METHOD FOR TREATING CARTILAGE DISORDERS
Field of the Invention
The present invention relates generally to the treatment of cartilage disorders, including stimulation of cartilage repair and treatment of degenerative cartilagenous disorders.
Background of the Invention
Degenerative cartilagenous disorders broadly describe a collection of diseases characterized by degeneration or metabolic abnormalities of the connective tissues that are manifested by pain, stiffness and limitation of motion of the affected body parts. The origin of these disorders can be pathological or as a result of trauma or injury.
Osteoarthritis (OA) , also known as osteoarthrosis or degenerative joint disease, is the result of a series of localized degenerative processes that affect the articular structure and result in pain and diminished function. The incidence of OA increases with age, and evidence of OA involvement can be detected in some joints in the majority of the population by age 65. OA is often also accompanied by a local inflammatory component that may accelerate joint destruction.
OA is characterized by disruption of the smooth articulating surface of cartilage, followed by formation of clefts 'and fibrillation, and ultimately by the full-thickness loss of the cartilage. Coincident with the cartilaginous changes are alterations of the periarticular bone. These include the development of palpable bone enlargements at the joint margins and deformity resulting from assymetric cartilage destruction. OA symptoms include local pain at the affected joints, especially after use. With disease progression, symptoms may develop into a continuous aching sensation, local discomfort, and cosmetic alterations of the affected joint.
In contrast to the localized disorder OA, rheumatoid arthritis (RA) is a systematic destructive and debilitating disease that is believed to begin in the synovium, the tissues surrounding the joint. The prevalance of RA is about 1/6 that of OA in the general population of the United States. It is a chronic autoimmune disorder characterized by symmetrical synovitis of the joint and typically affects small and large diarthrodial joints, leading to their progressive destruction. As the disease progresses, the symptoms of RA may also include fever, weight loss, thinning of the skin, multi-organ involvement, scleritis, corneal ulcers, the formation of subcutaneous or subperiosteal nodules, and premature death.
The response of normal patients (e.g., preinjury or predisease) to injury or arthritic degeneration is often sub-optimal. The biochemical and mechanical properties of this damaged cartilage differ from those of normal cartilage, resulting in inadeguate or altered function. This damaged
cartilage, termed herein "fibrocartilage, " does not approximate the durability and function of normal cartilage.
Since cartilage is avascular and mature chondrocytes have little intrinsic potential for replication, mature cartilage has limited ability for repair. Thus, damage to the cartilage layer that does not penetrate to the subchondral bone does not undergo efficient repair. In contrast, when the subchondral bone is penetrated, its vascular supply allows a triphasic repair to take place. The resulting tissue is usually mechanically sub- optimal fibrocartilage. The degradation associated with osteoarthritis usually initially appears as fraying and fibrillation of the surface. Loss of proteoglycan from the matrix also occurs. As the surface fibrillation progresses, the defects penetrate deeper into the cartilage, resulting in loss of cartilage cells and matrix. The subchondral bone thickens, is slowly exposed, and may appear polished. Bony nodules or osteophytes also often form at the periphery of the cartilage surface and occasionally grow over the adjacent eroded areas. If the surface of these bony outgrowths is permeated, vascular ■ outgrowth may occur and cause the formation of tissue plugs containing fibrocartilage. The transplantation of chondrocytes is known as a means of stimulating cartilage repair. However, the possibility of the host's immunogenic response as well as the possible transmission of viral and other infectious diseases makes this method less desirable. These risks can be minimized to some extent with allograft and autogenous transplants; however, the culturing and growth of patient-specific cells is cost prohibitive on a mass scale .
Other methods of stimulating cartilage repair include the antagonism of molecules that are associated with or aggravate cartilage destruction, for example, interleukin-1-alpha (IL-lα) and nitric oxide (NO) . The cytokine IL-lα has catabolic effects on cartilage, including the generation of synovial inflammation and up-regulation of matrix metalloproteinases and prostaglandin expression (Baragi et al . , J. Clin. Invest., 56: 2454-24'60 (1995); Baragi et al . r Osteoarthritis Cartilage, 5: 275-282 (1997); Evans et al . r J. Keukoc. Biol., 64: 55-61 (1998); Evans and Robbins, J. Rheumatol., 24: 2061-2063 (1997); Kang et al . , Biochem. Soc. Trans., 25: 533-537 (1997); Kang et al . , Osteoarthritis Cartilage, 5: 139-143 (1997)). One mean's of antagonizing IL-lα is through application of soluble IL-1 receptor antagonist (IL-lra), a naturally-occurring protein that inhibits the effects of IL-1 by preventing IL-1 from binding to and activating its receptor on chondrocytes and synoviocytes, thereby lowering the effective concentration of IL-1.
NO plays a substantial role in the destruction of cartilage (Ashok et al . , Curr. Opin. Rheum., 10: 263-268 (1998)). Cartilage obtained from osteoarthritic joints endogenously produces large amounts of NO. Normal cartilage does not produce NO unless stimulated with cytokines such as IL-1, while osteoarthritic cartilage explants continue to express NO synthase for up to 3 days in culture despite the absence of added stimuli. Moreover, the inhibition of NO has been shown to prevent IL-lα-mediated cartilage destruction and chondrocyte death as well as the progression of osteoarthritis . The ability of peptide growth factors to promote repair of damaged cartilage has also been examined. Peptide growth factors are very significant regulators of cartilage growth and cell behavior (i.e., differentiation, migration, division, or matrix synthesis and/or breakdown) (Chen et a_Z . , Am J. Orthop . , 26: 396-406 (1997)). These factors are under investigation for their potential to induce host cartilage repair without transplantation of cells, and are being incorporated into engineered devices for implantation.
Because growth factors are soluble proteins of relatively small molecular mass that are rapidly absorbed and/or degraded, a great challenge exists in making them available to cells in sufficient quantity and for sufficient duration. It is likely desirable to have different factors present at the repair site during different parts of the developmental cycle, and for varying lengths of time. The ideal delivery vehicle is biocompatible and resorbable, has the appropriate mechanical properties, and results in no harmful degradation products. Growth factors that previously have been proposed to stimulate cartilage repair include insulin-like growth factor-I (IGF-1) (Osborn, J. Orthop. Res., 7: 35-42 (1989); Florini and Roberts, J. Gerontol., 35: 23-30 (1980); U.S. Pat. No. 5,843,899), basic fibroblast growth factor (bFGF) , [Toolan et al . , J. Biomec. Mat. Res., 41: 244-50 (1998); Sah et al . , Arch. Biochem. Biophys., 308: 137-47 (1994)), bone morphogenetic protein (BMP) (Sato and Urist, Clin. Orthop. Relat. Res., 183: 180-187 (1984); Chin et al . , Arthritis Rheum. 34: 314-324 (1991)), and transforming growth factor beta (TGF-β) (Hill and Logan, Prog. Growth Fac. Res . , 4: 45-68 (1992); Guerne et al . , J. Cell Physiol., 158: 476-484 (1994); Van der Kraan et al . , Ann. Rheum. Pis., 51: 643-647 (1992)).
It has been well established that the GH/IGF/IGFBP system is involved in the regulation of anabolic and metabolic homeostasis and that defects in this system may adversely affect growth, physiology, and glycemic control (Jones et al . , Endocr. Rev., 16: 3-34 (1995); Davidson, Endocr. Rev., ,8: 115-131 (1987); Moses, Curr. Opin. Endo. Diab., 4: 16-25 (1997)). It has been proposed that IGF-1 could be useful for the treatment or prevention of
osteoarthritis, because of its ability to stimulate both matrix synthesis and cell proliferation in culture (Osborn, J. Orthop. Res., 1_: 35-42 (1989) ) . IGF-1 has been administered with sodium pentosan polysulfate (PPS) (a chondrocyte σatabolic activity inhibitor) to severely osteoarthritic canines with the effect of reducing the severity of the disease perhaps by lowering the levels of active neutral metalloprotemase m the cartilage. In the model of mildly osteoarthritic canines, therapeutic intervention with IGF-1 and PPS together appeared to successfully maintain cartilage structure and biochemistry, while IGF alone was ineffective, as described m Rogachefsky, Osteoarthritis and Cartilage, 1: 105-114 (1993), Rogachefsky et al . , Ann NY Acad. Sc ., 732: 889-895 (1994). The use of IGF-1 either alone or as an adjuvant with other growth factors to stimulate cartilage regeneration has been described in WO 91/19510, WO 92/13565, US 5,444,047, and EP 434,652. IGF-1 has also been found useful in the treatment of osteoporosis m mammals exhibiting decreased bone mineral density and those exposed to drugs or environmental conditions that result m bone density reduction and potentially osteoporosis, as described m EP 560,723 and EP 436,469.
IGF-1 insufficiency may have an etiologic role m the development of osteoarthritis (Coutts et al . , "Effect of growth factors on cartilage repair," Instructional Course Lect., 47 : 487-494 (Amer. Acad. Orthop. Surg. : Rosemont, IL 1997)). Some studies indicate that serum IGF-1 concentrations are lower m osteoarthπtc patients than control groups, while other studies have found no difference Nevertheless, it has been shown that both serum IGF-1 levels and chrondrocyte responsiveness to IGF-1 decrease with age, with the latter likely due to high levels of IGF binding proteins (IGFBPs) (Floπni and Roberts, J. Gerontol., 35: 23-30 (1980); Martin et al . , J. Orthop. Res., 15: 491-498 (1997); Fernihough et al Arthr . Rheum. 39- 1556- 1565 (1996)). Thus, both the decreased availability of IGF-1 as well as diminished chondrocyte responsiveness/disregulation of IGFBPs thereto may contribute to the impaired cartilage matrix homeostasis and tissue degeneration that occurs with advancing age and disease.
Of the IGFBPs, IGFBP-3 appears to be the most responsible for regulating the total levels of IGF-1 and IGF-2 m plasma. IGFBP-3 is a GH- dependent protein and is reduced m cases of ■■ GH-deficiency or resistance (Jones et al . , supra; Rosenfield et al . , "IGF-1 treatment of syndromes of growth hormone msens tivity" In: The msulm-l ke growth factors and their regulatory proteins, Eds Baxter RC, Gluckman PD, Rosenfield RG. Excerpta Medica, Amsterdam, 1994), pp 357-464; Scharf et al . , J Hepatology, 25- 689- 699 (1996) ) . IGFBPs are able to enhance or inhibit IGF activity, depending largely on their post-translational modifications and tissue localization
(reviewed in Jones and Clemmons, Endocr. Rev. _16:3-34 (1995); Collet-Solberg and Cohen, Endocrinol. Metabol. Clin. North Am. 25:591-614 (1996)). In addition, disregulation in IGFBPs (-3, -4 and/or -5) may play a key role in arthritic disorders (Chevalier and Tyler, Brit. J. Rheum. 35 : 515-522 (1996); Olney et al . , J. Clin. Endocrinol. Metab. 81: 1096-1103 (1996); Martel-Pelletier et al . , Inflamm. Res., 47: 90-100 (1998)). It has been reported that IGF-1 analogs with very low binding affinity for IGFBPs were more effective than wild-type IGF-1 in stimulating proteoglycan synthesis (Morales, Arch Biochem. Biophys. 324, 173-188 (1997)). More recent data, however, suggest that IGFBPs contribute to IGF binding to and transport through cartilage tissue, and IGFBPs may thus regulate bioavailability of IGF-1 within the joint (Bhakta et al . , J. Biol. Chem., 275: 5860-5866 (2000)) .
The biodistribution of IGF-1 critically depends on (a) the formation of long-lived high molecular weight complexes and (b) the absolute IGFBP concentrations. The majority of IGF-1 in the circulation is found in complex with IGFBP-3 and a third protein termed acid-labile subunit (ALS) (Bach and Rechler, Diabetes Reviews, 3: 38-61 (1995) ; Clemmons, Cytokine Growth Factor Rev., 8_: 45-62 (1997); Jones and Clemmons, Endocr. Rev., 16: 3-34 (1995) ) . This ternary complex of 150-kD molecular weight is unable to traverse the vasculature walls and acts as a circulating reservoir for IGF's. As a consequence, the serum half-life of IGF-1 in ternary complexes is reported to be 12-15 hours, as opposed to 30 minutes in binary complexes, or 10 minutes in the free form (Simpson et al . , Growth Horm IGF Res, 8_: 83- 95 (1998); Twigg and Baxter, J. Biol. Chem., 273: 6074-6079 (1998)).
IGFBP-3 and -5 are apparently unique in their ability to form a ternary complex with ALS. ALS association occurs only in the presence of IGF-1, and a basic motif in the carboxy-terminal domains of IGFBP-3 and -5 seems to mediate this interaction (Baxter et al . τ J. Biol. Chem., 267 : 60-65 (1992); Firth et al . , J. Biol. Chem., 273: 2631-2638 (1998); Twigg and Baxter, supra ) .
The second determinant of IGF-1 biodistribution is the total concentration of binding proteins: IGFBP-3 is the most abundant binding protein, followed by IGFBP-1 and -2 levels, whereas the serum concentrations of IGFBP-4, -5, and -6 are quite low (Clemmons, Cytokine Growth Factor Rev., 8_: 45-62 (1997)). IGFBP-3 therefore represents the main IGF-1 carrier in the blood. In contrast, a substantial portion of IGFBP-1 and -2 in the blood are unoccupied. Hence, they appear to be the major modulators of free IGF-1 levels (Clemmons, 1997, supra ) . WO 94/04569 discloses a specific binding molecule, other than a natural IGFBP, that is capable of binding to IGF-1 and can enhance the
biological activity of IGF-1. WO 98/45427 published October 15, 1998; Lowman et al . , Biochemistry, 37: 8870-8878 (1998); and Dubaquie and Lowman, Biochemistry, 38 : 6386 (1999) disclose IGF-1 agonists identified by phage display. Also, WO 97/39032 discloses ligand inhibitors of IGFBP' s and methods for their use. Further, U.S. Pat. No. 5,891,722 discloses antibodies having binding affinity for free IGFBP-1 and devices and methods for detecting free IGFBP-1 and a rupture in a fetal membrane based on the presence of amniotic fluid in a vaginal secretion, as indicated by the presence of free IGFBP-1 in the vaginal secretion. WO 00/23469 published April 27, 2000 discloses fragments of IGFBPs and analogs of IGF-1 for use in, e.g., cancer, ischemic injury, and diabetes treatment.
There exists a continuing need for an effective therapy for the treatment and repair of cartilage, including cartilage damaged as a result of injury and/or disease. Summary of the Invention
Accordingly, the present invention concerns a method of treating a cartilage disorder as claimed, comprising contacting cartilage with an effective amount of an active agent selected from an IGF-1 analog with a binding affinity preference for IGFBP-3 over IGFBP-1, an IGF-1 analog with a binding affinity preference for IGFBP-1 over IGFBP-3, or an IGFBP displacer peptide that prevents the interaction of IGF with IGFBP-3 or IGFBP-1 and does not bind to a human IGF receptor. Preferably, the cartilage is treated in vivo in a mammal and the active agent is administered to the mammal. Also, the active agent is optionally contacted with the cartilage in an extended-release form and/or administered locally to the joint alone or, if the active agent is an IGFBP displacer peptide or IGF-1 analog with a preference for IGFBP-3 over IGFBP-1, together with IGF-1 and/or ALS, preferably human, native-sequence IGF-1 if the mammal is human.
Preferably, the active agent is an IGF-1 variant wherein the amino acid residue at position 3, 7, 10, 16, 25, or 49, or the amino acid residues at positions 3 and 49 of native-sequence human IGF-1 are replaced with an alanine, a glycine, or a serine residue, or an IGF-1 variant wherein the amino acid residue at position 9, 12, 15, or 20 is replaced with a lysine or arginine residue, or an IGFBP-3 displacer peptide designated as: Y24LY31A IGF-1; 4D3.3P; BP3-4D3.il; BP3-4D3.11DEL; BP3-4B3.3; BP3-01-OX; BP3-02-OX; BP3-06; BP3-08; BP3-15; BP3-16; BP3-17; BP3-25; BP3-27; BP3-28; BP3-30; BP3- 39; BP3-40; BP3-41; BP3-107; or BP3-108; or an IGFBP-1 displacer peptide designated as: BP1-01; BP1-02; BP1-04; BP1-10; BP1-11; BP1-12; BP1-13; BP1- 14; BP1-15; BP1-16; BP1-17; BP1-18; BP1-19; BP1-20; BP1-21A; BP1-21B; BPl- 25; BP1-40; BP67; BP68; BP1-625; BP1-625-Z; BP1-625T; BP1027; BP1028; BP1029; BP1030; (i+7)D; (i+8)B; and (i+8)C.
The letter followed by a number followed by a letter indicates an IGF- 1 analog wherein the amino acid letter to the left of the number is the original amino acid in native-sequence human IGF-1, the number is the position where the amino acid is changed, and the amino acid letter to the right of the number is the substituted amino acid. Hence, for example, F49A indicates an IGF-1 variant wherein the phenylalanine residue at position 49 of native-sequence human IGF-1 is changed to an alanine residue, and E3AF49A indicates an IGF-1 variant wherein the glutamine residue at position 3 of native-sequence human IGF-1 is changed to an alanine residue, and the phenylalanine residue at position 49 of native-sequence human IGF-1 is changed to an alanine residue.
In another embodiment, the above method is for the treatment of cartilage damaged or diseased as a result of a degenerative cartilagenous disorder. Preferably, the disorder is an articular cartilage disorder, and most preferably is OA or RA.
In a further embodiment, the above method is for the treatment of joints damaged directly or indirectly by injury, preferably microdamage or blunt trauma, a chondral fracture, an osteochondral fracture.
Optionally, the invention concerns the above treatment method wherein the cartilage is contacted with an effective amount of the IGF-1 analog or IGFBP displacer peptide as defined above in combination with an effective amount of a cartilage growth factor or cartilage catabolism antagonist.
In another embo'diment, the invention concerns a method of maintaining, enhancing, or promoting the growth of chondrocytes in serum-free culture by contacting the chondrocytes with an effective amount of an IGF-1 analog or an IGFBP displacer peptide as identified above. Alternatively, the method concerns contacting the chondrocyte with an effective amount of an IGF-1 analog or an IGFBP displacer peptide in an extended-release formulation. Alternatively, the present invention concerns a method of stimulating the regeneration or preventing the degradation of cartilage resulting from injury or a degenerative cartilagenous disorder by transplantation of an effective amount of chondrocytes previously treated with an effective amount of an IGF-1 analog or an IGFBP displacer peptide as defined above.
In another embodiment, the present invention concerns an article of manufacture comprising a container holding an IGF-1 analog or an IGFBP displacer peptide as defined above in a pharmaceutically acceptable carrier with instructions for its use in treating a cartilage disorder.
Brief Description of the Drawings Figures 1A-1C depict the DNA sequence (SEQ ID N0:1) of plasmid pt4.g8 used as a template to construct a phage library. Also shown is the amino
acid sequence (SEQ ID NO: 2) of an antibody-recognizable (gD-tag) peptide fused to gδp of bacteriophage M13.
Figure 2 shows gene-8 naive phage library enrichments with a selection using four library pools each and the targets IGF-1, IGFBP-1, and IGFBP-3. Figure 3 shows an IGF-1 blocking assay using g8-phage peptides from IGFBP-3 selections, where the phage titration is with 100 nM IGF-1. In the Figure, the open circles are peptide A3.1, the open triangles are peptide 4B3.4, the open squares are peptide 4C3.2, the solid circles are peptide 4D3.3, the solid triangles are peptide 4D3.4, and the solid squares are peptide 4D3.5.
Figure 4 shows an IGF-1 blocking assay using g8-phage peptides from IGFBP-3 selections, where the phage titration is without IGF-1. The designations for the peptides are the same as those described above for Fig. 3. Figure 5 shows an IGF-1 blocking assay using g8-phage peptides from IGFBP-3 selections, where the peptides (4C3.2, 4D3.8, 4D3.9, 4D3.11, and
4D3.12) are from a NEϋTRAVIDIN™/DTT selection. The solid bars are with 100 μM IGF-1 and the open bars are without IGF-1.
Figure 6 shows an IGF-1 blocking assay using g8-phage peptides from IGFBP-3 selections where the peptides (indicated on the x axis) are from direct-coat/HCl selection. The solid bars are with 100 μM IGF-1 and the open bars are without IGF-1.
Figure 7 depicts a competition assay of IGFBP-3 inhibition by a peptide binding to IGFBP-3 (designated BP3-01) using a BIACORE™ surface-plasmon-resonance device to measure free binding protein. The circles indicate 800 response units (RU) of IGF-1 and the squares indicate 400 RU of immobilized IGF-1.
Figure 8 depicts a competition assay of IGFBP-3 inhibition by a peptide binding to IGFBP-3 (designated BP3-02) using a BIACORE™ surface-plasmon-resonance device to measure free binding protein. The circles indicate 800 RU of IGF-1 and the squares indicate 400 RU of immobilized IGF-1.
Figure 9 shows a radiolabeled IGF-1 plate assay of the ability of two peptides that bind to IGFBP-3 but not to the Type 1 IGF receptor (BP3-01-ox: circles, and BP3-02-ox: squares) to inhibit IGFBP-3.
Figure 10 shows a radiolabeled IGF-1 plate assay of the ability of the two IGFBP-3 binding peptides described for Fig. 9 to inhibit IGFBP-1 (symbols are the same) .
Figures 11A-11D depict KIRA assays of IGF-1 activity using three peptides (BP1-01: squares, BPl-02: circles, and BP03-ox: triangles). Fig.
11A depicts the peptides alone, Fig. 11B depicts the peptides plus IGF-1 plus IGFBP-1, Fig. 11C depicts the peptides plus IGF-1, and Fig. 11D depicts the peptides plus IGF-1 plus IGFBP-3.
Figure 12 depicts an IGF-2 competition assay of IGFBP-3 inhibition by four peptides, designated BP3-01-ox (open squares) , BP3-14 (open circles) ,
BP3-15 (closed circles), and BP3-17 (closed squares), using a BIACORE™ surface-plasmon-resonance device to measure free binding protein. Each peptide was tested using 20 nM IGFBP-3 and approximately 1500 RU of immobilized IGF-2. Figures 13A and 13B show a phage ELISA of the variant, G1S-A70V IGF-1, binding to IGFBP-1 (Fig. 13A) and IGFBP-3 (Fig. 13B) . Microtiter plates coated with 1 μg/ml IGFBP-1 (Fig. 13A) or IGFBP-3 (Fig. 13B) were incubated with phage particles displaying G1S-A70V m the presence of the indicated amounts of soluble competitor protein, IGFBP-1 (Fig. 13A) or IGFBP-3 (Fig. 13B) . The half-maximal inhibitory concentration (IC50) of competitor, i . e. , the inhibitory concentration of competitor that resulted m half-maximal binding of the phagemid m that particular experiment, is denoted for the respective IGFBP.
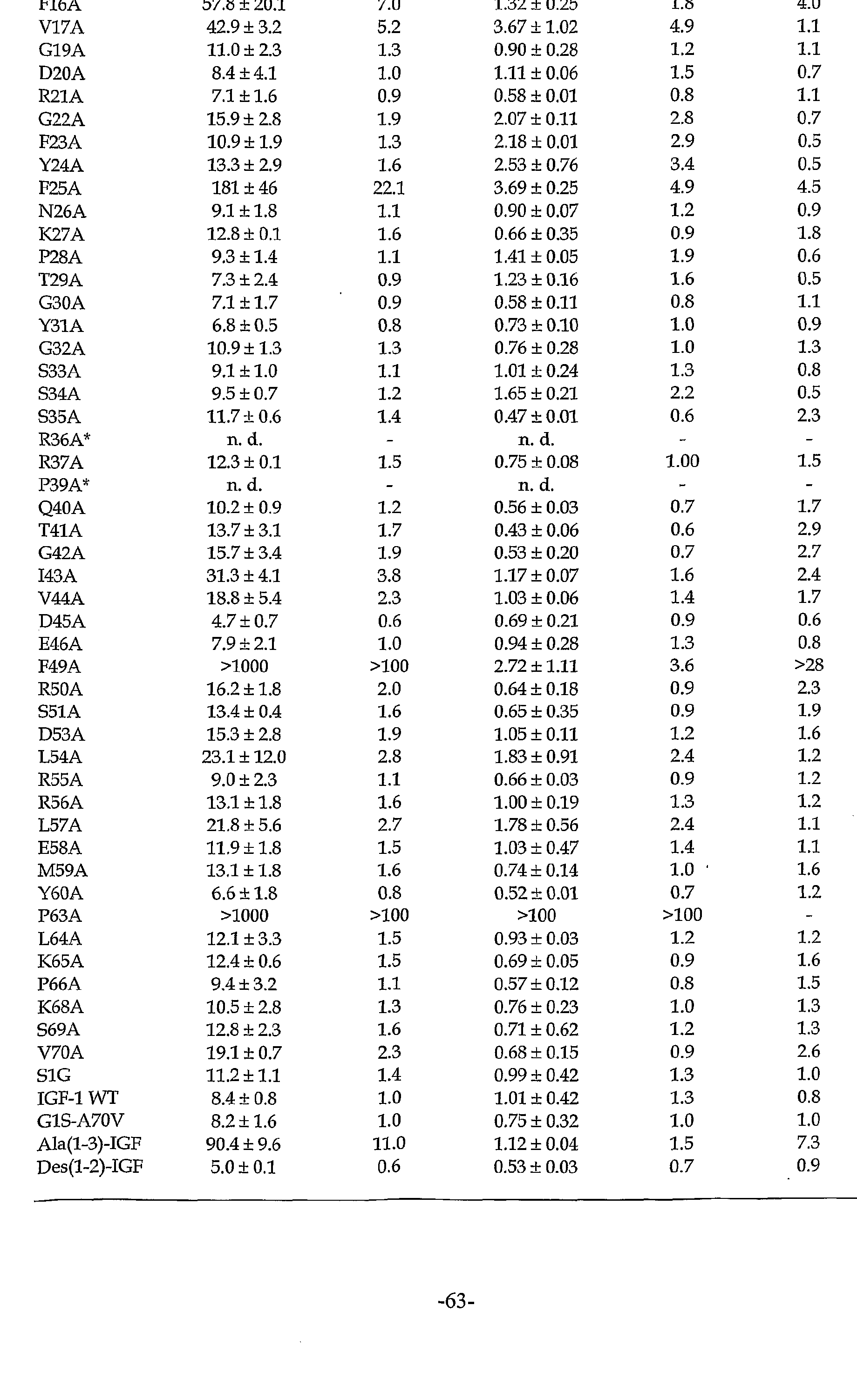
Figure 14 shows the loss or gam of IGFBP affinity for the IGF-1 mutants tested by phage ELISA. Relative IC50 values (ICsomu/iCso GIS-A7OV) of each IGF-1 alanine mutant (affinity changes of each mutant for the binding proteins with respect to IGF-1 G1S-A70V) are shown for IGFBP-1 (filled bars) and IGFBP-3 (open bars) . Data are taken from Table I below. Relative IC50 values <1 denote gain of affinity; values >1 denote loss of affinity. The asterisk indicates that these particular variants were not displayed on phage, as judged by antibody binding.
Figures 15A and 15B show binding specificity of the IGF-1 variant F49A displayed on phage to IGFBP-1 and -3, respectively, in competitive-phage ELISA. Phagemid particles displaying F49A (squares) were bound to plates coated with IGFBP-3 in the presence of the indicated amounts of soluble IGFBP-1 (Fig. 15A) or IGFBP-3 (Fig. 15B) . The same experiment was carried out m parallel with phage displaying the wild-type-like IGF-1 variant G1S- A70V (circles). See Tables I and II below for absolute IC5o values. Data points are mean + standard deviation, n=2. Immunosorbent plates were coated with 1 μg/ml IGFBP-3 and ELISA were carried out as described m the Examples below using wild-type IGF-1 phage (WT, circles) and IGF-F49A phage (F49A, squares) m parallel. Experiments were carried out m duplicate, and data points are shown as mean ± standard deviation.
Figure 16 discloses a sequence alignment of native-sequence human IGF- 1 (designated wtlGF) (SEQ ID NO:3), native-sequence human proinsulm (designated proinsulm) (SEQ ID NO:4), and native-sequence human insulin
(designated insulin (B chain) followed by insulin (A chain)) (SEQ ID NO: 5). The asterisks and dots indicate sequence identity and sequence similarity, respectively, at the indicated amino acid positions among the three sequences. Figures 17A-17D show a biosensor analysis of IGFBP binding to immobilized IGF-1 variants. Sensorgrams are shown for IGFBP-1 (Figs 17A, 17C) or IGFBP-3 (Figs. 17B, 17D) binding to immobilized wild-type IGF-1 (Figs. 17A, 17B) or F49A IGF variant (Figs. 17C, 17D) . The concentrations of ligand in each experiment were 1 μM, 500 nM, and 250 nM. See Table II for kinetic parameters.
Figures 18A-18B show a model of the functional binding epitopes for IGFBP-1 and IGFBP-3, respectively, on the surface of IGF-1. Amino acid side chains were classified according to their relative contribution in binding energy (Table I) and colored as follows: no effect (grey); 2-5 fold loss of apparent affinity (yellow) ; 5-10 fold (orange) ; 10-100 fold (bright red) ; > 100 fold (dark red) . If available, numbers from phage ELISA experiments in Table I below were used. BIACORE™ data were used instead for V11A, R36A, and P39A variants (Table II). The NMR structure of IGF-1 (Cooke et al . , Biochemistry, 30 : 5484, (1991) ) was represented using the program Insight II™ (MSI, San Diego, CA) . The binding epitope for IGFBP-1 (Fig. 18A) is located on the "upper" and "lower" face of the N-terminal helix (residues 8- 17), connected by the energetically-important residue F49. For IGFBP-3 (Fig. 18B) , individual IGF-1 side chains contribute very little binding energy. The binding epitope has shifted away from the N-terminus and newly includes G22, F23, Y24.
Figure 19 shows the amount of bound IGFBP-1, determined in a competitive BIACORE™ binding experiment, plotted against the IGF variant concentration for E3A/F49A (squares) and F49A (circles) .
Figures 20A and 20B show, respectively, the calculated IGF-1 activity in nM units for several IGF-1 variants at 13 nM (high) and 1.3 nM (low) variant concentrations using IGF-1 KIRA optical density analysis. The signal obtained for each IGF variant was compared to that of a standard- dilution series of wild-type IGF-1, and reported in terms of an apparent IGF-1 concentration corresponding to the observed activity. Figures 21A and 21B show IGF receptor activation curves for F49A IGF-1
(Fig. 21A) and E3A/F49A (Fig. 21B) as well as for wild-type IGF-1, as measured using serial dilutions in KIRA assays. The variants are represented by squares and the wild-type IGF-1 is represented by circles.
Figures 22A and 22B show an assessment of preliminary pharmacological properties of F49A and E3A/F49A IGF-1, radiolabeled and administered intravenously to rats. Figure 22A shows a time course of the rate at which
both molecules are cleared from the blood of the animals, where the squares represent wild-type IGF-1, the circles represent E3A/F49A IGF-1, and the diamonds represent F49A IGF-1. Figure 22B shows the tissue-to-blood ratio for these two IGF variants in different organs, namely, kidney, liver, spleen, heart, and pancreas, at 5, 15, and 30 minutes, where the solid bars represent wild-type IGF-1, the dotted bars represent E3A/F49A IGF-1, and the striped bars represent F49A IGF-1.
Figure 23 shows circular dichroism spectra of wild-type IGF-1 (circles), F49A IGF-1 (squares), and E3A/F49A IGF-1 (diamonds). Figure 24 is a bar graph showing the effect of control, wild-type IGF- 1, F49A, and E3A/F49A (at a concentration of 40 or 400 ng/ml) on cartilage matrix breakdown (proteoglycan release at 72 hours) .
Figure 25 is a bar graph showing the effect of wild-type IGF-1, F49A, and E3A/F49A (at a concentration of 40 or 400 ng/ml) on ILlα-induced cartilage breakdown at 72 hours.
Figure 26 is a bar graph showing the effect of control, wild-type IGF- 1, F49A, E3A/F49A (at a concentration of 40 or 400 ng/ml) on matrix synthesis .
Figure 27 is a bar graph showing the effect of wild-type IGF-1, F49A, and E3A/F49A (at a concentration of 40 or 400 ng/ml) on ILlα-induced inhibition of matrix synthesis.
Figure 28 is a bar graph showing the effect of control, wild-type IGF- 1, F49A and E3A/F49A (at a concentration of 40 or 400 ng/ml) on nitric oxide release. Figure 29 is a bar graph showing the effect of wild-type IGF-1, F49A, and E3A/F49A (at a concentration of 40 or 400 ng/ml) on ILlα-induced nitric oxide production.
Figures 30A and 30B show the binding curves for phage particles displaying either wild-type IGF-1 (circles) , D12K (squares) , or D12R (diamonds) bound to immobilized IGFBP-1 (Fig. 30A) or IGFBP-3 (Fig. 30B) .
Figures 31A-31D show the effects on porcine articular cartilage explants cultured in media {-) or media with D12K, D12R, or wild-type IGF-1 (at 10 nM) alone (Figs. 31A, 31C) or in the presence of IL-lα (+a) at 1 ng/ml (Figs. 31B, 31D) . Figure 32 shows the effect on articular cartilage matrix synthesis in human tissue from diseased joints cultured in media alone (-) or with F49, E3A/F49, F16/F49, D12K, D12R or wild-type IGF-1 (at 40 ng/ml) .
Figures 33A-33D show the effect on human articular cartilage explants cultured in media (-) or treated with wild-type IGF-1 by itself or in
combination with either BP3-40 (Figs. 33A, 33B) or BP3-15 (Figs. 33C, 33D) (at 0.1 mg/ml) .
Figures 34A-34C show the trimeric complex formation of F49A or E3A/F49A with IGFBP-3 and ALS. IGFBP-3 immobilized on a biosensor chip was saturated by including 1 μM wild-type IGF-1 (Fig. 34A) , F49A (Fig. 34B) , or E3A/F49A (Fig. 34C) in the running buffer. ALS was injected at 98 nM, 148 nM, and 33 nM, monitoring real-time association and dissociation to the preformed binary complex.
Figure 35 shows a BIAcore™ inhibition assay of IGF-I activity using seven different peptides (BP1-16: filled circles, (i+7)A: open circles, (i+7)B: open diamonds, (i+7)C: open triangles, (i+7)D: open squares, (i+8)B: filled squares, (i+8)C: filled triangles).
Figure 36 shows a KIRA assay of peptide activity using four different peptides (BP1-16: circles, BP1-02: squares, BP1-25: triangles, and BP1-40: diamonds) .
Figure 37 shows an analytical HPLC run of the trypsin-cleaved BP1-625- Z fusion. The major peaks were identified by mass spectrometry as (A) Z- do ain fragment and (B) BPl-625 peptide.
Figure 38 shows a BIAcore™ inhibition assay of IGF-I activity using four different peptides (BP1-01: circles, BPl-625: squares, BP1-21A: triangles, and BP1-25: diamonds) .
Figure 39 shows the effect on proteoglycan synthesis of articular cartilage explants from human joints removed from patients undergoing joint replacement cultured with IGF-1 alone (IGF) at 40 ng/ml, or IGF-1 with BP1- 17, BP3-15, or BP1-16 (0.1 mg/ml), or IGF-1 with buffer (HEPES). Detailed Description of the Invention I. Definitions
"IGF-1 analogs" are amino acid variants of native-sequence IGF-1, preferably variants of human wild-type IGF-1. The dissociation constant ( KΏ) of wild-type IGF-1 was determined to be 13 nM for IGFBP-1 and 1.5 nM for IGFBP-3. The difference in affinity for the IGFBP 's is due to a 10-fold
5 faster association rate {ka) of IGF-1 to IGFBP-3 (3.2 x 10 versus
^ _ι _ι 3.2 x 10 M s ) . Such analogs may have one or more amino acid alterations as compared to native IGF-1. As used herein, the term "IGF-1 analogs" refers either to an IGF-1 analog with a binding affinity preference for
IGFBP-3 over IGFBP-1 or an IGF-1 analog with a binding affinity preference for IGFBP-1 over IGFBP-3, as defined below.
An "IGF-1 analog with a binding affinity preference for IGFBP-3 over
IGFBP-1" refers to an IGF-1 analog that exhibits altered binding affinity for any one or more of the IGFBPs over that of native-sequence IGF-1, such
that the analog's relative binding affinity (R(3) ) for IGFBP-3 [defined as R(3)=KD(IGF-l:IGFBP-3) /KD (analog: IGFBP-3) ] is at least about 10-fold greater than its relative binding affinity (R(l)) for IGFBP-1 [defined as R(1)=KD (IGF-1: IGFBP-1) /KD(analog: IGFBP-1) ] , as shown by, for example, by kinetic analysis using a BIACORE™ instrument of the expressed and purified analogs .
Conversely, an "IGF-1 analog with a binding affinity preference for IGFBP-1 over IGFBP-3" refers to an IGF-1 analog that exhibits altered binding affinity for any one or more of the IGFBPs over that of native- sequence IGF-1, such that the analog's relative binding affinity (R(l)) for IGFBP-1 [defined as R(1)=KD(IGF-1 : IGFBP-1) /KD(analog: IGFBP-1) ] is at least about 10-fold greater than its relative binding affinity (R(3)) for IGFBP-3 [defined as R(3)= KD (IGF-1 : IGFBP-3) /KD (analog: IGFBP-3) ] , as shown by, for example, by kinetic analysis using a BIACORE™ instrument of the expressed and purified analogs.
"Peptides" have at least two amino acids and include polypeptides having at least about 50 amino acids. The definition includes peptide derivatives, their salts, or optical isomers.
An IGFBP displacer peptide that "inhibits" or "prevents" the interaction of an IGF with an IGFBP refers to a peptide that increases serum and tissue levels of biologically active IGF, no matter how this increase occurs. For instance, the peptide may partially or completely displace active IGF from a complex in which the IGF is bound to an IGFBP. The peptide under this definition may bind to an IGFBP, and possibly thereby act to displace an endogenous IGF formerly bound to the IGFBP. Alternatively, it may bind to IGF itself at a site remote from that involved in receptor interactions so as to inhibit or prevent the interaction of the IGF with IGFBP, but not inhibit or prevent the interaction of the IGF with any of its receptors. Further, while the peptide will occupy the IGFBP-3 binding site, the effect on the ternary complex with ALS will depend on whether the binary complexes can form ternary ones . Peptides that can form complexes with the ALS of the ternary complex will replace IGFs but not affect the concentration of IGFBP-3 or of ternary complexes . Peptides that cannot form complexes with ALS will occupy IGFBP-3, and the amount of ALS/IGFBP-3/IGF complex will be reduced. Preferably, the IGFBP displacer peptide is an IGFBP-3 or IGFBP-1 displacer peptide.
A peptide that "binds to IGFBP-3" or "binds to IGFBP-1" refers to a peptide that binds IGFBP-3 or IGFBP-1 to at least some degree, whether with high affinity or not. As used herein, "human IGF receptor" refers to any receptor for an IGF found in humans and includes the Type 1 and Type 2 IGF receptors in humans
to which both human IGF-1 and IGF-2 bind, such as the placental Type 1 IGF-1 receptor, etc.
A peptide that "does not bind to a human IGF receptor" does not bind at all to any such receptor, or binds to such receptor with an affinity more than about 200-fold less than wild-type human IGF-1 (hIGF-1) or wild-type human IGF-2 (hIGF-2) binds to such receptor. Preferably, the peptide binds to such receptor with an affinity of more than about 250-fold less than wild-type hIGF-1 or hIGF-2 binds to the same receptor or does not bind at all. The term "cartilage disorder" refers to any injury or damage to cartilage, and to a collection of diseases that are manifested by symptoms of pain, stiffness, and/or limitation of motion of the affected body parts. Included within the scope of "cartilage disorders" is "degenerative cartilagenous disorders", which is a colllection of disorders characterized, at least in part, by degeneration or metabolic derangement of connective tissues of the body, including not only the joints or related structures, including muscles, bursae (synovial membrane), tendons, and fibrous tissue, but also the growth plate, meniscal system, and intervertebral discs.
In one embodiment, the term "degenerative cartilagenous disorders" includes "articular cartilage disorders," which are characterized by disruption of the smooth articular cartilage surface and degradation of the cartilage matrix. Additional pathologies include nitric oxide production, and inhibition or reduction of matrix synthesis. Included within the scope of "articular cartilage disorder" are OA and RA. Examples of degenerative cartilagenous disorders include systemic lupus erythematosus and gout, amyloidosis or Felty's syndrome. Additionally, the term covers the cartilage degradation and destruction associated with psoriatic arthritis, kidney disorders, osteoarthrosis, acute inflammation (e.g., yersinia arthritis, pyrophosphate arthritis, gout arthritis (arthritis urica) , and septic arthritis) , arthritis associated with trauma, ulcerative colitis {e . g. , Crohn's disease), multiple sclerosis, diabetes (e.g., insulin- dependent and non-insulin dependent) , obesity, giant cell arthritis, and Sjδgren's syndrome. In one preferred embodiment, the disorder is microdamage or blunt trauma, a chondral fracture, or an osteochondral fracture.
"Osteoarthritis" or "OA" defines not a single disorder, but the final common pathway of joint destruction resulting from multiple processes. OA is characterized by localized assymetric destruction of the cartilage commensurate with palpable bone enlargements at the joint margins. OA typically affects the interphalangeal joints of the hands, the first carpometacarpal joint, the hips, the knees, the spine, and some joints in
the midfoot, while large joints, such as the ankles, elbows, and shoulders, tend to be spared. OA can be associated with metabolic diseases such as hemochromatosis and alkaptonuria, developmental abnormalities such as developmental dysplasia of the hips (congenital dislocation of the hips), limb-length descrepancies, including trauma and inflammatory arthritides such as gout, septic arthritis, and neuropathic arthritis. OA may also develop after extended mechanical instability, such as resulting from sports injury or obesity.
"Rheumatoid arthritis" or "RA" is a systemic, chronic, autoimmune disorder characterized by symmetrical synovitis of the joint and typically affects small and large diarthroid joints alike. As RA progresses, symptoms may include fever, weight loss, thinning of the skin, multiorgan involvement, scleritis, corneal ulcers, the formation of subcutaneous or subperiosteal nodules, and even premature death. The symptoms of RA often appear during youth and can include vasculitis, atrophy of the skin and muscle, subcutaneous nodules, lymphadenopathy, splenomegaly, leukopaenia, and chronic anaemia.
"Treatment" is an intervention performed with the intention of preventing the development or altering the pathology of a disorder. Accordingly, "treatment" refers to both therapeutic treatment and prophylactic or preventative measures, wherein the object is to prevent or slow down (lessen) the targeted pathological condition or disorder. Those in need of treatment include those already with the disorder as well as those in which the disorder is to be prevented. In treatment of a degenerative cartilagenous disorder, a therapeutic agent may directly decrease or increase the magnitude of response of a pathological component of the disorder, or render the disease more susceptible to treatment by other therapeutic agents, e.g., antibiotics, antifungals, anti-inflammatory agents, chemotherapeutics, etc. The term "treatment" includes a method for the prevention of initial or continued damage or disease of joints by degenerative cartilagenous disorders and/or injury.
The term "effective amount" is the minimum efficacious concentration of the IGF analog or IGFBP displacer peptide as set forth herein. This includes the minimum concentration of such protein or peptide that causes, induces, or results in either a detectable improvement or repair of damaged cartilage or a measurable protection from continued or induced cartilage destruction, such as the inhibition of synthesis or loss of proteoglycans from cartilage tissue.
"Cartilage growth factor" as used herein refers to agent (s) other than an IGF-1 analog or an IGFBP displacer peptide as identified herein that cause, induce, or result in an improvement in the condition of or protection
from initial or continued destruction of cartilage subject to damage by either injury or a degenerative cartilagenous disorder. Such cartilage growth factors include sulin-like growth factors { e . g. , IGF-1, IGF-2), platelet-derived growth factors (PDGFs) , bone orphogenic proteins (BMPs) , transforming growth factor-βs (1-3), members of the epidermal growth factor family (e.g., EGF, HB-EGF, TGF-α) , and fibroblast growth factors (FGFs).
"Cartilage catabolism antagonists" are those agents that inhibit, attenuate or otherwise block the activity or effect of molecules that are associated with or aggravate cartilage destruction. For example, IL-lα and nitric oxide (NO) are agents known to be associated with cartilage destruction. Thus, direct (ILlra) or indirect (IL-4 or IL-10) inhibitors of IL-lα or other inflammatory cytokmes (e.g., TNF-α) and NO production would be considered "cartilage catabolism antagonists." Moreover, antagonists of chondrocyte catabolism (e.g., sodium pentosan polysulfate, glucosam e (and variants thereof, such as mannosamme) or chondroit sulfate, tetracyclme, hyaluronan) would also be considered cartilage catabolism antagonists. Also included are agents that inhibit catabolism of cartilage indirectly, for example through their effects on the underlying, subchondral bone (e.g., bisphosphonates or osteoproteger (OPG) ) . "Chronic" administration refers to administration of the agent (s) m a continuous mode as opposed to an acute mode, so as to maintain the initial therapeutic effect (activity) for an extended period of time. "Intermittent" administration is treatment that is not consecutive without interruption, but rather is cyclic in nature. As used herein, "mammal" for purposes of treatment refers to any animal classified as a mammal, including humans, domestic, and farm animals, and zoo, sports, or pet animals, such as dogs, horses, cats, sheep, pigs, cows, etc. The preferred mammal herein is a human. The term "non-adult" refers to mammals that are from perinatal age (such as low-birth-weight infants) up to the age of puberty, the latter being those that have not yet reached full growth potential.
Administration "in combination with" one or more further therapeutic agents includes simultaneous (concurrent) and consecutive administration in any order. "Carriers" as used herein include pharmaceutically-acceptable carriers, excipients, or stabilizers that are non-toxic to the cell or mammal being exposed thereto at the dosages and concentrations employed. Often the physiologically-acceptable carrier is an aqueous pH-buffered solution. Examples of physiologically-acceptable carriers include buffers such as phosphate, citrate, and other organic acids; antioxidants including
ascorbic acid; low-molecular-weight (less than about 10 residues) polypeptides; proteins such as serum albumin, gelatin, or immunoglobulins; hydrophilic polymers such as polyvinylpyrrolidone; amino acids such as glycine, glutamine, asparagine, arginine, or lysine; monosaccharides, disaccharides, and other carbohydrates including glucose, mannose, or dextrins; chelating agents such as EDTA; sugar alcohols such as mannitol or sorbitol; salt-forming counter-ions such as sodium; hyaluronan; and/or nonionic surfactants such as TWEEN™, polyethylene glycol (PEG), and PLURONICS™. A "liposome" is a small vesicle composed of various types of lipids, phospholipids, and/or surfactants that is useful for delivery of a drug (such as the IGF-1 analog or IGFBP displacer peptide disclosed herein) to a mammal. The components of the liposome are commonly arranged in a bilayer formation, similar to the lipid arrangement of biological membranes.
The term "extended-release" or "sustained-release" formulations in the broadest possible sense means a formulation of active IGF-1 analog or IGFBP displacer peptide identified herein resulting in the release or activation of the active analog or peptide for a sustained or extended period of time— or at least for a period of time that is longer than if the analog or peptide were made available in vivo in the native or unformulated state. Optionally, the extended-release formulation occurs at a constant rate and/or results in sustained and/or continuous concentration of the active agent herein. Suitable extended-release formulations may comprise microencapsulation, semi-permeable matrices of solid hydrophobic polymers, biogradable polymers, biodegradable hydrogels, suspensions, or emulsions (e.g., oil-in-water or water-in-oil). Optionally, the extended-release formulation comprises poly-lactic-co-glycolic acid (PLGA) and can be prepared as described in Lewis, "Controlled Release of Bioactive Agents form Lactide/Glycolide polymer, " in Biodegradable Polymers as Drug Delivery Systems, M. Chasin and R. Langeer, Ed. (Marcel Dekker, New York), pp. 1-41. Optionally, the extended-release formulation is stable and the activity of the IGF-1 analog or IGFBP displacer peptide as identified herein does not appreciably diminish with storage over time. More specifically, such stability can be enhanced through the presence of a stabilizing agent such as a water-soluble polyvalent metal salt. As used herein, "IGF-1" refers to insulin-like growth factor-1 from any species, including bovine, ovine, porcine, equine, and human, preferably human, and, if referring to exogenous administration, from any source, whether natural, synthetic, or recombinant. "Native-sequence" human IGF-1, the sequence of which is shown in Fig. 16 (SEQ ID NO:3), is prepared, e . g. , by the process described in EP 230,869 published August 5, 1987; EP 128,733
published December 19, 1984; or EP 288,451 published October 26, 1988. More preferably, this native-sequence IGF-1 is recombinantly produced.
As used herein, "IGF-2" refers to insulin-like growth factor-2 from any species, including bovine, ovine, porcine, equine, and human, preferably human, and, if referring to exogenous administration, from any source, whether natural, synthetic, or recombinant. It may be prepared by the method described in, e . g. , EP 128,733.
An "IGFBP" or an "IGF binding protein" refers to a protein or pol peptide normally associated with or bound or complexed to IGF-1 or IGF- 2, whether or not it is circulatory (i.e., in serum or tissue). Such binding proteins do not include receptors. This definition includes IGFBP-
I, IGFBP-2, IGFBP-3, IGFBP-4, IGFBP-5, IGFBP-6, Mac 25 (IGFBP-7), and prostacyclin-stimulating factor (PSF) or endothelial cell-specific molecule
(ESM-1), as well as other proteins with high homology to IGFBPs. Mac 25 is described, for example, in Swisshelm et al . , Proc. Natl. Acad. Sci. USA, 92:
4472-4476 (1995) and Oh et al . , J. Biol. Chem., 271: 30322-30325 (1996).
PSF is described in Yamauchi et al . , Biochemical Journal, 303: 591-598
(1994). ESM-1 is described in Lassalle et al . , J. Biol. Chem., 271: 20458-
20464 (1996). For other identified IGFBPs, see, e.g., EP 375,438 published 27 June 1990; EP 369,943 published 23 May 1990; WO 89/09268 published 5
October 1989; Wood et al . , Molecular Endocrinology, 2: 1176-1185 (1988);
Brink an et al . , The EMBO . , 7: 2417-2423 (1988); Lee et al . , Mol.
Endocrinol., 2: 404-411 (1988); Brewer et al . , BBRC, 152: 1289-1297 (1988);
EP 294,021 published 7 December 1988; Baxter et al . , BBRC, 147: 408-415 (1987); Leung et al . , Nature, 330: 537-543 (1987); Martin et al . , J. Biol.
Chem., 261: 8754-8760 (1986); Baxter et al . , Comp. Biochem. Physiol., 91B:
229-235 (1988); WO 89/08667 published 21 September 1989; WO 89/09792 published 19 October 1989; and Binkert et al . , EMBO J., 8: 2497-2502 (1989). The term "acid-labile subunit" or "ALS" refers to an 85-kDa glycoprotein that forms a ternary complex with IGF-1 and IGFBP-3 or IGFBP-5. See, e.g., Bach and Rechler, Diabetes Reviews, 3: 38-61 (1995); Clemmons,
Cytokine Growth Factor Rev. , 8_: 45-62 (1997) ; and Jones and Clemmons,
Endocr. Rev., 16: 3-34 (1995).
I . Modes or Carrying Out the Invention The invention herein relates to the use of an IGF-1 analog or an IGFBP displacer peptide as defined above to treat cartilage disorders, preferably degenerative cartilagenous disorders, including regenerating and/or preventing the degradation of cartilage.
Examples of IGF-1 analogs with a binding affinity preference for IGFBP-3 over IGFBP-1 include an IGF-1 variant wherein the amino acid(s) of wild-type human IGF-1 at position 3, 7, 10, 16, 25, or 49 or at positions 3
and 49 of native-sequence human IGF-1 are replaced with an alanine, a glycine, and/or a serine residue. Preferably, one or both of the amino acids in question are substituted by an alanine or glycine residue, most preferably alanine. The more preferred IGF-1 analog with such binding affinity preference herein is F49A, F49G, F49S, E3A, E3G, E3S, E3AF49A,
E3AF49G, E3AF49S, E3GF49A, E3GF49G, E3GF49S, E3SF49A, E3SF49G, E3SF49S,
F16A, F16G, F16S, F16AF49A, F16GF49A, F16SF49A, F16AF49S, F16AF49G,
F16SF49S, F16SF49G, F16GF49S, or F16GF49G.
Examples of IGF-1 analogs with a binding affinity preference for IGFBP-1 over IGFBP-3 include an IGF-1 variant wherein the amino acid(s) of wild-type human IGF-1 at position 9, 12, 15, or 20 is/are replaced with a lysine or arginine residue. The more preferred IGF-1 analog with such binding affinity preference herein is D12K or D12R.
Examples of IGFBP-3 displacer peptides include a peptide selected from the group consisting of:
Y24LY31A (IGF-1 variant);
4D3.3P (ASEEVCWPVAEWYLCNMWGR) (SEQ ID NO: 6) ;
BP3-4D3.il (VAWEVCWDRHDQGYICTTDS) (SEQ ID NO:7);
BP3-4D3.11DEL (AWEVCWDRHQGYICTTDS) (SEQ ID NO:8); BP3-4B3.3 (EESECFEGPGYVICGLVG) (SEQ ID NO:9);
BP3-01-OX (SEEVCWPVAEWYLCNMWG) (SEQ ID NO:10);
BP3-02-OX (DMGVCADGPWMYVCEWTE) (SEQ ID NO: 11);
BP3-06 (TGVDCQC*GPVHC*VCMDWA) (SEQ ID NO:12);
BP3-08 (TVANCDC*YMPLC*LCYDSD) (SEQ ID NO:13); BP3-15 (SEEVCWPVAEWYLCN) (SEQ ID NO: 14 ) ;
BP3-16 (VCWPVAEWYLCNMWG) (SEQ ID NO:15);
BP3-17 (VCWPVAEWYLCN) (SEQ ID NO: 16);
BP3-25 (CWPVAEWYLCN) (SEQ ID NO:17);
BP3-27 (EVCWPVAEWYLCN) (SEQ ID N0:18); BP3-28 (EEVCWPVAEWYLCN) (SEQ ID N0:19);
BP3-30 (ASEEVCWPVAEWYLCN) (SEQ ID N0:20);
BP3-39 (SEEVCWPVAEWYLCN-nh2) (SEQ ID NO:21);
BP3-40 (ac-SEEVCWPVAEWYLCN-nh2) (SEQ ID NO:22);
BP3-41 (GPETCWPVAEWYLCN) (SEQ ID NO:23); BP3-107 (suc-CQLVRPDLLLCQ-nh2) (SEQ ID NO:24); and
BP3-108 (suc-IPVSPDWFVCQ-nh2) (SEQ ID NO:25); where the C* indicates a cysteine that has been linked to another cysteine in the peptide. The remaining Cys pairs are also oxidized as disulfides in each peptide. The more preferred IGFBP-3 displacer peptide herein is BP3- 15, BP3-39, BP3-40, BP3-01-OX, BP3-27, BP3-28, BP3-30, BP3-41, or 4D3.3P.
The most preferred IGFBP-3 displacer peptide herein is BP3-15, BP3-39, or
BP3-40.
Examples of IGFBP-1 displacer peptides include a peptide selected from the group consisting of: BP1-01 (CRAGPLQWLCEKYFG) (SEQ ID NO:26);
BP1-02 (SEVGCRAGPLQWLCEKYFG (SEQ ID NO:27);
BP1-04 (CRAGPLQWLCE) (SEQ ID NO: 28)
BP1-10 (CRKGPLQWLCELYF) (SEQ ID NO: 29)
BP1-11 (CRKGPLQWLCEKYF) (SEQ ID O:30); BP1-12 (CKEGPLQWLCEKYF) (SEQ ID NO:31);
BP1-13 (CKEGPLLWLCEKYF) (SEQ ID NO: 32) ;
BP1-14 (SEVGCRAGPLQWLCEKYFG-nh2) (SEQ ID NO:33);
BP1-15 (CAAGPLQWLCEKYF) (SEQ ID NO: 34) ;
BP1-16 (CRAGPLQWLCEKYF-nh2) (SEQ ID NO: 35) ; BP1-17 (CRAGPLQWLCEK-nh2) (SEQ ID NO: 36) ;
BP1-18 (CRAGPLQWLCEKAA) (SEQ ID NO:37);
BP1-19 (SEMVCRAGPLQWLCEIYF-nh2*) (SEQ ID NO: 38 ) ;
BP1-20 (EARVCRAGPLQWLCEKYF-nh2) (SEQ ID NO: 39)
BP1-21A (SEVGCRAGPLQWLCEKYFSTY-nh2) (SEQ ID NO: 40) 'BP1-21B (CRAGPLQWLCEKYFSTY-nh2) (SEQ ID NO:41)
BP1-25 (EARVCRAGPLQWLCEKYFSTY) (SEQ ID NO: 42)
BP1-40 (GQQSCRAGPLQWLCEKYFSTY) (SEQ ID NO:43)
BP67 (CRAGPLQWLCERYF) (SEQ ID NO:44);
BP68 (CRAGPLQWLCEKFF) (SEQ ID NO: 45); BPl-625 (GQQSCAAGPLQWLCEHYFSTYGR) (SEQ ID NO:46);
BP1-625-Z (GQQSCAAGPLQWLCEHYFSTYGRGGGSGGAQHDEAVDNKFNKE QQNAFYEILHLPNLNEEQRNAFIQSLKDDPSQSANLLAEAKKLN DAQAPNVDMN) (SEQ ID NO: 47);
BP1-625T (GQQSCAAGPLQWLCEHYFSTY) (SEQ ID NO:153);
BP1027 (CKAGPLLWLCERFF) (SEQ ID NO:48)
BP1028 (CRAGPLQWLCERFF) (SEQ ID NO: 49)
BP1029 (CREGPLQWLCERFF) (SEQ ID NO:50)
BP1030 (CKEGPLLWLCERFF) (SEQ ID NO: 51); (i+7)D (acRAGPLEWLAEKYEG) (SEQ ID NO: 52 ) ;
(i+8)B (acRPLEWLAEKYFΞ) (SEQ ID NO: 53) ; and
(i+8)C (acRAGPLEWLAEKYFE) (SEQ ID NO:54); where the C* indicates a cysteine that has been linked to another cysteine in the peptide, and the remaining Cys pairs are also oxidized as disulfides in each peptide. The more preferred IGFBP-1 displacer peptide herein is
BPl-16, BP1-20, BP1-21A, BP1-25, BP1-40, BP625, BP625-Z, and BP625T; and
most preferred are BPl-20, BP1-21A, BPl-25, BPl-40, BPl-625, BP1-625-Z, and BP1-625T.
The still more preferred active agents herein are F49A, E3A, F16A,
E3AF49A, F16AF49A, D12K, D12R, BP3-15, BP3-40, BP3-39, BP1-16, BPl-20, BP1- 21A, BPl-25, BPl-40, BPl-625, and BP1-625-Z; and the most preferred are
F49A, E3AF49A, F16AF49A, D12K, D12R, BP3-15, BP3-40, BP3-39, BPl-20, BP1-
21A, BPl-25, BPl-40, BPl-625, BP1-625-Z, and BP1-625T.
The IGF-1 analogs and IGFBP displacer peptides useful in accordance with this invention can be made by any means that are known in the art, including chemical synthesis or recombinant production. Chemical synthesis, especially solid phase synthesis, is preferred for short (e.g., less than 50 residues) peptides or those containing unnatural or unusual amino acids such as D-Tyr, Ornithine, amino adipic acid, and the like. Recombinant procedures are preferred for longer polypeptides. When recombinant procedures are selected, a synthetic gene may be constructed de nσvo or a natural gene may be mutated by, for example, cassette mutagenesis. Set forth below are exemplary general recombinant procedures .
From a -purified IGF-1 and its amino acid sequence, for example, an IGF variant that is a peptidyl mutant of an IGF-1 parent molecule may be produced using recombinant -DNA techniques. These techniques contemplate, in simplified form, taking the gene, either natural or synthetic, encoding the analog; inserting it into an appropriate vector; inserting the vector into an appropriate host cell; culturing the host cell to cause expression of the gene; and recovering or isolating the analog produced thereby. Preferably, the recovered analog is then purified to a suitable degree.
Somewhat more particularly, the DNA sequence encoding a peptidyl IGF variant is cloned and manipulated so that it may be expressed in a convenient host. DNA encoding parent polypeptides can be obtained from a genomic library, from cDNA derived from mRNA from cells expressing the parent polypeptide, or by synthetically constructing the DNA sequence (Sa brook et al . , Molecular Cloning: A Laboratory Manual (2d ed. ) , Cold Spring Harbor Laboratory, N.Y., 1989).
The parent DNA is then inserted into an appropriate plasmid or vector which is used to transform a host cell. In general, plasmid vectors containing replication and control sequences that are derived from species compatible with the host cell are used in connection with those hosts. The vector ordinarily carries a replication site, as well as sequences which encode proteins or peptides that are capable of providing phenotypic selection in transformed cells. For example, E. coli may be transformed using pBR322, a plasmid derived from an E. coli species (Mandel et al . , J. Mol. Biol. 53: 154
(1970)). Plasmid pBR322 contains genes for ampicillin and tetracycline resistance, and thus provides easy means for selection. Other vectors include different features such as different promoters, which are often important in expression. For example, plasmids pKK223-3, pDR720, and pPL- lambda represent expression vectors with the tac, trp, or PL promoters that are currently available (Pharmacia Biotechnology) .
A preferred vector is pB0475. This vector contains origins of replication for phage and E. coli that allow it to be shuttled between such hosts, thereby facilitating both mutagenesis and expression (Cunningham et al . , Science, 243: 1330-1336 (1989); U.S. Pat. No. 5,580,723). Other preferred vectors are pRlT5 and pRlT2T (Pharmacia Biotechnology) . These vectors contain appropriate promoters followed by the Z domain of protein A, allowing genes inserted into the vectors to be expressed as fusion proteins.
Other preferred vectors can be constructed using standard techniques by combining the relevant traits of the vectors described above. Relevant traits include the promoter, the ribosome binding site, the decorsin or ornatin gene or gene fusion (the Z domain of protein A and decorsin or ornatin and its linker) , the antibiotic resistance markers, and the appropriate origins of replication. The host cell may be prokaryotic or eukaryotic. Prokaryotes are preferred for cloning and expressing DNA sequences to produce the parent IGF-1 polypeptide, segment-substituted peptides, residue-substituted peptides, and peptide variants. For example, E. coli K12 strain 294 (ATCC No. 31446) may be used as well as E. coli B, E. coli X1776 (ATCC No. 31537), and E. coli c600 and c600h.fl, E. coli W3110 (F-, gamma-, prototrophic/ATCC
No. 27325), bacilli such as Bacillus subtilis, and other enterobacteriaceae such as Salmonella typhimurium or Serratia marcesans , and various
Pseudomonas species. The preferred prokaryote is E. coli W3110 (ATCC 27325).
When expressed by prokaryotes, the analogs or peptides typically contain an N-terminal methionine or a formyl methionine and are not glycosylated. In the case of fusion proteins, the N-terminal methionine or formyl methionine resides on the amino terminus of the fusion protein or the signal sequence of the fusion protein. These examples are, of course, intended to be illustrative rather than limiting. In addition to prokaryotes, eukaryotic organisms, such as yeast cultures, or cells derived from multicellular organisms may be used. In principle, any such cell culture is workable. However, interest has been greatest in vertebrate cells, and propagation of vertebrate cells in culture (tissue culture) has become a reproducible procedure. Tissue Culture, Academic Press, Kruse and Patterson, editors (1973) . Examples of such
useful host cell lines are VERO and HeLa cells, Chinese Hamster Ovary (CHO) cell lines, W138, 293, BHK, COS-7 and MDCK cell lines.
A variation on the above procedures contemplates the use of gene fusions, wherein the gene encoding the desired analog or peptide is associated, in the vector, with a gene encoding another protein or a fragment of another protein. This results in the desired analog or peptide being produced by the host cell as a fusion with another protein or peptide. The "other" protein or peptide is often a protein or peptide that can be secreted by the cell, making it possible to isolate and purify the desired analog or peptide from the culture medium and eliminating the necessity of destroying the host cells that arises when the desired analog or peptide remains inside the cell. Alternatively, the fusion protein can be expressed intracellularly. It is useful to use fusion proteins that are highly expressed. The use of gene fusions, though not essential, can facilitate the expression of heterologous analogs and peptides m E. coli as well as the subsequent purification of those gene products (Harris, Genetic Engineering, Williamson, R., Ed. (Academic Press, London, Vol. 4, 1983), p. 127; Ljungquist et al . , Eur. J. Biochem., 186: 557-561 (1989) and Ljungquist et al . , Eur. J. Biochem., 186: 563-569 (1989)). Protein A fusions are often used because the binding of protein A, or more specifically the Z domain of protein A, to IgG provides an "affinity handle" for the purification of the fused protein. It has also been shown that many heterologous proteins are degraded when expressed directly m E. coli, but are stable when expressed as fusion proteins (Marston, Biochem J. , 240 : 1 (1986) ) .
Fusion proteins can be cleaved using chemicals, such as cyanogen bromide, which cleaves at a methionine, or hydroxylamme, which cleaves between an Asn and Gly residue. Using standard recombinant DNA methodology, the nucleotide base pairs encoding these am o acids may be inserted just prior to the 5' end of the gene encoding the desired analog or peptide.
Alternatively, one can employ proteolytic cleavage of fusion protein (Carter, m Protein Purif cation: From Molecular Mechanisms to Large-Scale Processes, Ladisch et al . , eds . (American Chemical Society Symposium Series No. 427, 1990), Ch 13, pages 181-193). Proteases such as Factor Xa, thrombm, and subtilisin or its mutants, and a number of others have been successfully used to cleave fusion proteins. Typically, a peptide linker that is amenable to cleavage by the protease used is inserted between the "other" protein (e.g., the Z domain of protein A) and the desired analog or peptide. Using recombinant DNA methodology, the nucleotide base pairs encoding the linker are inserted between the genes or gene fragments coding for the other proteins.
Proteolytic cleavage of the partially purified fusion protein containing the correct linker can then be carried out on either the native fusion protein, or the reduced or denatured fusion protein.
The analog or peptide may or may not be properly folded when expressed as a fusion protein. Also, the specific peptide linker containing the cleavage site may or may not be accessible to the protease. These factors determine whether the fusion protein must be denatured and refolded, and if so, whether these procedures are employed before or after cleavage.
When denaturing and refolding are needed, typically the analog or peptide is treated with a chaotrope, such a guanidine HC1. Then it is treated with a redox buffer, containing, for example, reduced and oxidized dithiothreitol or glutathione at the appropriate ratios, pH, and temperature, such that the analog or peptide is refolded to its native structure . When analogs and peptides are not prepared using recombinant DNA technology, they are preferably prepared using solid-phase synthesis, such as that generally described by Merrifield, J. Am. Chem. Soc, 85: 2149 (1963), although other equivalent chemical syntheses known in the art are employable. In vitro protein synthesis may be performed using manual techniques or by automation. Automated synthesis may be accomplished, for instance, using an Applied Biosystems peptide synthesizer (Foster City, CA) following the manufacturer's instructions. Varous portions of the analog or peptide may be chemically synthesized separately and combined using chemical or enzymatic methods to produce the full-length analog or peptide. Solid-phase synthesis is initiated from the C-terminus of the analog or peptide by coupling a protected α-amino acid to a suitable resin. Such a starting material can be prepared by attaching an α-amino-protected amino acid by an ester linkage to a chloromethylated resin or a hydroxymethyl resin, or by an amide bond to a BHA resin or MBHA resin. The preparation of the hydroxymethyl resin is described by Bodansky et aJ . , Chem. Ind.
(London) , 38 : 1597-1598 (1966) . Chloromethylated resins are commercially available from BioRad Laboratories, Richmond, CA and from Lab. Systems, Inc.
The preparation of such a resin is described by Stewart et al . , "Solid
Phase Peptide Synthesis" (Freeman and Co., San Francisco 1969), Chapter 1, pp. 1-6. BHA and MBHA resin supports are commercially available and are generally used only when the desired analog or peptide being synthesized has an unsubstituted amide at the C-terminus .
The amino acids are coupled to the analog/peptide chain using techniques well known in the art for the formation of peptide bonds . One method involves converting the amino acid to a derivative that will render
the carboxyl group more susceptible to reaction with the free N-terminal amino group of the peptide fragment. For example, the amino acid can be converted to a mixed anhydride by reaction of a protected amino acid with ethylchloroformate, phenyl chloroformate, sec-butyl chloroformate, isobutyl chloroformate, pivaloyl chloride or like acid chlorides. Alternatively, the amino acid can be converted to an active ester such as a 2,4,5- trichlorophenyl ester, a pentachlorophenyl ester, a pentafluorophenyl ester, a p-nitrophenyl ester, a N-hydroxysuccinimide ester, or an ester formed from 1-hydroxybenzotriazole . Another coupling method involves use of a suitable coupling agent such as N,N' -dicyclohexylcarbodiimide or N,N' -diisopropyl-carbodiimide. Other appropriate coupling agents, apparent to those skilled in the art, are disclosed in E. Gross and J. Meienhofer, The Peptides: Analysis, Structure, Biology, Vol. I: Major Methods of Peptide Bond Formation (Academic Press, New York, 1979) .
It should be recognized that the -amino group of each amino acid employed in the analog/peptide synthesis must be protected during the coupling reaction to prevent side reactions involving their active α—amino function. It "should also be recognized that certain amino acids contain reactive side-chain functional groups (e.g., sulfhydryl, amino, carboxyl', and hydroxyl) and that such functional groups must also be protected with suitable protecting groups to prevent a chemical reaction from occurring at that site during both the initial and subsequent coupling steps. Suitable protecting groups, known in the art, are described in Gross and Meienhofer, The Peptides: Analysis, Structure, Biology, Vol.3: "Protection of Functional Groups in Peptide Synthesis" (Academic Press: New York, 1981).
In the selection of a particular side-chain protecting group to be used in synthesizing the analogs/peptides, the following general rules are followed. An α—amino protecting group (a) must render the α-amino function inert under the conditions employed in the coupling reaction, (b) must be readily removable after the coupling reaction under conditions that will not remove side-chain protecting groups and will not alter the structure of the analog/peptide fragment, and (c) must eliminate the possibility of racemization upon activation immediately prior to coupling. A side-chain protecting group (a) must render the side chain functional group inert under the conditions employed in the coupling reaction, (b) must be stable under the conditions employed in removing the α-amino protecting group, and (c) must be readily removable upon completion of the desired analog/peptide under reaction conditions that will not alter the structure of the analog/peptide chain.
It will be apparent to those skilled in the art that the protecting groups known to be useful for analog/peptide synthesis will vary in reactivity with the agents employed for their removal. For example, certain protecting groups such as triphenylmethyl and 2-(p- biphenylyl) isopropyloxycarbonyl are very labile and can be cleaved under mild acid conditions. Other protecting groups, such as t-butyloxycarbonyl (BOC) , t-amyloxycarbonyl, adamantyl-oxycarbonyl, and p- methoxybenzyloxycarbonyl are less labile and require moderately strong acids, such as trifluoroacetic, hydrochloric, or boron trifluoride in acetic acid, for their removal. Still other protecting groups, such as benzyloxycarbonyl (CBZ or Z) , halobenzyloxycarbonyl, p- nitrobenzyloxycarbonyl cycloalkyloxycarbonyl, and isopropyloxycarbonyl, are even less labile and require stronger acids, such as hydrogen fluoride, hydrogen bromide, or boron trifluoroacetate in trifluoroacetic acid, for their removal. Among the classes of useful amino acid protecting groups are included:
(1) for an α-amino group, (a) aromatic urethane-type protecting groups, such as fluorenylmethyloxycarbonyl (FMOC) CBZ, and substituted CBZ, such as, e.g., p-chl.orobenzyloxycarbonyl, p-6-nitrobenzyloxyσarbonyl, p- bromobenzyloxycarbonyl, and p-methoxybenzyloxycarbonyl, o- chlorobenzyloxycarbonyl, 2, 4-dichlorobenzyloxycarbonyl, 2,6- dichlorobenzyloxycarbonyl, and the like; (b) aliphatic urethane-type protecting groups, such as BOC, t-amyloxycarbonyl, isopropyloxycarbonyl, 2- (p-biphenylyl) -isopropyloxycarbonyl, allyloxycarbonyl and the like; (c) cycloalkyl urethane-type protecting groups, such as cyclopentyloxycarbonyl, adamantyloxycarbonyl, and cyclohexyloxycarbonyl; and d) allyloxycarbonyl. The preferred α-amino protecting groups are BOC or FMOC.
(2) for the side chain amino group present in Lys, protection may be by any of the groups mentioned above in (1) such as BOC, p- chlorobenzyloxycarbonyl, etc.
(3) for the guanidino group of Arg, protection may be by nitro, tosyl, CBZ, adamantyloxycarbonyl, 2, 2, 5, 7, 8-pentamethylchroman-6-sulfonyl or 2, 3, 6-trimethyl-4-methoxyphenylsulfonyl, or BOC.
(4) for the hydroxyl group of Ser, Thr, or Tyr, protection may be, for example, by C1-C4 alkyl, such as t-butyl; benzyl (BZL) ; substituted BZL, such as p-methoxybenzyl, p-nitrobenzyl, p-chlorobenzyl, o-chlorobenzyl, and 2, 6-dichlorobenzyl .
(5) for the carboxyl group of Asp or Glu, protection may be, for example, by esterification using groups such as BZL, t-butyl, cyclohexyl, cyclopentyl, and the like.
(6) for the imidazole nitrogen of His, the tosyl moiety is suitably employed.
(7) for the phenolic hydroxyl group of Tyr, a protecting group such as tetrahydropyranyl, tert-butyl, trityl, BZL, chlorobenzyl, 4-bromobenzyl, or 2, 6-dichlorobenzyl is suitably employed. The preferred protecting group is 2, 6-dichlorobenzyl.
(8) for the side-chain amino group of Asn or Gin, xanthyl (Xan) is preferably employed.
(9) for Met, the amino acid is preferably left unprotected. (10) for the thio group of Cys, p-methoxybenzyl is typically employed.
The C-terminal amino acid, e . g. , Lys, is protected at the N-amino position by an appropriately selected protecting group, in the case of Lys, BOC. The BOC-Lys-OH can be first coupled to the benzyhydrylamine or chloromethylated resin according to the procedure set forth in Horiki et al . , Chemistry Letters, 165-168 (1978) or using isopropylcarbodiimide at about 25°C for 2 hours with stirring. Following the coupling of the BOC- protected amino acid to the resin support, the α-amino protecting group is removed, as by using trifluoroacetic acid (TFA) in methylene chloride or TFA alone. The deprotection is carried out at a temperature between about 0°C and room temperature. Other standard cleaving reagents, such as HC1 in dioxane, and conditions for removal of specific a-amino protecting groups are described in the literature.
After removal of the α-amino protecting group, the remaining α—amino and side-chain protected amino acids are coupled stepwise within the desired order. As an alternative to adding each amino acid separately in the synthesis, some may be coupled to one another prior to addition to the solid-phase synthesizer. The selection of an appropriate coupling reagent is within the skill of the art. Particularly suitable as a coupling reagent is N, N' -dicyclohexyl carbodiimide or diisopropylcarbodiimide .
Each protected amino acid or amino acid sequence is introduced into the solid-phase reactor in excess, and the coupling is suitably carried out in a medium of dimethylformamide (DMF) or CH2C12 or mixtures thereof. If incomplete coupling occurs, the coupling procedure is repeated before removal of the N-amino protecting group prior to the coupling of the next amino acid. The success of the coupling reaction at each stage of the synthesis may be monitored. A preferred method of monitoring the synthesis is by the ninhydrin reaction, as described by Kaiser et al ■ , Anal . Biochem, 3_4: 595 (1970) . The coupling reactions can be performed automatically using well known methods, for example, a BIOSEARCH 9500™ peptide synthesizer.
Upon completion of the desired analog/peptide sequence, the protected analog/peptide must be cleaved from the resin support, and all protecting groups must be removed. The cleavage reaction and removal of the protecting groups is suitably accomplished simultaneously or stepwise. When the resin support is a chloro-methylated polystyrene resin, the bond anchoring the analog/peptide to the resin is an ester linkage formed between the free carboxyl group of the C-terminal residue and one of the many chloromethyl groups present on the resin matrix. It will be appreciated that the anchoring bond can be cleaved by reagents that are known to be capable of breaking an ester linkage and of penetrating the resin matrix.
One especially convenient method is by treatment with liquid anhydrous hydrogen fluoride. This reagent not only will cleave the analog/peptide from the resin but also will remove all protecting groups. Hence, use of this reagent will directly afford the fully deprotected analog/peptide. When the chloromethylated resin is used, hydrogen fluoride treatment results in the formation of the free peptide acids. When the benzhydrylamine resin is used, hydrogen fluoride treatment results directly in the free peptide amines. Reaction with hydrogen fluoride in the presence of anisole and dimethylsulfide at 0°C for one hour will simultaneously remove the side- chain protecting groups and release the analog/peptide from the resin.
When it is desired to cleave the analog/peptide without removing protecting groups, the protected analog/peptide-resin can undergo methanolysis to yield the protected analog/peptide in which the C-terminal carboxyl group is methylated. The methyl ester is then hydrolyzed under mild alkaline conditions to give the free C-terminal carboxyl group. The protecting groups on the analog/peptide chain then are removed by treatment with a strong acid, such as liquid hydrogen fluoride. A particularly useful technique for methanolysis is that of Moore et al . r Peptides, Proc. Fifth finer. Pept. Symp., M. Goodman and J. Meienhofer, Eds., (John Wiley, N.Y., 1977) , p. 518-521, in which the protected analog/peptide-resin is treated with methanol and potassium cyanide in the presence of crown ether.
Another method for cleaving the protected analog/peptide from the resin when the chloromethylated resin is employed is by ammonolysis or by treatment with hydrazine. If desired, the resulting C-terminal amide or hydrazide can be hydrolyzed to the free C-terminal carboxyl moiety, and the protecting groups can be removed conventionally.
It will also be recognized that the protecting group present on the N- ter inal α—amino group may be removed preferentially either before or after the protected analog/peptide is cleaved from the support . Purification of the analogs and peptides of the invention is typically achieved using conventional procedures such as preparative HPLC (including
reversed phase HPLC) or other known chromatographic techniques such as gel permeation, ion exchange, partition chromatography, affinity chromatography (including monoclonal antibody columns) or countercurrent distribution.
The analogs and peptides of this invention may be stabilized by polymerization. Polymerization may be accomplished by crosslinking monomer chains with polyfunctional crosslinking agents, either directly or indirectly, through multi-functional polymers. Ordinarily, two substantially identical analogs/peptides are crosslinked at their C- or N- termini using a bifunctional crosslinking agent. The agent is used to crosslink the terminal amino and/or carboxyl groups. Generally, both terminal carboxyl groups or both terminal amino groups are crosslinked to one another, although by selection of the appropriate crosslinking agent the alpha-amino group of one analog/peptide is crosslinked to the ter inal- carboxyl group of the other analog/peptide. Preferably, the analogs/peptides are substituted at their C-termini with cysteine. Under conditions well known in the art a disulfide bond can be formed between the terminal cysteines, thereby crosslinking the analog/peptide chains. For example, disulfide bridges are conveniently formed by metal-catalyzed oxidation of the free cysteines or by nucleophilic substitution of a 'suitably modified cysteine residue. Selection of the crosslinking agent will depend upon the identities of the reactive side chains of the amino acids present in the analogs/peptides. For example, disulfide crosslinking would not be preferred if cysteine were present in the analog/peptide at additional sites other than the C-terminus. Also within the scope hereof are analogs/peptides crosslinked with methylene bridges.
Suitable crosslinking sites on the analogs/peptides, aside from the N- terminal amino and C-terminal carboxyl groups, include epsilon amino groups found on lysine residues, as well as amino, imino, carboxyl, sulfhydryl and hydroxyl groups located on the side chains of internal residues of the analogs/peptides or residues introduced into flanking sequences.
Crosslinking through externally added crosslinking agents is suitably achieved, e.g., using any of a number of reagents familiar to those skilled in the art, for example, via carbodiirαide treatment of the analog or peptide. Other examples of suitable multi-functional (ordinarily bifunctional) crosslinking agents are found in the literature.
The analogs and peptides of this invention also may be conformationally stabilized by cyclization. The analogs/peptides ordinarily are cyclized by covalently bonding the N- and C-terminal domains of one analog/peptide to the corresponding domain of another analog/peptide of this invention so as to form cyclo-oligomers containing two or more iterated analog/peptide sequences, each internal analog/peptide having substantially
the same sequence. Further, cyclized analogs/peptides (whether cyclo- oligomers or cyclo-monomers) are crosslinked to form 1-3 cyclic structures having from 2 to 6 peptides comprised therein. The analogs/peptides preferably are not covalently bonded through α-amino and main chain carboxyl groups (head to tail), but rather are crosslinked through the side chains of residues located in the N- and C-terminal domains. The linking sites thus generally will be between the side chains of the residues .
Many suitable methods per se are known for preparing mono-or poly- cyclized analogs/peptides as contemplated herein. Lys/Asp cyclization has been accomplished using Nα-Boc-amino acids on solid-phase support with
Fmoc/9-fluorenylmethyl (OFm) side-chain protection for Lys/Asp; the process is completed by piperidine treatment followed by cyclization.
Glu and Lys side chains also have been crosslinked in preparing cyclic or bicyclic analogs/peptides: the analog/peptide is synthesized by solid- phase chemistry on a p-methylbenzhydrylamine resin. The analog/peptide is cleaved from the resin and deprotected. The cyclic analog/peptide is formed using diphenylphosphorylazide in diluted methylforma ide . For an alternative procedure, see Schiller et al . , Peptide Protein Res., 25: 171- 177 (1985). See also U.S. Pat. No. 4,547,489. Disulfide crosslinked or cyclized analogs/peptides are generated by conventional methods. The method of Pelton et al . (J. Med. Chem., 29: 2370-2375 (1986)) is suitable, except that a greater proportion of cyclo- oligomers are produced by conducting the reaction in more concentrated solutions than the dilute reaction mixture described by Pelton et al . , for the production of cyclo-monomers . The same chemistry is useful for synthesis of dimers or cyclo-oligomers or cyclo-monomers. Also useful are thio ethylene bridges. Lebl and Hruby, Tetrahedron Letters, 25: 2067-2068 (1984). See also Cody et al . , J. Med. Chem., 28: 583 (1985).
The desired cyclic or polymeric analogs/peptides are purified by gel filtration followed by reversed-phase high-pressure liquid chromatography or other conventional procedures. The analogs/peptides are sterile filtered and formulated into conventional pharmacologically acceptable vehicles .
The starting materials required for the processes described herein are known in the literature or can be prepared using known methods and known starting materials.
If in the analogs/peptides being created carbon atoms bonded to four nonidentical substituents are asymmetric, then the analogs/peptides may exist as diastereoisomers, enantiomers or mixtures thereof. The syntheses described above may employ racemates, enantiomers or diastereomers as starting materials or intermediates . Diastereomeric products resulting from
such syntheses may be separated by chromatographic or crystallization methods. Likewise, enantiomeric product mixtures may be separated using the same techniques or by other methods known in the art. Each of the asymmetric carbon atoms, when present, may be in one of two configurations R) or S) and both are within the scope of the present invention.
The analogs and peptides of this invention may be contacted with the cartilage by any suitable technique, and may be combined, analog with analog, analog with peptide, or peptide with peptide. If treatment is in vivo, the analog or peptide is administered to the mammal via, e.g., oral, parenteral (e.g., intramuscular, intraperitoneal, intravenous, intra- articular, or subcutaneous injection or infusion, or implant), nasal, pulmonary, vaginal, rectal, sublingual, or topical routes of administration, and can be formulated in dosage forms appropriate for each route of administration. The specific route of administration will depend, e.g., on the medical history of the patient, including any perceived or anticipated side effects using the analog or peptide, the type of analog or peptide being administered, and the particular type of disorder to be corrected.
• Most preferably, the administration is by continuous infusion (using, e.g., slow-release devices or minipumps such as osmotic pumps or skin patches) , or by injection (using, e.g., intravenous, intra-articular or subcutaneous means) . Preferably, the analog or peptide is administered locally, for example, directly to the joint where repair or prevention is needed.
The analog or peptide to be used in the therapy will be formulated and dosed in a fashion consistent with good medical practice, taking into account the clinical condition of the individual patient (especially the side effects of treatment with the analog or peptide) , the type of disorder, the site of delivery, the method of administration, the scheduling of administration, and other factors known to practitioners. The effective amounts of the analog or peptide for purposes herein are thus determined by such considerations and must be amounts that result in bioavailability of the drugs to the mammal and the desired effect.
A preferred administration is a chronic administration of about two times per day for 4-8 weeks to reproduce the effects of IGF-1. As an alternative to injection, chronic infusion may be employed using an infusion device for continuous subcutaneous (SC) or intra-articular infusions. An intravenous bag solution may also be employed. The key factor in selecting an appropriate dose for the disorder in question is the result obtained, as measured by criteria for measuring treatment of the cartilage disorder as are deemed appropriate by the medical practitioner. As a general proposition, the total pharmaceutically-effective amount of the analog or peptide administered parenterally per dose will be in a
range that can be measured by a dose-response curve. For example, IGFs bound to IGFBPs or in the blood can be measured in body fluids of the mammal to be treated to determine the dosing. Alternatively, one can administer increasing amounts of the analog or peptide to the patient and check the serum levels of the patient for IGF-1 and IGF-2. The amount of analog or peptide to be employed can be calculated on a molar basis based on these serum levels of IGF-1 and IGF-2.
Specifically, one method for determining appropriate dosing of the analog or peptide entails measuring IGF levels in a biological fluid such as a body or blood fluid. Measuring such levels can be done by any means, including RIA and ELISA. After measuring IGF levels, the fluid is contacted with the analog or peptide using single or multiple doses. After this contacting step, the IGF levels are re-measured in the fluid. If the fluid IGF levels have fallen by an amount sufficient to produce the desired efficacy for which the molecule is to be administered, then the dose of the molecule can be adjusted to produce maximal efficacy. This method may be carried out in vitro or in vivo . Preferably, this method is carried out in vivo, i . e. , after the fluid is extracted from a mammal and the IGF levels measured, the analog or peptide herein is administered to the mammal using single or multiple doses (that is, the contacting step is achieved by administration to a mammal) . Then the IGF levels are re-measured from fluid extracted from the mammal.
Another method for determining dosing is to use antibodies to the analog or peptide or another detection method for the analog or peptide in the LIFA format. This would allow detection of endogenous or exogenous IGFs bound to IGFBP and the amount of analog or peptide bound to the IGFBP.
Another method for determining dosing would be to measure the level of "free" or active IGF in blood. For some uses the level of "free" IGF would be a suitable marker of efficacy and effective doses or dosing. The amount of active IGF may also be measured in the synovial fluid.
For example, one method is described for detecting endogenous or exogenous IGF bound to an IGF binding protein or the amount of the analog or peptide herein or detecting the level of unbound IGF in a biological fluid. This method comprises: (a) contacting the fluid with 1) a means for detecting the analog or peptide that is specific for the analog or peptide (such as a first antibody specific for epitopes on the analog or peptide) attached to a solid-phase carrier, such that in the presence of the analog or peptide the IGF binding sites remain available on the analog or peptide for binding to the IGF binding protein, thereby forming a complex between the means and the IGF binding protein; and 2) the analog or peptide for a period of time
sufficient to saturate all available IGF binding sites on the IGF binding protein, thereby forming a saturated complex;
(b) contacting the saturated complex with a detectably labeled second means which is specific for the IGF binding protein (such as a second antibody specific for epitopes on the IGFBP) which are available for binding when the analog or peptide is bound to the IGF binding protein; and
(c) quantitatively analyzing the amount of the labeled means bound as a measure of the IGFBP in the biological fluid, and therefore as a measure of the amount of bound analog or peptide and IGF binding protein, bound IGF and IGF binding protein, or active IGF present in the fluid.
Given the above methods for determining dosages, in general, the amount of analog or peptide that may be employed can be estimated, i.e., from about 1 μg/kg/day to 10 mg/kg/day, preferably about 10 μg/kg/day to 1 mg/kg/day, more preferably about 10 - 200 μg/kg/day, might be used, based on kg of patient body weight, although, as noted above, this will be subject to a great deal of therapeutic discretion.
It is noted that dosages and desired drug concentrations of pharmaceutical compositions employable with the present invention may vary depending on the particular use envisioned. The determination of the appropriate dosage or route of administration is well within the skill of an ordinary physician. Animal experiments provide reliable guidance for the determination of effective doses for human therapy. Interspecies scaling of effective doses can be performed following the principles laid down by Mordenti and Chappell, "The use of interspecies scaling in toxicokinetics" in Toxicokinetics and New Drug Development, Yacobi et al . , Eds., (Pergamon Press: New York, 1989), pp. 42-96.
The analog or peptide is suitably administered by a sustained-release system. Suitable examples of sustained-release compositions include semi- permeable polymer matrices in the form of shaped articles, e.g., films, or microcapsules . Sustained-release matrices include polylactides (U.S. Pat. No. 3,773,919, EP 58,481), copolymers of L-glutamic acid and gamma-ethyl-L- glutamate (Sidman et al . , Biopolymers, 22, 547-556 (1983), poly (2- hydroxyethyl ethacrylate) (Langer et al . , J . Biomed. Mater. Res., 15: 167- 277 (1981), and Langer, Chem. Tech., 12: 98-105 (1982)), ethylene vinyl acetate (Langer et al . , supra ) or poly-D- (-) -3-hydroxybutyric acid (EP 133,988). Sustained-release compositions also include a liposomally entrapped analog or peptide. Liposomes containing the analog or peptide are prepared by methods known per se : DE 3,218,121; Epstein et al . , Proc. Natl. Acad. Sci. U.S.A., 82: 3688-3692 (1985); Hwang et al . , Proc. Natl. Acad. Sci. U.S.A., 77: 4030-4034 (1980); EP 52,322; EP 36,676; EP 88,046; EP 143,949; EP 142,641; Japanese Pat. Appln. 83-118008; U.S. Pat. Nos.
4,485,045 and 4,544,545; and EP 102,324. Ordinarily, the liposomes are of the small (from or about 200 to 800 Angstroms) unilamellar type in which the lipid content is greater than about 30 mol. percent cholesterol, the selected proportion being adjusted for the most efficacious therapy. PEGylated analogs or peptides having a longer life can also be employed, based on, e.g., the conjugate technology described in' O 95/32003 published November 30, 1995.
For parenteral administration, in one embodiment, the analog or peptide is formulated generally by mixing each at the desired degree of purity, in a unit dosage injectable form (solution, suspension, or emulsion) , with a pharmaceutically, or parenterally, acceptable carrier, i.e., one that is non-toxic to recipients at the dosages and concentrations employed and is compatible with other ingredients of the formulation. For example, the formulation preferably does not include oxidizing agents and other peptides that are known to be deleterious to polypeptides.
Generally, the formulations are prepared by contacting the analog or peptide uniformly and intimately with liquid carriers or finely divided solid carriers or both. Then, if necessary, the product is shaped into the desired formulation. Preferably the carrier is a parenteral carrier, more preferably a solution that is isotonic with the blood or synovial fluid of the recipient. Examples of such carrier vehicles include water, saline, Ringer's solution, a buffered solution, hyaluronan, and dextrose solution. Non-aqueous vehicles such as fixed oils and ethyl oleate are also useful herein. The carrier suitably contains minor amounts of additives such as substances that enhance isotonicity and chemical stability. Such materials are non-toxic to recipients at the dosages and concentrations employed, and include buffers such as phosphate, citrate, succinate, acetic acid, and other organic acids or their salts; antioxidants such as ascorbic acid; low molecular weight (less than about ten residues) polypeptides, e . g. , polyarginine or tripeptides; proteins, such as serum albumin, gelatin, or immunoglobulins; hydrophilic polymers such as polyvinylpyrrolidone; glycine; amino acids such as glutamic acid, aspartic acid, histidine, or arginine; monosaccharides, disaccharides, and other carbohydrates including cellulose or its derivatives, glucose, mannose, trehalose, or dextrins; chelating agents such as EDTA; sugar alcohols such as mannitol or sorbitol; counter- ions such as sodium; non-ionic surfactants such as polysorbates, poloxamers, or polyethylene glycol (PEG); and/or neutral salts, e.g., NaCl, KC1, MgCl2, CaCl2, etc. The analog or peptide typically formulated in such vehicles at a pH of from or about 4.5 to 8. It will be understood that use of certain of the
foregoing excipients, carriers, or stabilizers will result in the formation of salts of the analog or peptide. The final preparation may be a stable liquid or lyophilized solid.
Typically about 0.5 to 500 mg of the analog or peptide or mixture of analogs and/or peptides, as the free acid or base form or as a pharmaceutically acceptable salt, is compounded with a physiologically acceptable vehicle, carrier, excipient, binder, preservative, stabilizer, flavor, etc. , as called for by accepted pharmaceutical practice. The amount of active ingredient in these compositions is such that a suitable dosage in the range indicated is obtained.
The analog or peptide to be used for therapeutic administration must be sterile. Sterility is readily accomplished by filtration through sterile filtration membranes (e.g., 0.2 micron membranes). Therapeutic compositions generally are placed into a container having a sterile access port, for example, an intravenous solution bag or vial having a stopper pierceable by a hypodermic injection needle.
The analog or peptide ordinarily will be stored in unit or multi-dose containers, for example, sealed ampules or vials, as an aqueous solution or as a lyophilized formulation for reconstitution. As an example of a lyophilized formulation, 10-mL vials are filled with 5 mL of sterile- filtered 1% (w/v) aqueous solution of analog or peptide, and the resulting mixture is lyophilized. The infusion solution is prepared by reconstituting the lyophilized analog or peptide using bacteriostatic Water-for-Injection.
Combination therapy with the analog or peptide herein and one or more other appropriate reagents that enhance the effect of the analog or peptide is also part of this invention. These include antagonists to cytokines, NO, or IL-lra, a cartilage catabolism antagonist, or a cartilage growth factor, such as wild-type IGF-1 and/or ALS if the active agent is an IGFBP-3 displacer peptide or an IGF-1 analog with a binding affinity preference for IGFBP-3 over IGFBP-1. In addition, the IGFBP displacer peptide may be co- administered with an IGF-1 analog herein, preferably with the analog with a binding affinity preference for IGFBP-1 over IGFBP-3.
The active agent and reagent to enhance its effect may be administered concurrently or sequentially, and the reagent may be administered at the same or lower doses than they would otherwise be administered 'if given alone. The displacer peptide/IGF-1 analog with binding preference for IGFBP-3 can be administered separately from the IGF-1 and/or ALS, but preferably these agents are administered together as a binary or ternary complex where such a complex can be formed. Administration as a complex of ALS, IGF-I, and IGFBP-3 displacer peptide/IGF-1 analog results in the longest half-life for the active agent.
The invention herein also contemplates using gene therapy for treating a mammal, using nucleic acid encoding the analog or peptide. Generally, gene therapy is used to increase (or overexpress) IGF levels the mammal. Nucleic acids that encode the analog or peptide can be used for this purpose. Once the ammo acid sequence is known, one can generate several nucleic acid molecules using the degeneracy of the genetic code, and select which to use for gene therapy.
There are two major approaches to getting the nucleic acid (optionally contained m a vector) into the patient's cells for purposes of gene therapy: m vivo and ex v vo For in v vo delivery, the nucleic acid is injected directly into the patient, usually at the s te where the analog or peptide is required For ex vivo treatment, the patient's cells are removed, the nucleic acid s introduced into these isolated cells and the modified cells are administered to the patient either directly or, for example, encapsulated within porous membranes which are implanted into the patient See, e.g., U.S. Patent Nos. 4,892,538 and 5,283,187.
There are a variety of techniques available for introducing nucleic acids into viable cells. The techniques vary depending upon whether the nucleic acid is transferred into cultured cells m vitro, or n vivo the cells of the intended host. Techniques suitable for the transfer of nucleic acid into mammalian cells m vitro include the use of liposomes, electroporation, micromjection, cell fusion, DEAE-dextran, the calcium phosphate precipitation method, viral infection etc A commonly used vector for ex v vo delivery of the gene is an adeno- or retro- virus. The currently preferred in vivo nucleic acid transfer techniques include infection with viral vectors (such as adenovirus, Herpes simplex I virus, retrovirus, or adeno-associated virus) and lipid-based systems (useful lipids for lipid-mediated transfer of the gene are DOTMA, DOPE and DC-Choi, for example) . In some situations it is desirable to provide the nucleic acid source with an agent that targets the target cells, such as an antibody specific for a cell surface membrane protein or the target cell, a ligand for a receptor on the target cell, etc. Where liposomes are employed, proteins that bind to a cell-surface membrane protein associated with endocytosis may be used for targeting and/or to facilitate uptake. Examples include capsid proteins or fragments thereof tropic for a particular cell type, antibodies for proteins that undergo ternal zation m cycling, and proteins that target mtracellular localization and enhance mtracellular half-life. The technique of receptor-mediated endocytosis is described, for example, by Wu et al . , J. Biol. Chem., 262: 4429-4432 (1987); and Wagner et al . , Proc. Natl. Acad. Sci. USA, 87: 3410-3414 (1990). For review of the currently known gene marking and gene therapy protocols, see
Anderson et al . , Science, 256: 808-813 (1992). See also WO 93/25673 and the references cited therein.
Kits are also contemplated for this invention. The article of manufacture comprises a container and an instruction. Suitable containers include, for example, bottles, vials, syringes, and test tubes. The containers may be formed from a variety of materials such as glass or plastic. The container holds a composition which is effective for treating the cartilage disorder, e . g. , degenerative cartilagenous disorder, and may have a sterile access port (for example, the container may be an intravenous solution bag or a vial having a stopper pierceable by a hypodermic injection needle) . The active agent in the composition is an IGF-1 analog or an IGFBP displacer peptide as defined herein. The composition can comprise any or multiple ingredients disclosed herein. The instruction on, or associated with, the container indicates that the composition is used for treating a cartilage disorder. For example, the instruction could indicate that the composition is effective for the treatment of osteoarthritis, rheumatoid arthritis, or any other degenerative cartilagenous disorder. The article of manufacture may further comprise a second container comprising a pharmaceutically- acceptable buffer, such as phosphate-buffered saline, Ringer's solution, or dextrose solution. Alternatively, the composition may contain any of the carriers, excipients, and/or stabilizers mentioned hereinabove. It may further include other materials desirable from a commercial and user standpoint, including other buffers, diluents, filters, needles, syringes, and package inserts with instructions for use. The kit optionally includes a separate container, preferably a vial, for a co-agent to be administered along with the active agent, such as IGF-1.
The invention will be more fully understood by reference to the following examples. They should not, however, be construed as limiting the scope of the invention. All literature and patent citations mentioned herein are expressly incorporated by reference.
EXAMPLES Examples 1-4 below are taken from WO 98/45427 as describing IGFBP-3 displacer peptides defined herein. Based upon the results of in vitro and in vivo experiments using an IGFBP-3 displacer peptide with amino acid changes at residues 24 and 31 (Y24L,Y31A), also designated (Leu24,Ala31) hIGF-1 or IGF-M, disclosed in WO 98/45427, it is predicted that other peptides that inhibit the interaction of an IGF with an IGFBP, and bind poorly or not at all to the IGF-1 receptor, should increase active IGF levels in a subject being treated. In addition, it is possible that another class of molecules might bind IGF-1 itself at a site remote from that involved in receptor interactions in such a way as to inhibit or prevent the
interaction of IGF-1 with the IGFBPs, but not the interaction of IGF-1 with its receptor.
In the examples, common α-ammo acids may be described by the standard one- or three-letter amino acid code when referring to intermediates and final products . By common α-ammo acids is meant those ammo acids incorporated into proteins under RNA direction. Standard abbreviations are listed in The Merck Index, 10th Edition, pp Mιsc-2 - Misc-3. Unless otherwise designated the common α-amino acids have the natural or "L"-confιguratιon at the alpha carbon atom. If the code is preceded by a "D" this signifies the opposite enantiomer of the common α-amino acid. Modified or unusual α-amino acids such as norleucme (Nle) and ornithme (Orn) are designated as described m U.S. Patent and Trademark Office Official Gazette 1114 TMOG, May 15, 1990.
EXAMPLE 1 Phage-derived Peptides to Bind IGF-1 and Binding Proteins
Introduction:
It has been shown that peptides that bind specifically and with measurable affinity to target molecules, such as proteins, can be identified from an initial library of many binding and non-binding peptides through binding selections using bacteriophage coat-protein fusions (Smith, Science, 228: 1315 (1985); Scott and Smith, Science, 249: 386 (1990); Cw rla et al . , Proc. Natl. Acad. Sci. USA, 8_: 309 (1990); Devlin et al . , Science, 249: 404 (1990); reviewed by Wells and Lowman, Curr Opin. Struct. Biol., 2: 597 (1992); U.S. Pat. No. 5,223,409). In addition, both proteins and peptides displayed on phage can be affinity-enhanced through iterative cycles of mutations, selection, and propagation.
Libraries of peptides differing m sequence at particular residue positions can be constructed using synthetic oligodeoxynucleotides . Peptides are displayed as fusion proteins with a phage coat protein (such as g3p or g8p) on bacteriophage particles, each of which contains a single-stranded DNA genome encoding the particular peptide variant. After cycles of affinity purification, using an immobilized target molecule, individual bacteriophage clones are isolated, and the ammo acid sequence of their displayed peptides is deduced from their DNA sequences. Materials and Methods:
Construction of peptide-phage libraries
To identify a set of peptide molecules having the ability to bind to IGF-1 or to an IGF binding protein, such as IGFBP-1 or IGFBP-3, several diverse phage libraries of peptides, of length ranging from 18 to 20 residues, were constructed. Peptides of this size were chosen order to favor the selection of peptides capable of maintaining well-defined
structures m solution . Because natural-ammo acid peptides of this size have a potential sequence diversity of 20 18- ■20 20 (i.e. 2.6 x 10 23 to 1.0 x
10-26) variants, it is not practical to construct and test all such variants. Instead, certain residues were fixed or constant, which might be expected to allow or promote stable elements of peptide structure such as disulfide bonds or beta-turns, within each peptide.
Structural constraints or frameworks have previously been used for presentation of peptide libraries on phage and for subsequent, successive enhancement of binding affinities through mutation and selection. Such structured frameworks may favor stable binding conformations of peptide segments. By analogy, immunoglobulins provide a stable (and conserved) structural framework for presentation of a diversity of different peptide loops (CDR's, complementarity-determining regions) which can bind different antigens . Used as a template for library constructions was a plasmid, pt4.g8 (complete DNA sequence shown m Fig. 1) expressing an antibody-recognizable (gD-tag) peptide fused to g8p of bacteriophage M13. This plasmid contains s gle-stranded and double-stranded origins of DNA replication. The phoA promoter and STII secretion-signal sequences are upstream of the gD peptide (underlined below) , which is followed by a "linker" peptide (double underlined below), and then the g8p of bacteriophage M13:
SGTAMADPNRFRGKDLAGSPGGGSGGGAEGDDPAKAAFNSLOASATEYIGYAWAMVVVIVGATIGIKLFKKFTSKA S (SEQ ID NO:55)
Several random-sequence peptide libraries (Table I) were constructed using single-stranded template-directed mutagenesis (Kunkel et al . , Methods . Enzymol. , 20 : 125 (1991)), with the oligonucleotides described below.
TABLE I Large Naive Libraries for g8 Display
Library Oligonucleotide no. Peptide motif SEQ ID NO
A HL-300 SGTACX2GPX4CSLAGSP (SEQ ID NO: :56)
B HL-301 X4CX2GPX4CX4 (SEQ ID NO: :57)
C HL-302 x20 (SEQ ID NO: :58)
D HL-303 x7cx4cx7 (SEQ ID NO: :59)
D HL-304 x7cx5cx6 (SEQ ID NO: :60)
D HL-305 x6cx6cx6 (SEQ ID NO: :61)
D HL-306 x6cx cx5 (SEQ ID NO: 62)
D HL-307 x5cx8cx5 (SEQ ID NO: 63)
D HL-308 x5cx9cx4 (SEQ ID NO: 64)
D HL-309 x4cx10cx4 (SEQ ID NO: 65)
A. Beta-turn sequence motif
An example of a peptide of known three-dimensional structure is given by Wrighton et al . , who selected a peptide agonist for the erythropoiet receptor (EPO-R) by phage display (Wrighton et al . , Science, 273 : 458 (1996)). The peptide GGTYSCHFGP TWVCKPQGG (SEQ ID NO: 66) (having a disulfide bond joining the two Cys residues) forms a dimer of two beta hairpins, the crystallized complex with EPO-R (Livnah et al . , Science, 273: 464 (1996)). Although the structure of the unbound form of this peptide n solution has not been reported, the beta-turn structure formed by this peptide in complex with EPO-R suggested that similar structures might be formed by peptides of the form CX2GPX4C (SEQ ID NO: 67) .
As one type of structured peptide library, a portion of the gD peptide was replaced with the motif CX2GPX4C (SEQ ID NO: 67), leaving the upstream and downstream ("flanking") residues unchanged from that of the starting plasmid. Thus, this library was designed to display on phage particles the peptide SGTACX2GPX4CSLAGSP (SEQ ID NO:56), where X represents any of the 20 natural L-a ino acids, fused to the linker and gδp described above. This library was constructed using the oligonucleotide HL-300:
5 ' -GCC TAT GCA TCT GGT ACC GCC TGC NNS NNS GGT CCT NNS NNS NNS NNS TGT TCT CTG GCA GGT TCA CCA G-3' (SEQ ID NO:68), where N indicates a mixture of the nucleotides A, G, C, and T, and S represents a mixture of the nucleotides G and C.
An additional library was constructed to allow for further interactions within the peptide and/or with the target proteins by randomizing the flanking sequences as well. This library was constructed with the form X4C 2GPX4CX4 (SEQ ID NO: 57) by using oligonucleotide HL-301:
5 ' -GCT ACA AAT GCC TAT GCA NNS NNS NNS NNS TGC NNS NNS GGT CCT NNS NNS NNS NNS TGT NNS NNS NNS NNS GGT GGA GGA TCC GGA GGA G-3' (SEQ ID NO: 69) . B. Disulfide-loop motifs Because many additional peptide conformations might be productive for binding to a given target protein, it was desirable to test other types of peptide sequence motifs in phage-displayed libraries. For example, a single disulfide bond within a small peptide may favor stable structures that allow for relatively higher-affmity binding than m unconstrained structures (Geysen et al . , Mol . Immunol . , 23: 709 (1986); Wood et al . , Science, 232: 633 (1986); Oldenburg et al . , Proc. Natl. Acad. Sci. USA, 89: 5393 (1992); O'Neil et al . , Proteins, JL4: 509 (1992); McLafferty et al . , Gene, 128.: 29 (1993); Giebel et al . , Biochem. , 34: 15430 (1995)). Several peptide-phage libraries were therefore constructed, of the form XmCXnCXk, where m=4, n=10, and k=4, or where m=5, n=8-9, and k=4-5, or m=6, n=6-7, and k=5-6, or m=7,
n=4-5, and k=6-7 (SEQ ID NOS:59 to 65). In these peptides, a disulfide bond is predicted to form a stabilizing constraint for peptide conformation.
These peptide libraries (see Table I) were constructed as X CX4CX7
(SEQ ID NO: 59), using oligonucleotide HL-303: 5'-GCT ACA AAT GCC TAT GCA NNS NNS NNS NNS NNS NNS NNS TGC NNS NNS NNS NNS TGC NNS NNS NNS NNS NNS NNS NNS GGT GGA GGA TCC GGA GGA G-3' (SEQ ID NO: 70); X7CX5CX6 (SEQ ID NO: 60), using oligonucleotide HL-304:
5'-GCT ACA AAT GCC TAT GCA NNS NNS NNS NNS NNS NNS NNS TGC NNS NNS NNS NNS NNS TGC NNS NNS NNS NNS NNS NNS GGT GGA GGA TCC GGA GGA G-3' (SEQ ID NO: 71) ;
X6CX6CX6 (SEQ ID NO: 61), using oligonucleotide HL-305:
5'-GCT ACA AAT GCC TAT GCA NNS NNS NNS NNS NNS NNS TGC NNS NNS NNS NNS NNS NNS TGC NNS NNS NNS NNS NNS NNS GGT GGA GGA TCC GGA GGA G-3' (SEQ ID NO:72) ; X6CX7CX5 (SEQ ID NO: 62), using oligonucleotide H -306:
5'-GCT ACA AAT GCC TAT GCA NNS NNS NNS NNS NNS NNS TGC NNS NNS NNS NNS NNS
NNS NNS TGC NNS NNS NNS NNS NNS GGT GGA GGA TCC GGA GGA
G-3' (SEQ ID NO:73) ;
X5CX8CX5 (SEQ ID NO:63), using oligonucleotide HL-307: 5'-GCT ACA AAT GCC TAT GCA NNS NNS NNS NNS NNS TGC NNS NNS NNS NNS NNS NNS NNS NNS TGC NNS NNS NNS NNS NNS GGT GGA GGA TCC GGA GGA G-3' (SEQ ID NO:74); X5CX9CX4 (SEQ ID NO: 64), using oligonucleotide HL-308:
5 ' -GCT ACA AAT GCC TAT GCA NNS NNS NNS NNS NNS TGC NNS NNS NNS NNS NNS NNS NNS NNS NNS TGC NNS NNS NNS NNS GGT GGA GGA TCC GGA GGA G-3' (SEQ ID NO:75); and
X CX10CX4 (SEQ ID NO:65), using oligonucleotide HL-309:
5' -GCT ACA AAT GCC TAT GCA NNS NNS NNS NNS TGC NNS NNS NNS NNS NNS NNS NNS
NNS NNS NNS TGC NNS NNS NNS NNS GGT GGA GGA TCC GGA GGA
G-3' (SEQ ID NO:76) . C. Unconstrained peptides
Unconstrained libraries (i.e., having no fixed residues within the peptide) have also yielded specific binding molecules (Scott and Smith, supra; Cwirla et al . , supra; Devlin et al . , supra; Kay et al . , Gene, 128 : 59
(1993) ) . Such libraries may yield structured peptides, nevertheless, since noncovalent interactions may still induce structure in the bound, and/or unbound forms. An unconstrained peptide library, of the form 2Q (SEQ ID
NO: 58), was constructed using oligonucleotide HL-302:
5' -GCT ACA AAT GCC TAT GCA NNS NNS NNS NNS NNS NNS NNS NNS NNS NNS NNS NNS
NNS NNS NNS NNS NNS NNS NNS NNS GGT GGA GGA TCC GGA GGA G-3' (SEQ ID NO: 77) .
Polyvalent (g8) phage binding selections
The products of random mutagenesis reactions were transformed into
XL1-BLUE™ E. coli cells (Stratagene) by electroporation and amplified by growing 15-16h with M13K07 (Vieira and Messing, Methods Enzy ol., 153: 3-11 (1987)) or VCSM13 helper phage (Stratagene Corp.). Based upon plating of the initial transformations, the number of fransformants per library was approximately 1.8 x IO8 for library HL-300, 7.9 x IO8 for HL-301, 5.0 x IO8 for HL-302, 5.3 x IO8 for HL-303, 5.6 x IO8 for HL-304, 5.0 x 108 for HL-305, 6.3 x 108 for HL-306, 4.5 x 108 for HL-307, 1.9 x 108 for HL-308, and 2.1 x 108 for HL-309.
IGFBP-3 and IGF-1 were biotinylated with a 1.5:1 molar ratio of a cleavable biotin reagent, EZ-LINK™ NHS-SS-Biotin (Pierce), to protein, using the manufacturer's instructions.
The initial selection of peptides for binding to IGFBP-3 or IGF-1 was carried out using phage pools of approximately lO-^ phage/ml (100 μl total volume) . MAXISORP™ 96-well plastic plates (Nunc) were coated with a solution of 2 μg/ml of NEUTRAVIDIN™ brand avidin (Pierce) in 50 mM sodium carbonate buffer, pH 9.6, overnight at 4°C. The NEUTRAVIDIN™ solution was then removed, and the plates were incubated with a blocking solution of 5 g/1 of bovine serum albumin, or 5 g/1 of ovalbumin, or 5 g/1 of instant milk in 50 mM sodium carbonate buffer, for 1-2 h at room temperature. The blocking solution was then removed, and a solution of biotinylated target protein was added. After 1-2 h at room temperature, the target solution was removed, and the plates were washed ten times with PBS/TWEEN™ surfactant (0.05% TWEEN-20™ in PBS buffer).
Phage from the libraries described above were pooled as follows: pool A consisted of HL-300 phage, pool B of HL-301 phage, pool C of HL-302 phage, and pool D of phage from the HL-303, HL-304, HL-305, HL-306, HL-307, HL-308, and HL-309 libraries. Phage were added in PBS/TWEEN™/albumin/biotin (PBS/TWEEN™ buffer w th 1 μM biotin, 5 g/1 bovine serum albumin, or ovalbumin) to wells coated with each target, and with control wells that were coated with NEUTRAVIDIN™ or with albumin, but not biotinylated target.
The phage were allowed to bind 5-15 h at room temperature. The plates were then washed ten times with PBS/TWEEN™ buffer. Phage remaining bound to the plates were eluted by incubating with 50 mM DTT for 1-2 h at room temperature. The eluted phage were transfected into E. coli cells and allowed to grow overnight at 37 °C to amplify the phage .
The second and third cycles of binding selection were carried out as above, except that streptavidin (0.1 mg/ml) was included in the phage cocktails along with biotin. An aliquot was taken from each target-coated and control well incubated with each library, and serial dilutions of the diluted phage were performed to measure specific binding to target. The diluted phage were then transfected into E. coli cells and plated for colony counting .
The fourth round of binding selection was carried out on MAXISORP™ plates directly coated with 2 μg/ml of each target protein, or with albumin only. The results of phage-binding selections in cycles 2-4 are shown in Fig. 2.
The same initial phage libraries (A, B, C, D) were also used for binding selections to directly-coated IGFBP-3. In this case, MAXISORP™ 96-well plastic plates (Nunc) were coated with a solution of 2 μg/ml of IGFBP-3 in 50 mM sodium carbonate buffer, pH 9.6, overnight at 4°C. The target solution was then removed, and the plates were incubated with a blocking solution of 5 g/L of bovine serum albumin, for 1-2 h at room temperature. Phage were incubated with the plates as above, and non-binding phage washed away. The phage remaining bound were eluted by incubating with 20 mM HC1 for 10 min at room temperature. Thereafter, the acid-eluted phage were neutralized with one-fifth volume of 1 M Tris-HCl, pH 8.0. Phage were transfected for colony counting as described above. Screening of polyvalent phage clones ( IGF-blocking phage assay)
Peptide-phage clones were isolated by mixing phage pools with E. coli cells, and plating onto antibiotic-containing media. Colonies were isolated and grown with helper phage (as above) to obtain single-stranded DNA for sequencing. Peptide sequences selected for binding IGFBP-3 or IGF-1 were deduced from the DNA sequences of phagemid clones. A number of such clones are represented by the peptide sequences in Tables II and III, respectively. TABLE II
Peptide sequences from g8 display, IGFBP-3 selection
Name Peptide sequence
4A3.1 SGTACYGGPEWWCCSLAGSP (SEQ ID NO: 8)
4A3.3 SGTACYGGPEWWCCSLAGSP (SEQ ID NO: 79)
4A3.4 SGTACYGGPEWWCCSLAGSP (SEQ ID NO: 80)
4B3.1 DLAICAEGPEIWVCEETS (SEQ ID NO: 81)
4B3.2 DFWICLSGPGWEECLEWW (SEQ ID NO: 82) 4B3.3 EESECFEGPGYVICGLVG (SEQ ID NO: 83)
4B3.4 DMGVCADGPWMYVCEWTE (SEQ ID NO: 84)
4B3.5 DMGVCADGPWMYVCEWTE (SEQ ID NO: 85)
4C3.1 GSAGQGMTEEWAWIWEWWKE (SEQ ID NO: 86)
4C3.2 ELDGWVCIKVGEQNLCYLAE (SEQ ID NO: 87)
4C3.4 ELDGWVCIKVGEQNLCYLAE (SEQ ID NO: 88)
4C3.4 ELDGWVCIKVGEQNLCYLAE (SEQ ID NO: 88)
4C3.5 ELDGWVCIKVGEQNLCYLAE (SEQ ID NO: 88)
4D3.1 AIGGWCFIELDSLWCEEQIG (SEQ ID NO: 89)
4D3.2 SEDVECWQVWENLVCSVEHR (SEQ ID NO: 90)
44DD33..33 SEEVCWPVAEWYLCNMWGR (SEQ ID NO: 91)
4D3.4 RVGAYISCSETECWVEDLLD (SEQ ID NO: 92)
4D3.5 WFKTVCYEWEDEVQCYTLEE (SEQ ID NO: 93)
4D3.6 SEDVECWQVWENLVCSVEHR (SEQ ID NO: 94)
4D3.7 RLEEQCVEVNYEPSCSFTAN (SEQ ID NO: 95)
44DD33..88 SEEVCWPVAEWYLCNILGP (SEQ ID NO: 96)
4D3.9 ETVANCDCYMDLCLCYGSDR (SEQ ID NO: 97)
4D3.10 YHPISCMDHYYLIICDETVN (SEQ ID NO: 98)
4D3.11 VAWEVCWDRHDQGYICTTDS (SEQ ID NO: 99)
4D3.12 AEWAECWIAGDQLLCVGKDN (SEQ ID NO: 100)
2233AA33..11 EPWLCQYYEAAMLYLCWEEG (SEQ ID NO: 101)
23A3.2 AEEGMVWGWTGGWYNLDELC (SEQ ID NO: 102)
23A3.3 SGGAIYWPVEQFIAFMAVGK (SEQ ID NO: 103)
23A3.4 EPWLCQYYEAAMLYLCWEEG (SEQ ID NO: 104)
23A3.5 SGGAIYMPVEQFIAFMAVGK (SEQ ID NO: 105)
2233BB33..11 TGVDCQCGPVHCVCMDWA (SEQ ID NO: 12)
23B3.2 EVLLCSDGPQLYLCELYA (SEQ ID NO: 106)
23B3.4 SGVECVWGPQWGFCVEEY (SEQ ID NO: 107)
23B3.5 DKEVCYLGPETWLCFWWP (SEQ ID NO: 108)
23B3.6 EVLLCSDGPQLYLCELYA (SEQ ID NO: 109)
2233BB33..77 GDVECIEGPWGELCVWAD (SEQ ID NO:110)
23D3.1 FGGWSCQPTWVDVYVCNFEE (SEQ ID NO: 111)
23D3.2 AMWVCVSDWETVEECIQYMY (SEQ ID NO: 112)
23D3.3 AMWVCVSDWETVEECIQYMY (SEQ ID NO: 113)
23D3.4 AMWVCVSDWETVEECIQYMY (SEQ ID NO: 114)
2233DD33..55 AMWVCVSDWETVEECIQYMY (SEQ ID NO: 115)
23D3.6 TNWFFVCESGHQDICWLAEE (SEQ ID NO: 116)
TABLE III
Peptide sequences from g8 display, IGF-1 selection Clone Peptide sequence Library Frequency
HL-8 WVMECGAGPWPEGCTFML B 5/6
(SEQ ID NO: 117)
HL-26 RKTSQGRGQEMCWETGGCS C 1/6
(SEQ ID NO: 118)
HL-25 SWERGELTYMKLCEYMRLQQ C 4/6
(SEQ ID NO: 119)
HL-30 EHGRANCLITPEAGKLARVT C 1/6 (SEQ ID NO:120)
Such peptide-phage clones could represent specific target-binding peptides which either do or do not block ligand (IGF-1 to IGFBP-3) binding, or any of a number of non-bmdmg or background members of the selected pool. To distinguish among these possibilities, phage clones were tested for the ability to bind to IGFBP-3 in the presence and absence of IGF-1.
IGFBP-3 was coated directly onto MAXISORP™ plates as above. Phage from clonal cultures were mixed with IGF-1 (100 nM final concentration) , and incubated with the immobilized IGFBP-3 for 1 hour at room temperature. The plates were then washed ten times, as above, and a solution of rabbit anti-phage antibody mixed with a goat-anti-rabbit conjugate of horseradish peroxidase was added. After an incubation of 1 hour at room temperature, the plates were developed with a chromogenic substrate, o-phenylenediamine (Sigma). The reaction was stopped with addition of 1/2 volume of 2.5 M H2SO4. Optical density at 490 nm was measured on a spectrophotometric plate reader.
Titration of several IGFBP-3-selected peptide-phage clones showed all were inhibited by IGF-1 for binding to IGFBP-3 at some phage concentration (Figs. 3 and 4). These peptides are thus likely to occupy an overlapping site with the IGF-bmding epitope on IGFBP-3. Additional peptide-phage clones were screened similarly, at a low concentration of phage, with and without IGF-1.
Figure 5 shows the results of a blocking assay of several phagemid clones derived from three rounds of DTT elution, followed by one round of HC1 elution, as described above. In each case, the phagemid clone was grown from a single colony overnight at 37 °C in a culture volume of 5 ml. The phage particles were precipitated and resuspended 0.5 ml of PBS buffer. A 50-fold dilution of each phage solution was made into PBS/TWEEN™ buffer, and the phage were incubated with or without 100 nM IGF-1 on an IGFBP-3-
coated MAXISORP™ plate. As shown in Fig. 5, most clones were >40% inhibited for binding to IGFBP-3 at these phage concentrations, although clone 4D3.11 was only 5% inhibited under these conditions.
Figure 6 shows the results of a blocking assay of several phagemid clones derived from three rounds of HC1 elution, as described above. In each case, the phagemid clone was grown from a single colony overnight at 37°C in a culture volume of 5 ml. The phage particles were prepared as described above. In this case, as shown m Fig. 6, most clones were >80% inhibited for binding to IGFBP-3 at these phage concentrations, although clones 23A3.3 and 23A3.5 were only about 20% inhibited under these conditions .
The variation in the degree to which phage binding is blocked by a constant concentration of IGF-1, as a function of phage dilution (Fig. 3) , or as a function of peptide displayed (Figs. 5-6) s of interest because, without being limited to any one theory, it may be predictive of (1) the degree of overlap between IGF-1- and peptide-b ding epitopes on the IGFBP-3 molecule, and/or (2) the relative affinity of IGF-1 versus phage-displayed peptide for binding to IGFBP-3. Since all peptide-phage clones tested here showed some degree of inhibition with IGF-1, it is likely that the epitope for peptide-bmding on IGFBP-3 for each lies within an area occupied by bound IGF-1. Peptide assays (see below) support this conclusion (i.e., case 1). On the other hand, without being limited to any one theory, it s possible that some peptide epitopes could be simply within an area for which binding of the phage particle displaying such peptides is steπcally excluded by bound IGF-1.
The dependence of inhibition upon phage concentration, and the differences among phage clones (Fig. 3) may reflect case 2. In particular, phage clones whose binding to an IGFBP-3 coated plate was inhibited only at low phage concentrations (e.g., 4D3.3, 4B3.4, corresponding to peptides BP3-01-ox and BP3-02-OX, respectively) appear to yield higher-affinity peptides (see below) for IGFBP-3 than do those phage clones whose binding to an IGFBP-3 coated plate was inhibited both at high and at low phage concentrations (e.g., 4C3.2, 4D3.5, corresponding to peptides BP-23 and BP-24, respectively) . Thus, this type of phage-titration blocking assay may be generally useful as a means to predict the relative affinities and inhibitory potencies of peptides derived from phage displayed libraries. Monovalent (g3) display of lGFBP-3-bindmg peptides
Affinity maturation of a peptide or protein sequence by successive rounds of random mutagenesis, selection, and propagation can be efficiently accomplished when the copy number of displayed peptides or proteins is
limited (Bass et al . , Proteins, 8^: 309-314 (1990)). Such an affinity maturation process is illustrated by the affinity maturation of hGH (U.S.
Pat. No. 5,534,617). In this case, the copy number of displayed hGH was limited by fusing the displayed protein to g3, rather than to g8 of bacteriophage particles, restricting the expression level of hGH, and using a helper phage to supply wild-type g3p for phagemid packaging and propagation.
To select for higher affinity peptide variants from pools of phage displaying peptides on g8p, peptide cDNAs from two round 4 gδ library pools, 4B and 4D, were transferred to a g3 vector for monovalent phage display.
Binding selections were carried out for three rounds, as described above, with acid elution of binding phage.
Peptide sequences obtained after three rounds of selections are shown in Table IV. Two clones, 4B3.3 and 4D3.11, dominated the selected pools, and were seen in the earlier, g8 phage selections. A third clone, 3Ai.2, represents a new peptide sequence that was not identified from g8 display.
In phage-ELISA competition assays, the apparent affinity of the g3-4B3.3 and g3-4D3.11 clones ' was <100 nM; however, the corresponding peptides showed much weaker inhibition (see below) . TABLE IV
Peptide sequences from g3 display, IGFBP-3 selection
Clone Peptide sequence Library Frequency 3Ai.l= EESECFEGPGYVICGLVG 4B 6/10
4B3.3 (SEQ ID NO: 8)
3Ai.2 VEDECWMGPDWAVCWTWG 4B 4/10
(SEQ ID NO:121)
3Bi.l= VAWEVCWDRHDQGYICTTDS 4D 10/10
4D3.11 (SEQ ID NO: 6)
It is anticipated that affinity improvements can be obtained by iteratively mutating, selecting, and propagating peptide-phage libraries, as described for hGH. See, e.g., U.S. Pat. No. 5,534,617. Peptide assays
Peptides were synthesized corresponding to a number of phage-derived sequences . In cases where two Cys residues were found in the peptide sequence, the disulfide (oxidized or "ox" suffix) monomeric form of the peptide was prepared and purified. In cases where four Cys residues were found, the {1-4, 2-3}-disulfide form was prepared and purified.
The ability of these peptides to bind IGFBP-3 and block IGF-1 binding was tested in one or more of the following assays.
BIACORE™ competition assay (for IGFBPS binders)
IGF-1 was immobilized on a dextran chip for inhibition assays using a
BIACORE™™ 2000 surface-plasmon-resonance device (BIAcore, Inc., Piscataway, NJ) to measure free binding protein. IGF-1 was biotinylated as described above, and injected over a chip to which streptavidin had been coupled (BIAcore, Inc.) to give 400 to 800 RU (response units) of immobilized IGF-1. The IGF-1 showed no detectable dissociation over the time course of each experiment . Serial dilutions of peptide were mixed with a constant concentration (40 nM) of IGFBP-3. After incubation for >1 hour at room temperature, an aliquot of 20 μL was injected at a flow rate of 20 μL/min over the IGF-1 chip. Following the injection, a response reading was taken to measure the relative amount of IGFBP-3 bound to the IGF-1.
The results (Figs. 7-8) show a dose-response curve for each peptide 's inhibition of IGFBP-3 binding to the chip. In particular, the most effective inhibitors of IGFBP-3 binding tested were peptides BP3-01-ox (corresponding to phage clone 4D3.3), and a truncated form of this peptide, BP3-15 (see Table V) . In that table, a disulfide bond is formed between the two Cys residues of each 2-Cys containing peptide. For peptides containing four cysteines, the two Cys* residues form a disulfide and the remaining two form a second disulfide. These peptides showed IC50's of 2 μM and 0.75 μM, respectively. Other peptides such as BP3-4D3.il (phage clone 4D3.11 from g8 display and 3Bi.l from g3 display) showed inhibition with IC50's of <10 μM. IGFBP-1 did not show binding to IGF-1 immobilized in this manner.
TABLE V Inhibition of IGF-1 binding to IGFBP-3 by synthetic peptides
Peptide Seguence BIACORE™ AssayIC50 (μM)
BP-23 ELDGWVCIKVGEQNLCYLAEG-nh2 220
(SEQ ID NO:122)
BP-24 WFKTVCYEWEDEVQCYTLEEG-nh2 100-300
(SEQ ID NO:123)
BP-25 RVGAYISCSETECWVEDLLDG-nh2 >1000 (SEQ ID NO:124)
BP3-4D3.il VAWEVCWDRHDQGYICTTDS <10
(SEQ ID NO: 7) BP3-4D3.11DE AWEVCWDRHQGYICTTDS 80
(SEQ ID NO: 8)
BP3-13 CWDRHDQGYICTTDS >1000
(SEQ ID NO:125)
BP3--4B3.3 EESECFEGPGYVICGLVG (SEQ ID NO: 9)
BP3- -02-ox DMGVCADGPWMYVCEWTE 12
(SEQ ID NO:ll)
BP3- -01-ox SEEVCWPVAEWYLCNMWG (SEQ ID NO:10)
BP3- -15 SEEVCWPVAEWYLCN 0.75 (SEQ ID NO: 14)
BP3-16 VCWPVAEWYLCNMWG 30
(SEQ ID NO: 15)
BP3-17 VCWPVAEWYLCN 9
(SEQ ID NO: 16)
BP3-06 TGVDCQC*GPVHC*VCMDWA 5 (SEQ ID NO: 12)
BP3-08 TVANCDC*YMPLC*LCYDSD 15
(SEQ ID NO: 13) where nh2 means that the peptide has been blocked with an amide and where the C* indicates a cysteine that has been linked to another cysteine in the . peptide. The remaining Cys pairs are also oxidized as disulfides in each peptide. Radiolabeled IGF assay (for IGFBP-3 binders)
As an additional assay of peptide activity, several peptides were tested in an assay using 125I-labeled IGF-1 to measure inhibition of IGFBP binding, as described above (Assay 3) . Serial dilutions of peptide were added to an IGFBP-1 or an IGFBP-3 plate. Thereafter, 125I-labeled IGF-1 was added and the plates were incubated for 2 hours. The plates were then washed and counted to determine the amount of bound IGF-1.
Fig. 9 shows the inhibition of two lGFBP-3-selected peptides, BP3-01-ox and BP3-02-OX, for IGF-1 binding to an IGFBP-3 plate. In contrast, these peptides did not inhibit IGF-1 binding to an IGFBP-1 coated plate (Fig. 10) .
In vitro activation (KIRA)
The ability of several synthetic peptides to block IGF-1 binding to
IGFBPs and release functional IGF-1 was tested in a KIRA assay of IGF-1 activity, as described above. Cells were treated with peptide alone, peptide plus IGF-1 plus IGFBP-1, peptide plus IGF-1, and peptide plus IGF-1 plus IGFBP-3. The results are shown in Figs. 11A-D, respectively.
While BP3-01-ox has a lower affinity for the IGFBP than the other molecules tested in this assay, the fact that BP3-01-ox inhibits binding of
IGFBP-3 to IGF-1 is, in itself, useful for various purposes, including for the LIFA and other assays noted above. Further, the KIRA assay only used
IGF-1; it did not employ IGF-2, and BP3-01-ox was found to inhibit binding of IGFBP-3 to IGF-2 as noted in the competition assay described below.
BIACORE™ competition assay (for IGFBPS binders)
IGF-2 was immobilized on a dextran chip for inhibition assays using a BIACORE™ 2000 surface-plasmon-resonance device (BIAcore, Inc., Piscataway, NJ) to measure free binding protein. IGF-2 was biotinylated as described above, and injected over a chip to which streptavidin had been coupled (BIAcore, Inc.) to give approximately 1500 RU of immobilized IGF-2. The IGF-2 showed no detectable dissociation over the time course of each experiment. Serial dilutions of peptide were mixed with a constant concentration (20 nM) of IGFBP-3. After incubation for >1 hour at room temperature, an aliquot of 20 μl was injected at a flow rate of 20 μl/min over the IGF-2 chip. Following the injection, a response reading was taken to measure the relative amount of IGFBP-3 bound to the IGF-2. The results (e.g., see Fig. 12) show a dose-response curve for each peptide' s inhibition of IGFBP-3 binding to IGF-2. Peptides BP3-01-OX, BP3- 14, BP3-15, and BP3-17 showed IC50's of 0.92 μM, 1.0 μM, 0.78 μM, and 5.1 μM, respectively. Thus, these peptides inhibit the binding of IGFBP-3 both to IGF-1 and to IGF-2. EXAMPLE 2
Displacement of IGF-1 from IGFBPs using BP3-15 This Example tests an IGFBP-3-specific peptide, BP3-15, for its ability to block the binding of -^^I-IGF-l in human serum. Human serum was incubated with i25j_jQj_ + -j-he peptide and the amount of tracer bound to IGFBPs via size-exclusion chromatography was measured. Addition of the peptide resulted in an approximate 42% decrease in •'■25j_jgp_ associated with the 150-KD IGF/IGFBP-3/ALS complex and a 59% increase in the amount of free 125I-IGF-1. The peptide did not decrease ^-^1-lQΕ-l binding to the 44-KD IGFBPs (in fact, it slightly increased it) , indicating that the peptide only competes with IGF-1 for binding to IGFBP-3.
These results indicate that the analog (at 0.2 mM) can compete with IGF-1 for binding to IGFBP-3 in human serum.
EXAMPLE 3 Relative Affinity of IGFBP-3 Binding Peptide Variants The relative affinities of various BP3-01-ox variants were measured by the BIACORE™ competition assay. The results are shown in Table VI. It can be seen that 4D3.3P (SEQ ID NO: 6), BP3-30 (SEQ ID NO: 20), BP3-41 (SEQ ID NO:23), BP3-40 (SEQ ID NO:22), BP3-39 (SEQ ID NO:21), BP3-28 (SEQ ID NO:19), BP3-27 (SEQ ID NO: 18), and BP3-25 (SEQ ID NO: 17), have affinities similar to
or greater than that of BP3-01-ox and are expected to increase the availability of IGF-1 in an in vitro cell culture assay. The lack of measurable activity for peptide BP3-24 (SEQ ID NO: 126) indicates the critical role that the intact disulfide plays in maintaining a peptide conformation favorable for binding to IGFBP-3 for this series of peptides.
TABLE VI Relative affinities of BP3-01-ox variants by BIACORE™ competition assay
Variant Peptide IGF-1 Inhibition name sequence IC50 (μM) IC50 (mut ) /IC50 (wt )
4d3.3P ASEEVCWPVAEWYLCNMWGR 5.6 2.8
(SEQ ID NO: 6) BP3-30 ASEEVCWPVAEWYLCN 5.6 2.8
(SEQ ID NO:20)
BP3-41 GPETCWPVAEWYLCN 4.0 2.0
(SEQ ID NO:23)
BP3-01-OX SEEVCWPVAEWYLCNMWG 2.0 -1-
(SEQ ID NO:10)
BP3-40 ac-SEEVCWPVAEWYLCN-nh2 0.66 0.33 (SEQ ID NO:22)
BP3-39 SEEVCWPVAEWYLCN-nh2 0.66 0.33
(SEQ ID NO:21) BP3-15 SEEVCWPVAEWYLCN 0.72 0.36
(SEQ ID NO:14)
BP3-28 EEVCWPVAEWYLCN 5.4 2.7
(SEQ ID NO:19)
BP3-27 EVCWPVAEWYLCN 2.8 1.4
(SEQ ID NO:18)
BP3-25 CWPVAEWYLCN 46 23 (SEQ ID NO:17)
BP3-24 WPVAΞWYLCN >1000 >500
(SEQ ID NO:126)
EXAMPLE 4
Screening of additional libraries for binding to IGFBP-3 Additional polyvalent (g8) peptide-phage libraries were desgined and sorted that yielded two peptides that inhibited IGFBP-3 binding to IGF-1. The results, shown in Table VII, indicate that BP3-107 (SEQ ID NO: 24) and BP3-108 (SEQ ID NO: 25) are inhibitors and they are expected to increase the availability of IGF-1 in an in vi tro cell culture assay.
TABLE VII
Peptide inhibition of IGFBP-3 binding to IGF-1 by BIACORE™ competition
Peptide Phage parent Sequence IC50 (μM)
BP3-107 t4H3.6 suc-CQLVRPDLLLCQ-nh2 100 (SEQ ID NO:24)
BP3-108 t4H3.9 suc-IPVSPDWFVCQ-nh2 20 (SEQ ID NO:25)
EXAMPLE 5 Structure/Function of BP1-01 and Affinity Maturation
A. Kinetics of BP1 -01 Binding to IGFBP-1 WO 98/45427 published October 15, 1998 discloses the preparation and characterization of the IGFBP-1 displacer peptide BP1-01 (CRAGPLQWLCEKYFG) (SEQ ID NO: 26). The kinetics of BP1-01 peptide variants were examined in a BIAcore™ (BIAcore, Inc., Piscataway, NJ) assay using IGFBP-1 covalently coupled via EDC/NHS (as described by the manufacturer) to a dextran chip. Peptide BP1-01 displayed dissociation kinetics too rapid to measure.
However, BP1-02, the 19-mer variant (SEVGCRAGPLQWLCEKYFG) (SEQ ID NO: 27) displayed measurable kinetics. The association rate constant was 2.30 x 105 M"1 sec"1 and the dissociation rate constant was 5.03 x 10"2 sec"1. The latter implies a half-life for peptide dissociation from IGFBP-1 of approximately 28 sec. The association rate constant is moderately fast, consistent with the notion that the peptide may not undergo significant conformation change upon binding to IGFBP-1.
B. Scanning mutagenesis of BPl -01 peptides
Two series of synthetic peptide variants were generated to determine which side chains of the BPl-01 peptide might contribute directly to binding IGFBP-1. In the first series an alanine-scanning approach (Cunningham and Wells, Science, 244: 1081-1085 (1989)) was used to remove that portion of each side chain beyond the beta carbon. The contribution of these atoms to the free energy of binding of the peptide to IGFBP-1 was then assessed by measuring the potency (IC50) of the variant for inhibiting IGFBP-1 binding to IGF-I or IGF-II in a BIAcore™ competition assay, analogous to that described for IGFBP-3. The results are shown in Table VIII.
A second series of peptides made use of non-natural amino acids to probe whether other structural features such as an added methyl group at the alpha carbon, or an isomer (D-alanine) could affect peptide binding to
IGFBP-1. The potencies of these peptides were measured by biotinylated-
IGFBP-1 ELISA assay, with the results shown in Table IX. These results confirm the importance of side chains L6, 9, W8, and Y13 in the binding of BPl-01 to IGFBP-1. Structural contributions are also suggested by the effects of substitutions at R2 and A3. In contrast, some substitutions, such as aib substitutions at G4, Q7, Ell, K12, and F14, had little or no effect upon binding affinity. Peptides including one or more of these substitutions may nevertheless by useful because non-natural amino acids often confer upon a peptide greater resistance to proteolysis (see Schumacher et al . , Science, 271: 1854 (1996) and references therein) . Such peptides may achieve a longer half-life in serum than those having only natural amino acids .
In view of the results shown in Table IX, it is expected that peptides with a D-alanine substituted at position 2, 3, or 6 of BPl-01 or with an alpha-aminoisobutyrate substituted at position 7, 8, 9, 11, 12, 13, or 14 will increase the availability of IGF-I in an in vi tro cell culture assay.
Lastly, the relative affinities of various C-terminal BPl-01 variants were determined by ELISA, as shown in Table X. These data show that the C- terminal region of the peptide is important for binding. Only peptide BP1- 18 (SEQ ID NO: 37) retained measurable inhibitory activity for IGF-I: IGFBP-1 binding. It is expected that this peptide will increase the availability of IGF-I in an in vi tro cell culture assay.
Taken together, the structure-function data suggest that a smaller, including a non-peptidyl, compound could be designed to mimic the action of the BPl-01 peptide by including elements of the C-terminus of this peptide in combination with the side chains L6, L9, W8, and Y13.
TABLE VIII Relative affinities of BPl-01 Ala-scan peptide variants by BIAcore™
Variant IGF-I Inhibition IGF-II Inhibition IC50(mut)/lC50(wt) IC50 (mut ) /IC50 (wt)
Cl n.d. n.d.
R2A 0.9 0.9
A3 -1- -1-
G4 n.d. n.d. P5 n.d. n.d.
L6A 30.3 34.7
Q7A 0.7 0.6
W8A 7.4 6.4
L9A 33.2 29.7 CIO n.d. n.d.
"E11A 2.9 2.4
K12A 7.9 5.3
Y13A 12.5 14.6
F14A 6.2 5.8 (wt) -1- -1-
TABLE IX
Relative affinities of BPl-01 non-natural peptide variants by ELISA
(a= D-alanine; aib= alpha-aminoisobutyrate)
IGF-I Inhibition
Variant IC50 (mut) /IC50 (wt)
Cl n.d.
R2a 50
A3a 34 G4a 0.6
P5 n.d.
L6a 400
Q7aib 1.6
W8aib 24 L9aib 400
CIO n.d.
Ellaib 1.0
K12aib 2.0
Y13aib 7.1 Fl4aib 3.0
(wt) -1-
TABLE X Relative affinities of C-terminal BPl-01 variants by ELISA
Variant Peptide IGF-I Inhibition name seq . IC50 (mut ) / IC50 (wt )
BPl- 01 CRAGPLQWLCEKYFG -1-
( SEQ ID NO : 26 )
BP1-04 CRAGPLQWLCE >1000
(SEQ ID NO:28)
BP1-17 CRAGPLQWLCEK >1000 (SEQ ID NO:36)
BP1-18 CRAGPLQWLCEKAA 148
(SEQ ID NO: 37)
C. Polyvalen t (g8) selection of BPl -01 secondary libraries
NNS codons were used to generate diverse peptide libraries as described above. Affinity selections were performed by solution binding of phage to biotinylated IGFBP-1 (prepared as described above) in solution to minimize avidity effects. A similar strategy was used for antibody-phage selections by Hawkins et al . , J. Mol. Biol., 226: 889 (1992). For each round of selection, the target amount was reduced to select for enhanced affinity variants. Typically, IO9 - 1010 purified phage were preblocked with MPBST (5% skim milk in PBS + 0.05% TWEEN™ 20) for 1 hr at room temperature and screened for binding to biotinylated target. Binding conditions are described below. Phage that bound to target were captured by incubating with streptavidin-magnetic beads (Promega Corp., Madison, WI) for 2-5 minutes at room temperature. After binding, the beads were washed with PBS- TWEEN™/MPBST ten times before eluting with 0.1 M HC1. The eluate was immediately neutralized with 1/3 volume of 1 M TRIS, pH 8.0. The eluted phage were propagated by infecting XL1 for the next selection cycle. Rounds
1, 2, 3 were carried out with 400 nM, 200 nM, and 20 nM target, respectively, with 1-h incubations. Round 4 was carried out with 4 nM target overnight. All binding reactions were performed at room temperature.
The identified mutations are shown in Table XIII of WO 98/45427 and the relative affinities by ELISA plate assay or BIAcore™ are shown in Table XI below. It can be seen that BP1-10 (SEQ ID NO:29), BP1-11 (SEQ ID NO:30), BPl-12 (SEQ ID NO:31), BP1-13 (SEQ ID NO:32), BP1-15 (SEQ ID NO:34), BP68 (SEQ ID NO:45), BP1027 (SEQ ID NO:48), BP1028 (SEQ ID NO:49), BP1029 (SEQ ID NO:50), and BP1030 (SEQ ID NO:51) are of comparable or higher affinity than
BPl-02 and BPl-01, and are thus expected to increase the availability of IGF-I in an in vitro cell culture assay.
TABLE XI
Relative affinities of g8 BPl-01 selectants by ELISA plate assay or BIAcore™*
Variant Peptide IGF-I Inhibition name se . IC50(mut)/IC50(wt)
BPl-02 SEVGCRAGPLQWLCEKYFG 0.37
(SEQ ID NO:27)
BPl-01 CRAGPLQWLCEKYFG
(SEQ ID NO:26)
BP1-10 CRKGPLQWLCELYF 1.1*
(SEQ ID NO:29)
BP1-11 CRKGPLQWLCEKYF 1.9*
(SEQ ID NO:30)
BP1-12 CKEGPLQWLCEKYF 2.9*
(SEQ ID NO:31)
BP1-13 CKEGPLLWLCEKYF 2.5*
(SEQ ID NO:32)
BP1-14 SEVGCREGPLQWLCEKYF 0.26
(SEQ ID NO:33)
BP1-15 CAAGPLQWLCEKYF
(SEQ ID NO:34)
BP67 CRAGPLQWLCERYF 0.34
(SEQ ID NO:44)
BP68 CRAGPLQWLCEKFF 0.39
(SEQ ID NO:45)
BP1027 CKAGPLLWLCERFF
(SEQ ID NO:48)
BP1028 CRAGPLQWLCERFF 4.6
(SEQ ID NO: 49)
BP1029 CREGPLQWLCERFF 1.7
(SEQ ID NO: 50)
BP1030 CKEGPLLWLCERFF 4.3
(SEQ ID NO: 51)
D. Monovalent (g3) selection of BPl-01 secondary libraries
Monovalent (g3) selections of BPl-01 secondary libraries were carried out essentially as described in part C above. Templates contained either the TAA stop codon at the targeted sites for randomization or an entirely unrelated binding sequence from BPl-01. Selection conditions were as described below with BSA replacing milk in the blocking buffer. Phage- target complexes were captured by magnetic streptavidin beads (Promega Corp., Madison, WI) . Biotinylated target was preincubated with phage for 1- 3 h at room temperature in each round, with the target concentrations being reduced from 200-500 nM in round 1, to 50-100 nM in round 2, 10-50 nM in round 3, and 1-20 nM in round 4.
The identified mutations are shown in Table XV of WO 98/45427 and the relative affinities, as determined by BIAcore™ competition assay or by ELISA plate assay (carried out as above, except that 5% acetonitrile was μsed for peptide solubility) of several peptides selected are shown in Table XII below. BP1-16 (SEQ ID NO:35), a 13-residue version of BPl-01 (lacking the C-terminal Gly) , had similar affinity to that of BPl-01. Substitutions at the N-terminus or C-terminus yielded affinity improvements. For example, compared with BP1-16, addition of the STY sequence at the C-terminus yielded about a 3-fold affinity improvement for peptide BP1-21B. (A similar effect was seen in the context of the 18-mer: namely, a 3-fold improvement was observed between BP1-14 and BP1-21A. Substitution of the N-terminal S to G motif also improved affinity by 2- to 3-fold in peptides BP1-19 and BPl-20.
All of these peptides had similar or improved apparent affinity for IGFBP-1 as compared with BPl-01 and BPl-02 and are thus expected to increase the availability of IGF-I in an in vitro cell culture assay.
TABLE XII
Relative affinities of g3 BPl-01 selectants by BIAcore™ or ELISA plate assay*
Variant Peptide ' IGF-I IGF-I name seq. Inhibition Inhibition
IC50U16)/ IC50(114)/ C50(mut) IC50(mut)
BPl-14 SEVGCRAGPLQWLCEKYFG-nh2 4.8 -1-
(SEQ ID NO: 33)
BPl-16 CRAGPLQWLCEKYF-nh2 -1- 0.21 (SEQ ID NO: 35)
BPl-19 SEMVCRAGPLQWLCEIYF-nh2* 9.9 2.1
(SEQ ID NO:38) BPl-20 EARVCRAGPLQWLCEKYF-nh2 12 2.6
(SEQ ID NO:39)
BP1-21A SEVGCRAGPLQWLCEKYFSTY-nh2 15 3 . 2
( SEQ ID NO : 40 )
BP1-21B CRAGPLQWLCEKYFSTY-nh2 3 . 1 0 . 67
( SEQ ID NO : 41 )
EXAMPLE 6 Alanine-Scanning Mutagenesis of IGF-1 and Structural IGF-1 Analogs
Introduction:
An alanine-scanning mutagenesis approach (Cunningham and Wells, Science, 244: 1081-1085 (1989); US Pat. No. 5,834,250) was used to remove that portion of each side chain of IGF-1 beyond the beta carbon. The contribution of these atoms to the free energy of binding of the IGF-1 analog to IGFBP-1 or to IGFBP-3 was then assessed by competitive phage ELISA. In this assay, IGFBP-1 or IGFBP-3 is used to inhibit IGF-phage mutants from binding to an IGFBP-1- or IGFBP-3-coated immunosorbant plate. From a titration series of binding protein, binding (IC50) can be calculated. Some mutants were also assessed for direct binding in BIACORE™ assays. Materials and Methods : Construction of phagemid vector and mutagenesis
The gene encoding mature human IGF-1 was amplified from pBKIGF2B (U.S. Pat. No. 5,342,763) using PCR primers 5 ' -AGC TGC TTT GAT ATG CAT CTC CCG AAA CTC TGT GCG GT-3' (SEQ ID NO: 127) and 5 ' -GAG CGA TCT GGG TCT AGA CAG ATT TAG CGG GTT TCA G-3' (SEQ ID NO:128). The resulting fragment was cut with iVsil and Xbal , and ligated into pH0753 previously digested with Nsil and Xbal . pH0753 is a derivative of phGHam-g3 (Lowman et al . , Biochemistry, 30: 10832- 10838 (1991) ) in which the additional Xbal site in the alkaline phosphatase promoter (PhoA) region has been deleted using the oligonucleotide 5' -AAA AGG GTA TGT AGA GGT TGA GGT-3 ' (SEQ ID NO: 129). The ligated vector pH0753 containing the IGF-1 open reading frame was named pIGF-g3. It encodes for IGF-1 harboring the double mutation G1S-A70V fused- to a fragment of the gene III protein (residues 249-406) from the E. coll bacteriophage 13. Binding of this IGF-1 variant to IGFBP-1 and -3 was found to be indistinguishable from wild-type IGF-1. Alanine mutagenesis was performed using single- stranded plasmid pIGF-g3 as template (Kunkel et al . , Methods Enzy ol., 204 : 125-139 (1991) ) . All residues of IGF-1 with the exception of cysteines and alanines were singly replaced by alanine. The resulting constructs were verified by DNA sequencing.
Binding of IGF mutan ts displayed on phage to IGFBP-1 and -3 (phage ELISA)
Immunosorbent plates (Nunc, MAXISORP™, 96 wells) were coated with 100 μl/well of 1 μg/ml IGFBP-1 or IGFBP-3 in PBS buffer pH 7.2 at 4oC overnight.
The plates were then blocked with 0.5 % TWEEN 20™/PBS (also used as binding
buffer) for 2 hours at room temperature (prote aceous blocking agents like bovine serum albumin were avoided to prevent potential IGF or IGFBP contamination) . E. coli cells (XLl-Blue, Stratagene) freshly transformed with phagemid vector were grown overnight m 5 mL 2YT medium (Sambrook et al . , supra) the presence of M13-VCS helper phage (Stratagene) Phage particles were harvested and resuspended m PBS buffer as described in Lowman, H.B., "Phage Display of Peptide Libraries on Protein Scaffolds," m Cabilly, S. (ed.), Combinatorial Peptide Library Protocols (Humana Press Inc.: Totowa, NJ, 1998), pp. 249-264. Then phage concentrations were normalized to yield a maximal ELISA signal of 0.2-0.4 for each mutant (Lowman, m Cabilly, S. (ed.), supra) . Threefold serial dilutions of soluble competitor were prepared on non-absorbent microtiter plates (Nunc, F, 96 wells) with binding buffer (0.5% TWEEN 20™/PBS) containing phage at the previously determined concentrations. The dilution range of competitor protein extended over six orders of magnitude, starting at 5 μM for IGFBP-1 and 500 nM for IGFBP-3. After blocking, the plates containing immobilized target were washed with 0.05% TWEEN™/PBS buffer and subsequently incubated w th 80 μl/well of the premixed phage-competitor solutions for 1 hour at room temperature. After washing, bound phage was detected with 80 μl/well of a solution containing a primary rabbit anti-phage polyclonal antibody and a secondary goat anti-rabbit monoclonal antibody-horseradish peroxidase conjugate m 0.5% TWEEN 20™/PBS. o-Phenylenediamme (Sigma) and tetramethylbenzidme (Kirkegaard and Perry) were used as chromogenic substrates, resulting m product detection at 492 and 450 nm, respectively. IC5o values were determined by fitting the binding data to a generic saturation curve (Lowman, m Cabilly, S. (ed. ) , supra) . At least two individual clones of each IGF-1 mutant were assayed. Numbers Table XIII represent mean ± standard deviation of individually assessed IC50 values. Expression and purification of IGFBP-1 and IGFBP-3 Human IGFBP-1 was expressed m CHO cells and purified from the conditioned medium as described by Mortensen et al . , Endocrinology, 138 : 2073-2080 (1997) . Recombinant human IGFBP-3 has also been cloned and expressed in mammalian cells (Wood et al , Mol Endocrinology, 2: 1176-1185 (1988)). Purification from conditioned medium essentially followed the procedure described for IGFBP-1, with use of an IGF affinity column (Martin and Baxter, J. Biol. Chem., 261: 8754-8760 (1986)). Expression and purification of soluble IGF-1 mutants
Plasmid pBKIGF2B (U.S. Pat. No. 5,342,763) expresses human wild-type IGF-1 fused to the leader peptide of lamB under the control of the PphoA promoter. For ease of site-directed mutagenesis the phage fl origin of replication (fl on) was introduced into plasmid pBKIGF2B. For that purpose
a 466-BP BamHI fragment containing the fl ori was excised from pH0753 (Lowman et al . , supra, 1991), while plasmid pBKIGF2B was linearized with EcoRI. Vector and fragment were both treated with Klenow enzyme to fill m restriction-site overhangs prior to blunt-end ligation. Correct constructs were selected for the ability to produce single-stranded phagemid DNA m the presence of M13VCS helper phage. The resulting phagemid vector was named pBKIGF2B-fl-orι and was used as template to construct the IGF-1 ala-mutants of interest (see Table XIV) using the procedure of unkel et al . , Methods Enzymol. , 204: 125-139 (1991)). Every mutagenesis step was confirmed by DNA sequencing.
Expression of IGF-1 mutants was as described for the IGF-1 wild-type (Joly et al . , Proc. Natl. Acad. Sci. USA, 95: 2773-2777 (1998)), but without transient overexpression of oxidoreductases . The purification procedure was based on a previous protocol (Chang and Swartz, "Single-Step Solubilization and Folding of IGF-1 Aggregates from Escherichia coli" In Cleland, J. L. (ed.), Protein Folding In Vivo and In Vitro (American Chemical Society, Washington, DC, 1993), pp. 178-188), with minor adaptations. Typically, 6 g of wet cell paste (equivalent to 2 liters low phosphate medium grown for 24 hrs) was resuspended in 150 ml of 25 mM Tπs-HCl pH 7.5 containing 5 mM EDTA. Cells were lysed in a miσrofluid zer (Microfluidics Corp., Newton, MA) , and retractile particles containing accumulated IGF-1 aggregates were collected by centrifugation at 12,000 x g. Refractile particles were washed twice with lysis buffer, twice with lysis buffer containing 1 % N-lauroyl- sarcosine (Sigma) to extract membrane proteins, and twice with lysis buffer again. Washed retractile bodies were resuspended at approximately 2 mg/ml 50 mM CAPS (3- (cyσlohexylammo) -1-propanesulfonic acid; Sigma) buffer pH 10.4 containing 2 M urea, 100 mM NaCl, 20% MeOH, and 2 mM DTT. This procedure combines solubilization of refractile bodies and subsequent oxidative refolding of IGF-1 mutants (Chang and Swartz, supra) . After 3 hrs at room temperature the refolding solutions were filtered through microconcentrator membranes (Centπcon, Amicon) with a molecular weight cut off of 50 kDa. The majority of monomeric IGF-1 was recovered the eluate, while higher molecular weight contaminants were concentrated in the retentate. At this point IGF-1 fractions were > 95% pure, as judged from SDS-PAGE analysis. To separate correctly disulfide-bonded IGF-1 from IGF- swap (containing two non-native disulfides; Hober et al., Biochemistry, 31: 1749-1756 (1992); Miller et al . , Biochemistry, 32: 5203-5213 (1993)), refolding solutions were acidified with 5 % acetic acid and loaded on a Dynamax™ C18 semi-preparative HPLC column (Varian; 10.0 mm ID) at 4 ml/mm. Buffers were H2O/0.1 % TFA (A) and acetonιtrile/0.1 % TFA (B) . Separation of the disulfide isomers was achieved by applying the following gradient: 0-
30 % B in 20 min, 30-45 % B in 60 min. The ratio of native IGF-1 to IGF- swap was usually about 2:1 for each mutant, with IGF-swap eluting earlier in the gradient than native IGF-1. The molecular mass of each mutant was verified by mass spectrometry. After HPLC purification, samples were lyophilized and reconstituted at approximately 1 mg/ml in 100 mM HEPES buffer, pH 7.4. Biosensor kinetic measurements
The binding affinities of the IGF variants for IGFBP-1 and IGFBP-3 were determined using a BIACORE™-2000 real time kinetic interaction analysis system (Biacore, Inc., Piscataway, NJ) to measure association (Jra) and dissociation { kd) rates. Carboxymethylated dextran biosensor chips (CM5, BIAcore Inc.) were activated with EDC (N-ethyl-N' - (3-dimethylaminopropyl) - carbodiimide hydrochloride) and NHS (N-hydroxysuccinimide) according to the supplier's instructions. For immobilization, IGF mutants in 20 mM sodium acetate, pH 4.8, were injected onto the biosensor chip at a concentration of 50 μg/ml to yield approximately 450-600 RU's (resonance-responce units) of covalently-coupled protein. Unreacted groups were blocked with an injection of 1 M ethanola ine. Kinetic measurements were carried out by injecting two-fold serial dilutions (starting at 1 μM) of either IGFBP-1 or IGFBP-3 in running buffer (PBS, 0.05 % TWEEN 20™, 0.1 % ovalbumin, 0.1 % sodium azide) at 25DC using a flow rate of 20 μl/min. Association rates { ka) and dissociation rates { kd) were calculated separately using a 1:1 Langmuir association model in the BIACORE™ evaluation software v. 3.0. The equilibrium dissociation constant (.K ) was calculated as kd/ka - Results :
Monovalent phage display of IGF-1
For a rapid and comprehensive alanine scan of the 70 amino acid residues of IGF-1 it was first determined whether the protein could be monovalently displayed on the surface of phage M13 (Bass et al . , supra) . Phage display technology combines the advantage of rapid single-stranded DNA mutagenesis with an easy purification of the resulting mutant protein, simply by isolation of the corresponding phage particles (e.g., Cunningham et al . , EMBO . , 13: 2508-2515 (1994)). A vector was constructed in which mature human IGF-1 was fused to the carboxy-terminal domain of the M13 gene III product. This construct includes the stll signal sequence which directs the fusion protein to the periplasmic space of E. coli and allows monovalent display of the protein (Bass et al . , supra; Lowman et al . , supra, 1991) . For cloning purposes the first and the last amino acids of IGF-1 were changed; the resulting mutant G1S-A70V was used as the template construct for the subsequent alanine scanning mutagenesis.
When phage particles displaying IGF-1 G1S-A70V were isolated and assayed in a binding competition phage ELISA for their affinity to IGFBP' s, the IC50 determined in that experiment were 8.5 nM for IGFBP-1 and 0.5 nM for IGFBP-3 (Fig. 13) . These values are in good agreement with dissociation constants determined by BIACORE™ experiments using wild-type IGF-1 (Heding et al . , J. Biol. Chem., 271: 13948-13952 (1996)). Wild-type IGF-1 affinities determined by radioactive immunoassays (RIA) are ~ 2.8 nM for IGFBP-1 and ~ 0.8 nM for IGFBP-3, further supporting the IC50 values derived from phage ELISA. Additionally, phage particles displaying IGF-1 G1S-A70V were efficiently captured by 11 independent monoclonal mouse anti-IGF-1 antibodies immobilized on microtiter plates. These results together suggested that the displayed IGF-variant is folded correctly and accessible on the surface of the phage particles. Ala-scanning mutagenesis of IGF-1 binding to IGFBP-1 and IGFBP-3 All residues of G1S-A70V IGF-1 with the exception of the four native alanines and six cysteines were singly substituted by alanine, using the described G1S-A70V IGF-1 gill vector as a template. Additionally, the single mutants S1G and V70A and the double-mutation restoring wild-type IGF- 1 were constructed. Each of these constructs was expressed in E. coli and displayed on phage. IC50 values for binding to IGFBP-1 and IGFBP-3 were determined by competitive phage ELISA as shown in Fig. 13. At least two different clones of every mutant were tested. The resulting IC50 values are listed in Table XIII, and the loss or gain in IC50 for each mutant with respect to G1S-A70V is graphed in Fig. 14.
TABLE XIII
Apparent Affinities (ICS0) of IGF-1 Variants for IGFBP-1 and IGFBP-3 Determined by Phage Display3
IGFBP-1 IGFBP-3
IGF-1 Relative relative relative mutant IC50 (nM) IC50 IC50 (nM) IC50 specificity
S1A 5.2 ± 0.9 0.6 0.91 ± 0.32 1.2 0.5
P2A 11.0 + 3.7 1.3 0.81 ± 0.18 1.1 1.2
E3A 278 ± 86 33.9 1.05 ± 0.08 1.4 24.2
T4A 19.4 ± 6.4 2.4 0.80 ± 0.02 1.1 2.2
L5A 55.3 ± 11.6 6.7 1.53 + 0.22 2.0 3.3
G7A >1000 >100 4.58 + 0.28 6.1 >16
E9A 8.6 ± 0.6 1.0 1.32 ± 0.30 1.8 0.6 10A 311 + 87 37.9 3.55 ± 0.33 4.7 8.1
V11A* n. d. - n. d. - -
D12A 4.3 ± 0.8 0.5 1.49 ± 0.38 2.0 0.3
L14A 36.7 + 1.1 4.5 0.90 + 0.04 1.2 3.7
Q15A 13.9 ± 0.9 1.7 1.26 ± 0.41 1.7 1.0
a The variants noted with an asterisk were not successfully displayed on phage (n.d.), as judged by antibody experiments described in the text. Relative IC
50 is defined as IC
50 nut/ICso
GIS-
A7OV- Relative specificity is defined as relative IC
50
IC
50 IGFBP-
3 for each variant . .
The majority of the alanine mutants yielded only minor changes in IC50 values in the phage ELISA. Importantly, wild-type IGF-1 showed the same affinities for IGFBP-1 and IGFBP-3 as G1S-A70V in which background the alanine substitutions were performed (Table XIII, Fig. 14). Only a few residues caused considerable (> 10-fold) losses in affinity when changed to alanine: E3, G7, L10, Vll, F25, R36, P39, F49, and P63 for IGFBP-1 binding; Vll, R36, P39, and P63 for IGFBP-3 binding. It has been noted that ala- substitutions of glycines and prolines can lead to structural perturbations of the protein backbone (Di Cera, Chem. Rev., 98: 1563-1591 (1998)). Only a few modest improvements in binding affinity were found by alanine replacements. SIA, D12A, and D45A showed an approximately 2-fold increase in IGFBP-1 binding, while S35A and T41A showed a similar effect for IGFBP-3. However, 2-fold changes in IC50 values are at the limit of precision in these experiments. IGFBP- specificity determinants
E3A, G7A, L10A, F25A, and F49A showed a differential effect in binding IGFBP-1 versus IGFBP-3. For these five IGF-1 single alanine mutants the relative IC50 for IGFBP-1 differed by more than 4-fold from the one for IGFBP-3 (Fig. 14, Table XIII, relative specificity). E3A and F49A showed the biggest relative specificity factors in this group. Alanine substitution of E3 had virtually no effect on IGFBP-3 affinity (1.4 fold), while binding to IGFBP-1 was weakened 34-fold. Even more dramatic, the affinity of F49A was reduced more than 100-fold for IGFBP-1 but only 3.6- fold for BP-3. This result was illustrated in a direct comparison by phage ELISA. Phage particles displaying IGF-1 F49A were added to IGFBP-3 coated wells in the presence of soluble IGFBP-1 (Fig. 15A) or IGFBP-3 (Fig. 15B) . Compared to control phage displaying IGF-1 G1Ξ-A70V, the binding curve of F49A shifted by more than two orders of magnitude in the IGFBP-1 competition (Fig. 15A) . In contrast, the binding curves were similar in the IGFBP-3 competition, and the IC50 values differed by less than a factor of 4 (Fig. 15B) . Thus, E3 and F49 are two major specificity determinants for IGFBP-1 binding in the IGF-1 molecule .
Residues G7, L10, and F25 appeared to be important for binding of both IGFBP 's, although showing a more pronounced loss of affinity for IGFBP-1 than for IGFBP-3 when substituted by alanines . No significant specificity determinant for IGFBP-3 was identified, such as a mutant binding much tighter to IGFBP-1 than to IGFBP-3. However, mutations E9A, D12A, F23A,
Y24A, T29A, S34A, and D45A had slightly larger (about 2-fold) effects on
IGFBP-3 than on IGFBP-1 binding.
BIACORE™ measurements of purified soluble IGF mutants
For validation of the results obtained by phage ELISA, specific alanine mutants were expressed and purified for kinetic analysis using a BIACORE™ instrument. The dissociation constant (f ) of wild-type IGF-1 was determined to be 13 nM for IGFBP-1 and 1.5 nM for IGFBP-3 (Figs. 17A and 17B; Table XIV). The difference in affinity for the IGFBP's is due to a 10- fold faster association rate { ka) of IGF-1 to IGFBP-3 (3.2 x IO5 versus 3.2 x IO4 ~is_1) . These results correspond well with the absolute IC50 values determined by phage ELISA (Figs. 13A and 13B; Table XIII). As expected, the double-mutant G1S-A70V showed kinetic parameters essentially indistinguishable from wild-type (Table XIV) .
V11A, R36A, and P39A were tested because these variants had not been displayed correctly on phage, based upon the antibody recognition experiments (see above) . R36A and P39A showed wild-type kinetics for both binding proteins, whereas V11A showed a 5-fold reduction in affinity for both IGFBP-1 and IGFBP-3.
Furthermore, it was decided to examine the soluble IGF variant T A. This residue had been implicated in IGFBP binding in earlier publications (Bayne et al . , J. Biol. Chem., 263: 6233-6239 (1988); Clemmons et al . , J . Biol. Chem., 265: 12210-12216 (1990)), but had shown modest effects in the phage assays herein. The increase in the KD values of T4A relative to wild- type IGF-1 was approximately 2-3-fold higher than the IC50 ratios determined by phage ELISA (Table XIV) . A bigger discrepancy between the results obtained by phage and the biosensor analysis was seen for F16A. In this case the two methods differed by a factor of 4.
It has been shown that mutations in the first α-helical region have a destabilizing effect on the IGF-protein structure (Jansson et al . , Biochemistry, 36: 4108-4117 (1997)). Without being limited to any one theory, it is believed that the g3 fusion protein on the surface of the phage might be more stable than the refolded, purified soluble protein. This is supported by the BIACORE™ results obtained for F25A and F49A, two residues located outside the structurally sensitive N-terminal helix. The respective changes in KD and IC50 values are in excellent agreement for these two mutants (Table XIV) . The differential effect of F49A on binding to the IGFBP 's was confirmed by the BIACORE™ analysis. A 70-fold decrease in affinity was measured for IGFBP-1 binding (Fig. 17C; Table XIV), whereas IGFBP-3 binding was reduced only 4-fold (Fig. 17D; Table XIV) .
TABLE XIV
Kinetic Parameters for the Interaction of Purified IGF-1 Variants with
IGFBP-1 and -3 Determined by BIACORE™ Analysis8
Binding to IGFBP-1 Kd KD relative relative
KD IC
(x 10* ^s"1) (x 10" s"1) (nM)
IGF-1 WT 3.2 ± ° .2 4.1 + 0.2 13.0 + 1.0 1.0 1.0
G1S-A70V 3.2 ± ° .2 4.5 + 0.01 14.0 + 0.7 1.1 1.0
T4A 1.9 + 0. .2 16.7 + 1.6 90.0 +11.0 6.9 2.4
VI1A 1.9 ± °' .1 12.3 + 0.6 66.5 + 4.5 5.1
F16A 1.9 ± o. .6 60.3 + 4.5 321 + 98 25 6.0
F25A 1.5 ± °' .5 49.0 + 5.7 323 + 107 25 22
R36A 4.0 ± 0' .2 5.6 + 0.2 13.9 + 0.8 1.1
P39A 3.1 + 0. .2 4.2 + 0.1 13.6 +_0.8 1.0
F49A 1.26 + 115 + 1.5 913 + 551 70 >100
0.8
Binding to IGFBP-3 relative relative KD IC50
(x 10s M^s" (x IO4 s"1) (nM)
IGF-1 WT 3.2 + 0.5 4.7 + 0.8 1.5 + 0.3 1. .0 1 . 4
G1S-A70V 2.9 + 0.8 6.3 + 0.5 2.2 + 0.6 1, .5 1 . 0
T4A 1.8 + 0.6 5.5 + 0.1 3.1 + 1.0 2, ,1 1 . 1
VI1A 3.1 + 0.5 20.9 + 2.8 6.7 + 1.3 4. .5
F16A 1.1 + 0.4 11.4 + 2.7 10.3 + 4.7 6. .9 1 . 8
F25A 1.5 + 0.5 11.8 + 0.1 7.7 + 0.3 5. .1 4 . 9
R36A 4.0 + 0.1 4.7 + 0.2 1.2 + 0.1 0, .8
P39A 2.7 + 0.2 6.0 + 0.3 2.2 + 0.2 1. .5
F49A 2.7 + 0.7 17.1 + 0.9 6.3 + 1.7 4. .2 3 . 6
a The relative changes in dissociation constants (KDmut/KDwt) are compared to the relative ICSo values (IC50 ut ICS0 GIS-A OV) determined by phage display (Table XIII) .
Role of the N-terminal IGF-1 residues
Surprisingly, the IGFBP-3 interaction was generally much less affected by the alanine substitutions than was the interaction with IGFBP-1, despite the fact that IGFBP-3 binds IGF-1 with approximately 10-fold higher
affinity. Apart from P63A, no alanine mutant exhibited a > 6-fold reduction in IGFBP-3 affinity (Fig. 14; Table XIII).
It had previously been shown in biosensor experiments that des (1-3)- IGF-1 binds IGFBP-3 with 25-fold reduced affinity (Heding et al . , supra ) . This naturally-occurring form of IGF-1 lacks the first three N-terminal residues and shows increased mitogenic potency, presumably due to its reduction in IGFBP-binding (Bagley et al . , Biochem. J. , 259: 665-671 (1989)). Since none of the first three amino acid side chains seem to contribute any energy to the binding of IGFBP-3 (Table I) but nevertheless des (1-3) -IGF-1 is compromised in IGFBP-3 binding, without being limited to any one theory, it is hypothesized that backbone interactions might be involved.
This hypothesis was tested by displaying on phage a triple alanine mutant (Ala (1-3) -IGF-1) , substituting the first three N-terminal amino acids. If the backbone in that region contributes to the interaction with IGFBP-3 this mutant should be able to bind. Binding to IGFBP-1, however, should be reduced due to the lack of the E3 side chain (Table I) . As a control the des (1-2) -IGF-1 mutant was generated, testing for any potential backbone interactions with IGFBP-1 at positions 1 and 2. As expected, Ala (1-3) -IGF-1 showed a decreased IGFBP-1 affinity similar to E3A but no change in IGFBP-3 affinity (Fig. 14; Table XIII). For des (1-2) -IGF-1, no difference in affinity was observed for both binding proteins. Combined with the observations on des (1-3) -IGF-1 (Heding et aJ . , supra) , these results suggest, without limitation to any one theory, that the peptide backbone between residue 3 and 4 of IGF-1 mediates important interactions with IGFBP-3. Discussion:
The functional IGFBP-1 and IGFBP-3 binding epitopes on the surface of IGF-1 have been probed by alanine-scanning mutagenesis. Both binding epitopes are illustrated in Fig. 18. Individual IGF-1 side-chain interactions play a much more important role for binding to IGFBP-1 than to IGFBP-3. Two major binding patches are found for IGFBP-1 (Fig. 18A) . One is situated on the upper face of the N-terminal helix (composed of G7, L10, Vll, L14, F25, 143, and V44) and one the lower face (composed of E3, T4, L5, F16, V17, and L54). These two binding patches are bridged by F49 and R50. For IGFBP-3, the binding epitope is more diffuse and has shifted to include G22, F23, and Y24 (Fig. 18B) . Binding of IGFBP-3 is generally much less sensitive to alanine substitutions! In fact, the biggest reduction in affinity (apart from P63A, see below) is a 6-fold decrease seen for G7A. This result is intriguing since IGFBP-3 binds with 10-fold higher affinity to IGF-1 than does IGFBP-1. Most probably, without limitation to any one
theory, interactions originating from the IGF-1 main chain backbone are contributing to the binding of IGFBP-3. This hypothesis is further substantiated by the experiments with the Ala (1-3) -IGF mutant. While the single and triple alanine substitutions have no effect on IGFBP-3 binding, deletion of the first three amino acids resulted in a 25-fold decrease in affinity (Bagley et al . , supra; Clemmons et al . , Endocrinology, 131: 890-895 (1992); Heding et al . , supra ) . In summary, IGF-1 uses different binding modes to associate with IGFBP-1 and IGFBP-3: a few amino acid side-chain interactions are important for binding to IGFBP-1, while backbone interactions seem to play a major energetic role for bindilng to IGFBP-3.
A recent publication has investigated the binding epitope on IGF-1 for IGFBP-1 by heteronuclear NMR spectroscopy (Jansson et al . , J . Biol. Chem. , 273: 24701-24707 (1998)). The authors found that the IGF-1 residues 29, 30, 36, 37, 40, 41, 63, 65, and 66 amongst others experienced chemical shift perturbations upon complexation with IGFBP-1 at 30°C. Furthermore, Jansson and co-workers identified R36, R37, and R50 to be part of the functional binding epitope and tested those alanine mutants in BIACORE™ experiments. The largest change in affinity observed by these authors was a 3-fold decrease for R50A. However, due to the structural flexibility of IGF-1 already observed in the first NMR study of the hormone (Cooke et al :, supra) , Jansson et al . were unable to completely assign many residues in the NMR spectrum, including F49.
In similar studies of protein-protein interfaces it was found that only a few side-chain residues contribute to the bulk of free-binding energy (Clackson and Wells, Science, 267: 383-386 (1995); Kelley et al . , Biochemistry, 34: 10383-10392 (1995)). The same holds true for the IGF- 1GFBP-1 interaction. However, here, as it was noticed for tissue factor binding to factor Vila, the magnitude of the free energy of binding (ΔΔG) values derived from important side chains is smaller than in the case of growth hormone (Kelley et al . , supra ) . The residues with predominant ΔΔG contributions were not clustered on the IGF-1 surface like in the growth hormone-receptor interface (Clackson and Wells, supra ) , but still formed a continuous IGFBP-1 binding epitope (Fig. 18A) . In contrast, the IGFBP-3 binding epitope on IGF-1 was discontinuous, and side chains contributed very modest individual binding energies.
Substitution of P63 by alanine in IGF-1 results in a decreased affinity for both binding proteins that cannot be measured in the concentration range used in the competition phage ELISA' s. However, residue P63 is located on the opposite side of the IGF-1 molecule with respect to the main binding epitope. Furthermore, it has been noticed that alanine
substitutions of glycines and prolines can lead to structural changes (Di Cera, supra ) . In addition, Jansson et al . , 1998, supra, concluded that the C-terminal part of IGF-1 is not involved in direct IGFBP-1 contacts, but rather undergoes indirect conformational changes upon complex formation. An extensive characterization of antibody binding sites on IGF-1 has been carried out by Manes et al . , Endocrinology, 138: 905-915 (1997). They showed simultaneous binding of IGFBP-1 or -3 to IGF-1 in complex with antibodies recognizing the C-terminal D-domain. These results further support earlier observations that the D-domain, beginning with residue P63, is not involved in binding of IGFBP-1 or -3 (Bayne et al . , supra, 1988).
The major discrepancy between an IC0 ratio obtained by phage ELISA and a BIACORE™ result was observed with residue F16. As already mentioned substitution of this residue by alanine induced structural changes in the IGF-1 molecule (Jansson et al . , supra , 1997). The same effect was seen with the KD in the BIACORE™ results, but the affinity decrease was less pronounced in the phage ELISA experiments (see Table II) . Both BIACORE™ measurements used IGF-F16A that had been refolded during the purification procedure (Jansson et al . , supra, 1997). In phage display, however, the protein of interest is translocated naturally by the secretion machinery of E. coli . The low protein abundance in monovalent phage display (< 1 molecule per phage particle) may disfavor aggregation and misfolding. Additionally, fusing IGF-1 to the truncated g3 phage protein might exert a stabilizing effect on the native structure of the peptide.
The levels of IGFBP-3 are positively regulated by IGF-1. The role of IGFBP-1, in contrast, is less clear. This class of binding proteins is generally less abundant than IGFBP-3, and its levels are negatively regulated by insulin (Bach and Rechler, supra; Clemmons, supra, 1997; Jones and Clemmons, supra) .
Based on the results herein, IGFBP-specific variants of IGF-1 are obtained. Combination of several alanine mutations generates a variant that binds IGFBP-1 very weakly while retaining high-affinity binding of IGFBP-3.
The design of IGFBP-1 specific variants that no longer bind to IGFBP-3, can involve phage display of IGF-1 and the randomization of amino acids at specific positions (Cunningham et al . , 1994, supra; Lowman and Wells, J^ Mol. Biol., 243: 564-578 (1993)). Conclusion:
Residues in IGF-1 important for binding to IGFBP-1 and IGFBP-3 have been identified. Several residues were found that determine the binding specificity for a particular IGFBP. Recent publications (Loddick et al . , Proc. Natl. Acad. Sci. USA, 95: 1894-1898 (1998); Lowman et al . , supra, 1998) have reported animal studies where increased pools of bioavailable
"free" IGF-1 were generated by displacing endogenous IGF-1 from binding proteins. IGFBP-specific IGF-1 variants may be used diagnostically and therapeutically as described herein. EXAMPLE 7 Characterization of Certain IGF-1 Analogs
Materials and Methods:
Construction of IGF-1 Analogs
In Example 6 (and in Dubaguie and Lowman, supra ) IGF-1 analogs are identified in which binding affinity to IGFBP-1, IGFBP-3, or both binding proteins, was reduced. In particular, the total alanine-scanning mutagenesis of IGF-1 identified glutamic acid 3 (E3) and phenylalanine 49 (F49) , as well as phenylalanine 16 (F16) and phenylalanine 25 (F25) to some degree, as specificity determinants for binding to IGFBP-1. Phage display alanme-scannmg results suggested that both of the side chains at positions 3 and 49 selectively contribute considerable binding energy for complex formation with IGFBP-1 (~30-fold loss in affinity for E3A, -100 fold for F49A) , while their contribution in binding energy for IGFBP-3 is not detectable (E3A) or minor (~4-fold for F49A) (see Example 1 and Dubaguie and Lowman, supra) .
Further improved specificity for IGFBP-3 was likely to be attained by cumulative mutation of IGF-1, because the effects of point mutations are often additive with respect to their contribution to the free energy of binding (Wells, Biochemistry 29: 8509 (1990) ) . Therefore, a double mutant of IGF-1, E3A/F49A, was constructed by combining point mutations E3A and F49A in a single molecule. Although F16A showed a smaller IGFBP-specificity effect (Example 1 and Dubaguie and Lowman, supra) , the double mutant F16A/F49A was also constructed.
Also constructed was a new point mutant of IGF-1, Y31C, containing a single putative unpaired cystemyl thiol, to facilitate site-specific immobilization of IGF-1 for binding assays. Y31C was chosen because it is outside the binding epitopes for IGFBP-1 and IGFBP-3 (Dubaquie and Lowman, supra ) . This immobilization technique ensures a uniform ligand population (Cunningham and Wells, J. Mol. Biol., 234: 554 (1993)) for binding by the injected analyte ( .e., IGF binding protein). The advantage of this method over the previously-employed am e coupling is that the IGF-1 N-terminus is unblocked and free of any potential amme linkages to the chip matrix. This may be especially important for binding analysis of IGFBP-1, which is believed to interact with side chains of the IGF-1 N-terminus (Dubaquie and Lowman, supra) . Y31C displayed on phage showed wild-type-like affinities for both IGFBP-1 and IGFBP-3, supporting the notion that the region around
residue 31 is important in receptor binding, but forms no contact with the binding proteins (Bayne et al . , J. Biol. Chem., 264: 11004 (1988); Bayne et al . , J. Biol. Chem., 265: 15648 (1989)).
Single-alanine variants of IGF-1, including F49A, as well as the E3A/F49A double mutant, were expressed, purified, and refolded to give the appropriate disulfide isomer as judged by HPLC analysis (Example 1 herein and Dubaquie and Lowman, supra ) . These variants were tested in assays of specific binding-protein binding and receptor activation. Results : IGFBP-1 and IGFBPS Binding Affinity
The binding affinities of these variants for IGFBP-1 and IGFBP-3 were compared to that of wild-type IGF-1 using BIACORE™ analysis. Kinetic experiments with IGFBP-3 binding to immobilized IGF-1 or variants (Table III) were carried out as described in Example 1 and in Dubaquie and Lowman, supra, and compared with F49A TGF-1 and wild-type IGF-1. In this assay, the double mutant E3A/F49A was about 20-fold weaker in binding affinity to IGFBP-3 than wild-type, and the double mutant F16A/F49A was about 66-fold weaker (Table XV) .
TABLE XV
Kinetics of IGFBP-3 Binding to IGF-1 (*data from Table II of Example 1)
Immobilized IGFBP-3
Protein Ka (x 10s M'1) kd (x IO"4 s"1) KD (nM)
IGF-1* 3.2 ± 0.5 4.7 ± 0.8 1.5 ± 0.3 F49A IGF-1* 2.7 ± 0.7 17.1 + 0.9 6.3 + 1.7 E3A/F49A IGF-1 0.74 ± 0.4 13.3 + 0.6 22.2 ± 10.3 F16A/F49A IGF-1 0.4 + 0.1 38.6 + 2.7 99.0 ± 26.0
For measurements of IGFBP-1 binding to IGF-1, kinetics experiments were conducted using a single-cysteine IGF-1 variant, Y31C, that was immobilized onto the sensor chip surface via a disulfide linkage (BIACORE™ System Manual Supplement, 5a-l, Pharmacia (1991)). The results are consistent (Table XVI) with the binding affinity measured using wild-type IGF-1 immobilized via nonspecific amine coupling to the biosensor chip (Example 1 and Dubaquie and Lowman, supra) .
TABLE XVI Kinetics of IGFBP-1 Binding to IGF-1 (*data from Table XIV of Example 6)
IGFBP-1
Immobilized ka (x IO4 M"1) kd (x IO"4 s" KD (nM) Protein
Y31C IGF-1 3.9 ± 0.4 3.8 ± 0.1 10.0 + 1.1 IGF-1* 3.2 ± 0.2 4.1 ± 0.2 13.0 + 1.0
The binding of F49A and E3A/F49A to IGFBP-1 was too weak for accurate kinetic measurements. Therefore, a competitive binding assay (WO 98/45427 published October 15, 1998) was performed to estimate the corresponding affinities. The single-cysteine IGF-1 variant, Y31C, was used that was immobilized onto a BIACORE™ biosensor chip surface as described above. Competitive binding experiments yielding half-maximal inhibitory concentration values (IC50) were conducted as follows: 50 nM IGFBP-1 was incubated with a dilution series of the desired IGF variant. These protein mixture solutions were injected at 5 μL/min over a Bl chip containing cysteine-coupled IGF-1 Y31C (200 response units) . The amount of bound
IGFBP-1 was determined by subtracting non-specific binding after a 20-minute injection and plotted against the IGF variant concentration (Fig. 19) . The results are shown in Table XVII.
TABLE XVII Inhibition of IGFBP-1 Binding to Immobilized Y31C IGF-1
Immobilized Competing Protein IGFBP-1 Protein IC50 (μM)
Y31C IGF-1 F49A IGF-1 1.6 + 0.2
Y31C IGF-1 E3A/F49A IGF-1 64 ± 9
Compared to wild-type IGF-1, F49A and E3A/F49A had severely decreased binding affinities for IGFBP-1. F49A bound to IGFBP-1 with an IC50 of 1.6 ± 0.2 μM (Table XVII), while preserving a high-affinity dissociation constant (KD) of 6.3 ± 1.7 nM for IGFBP-3 (Table XV). Binding of E3A/F49A to IGFBP-1 was found to be even weaker, with an estimated IC50 of 64 ± 9 μM (Table XVII), while having only moderately reduced affinity (KD= 22.2 ± 10.3 nM) for IGFBP-3 (Table XV) . These in vi tro measurements suggest that neither IGF variant should stably associate with IGFBP-1 under physiological conditions. KIRA Assays of IGF Type I Receptor Activation The Kinase Receptor Activation Assay (KIRA) specifically and quantitatively monitors the extent of cytoplasmic IGF receptor phosphorylation upon extracellular stimulation by ligand (Sadick et al . , J. Pharm. B omed. Analysis, 19 (6): 883-891 (1998)). Several IGF variants, G1S/A70V, T4A, V11A, F16A, F25A, F16A/F49A, R36A, P39A, and F49A, were tested in single-concentration assays of receptor activation. The IGFBP-1 and IGFBP-3 binding affinities of these variants, except for F16A/F49A, are set forth in Table XIV and in Dubaquie and Lowman, supra . Table XVIII summarizes the relative affinities and specificities from BIACORE™ measurements . For the KIRA assay, variant concentrations were roughly estimated at 13 nM ("high concentration) or 1.3 nM ("low concentration"), based on optical density measurements. The signal obtained for each IGF variant was compared to that of a standard-dilution series of wild-type IGF-1, and reported in terms of an apparent IGF-1 concentration corresponding to the observed activity in the KIRA assay (Figs. 20A and 20B) . Although exact relative potencies were not measured, these results show that all tested mutants maintain the ability to activate the IGF type I receptor.
TABLE XVIII
Relative IGFBP-1 and IGFBP-3 Affinities of IGF-1 Variants. NDB, no detectable binding; ND, not determined; *data from Table XIV of Example 6)
IGFBP- 1 IGFBP- 3 Specificity
IGF-1 KD (mutant) / KD (mutant) / Relative BP-1/ Variant KD(IGF-1) ) KD(IGF-1)) Relative BP-3
G1S/A70V* 1.1 1.5 0.7
T4A* 6.9 2.1 3.3
VI1A* 5.1 4.5 1.1
F16A* 25 6.9 3.6
F25A* 25 5.1 4.9
R36A* 1.1 0.8 1.4
P39A* 1.0 1.5 0.7
F49A* 70 4.2 16.7
F16A/F49A ND 65.6 ND
Table XVIII shows that, in addition to F49A, F16A and F25A are both substantially reduced in affinity for IGFBP-1, but less so for IGFBP-3. Both still retain biological activity based on KIRA assays (Fig. 20) .
For determining relative potency of F49A and E3A/F49A, their ability to activate the type I IGF receptor was measured using serial dilutions in
KIRA assays. As shown in Figs. 21A-21B, both F49A and E3A/F49A display IGF receptor activation curves that are indistinguishable from wild-type IGF-1.
The half-maximal effective concentrations (EC50) were 20.0 ± 1.3 ng/ml for
F49A, 19.8 ± 0.5 ng/ml for E3A/F49A, and 18.9 ± 0.2 ng/ml and 19.8 ± 0.6 ng/ml for wild-type IGF-1. These results strongly suggest that both IGF mutants are fully biologically active.
Blood Clearance and Renal Accumulation of IGF-1 Variants in Rats
To assess preliminary pharmacological properties of F49A and E3A/F49A IGF-1, both proteins were radiolabeled and administered intravenously to rats. Figure 22A shows a time course of the rate at which both molecules are cleared from the blood of the animals. As expected due to their decreased IGFBP affinities, both variants were cleared at a faster rate compared to wild-type human IGF-1. Interestingly, the double mutant (E3A/F49A) was cleared faster than the single mutant (F49A) , correlating
well with the respective affinities for the major binding protein in the serum, IGFBP-3 (Table XV) . Figure 22B shows the tissue-to-blood ratio for the IGF variants in different organs. The majority of the radioactively- labeled IGF molecules were detected in the kidney, whereas radioactivity levels in the liver, spleen, heart, and pancreas were much lower. It is evident that the variants F49A and E3A/F49A accumulate at statistically significant higher levels in the kidney compared to wild-type IGF-1. Circular Dichroism Analysis of IGF-1 Variants
The circular dichroism spectra of F49A and E3A/F49A IGF-1 were analyzed to test whether the introduced mutations cause major changes to the protein structure. Structural destabilization could lead to increased proteolytic susceptibility, providing an alternative explanation for the faster blood clearance rates of the IGF variants. As shown in Fig. 23, however, both mutants have virtually identical spectra to the one recorded for wild-type IGF-1. The CD spectra reveal elements of both α-helix and random coil, as expected from NMR spectroscopy of IGF-1 (Cooke et al . , supra ) . The thermal stability of IGF-1 could not be determined accurately by circular dichroism, presumably due to the relatively high content (-30%) of random coil (Jansson et al . , supra , 1997) already present at room temperature. The fact that the CD spectra of both variants showed no significant deviation from wild-type IGF-1 is an indication that the introduced mutations do not alter the overall structure of IGF-1. Conclusion:
From the evidence presented above, it would be expected that the single and double mutants F16A, F16G, F16S, F25A, F25G, F25S, F49A, F49G, F49S, E3A/F49A, E3A/F49G, E3G/F49A, E3G/F49G, E3A/F49S, E3S/F49A, E3S/F49S, E3G/F49S, and E3S/F49G IGF-1 would be effective in treating cartilage disorders, since the alanine-substituted mutants exhibit a reduced affinity for IGFBP-1 without substantial loss of ability to bind to IGFBP-3 and are biologically active based on many tests. Further, such mutants are expected to be efficacious in treating cartilage disorders, since the alanine- substituted mutants only weakly bind to IGFBP-1 and there is disregulation in IGFBP-3 present in arthritic disorders (Martel-Pelletier et al . , supra) . It would also be expected that BP3-01 and BP3-15 would also be efficacious for this purpose in view of their role in displacing IGFBP-3 (Lowman et al . , supra , 1998; WO 98/45427).
EXAMPLE 8
Articular Cartilage Explant Assay of IGF-1 Analogs With Selectively Reduced Affinity for IGFBP-1 versus IGFBP-3 Introduction:
The experiments of this example examine both the synthetic and prophylactic potential of the IGF-1 analogs E3A/F49A and F49A on the cartilage matrix. Materials and Methods : Articular cartilage explants
The metacarpophalangeal joint of 18-24 month old cows was aseptically' dissected, and articular cartilage was removed by free-hand slicing, taking care so as to avoid the underlying bone. The cartilage was minced, washed, and cultured in bulk for at least 24 hours in a humidified atmosphere of 95% air and 5% C02 in serum-free (SF) LG DMEM/F12 medium with 0.1% BSA, 100 0/ml penicillin/streptomycin (Gibco), 2 mM L-Glutamine, 0.1 mM MEM sodium pyruvate (Gibco), 20 μg/ml GENTAMICIN™ (Gibco) antibiotic, and 1.25 mg/L AMPHOTERICIN B™ (Sigma) antibiotic. Articular cartilage was aliquoted into MICRONICS™ tubes (approximately 55 mg per tube) and incubated for at least 24 hours in the above media. Control (media alone) , wild-type IGF-1, E3A/F49A, or F49A was then added to each tube (to a final concentration of 40 or 400 ng/ml as indicated) . The media was harvested and changed at various time points (0, 24, 48 and 72 hours). Proteoglycan release Medium harvested at various time points was assayed for proteoglycan content using the 1, 9-dimethylmethylene blue (DMMB) colorimetric assay of Farndale and Buttle, Biochem. Bhiophys. Acta, 883: 173-177 (1985) . A standard curve was prepared of chondroitin sulfate ranging from 0.0 to 5.0 μg. Proteoglycan synthesis
After the media change at 48 hours, 35S-sul te (to a final concentration of 10 μCi/ml) was added to the cartilage explant cultures. After an additional 17 hours of incubation at 37 °C, the amount of proteoglycans in the media was measured using the DMMB assay. The cartilage pieces themselves were washed twice with explant media and digested overnight at 50°C in a 900 μL reaction volume of 10 mM EDTA, 0.1 M sodium phosphate, and 1 mg/ml proteinase K (Gibco BRL) . 600 μl of the digestion reaction was mixed (2:1) with 10% w/v cetylpyridinium chloride (Sigma) and centrifuged at 1000 x g for 15 minutes. The supernatant was removed and formic acid (500 μl, Sigma) was added to dissolve the pellets. The samples
were then transferred to scintillation vials containing 10 ml scintillation fluid (ICN) and read in a scintillation counter. Remaining proteoglycan in cartilage tissues
After 72 hours, the remaining articular cartilage explants were digested as described above under Proteoglycan synthesis and assayed for proteoglycan content using the DMMB colorimetric assay (referenced above under Proteoglycan release) . Nitric Oxide Assay
Articular cartilage media, saved from the cartilage explants at various times (24, 48, and 72 hours), was mixed with 10 μl 0.05 mg/ml 2,3- diaminonapthalene (DAN) in 0.62M HC1 and incubated at room temperature for 10-20 minutes in the dark. The reaction was terminated with 5 μl of 2.8N NaOH. The amount of fluorescence of 2, 3-diamionaphthotriazole was measured with a CYTOFLOR™ fluorescent plate reader at 365-nm excitation at 409-nm emission. Results:
The data presented in Figures 24-27 show that the two IGF-1 analogs tested' decrease cartilage matrix breakdown (as measured by proteoglycan release) , block the induction of cartilage matrix breakdown by IL-lα, induce cartilage matrix synthesis (as measured by proteoglycan synthesis) , and prevent inhibition of matrix synthesis by IL-lα.
These effects are similar to those of wild-type IGF-1. However, since human IGF-1 is employed with bovine tissue, the species differences may mask an inhibitory effect of endogenous binding proteins on wild-type IGF-1 activity.
The IGF-1 analogs significantly decreased nitric oxide release (Fig. 28) . In addition, the IGF-1 analogs blocked induction of nitric oxide by IL-lα (Fig. 29) . Discussion: It is shown herein that IGF-1 analogs are capable of inhibiting matrix breakdown, stimulating new matrix synthesis, and inhibiting nitric oxide release. In addition, these IGF-1 analogs inhibit the detrimental effects of interleukin 1, which is elevated in diseased joints.
The role of nitric oxide in breakdown of articular cartilage, especially the destruction associated with osteoarthritis has been described in Ashok et al . , Curr. Opin. Rheum., 10: 263-268 (1998). Since nitric oxide also has effects on other cells, the presence of nitric oxide within the joint could increase vasodilation and permeability, potentiate cytokine release by leukocytes, and stimulate angiogenic activity by monocyte- macrophages. Normal cartilage does not produce nitric oxide unless stimulated with cytokines such as IL-1, while osteoarthritic cartilage
explants continue to release nitric oxide for over 3 days in culture despite the absence of added stimuli. Thus, production of nitric oxide by cartilage correlates with a diseased state, and since nitric oxide appears to play a role in both the erosive and the inflammatory components of joint diseases, a protein or peptide that decreases nitric oxide production would likely be beneficial for the treatment of degenerative cartilagenous disorders. In fact, in vivo animal models suggest that inhibition of nitric oxide production reduces progression of arthritis (Pelletier et al . , Arthritis Rheum., 7: 1275-1286 (1998); van de Loo et al . , Arthritis Rheum., 41 : 634- 646 (1998); Stichtenoth and Frolich, Br. J. Rheumatol., 37: 246-257 (1998)).
The assay described herein is based on the principle that 2,3- diaminonapthalene (DAN) reacts with nitrite under acidic conditions to form
1- (H) -naphthotriazole, a fluorescent product. As nitric oxide is quickly metabolized into nitrite ( O^1) and nitrate (NO3 "1) , detection of nitrite is one means of detecting (albeit undercounting) the actual nitric oxide produced in cartilagenous tissue.
As described above, nitric oxide has detrimental effects on chondrocytes as well as other cell types within the joint. Since inhibition of nitric oxide has been shown to inhibit progression of arthritis in animals, the effect of the IGF analogs on nitric oxide further suggests that the tested IGF analogs would be protective for joint tissues in vivo . Finally, these analogs or the IGFBP displacer peptides are expected to have anabolic effects on tissues, such as arthritic cartilage, which are otherwise IGF-1 resistant. In summary, two IGFBP-selective variants (F49A and E3AF49A) demonstrated a 700-fold and 80, 000-fold apparent reduction in affinity for IGFBP-1, while preserving low nanomolar affinity for IGFBP-3, the major carrier of IGF-1 in plasma. Both variants displayed wild-type-like potency in cellular receptor kinase assays, stimulated human cartilage matrix synthesis, and retained their ability to associate with ALS in complex with IGFBP-3. Hence, the half-life of these variants is still determined by IGFBP-3, but their activity is no longer regulated by IGFBP-1. Furthermore, pharmacokinetic parameters and tissue distribution of these two IGF-1 variants in rats differed from wild-type IGF-1 as a function of their IGFBP affinities .
EXAMPLE 9
Generation of IGF-1 Analogs with Selectively Reduced Affinity for IGFBP-3 versus IGFBP-1 and Articular Cartilage Explant Assay Therefor
Introduction:
The IGFBPs are generally thought to inhibit the biological activity of IGF-1 by sequestering the growth factor into high-affmity complexes and thereby preventing its receptor association (Jones and Clemmons, Endocr . Rev. 16: 3-34 (1995)). The levels of IGFBP-3 (and IGFBP-4) were found to be increased m human inflammatory synovial fluid (Kanety et al , J. Rheumatol 23: 815-818 (1996) ) . This change IGFBP homeostasis is thought to contribute to the pathological condition by depriving cells from the IGF-1 survival signal. In this example IGF-1 molecules were generated with selectively reduced affinity for IGFBP-3, without altering activity on the IGF type I receptor. Such molecules would be predicted to be more biologically potent than wild-type IGF-1 in the presence of elevated pathophysiological IGFBP-3 levels. Furthermore, since IGFBP-3 is the major carrier of IGF-1 n serum, the half-life and biological distribution of such IGF-1 variants would presumably be drastically altered. The binding epitopes of IGFBP-1 and IGFBP-3 have been mapped by alan e-scannmg on the surface of IGF-1 (Dubaquie and Lowman, Biochemistry, 38: 6386-6396 (1999) ) . Each individual IGF-1 side-chain contributes only modest amounts of binding energy for IGFBP-3. Based on this observation it seems impossible to substantially decrease IGFBP-3 affinity by introducing only a few specific alanine mutations into IGF-1. A different strategy to disrupt protein-protein interactions involves the introduction of a charged residue into the binding interface, leading to electrostatic repulsion of the binding partners. It has been noted by Jansson et al . (Biochemistry, 36: 4108-4117 (1997) ) that IGF-1 is an electrostatically polarized protein w th a continuous negatively-charged patch at the N-termmus (including the B- region helix) , while the C-region is mainly positively charged. Based on these observations residue D12 was selected, since it does not contribute any binding energy for IGFBP-1 and seems to be part of the structural IGFBP- 3 binding epitope (Dubaquie and Lowman, supra) . It was reasoned that replacing residue 12 with a positive charge would disrupt the continuous negatively-charged patch that might possibly be involved the IGFBP-3 interaction. Materials and Methods : Generation of Analogs Phage particles displaying IGF-1 variants m which the aspartate residue at position 12 was mutated to lysme (D12K) or argmine (D12R) were
constructed as described m Dubaquie and Lowman, supra . Furthermore, these protein variants were expressed in E. coli and purified as described m Dubaquie and Lowman, supra . Articular Cartilage Explants The metacarpophalangeal joint of a six-month old pig was cultured as described above for bovine articular cartilage m Example 8. This explant was cultured in media alone or in media with D12K, D12R, or wild-type IGF-1 (at 10 nM) alone or m the presence of IL-lα at 1 ng/ml, as described above. Human explants were from patients undergoing joint replacement surgery. Articular cartilage was harvested, aliquoted (40-45 mg/tube) and cultured as above. Twenty-four hours later, explants were treated with 40 ng/ml IGF-1, F16A/F49A, E3A/F49A, or F49A, every day for five days. Fresh media was added on days 0, 1, 2, and 3. Matrix breakdown was determined by measuring the amount of proteoglycans the media using the DMMB assay as set forth above. Matrix (proteoglycan) synthesis was determined by measuring 3S- sulfate uptake as set forth above.
Further, for comparison purposes, F49A, E3A/F49A, F16A/F49A, D12K, D12R, or wild-type IGF-1 (at 40 ng/ml) were tested for cartilage matrix synthesis m human tissue as described above. Human articular cartilage from diseased joints was cultured media alone or with F49A, E3A/F49A, F16A/F49A, D12K, D12R or wild-type IGF-1 (at 40 ng/ml) and matrix synthesis was determined by measuring 35S-sulfate uptake as described above. Results :
Figure 30 shows the binding curves of phage particles displaying either wild-type IGF-1, D12K, or D12R bound to immobilized IGFBP-1 (Fig. 30A) or IGFBP-3 (Fig. 30B) . The wild-type IGF-1 phage particles bound to IGFBP-1 or IGFBP-3 generated similar detection signal intensities. D12K and D12R, however, displayed lower signal intensities when binding to IGFBP-3 compared to IGFBP-1. This result suggests that these variants have a selectively-reduced affinity for IGFBP-3 compared to wild-type IGF-1.
Like IGF-1, D12K and D12R inhibited cartilage matrix breakdown (Fig. 31A) and increased matrix synthesis (Fig. 31C) . In addition, l ke IGF-1, D12K or D12R inhibited the catabolic effects of IL-lα (Fig. 31B) and prevented IL-lα-induced inhibition of proteoglycan synthesis (Fig 31D) Thus, these mutants retain full activity, and are expected to be good therapeutic agents for cartilage disorders such as arthritis, which are characterized by increased matrix breakdown and decreased matrix synthesis. In addition, since high levels of IL-1 are found diseased joints, the ability of these mutants to inhibit the detrimental effects of IL-lα on cartilage further suggest utility of these IGF variants as treatments for arthritis .
Importantly, the IGF-1 variants have activity on human cartilage obtained from patients undergoing joint replacement, i . e . , with arthritis. As shown in Fig. 32, all five IGF variants tested with selective preferences for IGFBP-3 over IGFBP-1 or vice-versa (F49A, E3AF49A, F16/F49, D12K, and D12R) stimulated matrix synthesis in diseased human articular cartilage, and the activity of the variants was as good as, or better than, that of wild- type IGF-1. Unlike previous studies showing IGF-1 resistance in articular cartilage from osteoarthritic joints, cartilage from these particular patients remained responsive to IGF-1. Discussion:
The protein interface between IGF-1 and IGFBP-3 seems to be sensitive to mutations changing the charge distribution. Introducing positive charges into the N-terminal region of IGF-1 is expected to selectively reduce IGFBP- 3 affinity. The IGF-1 variants D12K and D12R are biologically active, as shown in the articular matrix synthesis stimulation experiments herein.
IGF-1 is a key regulator of matrix homeostasis in articular cartilage.
The metabolic imbalance in osteoarthritis that favors matrix breakdown over new matrix synthesis may be due, at least in part, to insensitivity of chondrocytes to IGF-1 stimulation. While the mechanism underlying this IGF- 1 resistance is not known, without being limited to any one theory, it is believed that IGFBPs, which are elevated in many arthritic patients, play a role. In these patients, IGF-1 analogs that do not bind to, and are thus not inhibited by, IGFBPs would likely stimulate cartilage repair in tissue that is otherwise IGF-1 resistant. Alternatively, peptides that block IGF-1 binding to inhibitory binding proteins may thus free IGF-1 to act on resident chondrocytes. Furthermore, based on the possible role of IGFBPs (especially IGFBP-1) in modifying the activity of IGF-1, the IGF-1 analogs described herein may have better clearance from, and/or transport through tissues within, human joints relative to that of wild-type IGF-1.
Since loss of cartilage matrix proteins is an early and continuous part of joint damage leading to joint failure, the ability of these IGF-1 analogs to inhibit cartilage catabolism and stimulate new matrix synthesis strongly suggests that these IGF analogs will have beneficial effects on diseased or damaged joints.
Il-lα has catabolic effects on cartilage, including the generation of synovial inflammation, up-regulation of matrix metalloproteinases, stimulation of matrix breakdown, and inhibition of proteoglycan and collagen synthesis. Furthermore, IL-1 protein is found in diseased, but not normal joints. Thus, the ability of the tested analogs to have positive effects on cartilage, as well as to counteract the deleterious effects of IL-lα,
strongly suggests that such molecules would have a protective effect on cartilage disorders, including damaged and/or diseased cartilage. In addition, such an activity predicts that the test IGF-1 analogs and peptides would inhibit the degradation that occurs m arthritic conditions, since antagonism of IL-lα function has been shown to reduce the progression of osteoarthritis (Arendt et al . , Ann Rev Immunol ■ , 16: 27-55 (1998)).
EXAMPLE 10 ,
Articular Cartilage Explant Assay of BP3-15 and BP3-40 Analogs Materials and Methods: Human articular cartilage explants were cultured n media or treated with IGF-1 by itself or in combination with either BP3-40 or BP3-15 at 0.1 mg/ml as set forth above. Matrix breakdown was determined by measuring the release of proteoglycans into media as described above Matrix synthesis was determined by counting the amount of 3S-sulfate incorporated into the cartilage tissue as described above Results and Discussion:
Since high levels of IGFBPs have been found in diseased cartilage, and these IGFBPs may inhibit IGF-1 activity, the effect of two IGFBP-3 displacer peptides on IGF-1 activity was tested. Human articular cartilage explants from arthritic patients were treated with IGF-1 alone or combination with an IGFBP displacer peptide (BP3-15 or BP3-40) . These displacer peptides appeared to enhance the ability of IGF-1 to decrease matrix breakdown (Fig. 33A, 33C) and to stimulate matrix synthesis (Fig. 33B, 33D) . Thus, these peptides are expected to be useful for conditions, such as arthritis, where high levels of inhibitory binding proteins are present. In such conditions, treatment with a displacer peptide alone, or combination with IGF-1, is expected to enhance the anabolic effects of endogenous or exogenous IGF-1 and to be useful in treating arthritis.
EXAMPLE 11 Complex formation with ALS
Materials and Methods :
Human ALS was expressed in CHO cells (Leong et al . , Mol Endocrinol., 6: 870-876 (1992)). The secreted ALS was enriched on DEAE-Sepharose, followed by affinity purification on an IGF-l/IGFBP-3 column, as described m Baxter et al . , J. Biol. Chem., 264: 11843-11848 (1989).
To monitor trimeπc complex formation, 300 RU's of biotinylated IGFBP- 3 (Sulfo-NHS-LC-LC-biot , Pierce, Rockford, IL) were immobilized on a streptavid -coupled chip (SA; Biacore, Inc., Piscataway, NJ) in PBS, 0.05% TWEEN™-20, and 0.01% sodium azide. The biosensor chip was primed with running buffer containing 1 μM of the respective IGF-1 analog (F49A or E3A/F49A). ALS was injected n amounts of 98, 148, and 333 nM at 50 μL/mm.
for 2 minutes, followed by a 2-minute dissociation period. The chip was regenerated with 10 μL of 2M KSCN (in 50 mM HEPES pH 7.2) at a flow rate of 20 μL/min., followed by a 3-minute buffer flow for baseline stabilization. Results : F49A and E3A/F49A form ternary complexes with IGFBPS and ALS
The majority of IGF-1 in serum is found in a 150-kDa ternary complex composed of IGFBP-3 and a glycoprotein termed ALS. The half-life of this ternary complex is an order of magnitude longer than that of free IGF-1 (Ferry et al . , Horm. Metab. Res., 31: 192-202 (1999)). Hence, ternary complex formation is a major determinant for IGF-1 biodistribution. To test whether the engineered IGF-1 variants were still able to form trimeric complexes with ALS, a biosensor binding experiment was performed. Biotinylated IGFBP-3 was immobilized on a biosensor chip, and ALS was injected having either wild-type IGF-1 (Fig. 34A) , F49A (Fig. 34B) , or E3A/F49A (Fig. 34C) included in the running buffer at a concentration of 1 μM. As shown in Figure 34, the ALS binding curves were essentially identical for the wild-type IGF-1 and the two IGF-1 variants. The dissociation rate for wild-type IGF-1 and each IGF-1 variant is listed in Table XIX, ranging from 4.0 x lO^s"1 to 6.3 x 10~4sN In the absence of any IGF-1 in the running buffer, ALS did not associate with IGFBP-3 on the chip. This experiment indicates that the IGF-1 variants retain the ability to form ternary complexes, and that their terminal half-life in serum therefore should not be drastically shorter than for wild-type IGF-1.
TABLE XIX ALS Dissociation Rates from IGFBP-3/IGF-1 complexes determined by Biosensor
Experiments
Clearance and distribution of F49A and E3A/F49A in rats .
Human wild-type IGF-1, F49A, and E3A/F49A were radiolabeled and administered intravenously to rats to assess their pharmacokinetic properties . Both IGF-1 variants cleared significantly faster when compared to wild-type human IGF-1 (Table XX) . E3A/F49A cleared at the fastest rate
(1107 + 45 ml/hr/kg), followed by F49A (427 ± 125 ml/hr/kg) and wt IGF-1 (151 ± 24.7 ml/hr/kg). A similar trend was observed for the intermediate half-life t1 2 β: E3A/F49A had the shortest, F49A intermediate, and wt IGF-1 the longest tι2 β (Table XX) . These findings correlate well with the corresponding in vitro affinities for the major binding protein in the serum, IGFBP-3. Interestingly, the steady-state distribution volumes were much greater for the IGF-1 variants, indicating that they are less confined to the plasma compartment than is wild-type IGF-1 (Table XX) . The greatest tissue-to-blood ratios for the IGF-1 molecules were detected in the kidney, whereas ratios in the liver, spleen, heart, and pancreas were much lower. It is evident from this experiment that F49A and E3A/F49A accumulate at statistically significant higher ratios in the kidney compared to wild-type IGF-1.
TABLE XX
Pharmacokinetic parameters following a single IV dose in rats.
Group Half-life (hr) clearance steady- (ml/hr/kg) state distribu
(ml/kg)
12SI-wt 0.033 + 1.22 ± 5.88 + 151 + 24.7 936 ± 94.8
IGF-1 0.004 0.237 0.374
125I-F49A 0.064 ± 0.971 ± 8 .97+ 4.50 427 + 3201 +
0.052 0.336+ 125*+ 488*+
125-p_ 0.032 + 0.321 + 6.23 + • 1107 ± 6520 +
E3A/F49A 0.005 0.091* 1.47 45.0* 874*
P < 0.05 compared to 125I-wt IGF-1 P < 0.05 compared to 125I-E3A/F49A
Discussion:
The ability of the analogs F49A and E3A/F49A to form ternary complexes with ALS and IGFBP-3 has important consequences for the serum half-life of these variants. How ALS interacts with the IGF-l/IGFBP-3 complex is unknown due to a lack of structural information on IGFBP-3 and ALS. A recent model of ALS postulates a donut-shaped structure whose internal cavity is lined with negative charges; these regions could potentially interact with positive charges on IGFBP-3 and IGFBP-5 (Janosi et al., J. Biol. Chem., 274 : 5292-5298 (1999)). The affinity of ALS for a crosslinked IGF-l/IGFBP-3 complex is on the order of 0.1 to 0.3 nM, depending on the carbohydrate content of ALS (Janosi et al., supra). In the biosensor measurements that
analyzed ALS binding to a non-covalent IGF-l/IGFBP-3 complex (by including saturating amounts of IGF-1 in the running buffer) , the dissociation rates of the wild-type IGF-1, F49A, and E3A/F49A remained constant between 4.0 x 10'V1 to 6.3 x lO'V1 (Table XIX). The experiment confirms that both IGF-1 variants tested form ternary complexes with ALS.
The pharmacokmetics of the IGF-1 variants m rats showed different characteristics from wild-type IGF-1 (Table XX) . It appears that the majority of differences are observed in the first 60-100 minutes of the experiment. In the pharmacokinetic analysis model, this phase is characterized by the initial and intermediate half-lives t1/2 α and tιz β, which likely describe the distribution of free and total radiolabeled molecules to extravascular tissues and their clearance (Table XX) . Following this initial phase, all three IGF-1 molecules are cleared at comparable rates, as suggested by their similar terminal half-life values t1/2 Y, which likely reflect the elimination of 15-ι from the rat (Table XX) . The trends t1/2 01 and tj2 β observed across the three molecules are generally consistent with a more rapid rate of distribution and clearance for the IGF-1 variants that are less bound to IGFBPs. Furthermore, the calculated steady-state distribution volumes and clearance rates also support the interpretation that IGFBP association plays a major role in the distribution and clearance of the IGF-1 variants. Therefore, without being limited to any one theory, this suggests that the initial differences in the pharmacokinetic profile reflect the IGF-l/IGFBP-3 binding equilibrium, since they correlate well with the corresponding affinities of the IGF-1 variants for IGFBP-3 m vitro .
Because IGFBP-3 in the blood seems to be saturated under normal conditions (Jones and Clemmons, Endocr. Rev., 16: 3-34 (1995)), it is believed, without limitation to any one theory, that the injected IGF-1 variants have to compete with endogenous IGF-1 for IGFBP-3 binding. The IGF- 1 molecules that are unable to associate with IGFBP-3 could diffuse out of the vasculature, and either associate with other binding proteins or be cleared at a rapid rate (tι/2 for free IGF-1 = 10-15 min11) . Those molecules that achieve ternary complex formation with IGFBP-3 and ALS, however, experience a more prolonged serum half-life and become "trapped" m the vasculature. Ternary complex formation of the IGF-1 variants occurs with the same efficiency as wild-type IGF-1 in vitro (Fig. 33, Table XIX), and this property appears to be reflected n the later phase (> 100 minutes) of the pharmacokinetic profile. The fact that both human IGF-1 variants and wild- type human IGF-1 display terminal half-lives in the order of 6-9 hours strongly suggests that complex formation with rat IGFBP-3 and ALS occurs in vivo. This is important, since all in vi tro experiments were done
exclusively with human IGFBP-3 and ALS, whereas the in vivo experiments rely on preservation of the engineered specificities for the homologous rat binding proteins.
EXAMPLE 12 Substitutions in BP1-16
Several single-residue substitutions in BP1-16 were tested for their effect on IGFBP-1 binding affinity by synthesizing peptides and measuring inhibition of IGFBP-1 binding to IGF-I. Sites for substitution were chosen based upon the known effect of an alanine or other substituted residue at the site.
G4 was previously found to be substitutable by D-alanine. Because the conformational effects of D-alanine are different from those of L-alanine, L-alanine was substituted for G4 in peptide BP1-29. Inhibition assays showed a 50-fold loss in binding affinity with this substitution (Table XXI) .
P5 was previously found to be highly conserved in phage-displayed peptide libraries; however, some substitutions were observed. For example, three different peptide-phage clones were found with arginine at this position. Therefore, the L-alanine substitution for proline was tested, as well as several alternative -substitutions (BP1-30, BP1-31, BP1-34). The results (Table XXI) show that P5A, P5N, and P5R are well tolerated.
L6 and L9 were completely conserved in 40 of 40 sequenced clones and 61 of 61 sequenced clones, respectively, from two different IGFBP-1 selected peptide-phage libraries. In addition, substitution of either of these residues with L-alanine or aib (alpha-aminoisobutyrate) side- chains resulted in a significant loss in IGFBP-1 binding affinity. Two further substitutions were tested at each position: norleucine (Nle) , an isomer of leucine, or arginine (the aliphatic portion of the side-chain of which might still be able to pack into the peptide structure) . While the Arg substitutions resulted in peptides having undetectable IGFBP-1 affinity (BP1-32 and BP1-26) , the Nle substitutions were well-tolerated (BPl-36 and BP1-37) . The non-natural substitutions L6(Nle) and L9(Nle) are therefore the only substitutions at these positions known to preserve moderate- affinity binding to IGFBP-1. W8 was also completely conserved in IGFBP-1 selected peptide-phage libraries, although the alanine substitution had a smaller effect on binding than in the case of L6 or L9. Therefore, several large side-chain substitutions were tested at this position. Interestingly, arginine, 1- naphthylalanine (Nal(l)), or histidine substitutions (BP1-22, BP1-23, and BPl-24, respectively) each had modest (<10-fold) effects on IGFBP-1 binding
affinity (Table XXI) . From these experiments, a new consensus sequence for IGFBP-1 binding may be formulated as follows:
CysXaa(6)Xaa(7)GlyXaa(g)LeuXaa(ii)TrpLeuCysXaa(i5)Xaa(i6)Xaa(i )Xaa(i8) (SEQ ID
NO: 130), where Xaa(6), Xaa< ), Xaa(g>, Xaa(n), Xaa(is), and Xaa(i6) are independently any amino acid, and Xaa(i2), Xaa(i7), and Xaa<ιg) are independently Nal(l), His, Phe, Trp, Tyr, Pro, Gin, or Met.
Table XXI Relative affinities of BP1-16 variants measured by ELISA or BIAcore™(*) inhibition assays
BP1-16 Peptide Fold potency reduction
Variant Sequence IC50 (mut) /IC50 (BP1-16)
BP1-16 CRAGPLQWLCEKYF (SEQ ID NO: 35) -1-
BP1-29 CRAAPLQWLCEKYF (SEQ ID NO: 131) 50
BP1-30 CRAGALQWLCEKYF (SEQ ID NO: 132) 1.5
BP1-31 CRAGRLQWLCEKYF (SEQ ID NO: 133) 2.0
BP1-34 CRAGNLQWLCEKYF (SEQ ID NO: 134) 3.1
BP1-32 CRAGPRQWLCEKYF (SEQ ID NO: 135) >1000
BP1-36 CRAGPLQWLCEKYF (SEQ ID N0.136), 6.9 where the underlined L is Nle
BP1-26 CRAGPLQWRCEKYF (SEQ ID NO: 137) >570
BP1-37 CRAGPLQWLCEKYF (SEQ ID N0.138), 1.7 where the underlined L is Nle
BP1-22 CRAGPLQRLCEKYF (SEQ ID NO: 139) 3.3*
BP1-23 CRAGPLQALCEKYF (SEQ ID NO: 140) , 4.8* where the underlined A is
Nal(l)
BPl-24 CRAGPLQHLCEKYF (SEQ ID NO: 141) 7.5
EXAMPLE 13 Minimization of the BPl-01 peptide via "locked helix" It was previously shown that removal of the disulfide bond in BPl-01 is destabilizing to both structure and function of the peptide. The possibility has been investigated of replacing the disulfide bond of BPl-01 with a chemically distinct structural constraint, while maintaining moderate binding affinity to IGFBP-1. These constraints were designed to link side- chain positions separated by 7 (from position i to position i+7) or 8 (from i to i+8) residues in the BPl-01 peptide.
The i+7 locked helix strategy, one of the approaches used herein, has been described by Phelan et al . , J. Am. Chem. Soc, 119: 455-460 (1997); WO 98/20036 published May 14, 1998, as have other i+7, i+3, and i+4 linkages (reviewed in Phelan et al . , supra ) . In addition, other side-chain substitutions, allowing for ionic or hydrophobic interactions or metal chelation, have been used for the purpose of stabilizing a helical structure (reviewed by Phelan et al . , supra) . Herein is described a novel i+8 locked
helix strategy, which is particularly useful for stabilization of the helical structure found in the BPl-01 peptide family.
Mutagenesis studies indicated that major determinants for IGFBP-1 binding reside primarily in the helical segment of BPl-01. These important binding determinants segregate mainly to one face of the helix, and include Leu6 and Leu9, and the aromatic residues Trp8, Tyrl3, and Phel4. Without being limited to any one theory, the remainder of the peptide might act primarily to stabilize the helix and to ensure appropriate presentation of the major side-chain binding determinants. Therefore, other methods for constraining the binding segment of the peptide to a helical conformation might yield potent BPl-binding peptides. Side-chain-side-chain crosslinks on the opposite helical face from the major BPl-binding determinants were chosen for use. This method has been described in WO 98/20036, supra . In the present case, the i+7 crosslinking connects residues replacing Gln7 to Phel4, which replaces one of the hydrophobia IGFBP-1 binding determinants. The i+8 crosslinking connects residues replacing Gln7 to Glyl5.
The crosslinking chemistry involves replacement of the appropriate two residues with glutamic acid residues (the first and last Glu (E) residues shown in Table XXII), where the two Glu residues are joined by forming amides with 1, 5-diaminopentane. This cross-linking method has been described in WO 98/20036, supra .
To develop active peptides shorter than BPl-01, it was also decided to delete the disulfide (Cysl-CyslO) and truncate the N-terminal loop region in constrained helical peptides. The CyslO was changed to Ala and Cysl replaced with an acetyl group (ac in Table XXII). Several shorter variants lacked one or more of the other loop residues. Thus, these peptides were cyclized only through the 1, 5-diaminopentane linkage. Such peptides, lacking disulfide bonds, may be more stable to degradation in vitro and in vivo. They may also be reduced in immunogenicity compared to disulfide- containing analogs .
Functional analysis of locked helices
Peptides were assayed in a BIAcore™ assay as described in WO 98/45427, supra . These inhibition assays (Fig. 35) compared the relative potency of these peptides for blocking the interaction of IGFBP-1 with IGF-1. Adding the "i+7 helical lock" to a variant of BPl-01 reduced relative potency (Table XXII) by 6-fold (peptide (i+8)C) to 8-fold (peptide (i+7)D or (i+8)B) in the best locked-helix variants. These peptides demonstrate that a disulfide bond is not necessary to obtain structured, functional peptides of the BPl-01 family. In contrast to the locked helix variants described above, a locked helix variant in which two of the key IGFBP-1 binding determinants were lost
((i+7) ; Table XXII) exhibited significant loss in binding activity relative to BPl-01. In this peptide, W8 is replaced with the first cross-linking residue and Glyl5 is replaced with the second cross-linking residue; F14 is replaced by alanine in this peptide. The disulfide bond is still present in this peptide.
Certain additions to the N-terminus and C-terminus of these peptides (see Example 3) are predicted to improve their binding affinity and potency, as in the case of disulfide-constrained peptide variants discussed below. Hence, a consensus sequence can be formulated as follows: Xaa(i- )Xaa(5)Xaa(6-7)ProLeuGluXaa(n)LeuAlaXaa(i )Xaa(i5)Xaa(i5) aa(χ7) GluXaa (igj (SEQ ID NO:142), wherein Xaa(i- j is absent or is between 1 and 4 amino acids of any kind; Xaa(S) is any amino acid, Xaa(6- ) is absent or is between 1 and 2 amino acids, Xaa(i4) and Xaa(is) are independently any amino acid, Xaajn; and Xaa(i6) are independently Nal(l), His, Phe, Trp, Tyr, Pro, Gin, or Met, X a(i7) is absent or is 1-napthyl-Ala, His, Phe, Trp, Tyr, Pro, Gin, or Met, and aa(ig) is absent or is Gly. NMR analysis of locked helices
H NMR spectroscopy was used to ascertain that the locked helix variants of BPl-01 did have the desired three-dimensional helical structure. 1-dimensional spectra and 2-dimensional COSY, TOCSY, and ROESY spectra were acquired for peptides (i+7)A, (i+7)B, (i+7)C, (i+7)D and (i+8)C; experimental details were similar to those described for BPl-01 in Lowman et al . , supra, 1998. Preliminary analysis of backbone ^JHN-Ha scalar coupling constants (derived from 2D COSY spectra) and short H
a (i)
(i+3) distances (derived from ROESY spectra), indicated that for (i+7)A, (i+7)C, (i+7)D, and
(i+8)C, the designed helix was present. In the case of (i+7)B, the NMR data were not consistent with a helical structure. The lack of a well-folded structure presumably explains the low affinity of this peptide for IGFBP-1
(>360 fold weaker than BPl-01) The scalar coupling and ROESY data for (i+7) A, (i+7)D, and (i+8)C were analyzed in more detail to generate input restraints for the calculation of three-dimensional structures as described previously for BPl-01 (Lowman et al . , supra, 1998). Comparison of the minimized mean structures of the locked helix variants to that of BPl-01 yielded RMSDs (N,Ca,C atoms of Leu6- Phel4) of 1.02 A and 0.22 A for (i+7)D and (i+8)C, respectively. Further, the packing of hydrophobic side-chains Leu6, Trp8, Leu9, and Tyrl3 in these two locked helix variants was also very similar to the packing in BPl-01.
Thus, the (i,i+7) and (i,i+8) locked helix scaffolds have successfully maintained many aspects of the BPl-01 structure without the need for a
disulfide bond. Although the covalent tethers in (i+7) did produce the desired two turns of helix (the N,Ca,C RMSD between minimized means of BPl-01 and (i+7)A is 1.06 A), some side- chain rotamers differed significantly from those of BPl-01. The structural analyses described above suggest that covalent tethers (other than the disulfide bond observed in BPl-01) may be used to control peptide structure. The use of i,i+7 or i,i+8 tethers produced peptides (i+7)D and (i+8)C that retained high affinity towards IGFBP-1 in the absence of a disulfide bond. Presumably, the affinity derives from stabilization of a structure that maintains both the backbone helical fold and the side-chain packing arrangement of the key binding determinants observed in BPl-01. Although the peptide (i+7) maintains the backbone fold, two of the key determinants (Trp8 and Phel4) are missing, and the orientation of others (e.g. Tyrl3) is perturbed; as a result, this peptide has reduced affinity. The peptide (i+7)B fails to adopt the desired fold, and hence has no measurable affinity for IGFBP-1.
Table XXII
Locked-helix variants of BPl-01
(The first and last Glus (Es) are sites of cyclizing "lock")
BP1-16 Peptide Fold potency reduction Variant Sequence IC50 (BPl-01) /IC50(mut)
BPl-01 CRAGPLQWLCEKYFG (SEQ ID NO: 26) -1-
(i+7)A acCRAGPLQELCEKYAE ( SEQ ID NO: 143) 40
(i+7)B AcLEWLAEKYEG (SEQ ID NO: 144) >360
(i+7)C AcPLEWLAEKYEG (SEQ ID NO:145) 20
(i+7)D AcRAGPLEWLAEKYEG (SEQ ID NO: 52) 7 . 7
(i+8) AcLEWLAEKYFE (SEQ ID NO: 146) >200
(i+8)B AcRPLEWLAEKYFE (SEQ ID NO: 53) 7 . 7
(i+8)C AcRAGPLEWLAEKYFE (SEQ ID NO: 54) 5. 9
EXAMPLE 14 N-terminal variants of BP1-16 Previous affinity-maturation experiments led to a peptide addition to the C-terminus of BPl-02, including a number of peptide-phage clones (Table XXIII), and the synthetic peptide BP1-21A, the sequence of which is shown in Table XXIII. Table XXIII illustrates the C-terminal substitutions in the background of BPl-02.
Table XXIII
C-terminal substitutions derived from round 3 of monovalent phage selections m the BPl-02 peptide background
BPl-02 Peptide SEQ Number of clones
Variant Sequence ID NO: sequenced
Y135C (BP1-21A) SEVGCRAGPLQWLCEKYFSTY 13 2
Y135D SEVGCRAGPLQWLCEKYFATY 14 3 Y135F SEVGCRAGPLQWLCEKYFQTY 15 1
Y135B SEVGCRAGPLQWLCEKYFQTYT 16 1
Y135A SEVGCRAGPLQWLCEKYFDTY 17 1
Y135E SEVGCRAGPLQWLCEKYFETY 18 1
Y135K SEVGCRAGPLQWLCEKYFKTY 19 1
It is sought herein to improve affinity further by two methods: substitution of the first four N-termmal ammo acid residues from BPl-20 into BP1-21A, and re-randomization of the N-termmal ammo acid residues of BP1-21A (m the context of the previously improved C-termmus) . Peptide BPl-25 (Table XXV) was synthesized to test the additivity (Wells, Biochemistry, 29: 8509-8517 (1990)) for the N-termmal and C- term al maximally-preferred substitutions. Compared with BP1-16 inhibition assays, BPl-25 showed about a 20-fold affinity improvement. However, the affinity of BPl-25 was not significantly improved over BP1-21A. This affinity improvement was confirmed m other assays described below.
In the second approach, a monovalent-display peptide-phage library, presenting BP1-21A as a fusion to g3p, was randomized (Lowman, Methods Mol . Biol. , 87: 249-264 (1998)) at the N-termmal four residues. Binding selection to IGFBP-1 was carried out by first allowing library phage to bind to solution biotinylated IGFBP-1, with an initial concentration of 50 nM, followed by 28 nM for the subsequent four rounds of selection. Peptide- phage capable of binding IGFBP-1 were captured by incubating with streptavidin magnetic beads (Promega) for 10 minutes at room temperature. For each round of selection, the washing was gradually modified to be more stringent. Off-rate selection was performed by adding 2.5 - 5 μM IGF m solution to prevent reb dmg of phage with faster off-rates. It is of interest to note that for the last round of selection (round 5), with an overnight incubation at 4°C the presence of 2.5 μM IGF, there were still phage remaining bound to the beads (2.2 x 10 total phage were eluted). Subsequent sequencing data revealed that 14 out of 20 selected clones had converged to a single DNA sequence (clone Y0791A; Table XXIV) . A peptide
corresponding to this sequence, BPl-40, was synthetically produced for analysis .
Table XXIV N-terminal substitutions derived from round 5 of monovalent phage selections in the BP1-21A peptide background
BP1-16 Peptide SEQ ID Number of clones
Variant Sequence NO: sequenced
Y0791A GQQSCRAGPLQWLCEKYFSTY 21 14 (BPl-40)
Y0791D ASSMCRAGPLQWLCEKYFSTY 22 1
Y0791H QGPDCRAGPLQWLCEKYFSTY 23 1
Y0791K QASECRAGPLQWLCEKYFSTY 24 1
Y0791L AETLCRAGPLQWLCEKYFSTY 25 1
Y0791S NSLLCRAGPLQWLCEKYFSTY 26 1
Y0791T AQWVCRAGPLQWLCEKYFSTY 27 1
Inhibition assays for measuring relative potencies of peptides for inhibiting IGFBP-1 binding to IGF-I have been described (e.g., WO 98/45427, supra ) . Peptides described herein were of sufficient binding affinity to allow for direct measurement of binding affinities by surface plasmon resonance (SPR) using a BIAcore™ system. The direct binding kinetics of IGFBP-1 peptides were measured by injecting a series of 2-fold diluted peptides in running buffer (0.05% TWEEN 20™ in PBS) over a carboxy-methyl (CM) biosensor chip coupled with about 590-1000 RU of IGFBP-1 at a flow rate of 50 μl/min on a BIAcore-2000™ or BIAcore-3000™ instrument. The immobilization of IGFBP-1 was performed through EDC/NHS chemistry as described by the manufacturer. Peptides were also injected through a flow cell containing IGFBP-3 as background control. Since the off-rate for most
-2 -1 of the peptides is relatively fast (in the range of 2 x 10 s ) , off-rate measurement was set for 30 minutes. This allowed for regeneration of IGFBP- 1 on the chip by simple dissociation, rather than by addition of eluent . For each dilution of peptides, a global fit of the sensorgram data was performed using a 1:1 Langmuir binding model. On-rates ranged from 4 x 10
6 —1 —1 to 1.9 x 10 M s . The binding affinities, KD, calculated as k0ff/kon are summarized in Table XXV. Peptides BPl-20, BP1-21A, BPl-25, and BPl-40 were all found to have similar binding affinities (KD) of about 20 nM to 40 nM.
The conclusion from these experiments is that N-terminal extensions to the BPl-01 peptide can improve binding affinity (as in BPl-02, BPl-20, BP1- 21A, BPl-25, BPl-40, and other variants identified in Table XXIV) . Some substitutions may alter expression levels in E. coli, since GQQS (SEQ ID NO: 147) was clearly selected from phage-displayed peptide libraries. However, peptides having the sequences SEVG (SEQ ID NO:148), SEMV (SEQ ID
NO: 149), EARV (SEQ ID NO: 150), or GQQS (SEQ ID NO: 151) at their N-termini all had similar binding affinities. Therefore, the nature of added side- chains at the N-terminus appears to have little effect upon peptide binding affinity. This suggests that main-chain interaction of the peptide in this region may contribute to binding affinity for IGFBP-1.
An improved consensus sequence for IGFBP-1 binding peptides is expected therefore to be:
Xaa(i_4) CysXaa (6) Xaa (7,GlyXaa(S)LeuXaa(11)Xaa (12)LeuCysXaa(ι5)Xaa (16) Xaa(17) Xaa (ι8) (SEQ ID N0.152), wherein Xaa(1-4) is absent or is between 1 and 4 amino acids of any kind, Xaa(6), Xaa(7), Xaa(9), Xaa(n), Xaa(15), and Xaa(16) are independently any amino acid, and Xaa(ι2), Xaa(17), and Xaa(1B) are independently Nal(l), His, Phe, Trp, Tyr, Pro, Gin, or Met. As noted in Example 1, truncation of the amino-terminal 4 residues (Xaa(ι-4)) has only a small effect on activity, giving a shorter consensus that still retains binding:
CysXaa(6)Xaa(7)GlyXaa(9)LeuXaa(ιι)TrpLeuCysXaa(i5)Xaa(16)Xaa[17)Xaa(1S) (SEQ ID NO:130) .
Table XXV Peptide affinity determinations by BIAcore™ kinetics
BP1-16 Peptide KD + (SD or SE)
Variant Sequence (nM)
BPl-02 SEVGCRAGPLQWLCEKYFG (SEQ ID 210 + 46
NO:50) BPl-20 EARVCRAGPLQWLCEKYF (SEQ ID 33 ± 15
NO:39) BP1-21A SEVGCRAGPLQWLCEKYFSTY (SEQ ID 41 ± 17
NO: 40) BPl-25 EARVCRAGPLQWLCEKYFSTY (SEQ ID 42 ± 11
NO:42) BPl-40 GQQSCRAGPLQWLCEKYFSTY (SEQ ID 27 ± 21
NO : 43)
EXAMPLE 15 Cell-based Assay of Peptide Activity A cell-based (KIRA) assay was previously described for measuring the amount of IGF-like activity displaced by peptides from mixtures of
IGF-I and binding proteins (Lowman et al . , supra , 1998; WO 98/45427,
supra ) . The KIRA assay was used to compare in vitro bioactivity of BPl- 16 , BPl- 02 , BPl-25 , and BPl-40 . In this example, very low concentrations of IGF-I and IGFBP-1 were used, i . e . , below the KD of the peptide : 2 nM [IGF-I] and 1 . 5 nM [IGFBP-1] , with a titration series of [peptide] = 0 . 1 to 200 nM . IGF-I and peptide were mixed and added to cells expressing IGF receptor for 30 min, then .IGFBP-1 was added for an additional 1 h .
Increased potency was observed for both peptides BPl-25 and BPl-40 over peptides BPl-16 and BPl-02 (Fig . 36 ) . However, under these conditions , BPl-02 was not significantly more active than BPl-16; and BPl-40 was not significantly more active than BPl-25. The EC2o ( concentration at which 20% of maximal IGF-I activity is observed) values were 10-20 nM for BPl-25 and BPl-40 , and 150-200 nM for BPl-16 and BPl-02 .
EXAMPLE 16
Biosynthesis of a BPl-01 peptide variant
An additional variant of BP1-21A was designed for peptide biosynthesis in E . coli . Tor this approach, a DNA sequence encoding the peptide was fused by site-directed mutagenesis to the gene for a consensus domain of protein-A known as Z-domain (Nilsson et al . , supra,_1987) . After expression and secretion from E. coli, the fusion protein was enzymatically cleaved with trypsin to yield free peptide, which can be purified from the enzymatic reaction mix (see, e.g., Varadarajan et al . , supra; Castellanos-Serra et al . , supra; Nilsson et al . , supra, 1996). A detailed procedure for trypsin digestions has been described in Smith, supra . Because this protease is highly specific for Arg and Lys residues, the BPl-40 peptide was modified by mutation of these residues for construction of the fusion. From previous mutagenesis and phage-library results, it was known that Arg and Lys residues of BPl-01 could be substituted without significant loss of binding affinity. Therefore, a fusion protein was designed with substitutions R2A and K12H (numbering is according to the BPl-01 sequence) . Furthermore, BPl-01, having a Gly residue following the C-terminal F14 of BPl-16, was known to have no significant effect on binding affinity. Therefore, a Gly-Arg sequence was added at the end of the peptide to allow for trypsin cleavage. The sequences of the BP1-625-Z fusion protein and the BPl-625 peptide (as cleaved by trypsin) are given in Table XXVI.
Table XXVI Peptide sequences for E. coli biosynthesis
Construct Peptide sequence
BP1-625-Z GQQSCAAGPLQWLCEHYFSTYGRGGGSGGAQHDEAVDNKFNKE QQNAFYEILHLPNLNEEQRNAFIQSLKDDPSQSANLLAEAKKLN DAQAPNVDMN (SEQ ID NO: 47)
BPl-625 GQQSCAAGPLQWLCEHYFSTYGR (SEQ ID NO: 46)
The fusion protein BP1-625-Z was produced from E. col shake-flask cultures. Culture supernatants were sterile-filtered, then applied to an IgG-Sepharose™ column (Pharmacia) . The bound fraction was eluted with 1M acetic acid, then lyophilized and resuspended in trypsin-digest buffer: 10 mM Tris (pH 8.0), 100 mM NaCl, 1 mM CaCl2. TPCK-treated trypsin (Sigma) was added at a weight/weight ratio of 1:100 to 1:200 (trypsin to substrate) and digestion was carried out at 25 °C for 1-2 hours. Thereafter, PMSF was added to 1 mM to stop the reaction. Samples were adjusted to 1 mM TFA and run on an analytical HPLC column with a 0-60% acetonitrile gradient in 0.1% TFA. The two predominant peaks were collected (Fig. 37) and shown by mass spectrometry to correspond to a Z-domam fragment, and the peptide BPl-625. The peptide BPl-625 fraction was lyophilized and resuspended 100 mM HEPES buffer, pH 7.2. Inhibition experiments were carried out a BIAcore™ assay as previously described, except that limiting amounts (9-10 nM IGFBP- 1) were used to make the assay sensitive with respect to affinities n the 10~8 M range. These assays showed that the BPl-625 peptide blocked IGFBP-1 binding to immobilized IGF-1 and was similar activity to BPl-25, having about 20-fold improved potency over BPl-01 (Fig. 38).
It may be predicted that BPl-625 will block IGF-I binding to IGFBP-1 and produce IGF-like activity on cells, with similar potency to BP1-21A, BPl-25, or BPl-40. It would also be expected that a peptide, BP1-625T, comprising the sequence:
GlyGlnGlnSerCysAlaAlaGlyProLeuGlnTrpLeuCysGluHisTyrPheSerThrTyr (SEQ ID NO: 153) would act similarly to BPl-625.
The BP1-625-Z fusion is useful for producing IGFBP-b d g peptides from E. coli, and the Z part of the fusion can be advantageously attached to other peptides herein than ust BPl-625.
EXAMPLE 17 Articular Cartilage Explants from Human Joints Materials and Methods:
The same Material and Methods (for human tissue) were used as described above, using articular cartilage explants from human joints removed from patients undergoing joint replacement. These explants were cultured with IGF-1 alone at 40 ng/ml, or IGF-1 with BPl-17, BP3-15, or BPl- 16 (0.1 mg/ml), or IGF-1 with buffer (HEPES).
Proteoglycan breakdown and synthesis were measured as described above. Results and Discussion:
The role of specific IGFBPs in IGF-1 activity was tested by treating articular cartilage explants from patients undergoing joint replacement with IGF-1 in the presence of peptides that inhibit IGF-1 binding to particular IGFBPs. In particular, BPl-16 inhibits IGF-1 binding to IGFBP-1, and BP3-15 inhibits IGF-1 binding to IGFBP-3. BPl-17 binds with much lower affinity to IGFBP-1. In addition, buffer alone (100 mM HEPES) was included as a negative control.
As shown in Figure 39, both BP3-15 and BPl-16, and also BPl-17 to a lesser extent, enhance the protective effect of IGF-1. Namely, BP3-15 and BPl-16, and to some extent BPl-17, enhance anabolism, as shown by the increase in matrix synthesis. Thus, these three peptides, and especially BP3-15 and BPl-16, are expected to be useful therapeutics for the treatment of arthritis.
The present invention has of necessity been discussed herein by reference to certain specific methods and materials . It is to be understood that the discussion of these specific methods and materials in no way constitutes any limitation on the scope of the present invention, which extends to any and all alternative materials and methods suitable for accomplishing the objectives of the present invention.