorporated into liposomes for administration in vivo to a subject. Further, the drug in the form of the conjugate is not toxic, as will also be shown below. After administration and upon exposure to endogeneous reducing agents or exposure to an exogeneous reducing agent, the conjugate decomposes to yield the drug in its native state and with biological activity.
- Fig. 3 A a synthetic reaction scheme for preparation of a conjugate of 5- fluorouracil is illustrated.
- Compound IV /r ⁇ ra-diacyl-diglycerol-dithiobenzalalcohol
- /r ⁇ ra-toluenesulfonyl chloride to form the intermediate compound IX.
- Reaction with 5-fluorouracil anion or sodium salt (Compound X) yields the desired lipid-DTB-5-fluorouracil conjugate (Compound XI).
- Decomposition of the lipid-DTB- 5-fluorouracil conjugate (Compound XI) upon exposure to a reducing agent, R'-SH is shown in Fig. 3B. Thiolytic cleavage of the conjugate results in regeneration of 5- fluorouracil in an unmodified form.
- Fig. 4 shows an alternative synthetic reaction scheme for preparation of a diacyldiglycerol-DTB-5-fluorouracil conjugate.
- 1-chloroethyl chloroformate (Compound XII) is reacted with /? ⁇ r ⁇ -diacyl-diglycerol-dithiobenzalalcohol (Compound IV) to form a reactive chloroethyl carbonate-DTB-diacyldiglycerol intermediate (Compound XII).
- the intermediate is subsequently reacted with 5-fluorouracil in the presence of ttiemanolamine (TEA) to yield a lipid-DTB-5-fluorouracil conjugate (Compound XIV).
- TAA ttiemanolamine
- a drug containing a reactive carboxyl moiety chlorambucil (Compound XV)
- Compound IV ⁇ ra-diacyl-diglycerol-dithiobenzalalcohol
- DCC 1,3- dicyclohexycarbodiimide
- DMAP dimethylaminopyridine
- a lipid- DTB-chlorambucil conjugate Compound XVI
- DCC 1,3- dicyclohexycarbodiimide
- DMAP dimethylaminopyridine
- the conjugate thiolytically decomposes to the products shown. Chlorambucil is subsequently regenerated in an unmodified state.
- Fig. 6 A shows the synthesis of a conjugate in accord with another embodiment of the invention.
- mitomycin C Compound XVII, Fig. 6B
- a drug containing a reactive amine moiety is reacted with /r ⁇ ra-diacyl-diglycerol- dithiobenzalalcohol (Compound IV) in the presence of phosgene to form a diacyldiglycerol-dithiobenzy 1-mitomycin-C conjugate (Compound XVIII).
- Compound IV /r ⁇ ra-diacyl-diglycerol- dithiobenzalalcohol
- phosgene diacyldiglycerol-dithiobenzy 1-mitomycin-C conjugate
- Fig. 6B shows the thiolytic decomposition of a diacyldiglycerol-DTB-mitomycin-
- hydrophobic moiety in the conjugate can be selected from any number of hydrophobic moieties, e.g. , lipids.
- lipids e.g. lipids
- a diacyldiglycerol lipid was used to form conjugates having the structure:
- R 2 and R 3 are hydrocarbons having between about 8 to about 24 carbon atoms.
- Fig. 7 shows another embodiment where cholesterol is used as the hydrophobic moiety in the conjugate.
- Cholesterol (Compound XIV) is reacted with methanesulfonyl chloride in dichloromethane in the presence of triemylamine (TEA). The resulting intermediate is then converted into the thiol derivative and ultimately into the principal dithiobenzyl alcohol, which is used to link mitomycin C in a similar fashion as described above for diacylglycerol.
- the invention includes a liposome composition comprised of a vesicle-forming lipid and a conjugate as described above.
- Liposomes are closed lipid vesicles used for a variety of therapeutic purposes, and in particular, for carrying therapeutic agents to a target region or cell by systemic administration of liposomes.
- liposomes having a surface coating of hydrophilic polymer chains, such as polyethylene glycol (PEG), are desirable as drug carries as these liposomes offer an extended blood circulation lifetime over liposomes lacking the polymer coating.
- the polymer acts as a barrier to blood proteins thereby preventing binding of the protein and recognition of the liposomes for uptake and removal by macrophages and other cells of the reticuloendothelial system.
- Liposomes include a conjugate in combination with a lipid, which in one embodiment is a vesicle-forming lipid, and, optionally, other bilayer components.
- a lipid which in one embodiment is a vesicle-forming lipid, and, optionally, other bilayer components.
- "Vesicle-forming lipids” are lipids that spontaneously form bilayer vesicles in water.
- the vesicle-forming lipids preferably have two hydrocarbon chains, typically acyl chains, and a polar head group.
- Examples include the phospholipids, such as phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidic acid (PA), phosphatidylinositol (PI), and sphingomyelin (SM).
- a preferred lipid for use in the present invention is hydrogenated soy phosphatidylcholine (HSPC).
- Another preferred family of lipids are diacylglycerols. These lipids can be obtained commercially or prepared according to published methods.
- the vesicle-forming lipid may be selected to achieve a degree of fluidity or rigidity, to control the stability of the liposome in serum, and to control the rate of release of an entrapped agent in the liposome.
- Liposomes having a more rigid lipid bilayer, or a liquid crystalline bilayer can be prepared by incorporation of a relatively rigid lipid, e.g., a lipid having a relatively high phase transition temperature, e.g. , up to about 80°C.
- Rigid lipids i.e. , saturated, contribute to greater membrane rigidity in the lipid bilayer.
- Other lipid components, such as cholesterol are also known to contribute to membrane rigidity in lipid bilayer structures.
- Lipid fluidity is achieved by incorporation of a relatively fluid lipid, typically one having a lipid phase with a relatively low liquid to liquid-crystalline phase transition temperature, e.g. , at or below room temperature (about 20-25°C).
- the liposome can also include other components that can be inco ⁇ orated into lipid bilayers, such as sterols. These other components typically have a hydrophobic moiety in contact with the interior, hydrophobic region of the bilayer membrane, and a polar head group moiety oriented toward the exterior, polar surface of the membrane.
- Another lipid component in the liposomes of the present invention is a vesicle- forming lipid derivatized with a hydrophilic polymer. In this lipid component, a derivatized lipid results in formation of a surface coating of hydrophilic polymer chains on both the inner and outer lipid bilayer surfaces. Typically, between about 1-20 mole percent of the derivatized lipid is included in the lipid composition.
- Hydrophilic polymers suitable for derivatization with a vesicle-forming lipid include polyvinylpyrrolidone, polyvinylmethylether, polymethyloxazoline, polyethyloxazoline, polyhydroxypropyloxazoline, polyhydroxypropylmethacrylamide, polymethacrylamide, polydimethylacrylamide, poly hydroxypropylmethacry late, polyhydroxyethylacrylate, hydroxymethylcellulose, hydroxyethylcellulose, polyethyleneglycol, and polyaspartamide.
- the polymers may be employed as homopolymers or as block or random copolymers.
- a preferred hydrophilic polymer chain is polyethyleneglycol (PEG), preferably as a PEG chain having a molecular weight between about 500 to about 10,000 Daltons, preferably between about 1,000 to about 5,000 Daltons.
- PEG polyethyleneglycol
- Methoxy or ethoxy-capped analogues of PEG are also preferred hydrophilic polymers. These polymers are commercially available in a variety of polymer sizes, e.g. , from about 12 to about 220,000 Daltons.
- Liposomes of the present invention include typically between about 1 and about 30 mole percent of the lipid-DTB-drug conjugate, preferably between about 5 and about 30 mole percent, more preferably between about 5 and about 20 mole percent.
- liposomes comprised of the vesicle- forming lipid hydrogenated soy phosphatidylcholine (HSPC), distearoyl phosphatidylethanolamine derivatized with methoxy-polyethylene glycol (mPEG-DSPE) and the conjugate shown in Fig. 9A, ⁇ ra-distearoyl-DTB-mitomycin C (Compound XVIII) were prepared as described in Examples 4A-4B.
- HSPC vesicle- forming lipid hydrogenated soy phosphatidylcholine
- mPEG-DSPE distearoyl phosphatidylethanolamine derivatized with methoxy-polyethylene glycol
- Fig. 9A ⁇ ra-d
- One of the liposome formulations included cholesterol (Example 4A), with the lipids HSCP/cholesterol/mPEG-DSPE//r ⁇ r ⁇ -distearoyl-DTB-mitomycin C (Compound XVIII) present at a molar ratio of 60/30/5/5.
- the lipids HSCP/mPEG-DSPE/ ⁇ ra-distearoyl-DTB-mitomycin C (Compound XVII) were present at a molar ratio of 90/5/5.
- the liposome formulations e.g., HSPC/cholesterol/mPEG-DSPE/conjugate Compound XVffl (hereinafter the "cholesterol-containing formulation”) and HSPC/mPEG- DSPE/conjugate Compound XVffl (hereinafter the "cholesterol-free liposome formulation”) were incubated at 37°C in the presence of 150 ⁇ M cysteine for 24 hours. Samples were withdrawn at selected time points and analyzed by high performance liquid chromatography (HPLC) to quantify the amount of conjugate and of free mitomycin C. The HPLC conditions are described in Example 5.
- HPLC high performance liquid chromatography
- Figs. 10A-10B show HPLC chromatograms for two liposome formulations.
- Fig. 10A the results for the cholesterol-free liposome formulation are shown.
- time zero there is no detectable free mitomycin C and all measurable drug is in the form of a lipid-DTB-drug conjugate that is liposome bound.
- Fig. 10B shows the results for the liposome formulation containing cholesterol.
- Fig. 11 is a plot showing the percent of mitomycin C released from the two liposome formulations, as determined from the chromatograms in Figs. 10A-10B.
- the cholesterol-free liposomes (closed diamonds) had a higher rate of release than the liposomes containing cholesterol (closed circles). More than 50% of the mitomycin C was released from the liposome-bound conjugate after 2 hours for the cholesterol-free formulation. For both formulations, greater than 80% of the drug was released at the end of the 24 hour incubation period.
- Figs. 12A-12B show the results of mitomycin C released from the lipid-DTB-drug conjugate inco ⁇ orated into the cholesterol-free liposomes (HSPC/PEG- DSPE/lipid-DTB-mitomycin C). The percent release during incubation with 150 ⁇ M are also shown (closed diamonds) for comparison. As seen, incubation at a higher concentration of reducing agent (1.5 mM, open diamonds) causes an increase in the rate of conjugate decomposition and rate of drug release.
- Fig. 12B shows the results for the liposome formulation containing cholesterol. Liposomes incubated in 1.5 mM (open circles) have a significantly higher decomposition rate than the same liposomes incubated in 150 ⁇ M cysteine (closed circles).
- Liposomes prepared as described in Examples 4A-4B with the molar ratios specified in Example 6A were tested. Cysteine at concentrations of 150 ⁇ M, 500 ⁇ M and 1000 ⁇ m was added to some of the test cells to effect thioytic decomposition of the conjugate and release of mitomycin C.
- IC50 values were taken as the drug concentration which caused a 50% inhibition of the control growth rate (IC 50 ), as described in Example 6. The results are shown in Table 1.
- the percent growth rate of M109 mouse carcinoma cells determined from the cytotoxicity studies is shown in Fig. 13.
- the percent growth rate is expressed as a percentage based on growth rate of Ml 09 cells in the absence of mitomycin C and of cysteine and is shown as a function of mitomycin C concentration, in nM.
- the growth rate of cells was determined as described in Example 6. As seen, the percent of cell growth rate decreases as the cysteine concentration is increased for both the liposomes containing cholesterol (open circles) and the cholesterol-free liposome formulation (closed squares). It can also be seen that cysteine has no effect on the activity of free mitomycin c and that mitomycin C is released from the conjugate to effectively inhibit cell growth.
- Figs. 14A-14B the results for the liposome formulation containing no cholesterol are shown.
- the growth rate of M109 cells is expressed as a percentage based on growth of M109 cells in the absence of drug and cysteine and is shown as a function of mitomycin C concentration in nM.
- the cells treated with mitomycin C in free form (open triangles) and with mitomycin C in free form plus 1000 ⁇ M cysteine (closed triangles) exhibit a decrease in growth rate due tl e toxicity of the drug in free form.
- Fig. 14B is a similar plot for the liposome formulation containing cholesterol.
- Cytotoxicity of free mitomycin C (closed squares) is not effected by the presence of cysteine.
- the cytotoxicity data shows that the cholesterol-free liposome formulation is more affected by cysteine.
- the IC50 of the cholesterol-free liposome formulation at certain cysteine concentrations is only 2-fold lower than that of the free drug alone.
- the liposome formulation containing cholesterol is less cytotoxic than the cholesterol-free liposome formulation.
- the data also shows that cysteine has no cytotoxic effect of the tumor cells and no effect on the cytotoxicity of free mitomycin C. It is also apparent from the data that cysteine increases in a dose-dependent fashion the cytotoxcity of liposome-bound mitomycin C. Thus, the cytotoxic effects observed for the liposomal formulations are mostly accounted for by cysteine-mediated release of mitomycin C from the lipid-DTB-drug conjugate.
- the in vivo pharmacokinetics of the liposomes containing cholesterol and the cholesterol-free liposome formulation was determined in rats. As described in Example 7, the animals were treated with a single bolus intravenous injection of approximately 0.1 mg/mL mitomycin C in free form or inco ⁇ orated into liposomes in the form of the lipid-DTB-mitomycin C conjugate in accord with the invention. After injection, blood samples were taken and analyzed for amount of mitomycin C. The results are shown in Figs. 16A-16B.
- Fig. 16A shows the concentration ( ⁇ g/mL) of mitomycin C in the blood of rats as a function of time in hours following intravenous injection.
- free mitomycin C (open squares) administered intravenously in free form is rapidly cleared from the blood.
- Mitomycin C in the form of a liposome-bound lipid-DTB-drug conjugate remains in circulation for a substantially longer period of time.
- Mitomycin C associated with liposomes containing cholesterol (closed diamonds) and with cholesterol-free liposomes (closed circles) was detected in the blood at greater than 10 ⁇ g/mL for 20-25 hours.
- Fig. 16B shows the percent of injected dose remaining in the blood as a function of time in hours following intravenous injection of the test formulations.
- the rac-3,3'-dithiobis(l,2-propanediol) product (Compound U) was acylated by adding the compound (980 mg, 4.6 mmol) to an oven-dried 100 mL round bottom flask and dissolving in dry methylene chloride (40 mL). To this, stearic acid (4.92 g, 17.1 mmol) and 4-dimethylamino)pyridinium 4-toluenesulfonate (1.38 g, 4.6 mmol) as the catalyst was and stirred at room temperature (25 °C) for 20 minutes.
- the mitomycin C solution was added drop-wise the acyl chloride solution. After 1 hour, the toluene was evaporated off and the crude product was chromatographed (1: 1 hexane: ethyl acetate) on silica. The purified product was then taken up in t-BuOH (50 mL) and lyophilized. The product was a pu ⁇ le solid (183 mg, 53%).
- Liposomes Containing Cholesterol 1. Liposome Preparation 59 mg HSPC, 14.4 mg cholesterol, 17.4 mg mPEG-DSPE, and 7.4 ⁇ ngpara- distearoyl-DTB-mitomycin C (molar ratio of 60/30/5/5) were added to 1 mL dehydrated ethanol at 60-65 °C and mixed until dissolved, approximately 10 minutes. A hydration medium composed of 10 mM histidine and 150 mM NaCl in distilled water was warmed to 70 °C.
- the warm lipid solution was rapidly added to the warm (63-67 °C) hydration medium, with mixing, to form a suspension of liposomes having heterogeneous sizes.
- the suspension was mixed for one hour at 63-67 °C.
- the liposomes were sized to the desired mean particle diameter by controlled extrusion through polycarbonate filter cartridges housed in Teflon-lined stainless steel vessels.
- the liposome suspension was maintained at 63-65 °C throughout the extrusion process, a period of 6-8 hours.
- Ethanol was removed from the liposome suspension by diafiltration.
- a histidine/sodium chloride solution was prepared by dissolving histidine (10 mM) and sodium chloride (150 mM) in sterile water. The pH of the solution was adjusted to approximately 7. The solution was filtered through a 0.22 ⁇ m Durapore filter. The liposome suspension was diluted in approximately a 1 : 1 (v/v) ratio with the histidine/sodium chloride solution and diafiltered through a polysulfone hollow-fiber ultrafilter. Eight volume exchanges were performed against the histidine/sodium chloride solution to remove the ethanol. The process fluid temperature was maintained at about 20-30 °C. Total diafiltration time was approximately 4.5 hours.
- lipid concentration and conjugate/drug concentration were determined by HPLC.
- Liposome particle size was measured by dynamic light scattering and the amount of "free" , unbound mitomycin C in the external suspension medium was measured by HPLC.
- Liposomes were prepared as described above with a lipid composition of HSPC, mPEG-DSPE and ⁇ ra-distearoyl-DTB-mitomycin C in a molar ratio of 90/5/5. Specifically, 88.5 mg HPSC, 17.9 mg mPEG-DSPE (PEG MW 2000 Daltons) and 7.3 mg of the conjugate were dissolved in 1 mL ethanol. Liposome size, lipid and drug concentration and free mitomycin C concentration in the external suspension medium were determined after each processing step.
- Liposomes prepared as described in Examples 4A-4B were diluted in 0.6 M octaylglucopyranoside. The liposomes were incubated in the presence of 150 mM cysteine at 37 °C. Samples with withdrawn at time zero, 30 minutes, 1 hour, 2 hours, 4 hours and 24 hours. A 20 ⁇ L volume was analyzed by HPLC using a Water Symmetry C 8 3.5 x 5 cm column. The flow rate was 1 mL/min and the mobile phase gradient as follows:
- Liposomes prepared as described in Example 4A-4B, were composed of
- HSPC/cholesterol/mPEG-DSPE/distearoyl-DTB-mitomycin C (90/45/5/5).
- the liposome preparations were sterile filtered through 0.45 ⁇ m cellulose membranes and were not downsized via extrusion. After liposome formation, mitomycin C concentration was determined by absorbance at 360 nm in liposomes solubilized by 10- 20 fold dilution in isopropanol and the phospholipid concentration was determined by inorganic phosphate assay.
- the liposomes containing cholesterol had an average diameter of 275 ⁇ 90 nm.
- the cholesterol-free liposomes had an average diameter of 150 + 50 nm.
- the phospholipid concentration in both liposome formulations was 10 ⁇ M/mL and the concentration of mitomycin C in both formulations was 120 ⁇ g/mL.
- the cytotoxic effect of free mitomycin C or mitomycin C in the form of a distearoyl-DTB-mitomycin C conjugate inco ⁇ orated into liposomes was assayed colorimetrically by a methylene blue staining method described previously (Horowitz, A.T. et l , Biochim. Biophys. Acta, 1 109:203-209 (1992)) with slight modifications. Upon completion of the assay, the cells were fixed and evaluated using the methylene blue staining assay.
- the plates were washed three times with deionized water, once with 0.1 M borate buffer (pH 8.5) and then stained for 60 minutes with 100 ⁇ l methylene blue (1 % in 0.1 M buffer borate, pH 8.5) at room temperature (20-25°C) .
- the plates were rinsed in five baths of deionized water to remove non-cell bound dye and then dried.
- the dye was extracted with 200 ⁇ l 0.1 N HC1 for 60 minutes at 37°C and the optical density was determined using a microplate spectrophotometer .
- the cell number determined by counting cells with a hemocytometer correlated well with the spectrophotometric absorbance.
- the percent growth inhibition or percent of control growth rate was obtained by dividing the growth rate of drug-treated cells by the growth rate of the untreated, control cells.
- the drug concentration which caused a 50% inhibition of the control growth rate (IC 50 ) was calculated by inte ⁇ olation of the two closest values of the growth inhibition curve.
- Mitomycin C was assayed in the range 10 "8 -10 "5 M.
- the liposomal formulations with conjugate-bound were assayed in the range 10 "8 - 3 x 10 "5 M.
- For interaction stodies cysteine (SIGMA, St. Louis, MO) was added together with the mitomycin C or liposome formulations to final concentration of 150, 500, or 1000 ⁇ M. The results are shown in Table 1 and in Figs. 13, 14 and 15A-15B.
- Liposomes containing cholesterol and cholesterol-free liposomes were prepared as described in Example 5 A and 5B.
- a solution of mitomycin C in free form was prepared by dissolving 11.9 mg of mitomycin C in 119 ⁇ L ethanol. After dissolution, approximately 11.8 ⁇ L of a solution of 10 mM histidine/ 150 mM saline was added. Prior to use, the mitomycn C solution was diluted to 100 ⁇ g/mL with the histidine/saline solution and filtered.
- a single intravenous injection of the test formulation was administered as a bolus dose.
- Blood samples were taken from each animal at the following times after injection: 30 seconds, 15 minutes, 30 minutes, 1 hour, 2 hours, 4 hours, 8 hours, 24 hours, 48 hours, 72 hours and 96 hours.
- the quantity of mitomycin C in the blood samples was determined by the HPLC procedure given below.
- a 200 mM iodoacetamine solution was prepared by placing 199.3 mg of iodoacetamide in 5.1 mL of 7.5% EDTA. 15 ⁇ L of the 200 mM iodoacetamide solution was placed in each 1 ⁇ L of blood sample.
- a mobile phase of methanol and the aqueous buffer were mixed via a gradient program using a Waters Alliance binary pump.
- the concentration ranges were 0.05-5.0 ⁇ g/mL and 0.1-5 ⁇ g/mL for mitomycin C and mitomycin C conjugate, respectively.
- the final volume was adjusted to 1 mL with methanol.
- a similar procedure was followed to prepare quality control samples.
- the concentrations of quality control samples was 0.1, 0.5 and 5 ⁇ g/mL for mitomycin C and 0.1 , 1 and 5 ⁇ g/mL for mitomycin C conjugate in rat plasma.
- the samples were spun down at 3,000 ⁇ m for 10 minutes at room temperature. 300 ⁇ L of supernatant was transferred to HPLC vials containing 300 ⁇ L insert for injection. 3.
- a Supelco ® C-8, 5 ⁇ , 4.6mm x 5 cm column was used.
- the mobile phase A was 10 mM ammonium phosphate, pH 7.
- Mobil phase B was methanol.
- the flow rate was 1 mL/min and detection was by UV at 360 nm.
- the injection volume was 40 ⁇ L and the typical run time was 15 minutes.
- the gradient program was as follows:
Abstract
Description












Claims


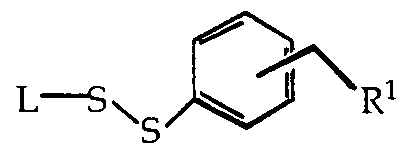




Priority Applications (13)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| HU0201425A HUP0201425A3 (en) | 1999-04-23 | 2000-04-21 | Conjugate having a cleavable linkage for use in a liposome |
| CA2369595A CA2369595C (en) | 1999-04-23 | 2000-04-21 | Conjugate having a cleavable linkage for use in a liposome |
| DE60030965T DE60030965T2 (en) | 1999-04-23 | 2000-04-21 | CONJUGATE CONTAINS A SPLICABLE BINDING FOR USE IN A LIPOSOM |
| DK00928321T DK1173222T3 (en) | 1999-04-23 | 2000-04-21 | Conjugate with a cleavable bond for use in a liposome |
| JP2000613474A JP4558952B2 (en) | 1999-04-23 | 2000-04-21 | Complexes with cleavable bonds for use in liposomes |
| KR1020017013571A KR100669053B1 (en) | 1999-04-23 | 2000-04-21 | Conjugate having a cleavable linkage for use in a liposome |
| EP00928321A EP1173222B1 (en) | 1999-04-23 | 2000-04-21 | Conjugate having a cleavable linkage for use in a liposome |
| IL14605500A IL146055A0 (en) | 1999-04-23 | 2000-04-21 | Conjugate having a cleavable linkage for use in a liposome |
| AU46577/00A AU769425B2 (en) | 1999-04-23 | 2000-04-21 | Conjugate having a cleavable linkage for use in a liposome |
| MXPA01010750A MXPA01010750A (en) | 1999-04-23 | 2000-04-21 | Conjugate having a cleavable linkage for use in a liposome. |
| IL146055A IL146055A (en) | 1999-04-23 | 2001-10-18 | Conjugate having a cleavable linkage for use in a liposome |
| NO20015144A NO20015144L (en) | 1999-04-23 | 2001-10-22 | Conjugate with a cleavable bond for use in a liposome |
| HK02103601.3A HK1041820B (en) | 1999-04-23 | 2002-05-13 | Conjugate having a cleavable linkage for use in a liposome |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US13089799P | 1999-04-23 | 1999-04-23 | |
| US60/130,897 | 1999-04-23 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2000064484A2 true WO2000064484A2 (en) | 2000-11-02 |
| WO2000064484A3 WO2000064484A3 (en) | 2001-11-15 |
Family
ID=22446872
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2000/010922 WO2000064484A2 (en) | 1999-04-23 | 2000-04-21 | Conjugate having a cleavable linkage for use in a liposome |
Country Status (18)
| Country | Link |
|---|---|
| US (8) | US6365179B1 (en) |
| EP (3) | EP1173222B1 (en) |
| JP (1) | JP4558952B2 (en) |
| KR (2) | KR100642955B1 (en) |
| CN (2) | CN100512879C (en) |
| AT (1) | ATE340592T1 (en) |
| AU (1) | AU769425B2 (en) |
| CA (1) | CA2369595C (en) |
| DE (1) | DE60030965T2 (en) |
| DK (1) | DK1173222T3 (en) |
| ES (1) | ES2272280T3 (en) |
| HK (1) | HK1041820B (en) |
| HU (2) | HUP0200797A3 (en) |
| IL (2) | IL146055A0 (en) |
| MX (2) | MXPA01010750A (en) |
| NO (1) | NO20015144L (en) |
| WO (1) | WO2000064484A2 (en) |
| ZA (2) | ZA200108726B (en) |
Cited By (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1385479A1 (en) * | 2001-03-26 | 2004-02-04 | Alza Corporation | Liposome composition for improved intracellular delivery of a therapeutic agent |
| WO2004110497A2 (en) | 2003-04-30 | 2004-12-23 | Alza Corporation | Mitomycin conjugates cleavable by thiols |
| WO2005053749A2 (en) * | 2003-11-26 | 2005-06-16 | Alza Corporation | Thiol-cleavable linkage between polymer and ligand |
| US7211440B2 (en) | 2002-03-08 | 2007-05-01 | Wallac Oy | Dissociative fluorescence enhancement assay |
| US7276248B2 (en) | 1999-04-23 | 2007-10-02 | Alza Corporation | Conjugate having a cleavable linkage for use in a liposome |
| CN101284136A (en) * | 2002-12-03 | 2008-10-15 | 布朗歇特洛克菲勒神经科学研究所 | Artificial low-density lipoprotein carriers for transport of substances across the blood-brain barrier |
| US9181321B2 (en) | 2013-03-14 | 2015-11-10 | Shire Human Genetic Therapies, Inc. | CFTR mRNA compositions and related methods and uses |
| US9308281B2 (en) | 2011-06-08 | 2016-04-12 | Shire Human Genetic Therapies, Inc. | MRNA therapy for Fabry disease |
| US9522176B2 (en) | 2013-10-22 | 2016-12-20 | Shire Human Genetic Therapies, Inc. | MRNA therapy for phenylketonuria |
| US9629804B2 (en) | 2013-10-22 | 2017-04-25 | Shire Human Genetic Therapies, Inc. | Lipid formulations for delivery of messenger RNA |
| US9717690B2 (en) | 2011-06-08 | 2017-08-01 | Rana Therapeutics, Inc. | Cleavable lipids |
| US9850269B2 (en) | 2014-04-25 | 2017-12-26 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| GB2552301A (en) * | 2016-07-11 | 2018-01-24 | Evox Therapeutics Ltd | Metabolic drug loading of EVs |
| US9877919B2 (en) | 2012-03-29 | 2018-01-30 | Translate Bio, Inc. | Lipid-derived neutral nanoparticles |
| US9957499B2 (en) | 2013-03-14 | 2018-05-01 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US10022455B2 (en) | 2014-05-30 | 2018-07-17 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US10138213B2 (en) | 2014-06-24 | 2018-11-27 | Translate Bio, Inc. | Stereochemically enriched compositions for delivery of nucleic acids |
| WO2019016208A1 (en) * | 2017-07-17 | 2019-01-24 | Technische Universiteit Eindhoven | Applicable chemical composition comprising an agent conjugated to a hydrophobic moiety and a carrier |
| US10576166B2 (en) | 2009-12-01 | 2020-03-03 | Translate Bio, Inc. | Liver specific delivery of messenger RNA |
| US11173190B2 (en) | 2017-05-16 | 2021-11-16 | Translate Bio, Inc. | Treatment of cystic fibrosis by delivery of codon-optimized mRNA encoding CFTR |
| US11174500B2 (en) | 2018-08-24 | 2021-11-16 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US11224642B2 (en) | 2013-10-22 | 2022-01-18 | Translate Bio, Inc. | MRNA therapy for argininosuccinate synthetase deficiency |
| US11253605B2 (en) | 2017-02-27 | 2022-02-22 | Translate Bio, Inc. | Codon-optimized CFTR MRNA |
| US11254936B2 (en) | 2012-06-08 | 2022-02-22 | Translate Bio, Inc. | Nuclease resistant polynucleotides and uses thereof |
| US11951181B2 (en) | 2023-04-03 | 2024-04-09 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
Families Citing this family (69)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7422902B1 (en) * | 1995-06-07 | 2008-09-09 | The University Of British Columbia | Lipid-nucleic acid particles prepared via a hydrophobic lipid-nucleic acid complex intermediate and use for gene transfer |
| US5981501A (en) * | 1995-06-07 | 1999-11-09 | Inex Pharmaceuticals Corp. | Methods for encapsulating plasmids in lipid bilayers |
| US6287591B1 (en) | 1997-05-14 | 2001-09-11 | Inex Pharmaceuticals Corp. | Charged therapeutic agents encapsulated in lipid particles containing four lipid components |
| US7112337B2 (en) * | 1999-04-23 | 2006-09-26 | Alza Corporation | Liposome composition for delivery of nucleic acid |
| GB9915074D0 (en) * | 1999-06-28 | 1999-08-25 | Cortecs Plc | Ligand-binding composition |
| US7094423B1 (en) * | 1999-07-15 | 2006-08-22 | Inex Pharmaceuticals Corp. | Methods for preparation of lipid-encapsulated therapeutic agents |
| DE60231868D1 (en) * | 2001-04-24 | 2009-05-20 | Purdue Research Foundation | FOLAT MIMETICS AND THEIR FOLAT RECEPTOR BINDING CONJUGATES |
| CA2472055A1 (en) * | 2001-11-07 | 2003-05-15 | Inex Pharmaceuticals Corporation | Improved mucosal vaccines and methods for using the same |
| US7842498B2 (en) * | 2001-11-08 | 2010-11-30 | Bio-Rad Laboratories, Inc. | Hydrophobic surface chip |
| US6737524B2 (en) * | 2002-03-25 | 2004-05-18 | Paul K. Smith | Activated polyethylene glycol compounds |
| US20040009944A1 (en) * | 2002-05-10 | 2004-01-15 | Inex Pharmaceuticals Corporation | Methylated immunostimulatory oligonucleotides and methods of using the same |
| AU2003243226A1 (en) * | 2002-05-15 | 2003-12-02 | Endocyte, Inc. | Vitamin-mitomycin conjugates |
| US8496961B2 (en) | 2002-05-15 | 2013-07-30 | Sutter West Bay Hospital | Delivery of nucleic acid-like compounds |
| EP2517730A3 (en) | 2003-01-27 | 2013-01-02 | Endocyte, Inc. | Vitamin receptor binding drug delivery conjugates |
| WO2005034979A2 (en) * | 2003-10-11 | 2005-04-21 | Inex Pharmaceuticals Corporation | Methods and compositions for enhancing innate immunity and antibody dependent cellular cytotoxicity |
| JP2007520481A (en) * | 2004-01-15 | 2007-07-26 | アルザ・コーポレーシヨン | Liposome composition for delivering therapeutic agents |
| US7282590B2 (en) * | 2004-02-12 | 2007-10-16 | The Research Foundation Of State University Of New York | Drug conjugates |
| US7931693B2 (en) * | 2004-02-26 | 2011-04-26 | Endosphere, Inc. | Method and apparatus for reducing obesity |
| US9592277B2 (en) * | 2004-04-14 | 2017-03-14 | Avirid, Inc. | Compositions with modified nucleases targeted to viral nucleic acids and methods of use for prevention and treatment of viral diseases |
| JP2007533750A (en) * | 2004-04-21 | 2007-11-22 | アルザ コーポレイション | Polymer conjugates releasable under mild thiol degradation conditions |
| CN1997399A (en) * | 2004-04-21 | 2007-07-11 | 阿尔扎公司 | Polymer conjugate releasable under mild thiolytic conditions |
| JP5149620B2 (en) | 2004-07-23 | 2013-02-20 | エンドサイト,インコーポレイテッド | Bivalent linker and conjugate thereof |
| AU2005282463A1 (en) * | 2004-09-03 | 2006-03-16 | Alza Corporation | Endogenously-formed conjugate of albumin |
| TW200612993A (en) * | 2004-10-08 | 2006-05-01 | Alza Corp | Lipopolymer conjugates |
| JP2008518951A (en) * | 2004-10-28 | 2008-06-05 | アルザ コーポレイション | Lyophilized liposome formulations and methods |
| JP5289935B2 (en) * | 2005-03-16 | 2013-09-11 | エンドサイト,インコーポレイテッド | Synthesis and purification of pteroic acid and its conjugates |
| JP2009504783A (en) * | 2005-08-19 | 2009-02-05 | エンドサイト,インコーポレイテッド | Ligand conjugates of vinca alkaloids, analogues and derivatives |
| RU2470668C2 (en) * | 2005-08-19 | 2012-12-27 | Эндосайт, Инк. | Conjugates of ligand and drugs |
| EP2046389A2 (en) * | 2006-07-19 | 2009-04-15 | The Board of Regents of the University of Texas System | Preparations of phospholipids and pharmaceuticals containing 5-amino salicylic acid for the treatment of inflammatory bowel disease |
| WO2008091655A2 (en) * | 2007-01-23 | 2008-07-31 | The Regents Of The University Of California | Methods, compositions and device for directed and controlled heating and release of agents |
| US20100104626A1 (en) | 2007-02-16 | 2010-04-29 | Endocyte, Inc. | Methods and compositions for treating and diagnosing kidney disease |
| CN103948937A (en) * | 2007-03-14 | 2014-07-30 | 恩多塞特公司 | Binding ligand linked drug delivery conjugates of tubulysins |
| ITRM20070327A1 (en) * | 2007-06-11 | 2008-12-12 | Univ Palermo | COLLOIDAL VECTORS WITH POLYAMINOACIDIC STRUCTURE FOR THE ORAL RELEASE OF PEPTIDES AND PROTEINS AND ITS RELATED PRODUCTION METHOD. |
| US9877965B2 (en) | 2007-06-25 | 2018-01-30 | Endocyte, Inc. | Vitamin receptor drug delivery conjugates for treating inflammation |
| RU2523909C2 (en) | 2007-06-25 | 2014-07-27 | Эндосайт, Инк. | Conjugates, containing hydrophilic spacers of linkers |
| US20100210575A1 (en) * | 2007-06-29 | 2010-08-19 | Wisconsin Alumni Research Foundation | Structuring effect of cholesterol in peg-phospholipid micelles, drug delivery of amphotericin b, and combination antifungals |
| EP2180881A2 (en) * | 2007-07-20 | 2010-05-05 | Basf Se | Vesicles comprising a transmembrane transport trigger system |
| CA2702945C (en) | 2007-10-23 | 2016-08-23 | Nektar Therapeutics Al, Corporation | Hydroxyapatite-targeting multiarm polymers and conjugates made therefrom |
| US9187521B2 (en) | 2007-10-25 | 2015-11-17 | Endocyte, Inc. | Tubulysins and processes for preparing |
| US9259398B1 (en) * | 2007-11-26 | 2016-02-16 | Abbott Cardiovascular Systems Inc. | Bioactive agent-loaded targeting micelles |
| KR101919093B1 (en) * | 2008-05-23 | 2018-11-16 | 더 유니버시티 오브 브리티쉬 콜롬비아 | Modified drugs for use in liposomal nanoparticles |
| CN102388073B (en) | 2009-02-04 | 2015-11-25 | 布里格姆及妇女医院股份有限公司 | Nano level platinic compound and using method thereof |
| BRPI1012036A2 (en) | 2009-05-27 | 2017-10-10 | Selecta Biosciences Inc | nanocarriers that have components with different release rates |
| IN2012DN02589A (en) * | 2009-08-26 | 2015-08-28 | Selecta Biosciences Inc | |
| US20120219538A1 (en) * | 2009-11-02 | 2012-08-30 | Therapeomic Ag | Stabilized protein formulations and use thereof |
| WO2011119995A2 (en) | 2010-03-26 | 2011-09-29 | Cerulean Pharma Inc. | Formulations and methods of use |
| DE102010042338A1 (en) * | 2010-10-12 | 2012-04-12 | Bayer Technology Services Gmbh | A composition for the treatment of controlled release cancer of the active ingredient |
| WO2012061717A1 (en) | 2010-11-05 | 2012-05-10 | Selecta Biosciences, Inc. | Modified nicotinic compounds and related methods |
| CA2834571A1 (en) | 2011-04-29 | 2012-11-01 | Selecta Biosciences, Inc. | Tolerogenic synthetic nanocarriers for inducing regulatory b cells |
| EP2606884A1 (en) | 2011-12-21 | 2013-06-26 | Ecole Polytechnique Fédérale de Lausanne (EPFL) | Inhibitors of notch signaling pathway and use thereof in treatment of cancers |
| US10080805B2 (en) | 2012-02-24 | 2018-09-25 | Purdue Research Foundation | Cholecystokinin B receptor targeting for imaging and therapy |
| US20140080175A1 (en) | 2012-03-29 | 2014-03-20 | Endocyte, Inc. | Processes for preparing tubulysin derivatives and conjugates thereof |
| WO2014062697A2 (en) | 2012-10-16 | 2014-04-24 | Endocyte, Inc. | Drug delivery conjugates containing unnatural amino acids and methods for using |
| US9827552B2 (en) | 2013-07-17 | 2017-11-28 | Clemson University | Functionalized lipid modification of solid phase surfaces for use in chromatography |
| US9937261B2 (en) * | 2014-06-09 | 2018-04-10 | Lipomedix Pharmaceuticals Ltd. | Combination therapy comprising a liposomal prodrug of mitomycin C and radiotherapy |
| AU2016233143B2 (en) | 2015-03-17 | 2021-03-25 | Alberto Gabizon | Methods for the treatment of bladder cancer |
| CN106519221B (en) * | 2015-09-10 | 2019-04-26 | 中国科学院高能物理研究所 | A kind of polyethylene glycol/daiamid copolymer, preparation method and the amphipathic siRNA carrier comprising the copolymer |
| CN106519211B (en) * | 2015-09-10 | 2018-10-09 | 中国科学院高能物理研究所 | A kind of amphipathic nature polyalcohol and the magnetic micella nano-carrier formed by it and its purposes |
| CN105866311B (en) * | 2016-05-25 | 2017-05-31 | 福建出入境检验检疫局检验检疫技术中心 | Determine the UPLC MS/MS methods of antiviral drugs residual in chicken |
| WO2018089481A1 (en) | 2016-11-08 | 2018-05-17 | Mallinckrodt Llc | Mitomycin c prodrug liposome formulations and uses thereof |
| US10618896B2 (en) | 2017-08-22 | 2020-04-14 | Dynavax Technologies Corporation | Alkyl chain modified imidazoquinoline TLR7/8 agonist compounds and uses thereof |
| WO2019099412A1 (en) | 2017-11-14 | 2019-05-23 | Dynavax Technologies Corporation | Cleavable conjugates of tlr7/8 agonist compounds, methods for preparation, and uses thereof |
| WO2020144657A1 (en) | 2019-01-11 | 2020-07-16 | Lipomedix Pharmaceuticals Ltd. | Liposome composition comprising liposomal prodrug of mitomycin c and method of manufacture |
| CA3134791A1 (en) | 2019-04-10 | 2020-10-15 | Cellestia Biotech Ag | Inhibitors of notch signalling pathway and use thereof in treatment of cancers |
| WO2022122667A1 (en) | 2020-12-07 | 2022-06-16 | Cellestia Biotech Ag | Pharmaceutical combinations for treating cancer |
| EP4008324A1 (en) | 2020-12-07 | 2022-06-08 | Cellestia Biotech AG | Combinations comprising an inhibitor of an anti-apoptotic protein, such as bcl-2, bcl-xl, bclw or mcl-1, and a notch signaling pathway inhibitor for treating cancer |
| WO2022253794A2 (en) | 2021-06-02 | 2022-12-08 | Cellestia Biotech Ag | Method for treating an autoimmune and inflammatory disease |
| WO2023079132A1 (en) | 2021-11-08 | 2023-05-11 | Cellestia Biotech Ag | Pharmaceutical combinations for treating cancer |
| EP4223292A1 (en) | 2022-02-07 | 2023-08-09 | Cellestia Biotech AG | Pharmaceutical combinations for treating cancer |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2254336A1 (en) * | 1973-12-17 | 1975-07-11 | Kyowa Hakko Kogyo Kk | |
| EP0317957A2 (en) * | 1987-11-23 | 1989-05-31 | Bristol-Myers Squibb Company | Drug-monoclonal antibody conjugates |
| EP0317956A2 (en) * | 1987-11-23 | 1989-05-31 | Bristol-Myers Squibb Company | Anti-tumor prodrugs |
| EP0510197A1 (en) * | 1990-01-11 | 1992-10-28 | Nippon Shinyaku Company, Limited | Fat emulsion |
| US5169934A (en) * | 1990-05-14 | 1992-12-08 | Anergen, Inc. | Intracellularly cleavable compounds |
| WO1997036904A1 (en) * | 1996-04-02 | 1997-10-09 | Sagami Chemical Research Center | Mitomycin c derivative and non-receptor tyrosine kinase inhibitor |
| WO1999029302A1 (en) * | 1997-12-05 | 1999-06-17 | Katarina Edwards | Drug delivery system with two-step targeting |
Family Cites Families (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4179337A (en) | 1973-07-20 | 1979-12-18 | Davis Frank F | Non-immunogenic polypeptides |
| GB8430252D0 (en) | 1984-11-30 | 1985-01-09 | Beecham Group Plc | Compounds |
| US4766106A (en) | 1985-06-26 | 1988-08-23 | Cetus Corporation | Solubilization of proteins for pharmaceutical compositions using polymer conjugation |
| US5059421A (en) | 1985-07-26 | 1991-10-22 | The Liposome Company, Inc. | Preparation of targeted liposome systems of a defined size distribution |
| US4917888A (en) | 1985-06-26 | 1990-04-17 | Cetus Corporation | Solubilization of immunotoxins for pharmaceutical compositions using polymer conjugation |
| JPH0615532B2 (en) * | 1986-02-03 | 1994-03-02 | テルモ株式会社 | 5-Fluorouracil derivative and pharmaceutical preparation containing the same |
| US4766105A (en) * | 1986-10-31 | 1988-08-23 | Shell Oil Company | Ethylene oxide catalyst and process for preparing the catalyst |
| JPH01113391A (en) * | 1987-10-24 | 1989-05-02 | Kyowa Hakko Kogyo Co Ltd | Mitomycin derivative |
| US5103556A (en) | 1988-05-05 | 1992-04-14 | Circon Corporation | Method of manufacturing an electrohydraulic probe |
| US4902502A (en) | 1989-01-23 | 1990-02-20 | Cetus Corporation | Preparation of a polymer/interleukin-2 conjugate |
| US5585112A (en) | 1989-12-22 | 1996-12-17 | Imarx Pharmaceutical Corp. | Method of preparing gas and gaseous precursor-filled microspheres |
| WO1993018751A1 (en) | 1992-03-23 | 1993-09-30 | Georgetown University | Liposome encapsulated taxol and a method of using the same |
| JP2813511B2 (en) * | 1992-06-10 | 1998-10-22 | 大日本スクリーン製造株式会社 | Method of applying coating liquid to substrate surface using roll coater |
| WO1994005259A1 (en) | 1992-09-02 | 1994-03-17 | Georgetown University | Method of encapsulating anthracycline glycosides in liposomes |
| US5395619A (en) | 1993-03-03 | 1995-03-07 | Liposome Technology, Inc. | Lipid-polymer conjugates and liposomes |
| US5820873A (en) * | 1994-09-30 | 1998-10-13 | The University Of British Columbia | Polyethylene glycol modified ceramide lipids and liposome uses thereof |
| RU2166512C2 (en) * | 1995-01-16 | 2001-05-10 | Коммонвелт Сайентифик Энд Индастриал Рисерч Организейшн | Conjugates of therapeutic compound with fatty acid |
| AU4857796A (en) | 1995-01-20 | 1996-08-07 | Florida State University | Sex-specific dna probe for parrots, methods and kits |
| TW520297B (en) | 1996-10-11 | 2003-02-11 | Sequus Pharm Inc | Fusogenic liposome composition and method |
| EP0932390A1 (en) * | 1996-10-11 | 1999-08-04 | Sequus Pharmaceuticals, Inc. | Therapeutic liposome composition and method |
| JPH1160499A (en) | 1997-08-22 | 1999-03-02 | Hiroshi Maeda | Antitumor agent |
| US6180095B1 (en) | 1997-12-17 | 2001-01-30 | Enzon, Inc. | Polymeric prodrugs of amino- and hydroxyl-containing bioactive agents |
| NZ507456A (en) | 1998-04-28 | 2003-10-31 | Applied Research Systems | Process and conjugated forms of PEGylated interferon- beta with polyethylene glycol (PEG) wherein the thiol reactive polyol agent is mono-methoxylated |
| US7112337B2 (en) | 1999-04-23 | 2006-09-26 | Alza Corporation | Liposome composition for delivery of nucleic acid |
| US7303760B2 (en) | 1999-04-23 | 2007-12-04 | Alza Corporation | Method for treating multi-drug resistant tumors |
| JP4558952B2 (en) | 1999-04-23 | 2010-10-06 | アルザ コーポレイション | Complexes with cleavable bonds for use in liposomes |
| IL146047A0 (en) | 1999-04-23 | 2002-07-25 | Alza Corp | Releasable linkage and compositions containing same |
| US7238368B2 (en) | 1999-04-23 | 2007-07-03 | Alza Corporation | Releasable linkage and compositions containing same |
| AU7868400A (en) | 1999-10-08 | 2001-04-23 | Alza Corporation | Neutral-cationic lipid for nucleic acid and drug delivery |
| ES2367891T3 (en) | 2000-09-29 | 2011-11-10 | Schering Corporation | INTERLEUCINA-10 PEGILADA. |
| US7260330B2 (en) | 2002-11-04 | 2007-08-21 | The Boeing Company | Optical communication system using correlation receiver |
-
2000
- 2000-04-21 JP JP2000613474A patent/JP4558952B2/en not_active Expired - Fee Related
- 2000-04-21 CN CNB008079013A patent/CN100512879C/en not_active Expired - Lifetime
- 2000-04-21 US US09/556,610 patent/US6365179B1/en not_active Expired - Lifetime
- 2000-04-21 DK DK00928321T patent/DK1173222T3/en active
- 2000-04-21 WO PCT/US2000/010922 patent/WO2000064484A2/en active IP Right Grant
- 2000-04-21 EP EP00928321A patent/EP1173222B1/en not_active Expired - Lifetime
- 2000-04-21 DE DE60030965T patent/DE60030965T2/en not_active Expired - Lifetime
- 2000-04-21 CN CNB00807707XA patent/CN1244376C/en not_active Expired - Fee Related
- 2000-04-21 EP EP05008357A patent/EP1579874A3/en not_active Withdrawn
- 2000-04-21 AT AT00928321T patent/ATE340592T1/en not_active IP Right Cessation
- 2000-04-21 MX MXPA01010750A patent/MXPA01010750A/en active IP Right Grant
- 2000-04-21 MX MXPA01010751A patent/MXPA01010751A/en not_active Application Discontinuation
- 2000-04-21 EP EP07018132A patent/EP1880736A1/en not_active Withdrawn
- 2000-04-21 AU AU46577/00A patent/AU769425B2/en not_active Ceased
- 2000-04-21 IL IL14605500A patent/IL146055A0/en active IP Right Grant
- 2000-04-21 US US09/556,056 patent/US6342244B1/en not_active Expired - Lifetime
- 2000-04-21 KR KR1020017013588A patent/KR100642955B1/en not_active IP Right Cessation
- 2000-04-21 HU HU0200797A patent/HUP0200797A3/en unknown
- 2000-04-21 CA CA2369595A patent/CA2369595C/en not_active Expired - Lifetime
- 2000-04-21 ES ES00928321T patent/ES2272280T3/en not_active Expired - Lifetime
- 2000-04-21 HU HU0201425A patent/HUP0201425A3/en unknown
- 2000-04-21 KR KR1020017013571A patent/KR100669053B1/en not_active IP Right Cessation
-
2001
- 2001-10-15 US US09/982,336 patent/US6605299B2/en not_active Expired - Lifetime
- 2001-10-18 IL IL146055A patent/IL146055A/en not_active IP Right Cessation
- 2001-10-22 NO NO20015144A patent/NO20015144L/en not_active Application Discontinuation
- 2001-10-23 ZA ZA200108726A patent/ZA200108726B/en unknown
- 2001-10-23 ZA ZA200108724A patent/ZA200108724B/en unknown
-
2002
- 2002-01-25 US US10/057,839 patent/US6984396B2/en not_active Expired - Lifetime
- 2002-05-13 HK HK02103601.3A patent/HK1041820B/en not_active IP Right Cessation
-
2003
- 2003-02-21 US US10/371,169 patent/US6849270B2/en not_active Expired - Lifetime
-
2005
- 2005-01-14 US US11/035,707 patent/US7285622B2/en not_active Expired - Lifetime
- 2005-08-12 US US11/202,913 patent/US7276248B2/en not_active Expired - Lifetime
-
2007
- 2007-09-19 US US11/903,199 patent/US7608687B2/en not_active Expired - Fee Related
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2254336A1 (en) * | 1973-12-17 | 1975-07-11 | Kyowa Hakko Kogyo Kk | |
| EP0317957A2 (en) * | 1987-11-23 | 1989-05-31 | Bristol-Myers Squibb Company | Drug-monoclonal antibody conjugates |
| EP0317956A2 (en) * | 1987-11-23 | 1989-05-31 | Bristol-Myers Squibb Company | Anti-tumor prodrugs |
| EP0510197A1 (en) * | 1990-01-11 | 1992-10-28 | Nippon Shinyaku Company, Limited | Fat emulsion |
| US5169934A (en) * | 1990-05-14 | 1992-12-08 | Anergen, Inc. | Intracellularly cleavable compounds |
| WO1997036904A1 (en) * | 1996-04-02 | 1997-10-09 | Sagami Chemical Research Center | Mitomycin c derivative and non-receptor tyrosine kinase inhibitor |
| WO1999029302A1 (en) * | 1997-12-05 | 1999-06-17 | Katarina Edwards | Drug delivery system with two-step targeting |
Non-Patent Citations (3)
| Title |
|---|
| DATABASE WPI Section Ch, Week 198923 Derwent Publications Ltd., London, GB; Class B02, AN 1989-170530 XP002159819 & JP 01 113391 A (KYOWA HAKKO KOGYO KK), 2 May 1989 (1989-05-02) * |
| DIAZ C ET AL: "Synthesis of disulfide-containing phospholipid analogs for the preparation of head group-specific lipid antigens: generation of phosphatidylserine antibodies" BIOCONJUGATE CHEMISTRY,US,AMERICAN CHEMICAL SOCIETY, WASHINGTON, vol. 9, no. 2, March 1998 (1998-03), pages 250-254, XP002125967 ISSN: 1043-1802 * |
| VAAGE J ET AL: "THERAPY OF PRIMARY AND METASTATIC MOUSE MAMMARY CARCINOMAS WITH DOXORUBICIN ENCAPSULATED IN LONG CIRCULATING LIPOSOMES" INTERNATIONAL JOURNAL OF CANCER, vol. 51, no. 6, 1992, pages 942-948, XP000979548 ISSN: 0020-7136 * |
Cited By (81)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7608687B2 (en) | 1999-04-23 | 2009-10-27 | Alza Corporation | Releasable linkage and compositions containing same |
| US7285622B2 (en) | 1999-04-23 | 2007-10-23 | Alza Corporation | Releasable linkage and compositions containing same |
| US7238368B2 (en) | 1999-04-23 | 2007-07-03 | Alza Corporation | Releasable linkage and compositions containing same |
| US7303760B2 (en) | 1999-04-23 | 2007-12-04 | Alza Corporation | Method for treating multi-drug resistant tumors |
| US7592307B2 (en) | 1999-04-23 | 2009-09-22 | Alza Corporation | Releasable linkage and compositions containing same |
| US7276248B2 (en) | 1999-04-23 | 2007-10-02 | Alza Corporation | Conjugate having a cleavable linkage for use in a liposome |
| US7108863B2 (en) | 2001-03-26 | 2006-09-19 | Alza Corporation | Liposome composition for improved intracellular delivery of a therapeutic agent |
| EP1385479A4 (en) * | 2001-03-26 | 2006-12-06 | Alza Corp | Liposome composition for improved intracellular delivery of a therapeutic agent |
| EP1385479A1 (en) * | 2001-03-26 | 2004-02-04 | Alza Corporation | Liposome composition for improved intracellular delivery of a therapeutic agent |
| US7211440B2 (en) | 2002-03-08 | 2007-05-01 | Wallac Oy | Dissociative fluorescence enhancement assay |
| US7682627B2 (en) * | 2002-12-03 | 2010-03-23 | Blanchette Rockefeller Neurosciences Institute | Artificial low-density lipoprotein carriers for transport of substances across the blood-brain barrier |
| JP2012131799A (en) * | 2002-12-03 | 2012-07-12 | Blanchette Rockefeller Neurosciences Inst | Artificial low-density lipoprotein carrier for transport of substance across blood-brain barrier |
| CN101284136A (en) * | 2002-12-03 | 2008-10-15 | 布朗歇特洛克菲勒神经科学研究所 | Artificial low-density lipoprotein carriers for transport of substances across the blood-brain barrier |
| US7576055B2 (en) * | 2002-12-03 | 2009-08-18 | Blanchette Rockefeller Neurosciences Institute | Artificial low-density lipoprotein carriers for transport of substances across the blood-brain barrier |
| AU2004247004B2 (en) * | 2003-04-30 | 2010-10-14 | Alza Corporation | Mitomycin conjugates cleavable by thiols |
| WO2004110497A2 (en) | 2003-04-30 | 2004-12-23 | Alza Corporation | Mitomycin conjugates cleavable by thiols |
| WO2004110497A3 (en) * | 2003-04-30 | 2005-03-24 | Alza Corp | Mitomycin conjugates cleavable by thiols |
| WO2005053749A3 (en) * | 2003-11-26 | 2006-11-09 | Alza Corp | Thiol-cleavable linkage between polymer and ligand |
| WO2005053749A2 (en) * | 2003-11-26 | 2005-06-16 | Alza Corporation | Thiol-cleavable linkage between polymer and ligand |
| US10576166B2 (en) | 2009-12-01 | 2020-03-03 | Translate Bio, Inc. | Liver specific delivery of messenger RNA |
| US10507183B2 (en) | 2011-06-08 | 2019-12-17 | Translate Bio, Inc. | Cleavable lipids |
| US10238754B2 (en) | 2011-06-08 | 2019-03-26 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for MRNA delivery |
| US9597413B2 (en) | 2011-06-08 | 2017-03-21 | Shire Human Genetic Therapies, Inc. | Pulmonary delivery of mRNA |
| US11234936B2 (en) | 2011-06-08 | 2022-02-01 | Translate Bio, Inc. | Cleavable lipids |
| US11185595B2 (en) | 2011-06-08 | 2021-11-30 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US9717690B2 (en) | 2011-06-08 | 2017-08-01 | Rana Therapeutics, Inc. | Cleavable lipids |
| US11291734B2 (en) | 2011-06-08 | 2022-04-05 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US11338044B2 (en) | 2011-06-08 | 2022-05-24 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US11052159B2 (en) | 2011-06-08 | 2021-07-06 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US10888626B2 (en) | 2011-06-08 | 2021-01-12 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US10702478B2 (en) | 2011-06-08 | 2020-07-07 | Translate Bio, Inc. | Cleavable lipids |
| US10507249B2 (en) | 2011-06-08 | 2019-12-17 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US10413618B2 (en) | 2011-06-08 | 2019-09-17 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US10350303B1 (en) | 2011-06-08 | 2019-07-16 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US11547764B2 (en) | 2011-06-08 | 2023-01-10 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for MRNA delivery |
| US9308281B2 (en) | 2011-06-08 | 2016-04-12 | Shire Human Genetic Therapies, Inc. | MRNA therapy for Fabry disease |
| US11730825B2 (en) | 2011-06-08 | 2023-08-22 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US10786455B2 (en) | 2012-03-29 | 2020-09-29 | Translate Bio, Inc. | Lipid-derived neutral nanoparticles |
| US10137086B2 (en) | 2012-03-29 | 2018-11-27 | Translate Bio, Inc. | Lipid-derived neutral nanoparticles |
| US9877919B2 (en) | 2012-03-29 | 2018-01-30 | Translate Bio, Inc. | Lipid-derived neutral nanoparticles |
| US11497716B2 (en) | 2012-03-29 | 2022-11-15 | Translate Bio, Inc. | Lipid-derived neutral nanoparticles |
| US10137087B2 (en) | 2012-03-29 | 2018-11-27 | Translate Bio, Inc. | Lipid-derived neutral nanoparticles |
| US11254936B2 (en) | 2012-06-08 | 2022-02-22 | Translate Bio, Inc. | Nuclease resistant polynucleotides and uses thereof |
| US10420791B2 (en) | 2013-03-14 | 2019-09-24 | Translate Bio, Inc. | CFTR MRNA compositions and related methods and uses |
| US10876104B2 (en) | 2013-03-14 | 2020-12-29 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US11510937B2 (en) | 2013-03-14 | 2022-11-29 | Translate Bio, Inc. | CFTR MRNA compositions and related methods and uses |
| US9713626B2 (en) | 2013-03-14 | 2017-07-25 | Rana Therapeutics, Inc. | CFTR mRNA compositions and related methods and uses |
| US11820977B2 (en) | 2013-03-14 | 2023-11-21 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US11692189B2 (en) | 2013-03-14 | 2023-07-04 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US9181321B2 (en) | 2013-03-14 | 2015-11-10 | Shire Human Genetic Therapies, Inc. | CFTR mRNA compositions and related methods and uses |
| US9957499B2 (en) | 2013-03-14 | 2018-05-01 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US11890377B2 (en) | 2013-10-22 | 2024-02-06 | Translate Bio, Inc. | Lipid formulations for delivery of messenger RNA |
| US9522176B2 (en) | 2013-10-22 | 2016-12-20 | Shire Human Genetic Therapies, Inc. | MRNA therapy for phenylketonuria |
| US10959953B2 (en) | 2013-10-22 | 2021-03-30 | Translate Bio, Inc. | Lipid formulations for delivery of messenger RNA |
| US10052284B2 (en) | 2013-10-22 | 2018-08-21 | Translate Bio, Inc. | Lipid formulations for delivery of messenger RNA |
| US11377642B2 (en) | 2013-10-22 | 2022-07-05 | Translate Bio, Inc. | mRNA therapy for phenylketonuria |
| US10208295B2 (en) | 2013-10-22 | 2019-02-19 | Translate Bio, Inc. | MRNA therapy for phenylketonuria |
| US10493031B2 (en) | 2013-10-22 | 2019-12-03 | Translate Bio, Inc. | Lipid formulations for delivery of messenger RNA |
| US11224642B2 (en) | 2013-10-22 | 2022-01-18 | Translate Bio, Inc. | MRNA therapy for argininosuccinate synthetase deficiency |
| US9629804B2 (en) | 2013-10-22 | 2017-04-25 | Shire Human Genetic Therapies, Inc. | Lipid formulations for delivery of messenger RNA |
| US11884692B2 (en) | 2014-04-25 | 2024-01-30 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US10155785B2 (en) | 2014-04-25 | 2018-12-18 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US11059841B2 (en) | 2014-04-25 | 2021-07-13 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US9850269B2 (en) | 2014-04-25 | 2017-12-26 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US10286083B2 (en) | 2014-05-30 | 2019-05-14 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US10286082B2 (en) | 2014-05-30 | 2019-05-14 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US10022455B2 (en) | 2014-05-30 | 2018-07-17 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US10293057B2 (en) | 2014-05-30 | 2019-05-21 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US10493166B2 (en) | 2014-05-30 | 2019-12-03 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US11433144B2 (en) | 2014-05-30 | 2022-09-06 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US10912844B2 (en) | 2014-05-30 | 2021-02-09 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US11104652B2 (en) | 2014-06-24 | 2021-08-31 | Translate Bio, Inc. | Stereochemically enriched compositions for delivery of nucleic acids |
| US10138213B2 (en) | 2014-06-24 | 2018-11-27 | Translate Bio, Inc. | Stereochemically enriched compositions for delivery of nucleic acids |
| GB2552301A (en) * | 2016-07-11 | 2018-01-24 | Evox Therapeutics Ltd | Metabolic drug loading of EVs |
| US11253605B2 (en) | 2017-02-27 | 2022-02-22 | Translate Bio, Inc. | Codon-optimized CFTR MRNA |
| US11173190B2 (en) | 2017-05-16 | 2021-11-16 | Translate Bio, Inc. | Treatment of cystic fibrosis by delivery of codon-optimized mRNA encoding CFTR |
| WO2019016208A1 (en) * | 2017-07-17 | 2019-01-24 | Technische Universiteit Eindhoven | Applicable chemical composition comprising an agent conjugated to a hydrophobic moiety and a carrier |
| US11174500B2 (en) | 2018-08-24 | 2021-11-16 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US11951181B2 (en) | 2023-04-03 | 2024-04-09 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US11951180B2 (en) | 2023-04-03 | 2024-04-09 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for MRNA delivery |
| US11951179B2 (en) | 2023-04-03 | 2024-04-09 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for MRNA delivery |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6365179B1 (en) | Conjugate having a cleavable linkage for use in a liposome | |
| US7238368B2 (en) | Releasable linkage and compositions containing same | |
| AU2002305094B2 (en) | Liposome composition for improved intracellular delivery of a therapeutic agent | |
| KR100617921B1 (en) | Liposome composition and method for administering a quinolone | |
| US20040022842A1 (en) | Liposome preparations containing oxaliplatin | |
| Karathanasis et al. | Preparation of in vivo cleavable agglomerated liposomes suitable for modulated pulmonary drug delivery | |
| EP1426044A1 (en) | Use of esters of L-carnitine or alkanoyl L-carnitines as cationic lipids for the intracellular delivery of pharmacologically active compounds | |
| AU2004247004B2 (en) | Mitomycin conjugates cleavable by thiols | |
| AU770390B2 (en) | Releasable linkage and compositions containing same | |
| JPH06228012A (en) | Liposome preparation | |
| WO2005069750A2 (en) | Therapeutic composition with nanoscale activation agents |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 00807901.3 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 2369595 Country of ref document: CA Ref document number: 2369595 Country of ref document: CA Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2000 613474 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2000928321 Country of ref document: EP Ref document number: 2001/08726 Country of ref document: ZA Ref document number: 514991 Country of ref document: NZ Ref document number: PA/a/2001/010750 Country of ref document: MX Ref document number: 46577/00 Country of ref document: AU Ref document number: 1020017013571 Country of ref document: KR Ref document number: 200108726 Country of ref document: ZA |
|
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): GH GM KE LS MW SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020017013571 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2000928321 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWG | Wipo information: grant in national office |
Ref document number: 46577/00 Country of ref document: AU |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2000928321 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1020017013571 Country of ref document: KR |