WO2004074246A2 - Biphenyl derivatives having beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity - Google Patents
Biphenyl derivatives having beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity Download PDFInfo
- Publication number
- WO2004074246A2 WO2004074246A2 PCT/US2004/004449 US2004004449W WO2004074246A2 WO 2004074246 A2 WO2004074246 A2 WO 2004074246A2 US 2004004449 W US2004004449 W US 2004004449W WO 2004074246 A2 WO2004074246 A2 WO 2004074246A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hydroxy
- piperidin
- ester
- ethylamino
- oxo
- Prior art date
Links
- 0 *C(Nc(cccc1)c1-c1ccccc1)=O Chemical compound *C(Nc(cccc1)c1-c1ccccc1)=O 0.000 description 5
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/40—Oxygen atoms
- C07D211/44—Oxygen atoms attached in position 4
- C07D211/46—Oxygen atoms attached in position 4 having a hydrogen atom as the second substituent in position 4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/452—Piperidinium derivatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/08—Bronchodilators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/12—Mucolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/38—Drugs for disorders of the endocrine system of the suprarenal hormones
- A61P5/40—Mineralocorticosteroids, e.g. aldosterone; Drugs increasing or potentiating the activity of mineralocorticosteroids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/40—Oxygen atoms
- C07D211/44—Oxygen atoms attached in position 4
- C07D211/48—Oxygen atoms attached in position 4 having an acyclic carbon atom attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/56—Nitrogen atoms
- C07D211/58—Nitrogen atoms attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D453/00—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids
- C07D453/02—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids containing not further condensed quinuclidine ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/08—Bridged systems
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/94—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving narcotics or drugs or pharmaceuticals, neurotransmitters or associated receptors
- G01N33/9406—Neurotransmitters
- G01N33/9433—(Nor)adrenaline
Definitions
- the present invention relates to novel biphenyl derivatives that are useful for treating pulmonary disorders.
- This invention also relates to phannaceutical compositions comprising such biphenyl derivatives, processes and intermediates for preparing such biphenyl derivatives and methods of using such biphenyl derivatives to treat pulmonary disorders.
- bronchodilators Pulmonary disorders, such as asthma and chronic obstructive pulmonary disease (COPD), are commonly treated with bronchodilators.
- bronchodilator in widespread use consists of ⁇ 2 adrenergic receptor (adrenoceptor) agonists, such as albuterol, formoterol and salmeterol. These compounds are generally administered by inhalation.
- adrenoceptor ⁇ 2 adrenergic receptor
- albuterol albuterol
- formoterol formoterol and salmeterol
- adrenoceptor ⁇ 2 adrenergic receptor
- adrenoceptor ⁇ 2 adrenergic receptor
- albuterol albuterol
- formoterol formoterol
- salmeterol adrenoceptor
- muscarinic receptor antagonists anticholinergic compounds
- compositions containing both a ⁇ 2 adrenergic receptor agonist and a muscarinic receptor antagonist are also known in the art for use in treating pulmonary disorders.
- U.S. Patent No. 6,433,027 discloses medicament compositions containing a muscarinic receptor antagonist, such as tiotropium bromide, and a ⁇ 2 adrenergic receptor agonist, such as formoterol fumarate.
- the present invention provides novel biphenyl derivatives that are useful for treating pulmonary disorders.
- compounds of this invention have been found to possess both ⁇ adrenergic receptor agonist and muscarinic receptor antagonist activity.
- the present invention is directed to a compound of formula I:
- each R 1 is independently selected from (l-4C)alkyl, (2-4C)alkenyl, (2-4C)alkynyl, (3-6C)cycloalkyl, cyano, halo, -OR la , -C(O)OR lb , -SR lc , -S(O)R ld , -S(O) 2 R le and -NR lf R lg ; each of R la , R lb , R lc , R ld , R le , R lf and R l is independently hydrogen, (l-4C)alkyl or phenyl-(l-4C)alkyl; b is 0 or an integer of from 1 to 3; each R 2 is independently selected from ( 1 -4C)alkyl, (2-4C)alkenyl, (2-4C)alkynyl,
- W is attached to the 3- or 4-position with respect to the nitrogen atom in the piperidine ring and represents O or NW a ;
- W a is hydrogen or (l-4C)alkyl;
- c is 0 or an integer of from 1 to 4;
- each R 3 is independently selected from (l-4C)alkyl, (2-4C)alkenyl, (2-4C)alkynyl, (3-6C)cycloalkyl, cyano, halo, -OR 3a , -C(O)OR 3b , -SR 3c , -S(O)R 3d , -S(O) 2 R 3e and -NR 3 R 3g ; or two R 3 groups are joined to form (l-3C)alkylene, (2-3C)alkenylene or oxiran- 2,3-diyl; each of R 3a , R 3b , R 3c , R 3d , R 3e , R 3f and R 3g is independently hydrogen or (1
- R 4 is a divalent group ofthe formula:
- d, e, f, g, h and i are each independently selected from 0 and 1 ;
- R 4a , R 4b , R 4 ° and R 4d are each independently selected from (l-lOC)alkylene, (2- 10C)alkenylene and (2-10C)alkynylene, wherein each alkylene, alkenylene or alkynylene group is unsubstituted or substituted with from 1 to 5 substituents independently selected from (l-4C)alkyl, fluoro, hydroxy, phenyl and phenyl-(l-4C)alkyl;
- a 1 and A 2 are each independently selected from (3-7C)cycloalkylene, (6- 10C)arylene, -O-(6-10C)arylene, (6-10C)arylene-O-, (2-9C)heteroarylene, -O-(2- 9C)heteroarylene, (2-9C)heteroarylene-O- and (3-6C)heterocyclene, wherein each cycloalkylene is unsubstituted or substituted with from 1 to 4 substitutents selected independently from (l-4C)alkyl, and each arylene, heteroarylene or heterocyclene group is unsubstituted or substituted with from 1 to 4 substituents independently selected from halo, (l-4C)alkyl, (l-4C)alkoxy, -S-(l-4C)alkyl, -S(O)-(l-4C)alkyl, -S(O) 2 -(l-4C)alkyl, -C(O)O(l-4
- a 3 and A 4 are each independently selected from (3-6C)cycloalkyl, (6-10C)aryl, (2-9C)heteroaryl and (3-6C)heterocyclyl, wherein each cycloalkyl is unsubstituted or substituted with from 1 to 4 substitutents selected independently from (l-4C)alkyl and each aryl, heteroaryl or heterocyclyl group is unsubstituted or substituted with from 1 to 4 substituents independently selected from halo, (l-4C)alkyl and (l-4C)alkoxy; provided that the number of contiguous atoms in the shortest chain between the two nitrogen atoms to which R 4 is attached is in the range of from 4 to 16;
- R 5 represents hydrogen or (l-4C)alkyl
- this invention is directed to a compound of formula II:
- R 4 is as defined herein (including any specific or preferred embodiments); W represents O or NH; or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- this invention is directed to a compound of formula III:
- R 4 is as defined herein (including any specific or preferred embodiments);
- W represents O or NH; or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- this invention is directed to a compound of formula IN:
- R 4 is as defined herein (including any specific or preferred embodiments);
- W represents O or NH; or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- this invention is directed to a pharmaceutical composition
- a pharmaceutical composition comprising a pharmaceutically acceptable carrier and a therapeutically effective amount of a compound of formula I or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- Such pharmaceutical compositions may optionally contain other therapeutic agents.
- this invention is directed to such a pharmaceutical composition wherein the composition further comprises a therapeutically effective amount of a steroidal anti-inflammatory agent, such as a corticosteroid.
- Compounds of this invention possess both ⁇ 2 adrenergic receptor agonist activity and muscarinic receptor antagonist activity. Accordingly, the compounds of formula I are useful for treating pulmonary disorders, such as asthma and chronic obstructive pulmonary disease.
- this invention is directed to a method for treating a pulmonary disorder, the method comprising administering to a patient in need of treatment a therapeutically effective amount of a compound of formula I or a pharmaceutically acceptable salt or solvate or stereoisomer thereof. Additionally, in another of its method aspects, this invention is directed to a method of providing bronchodilation in a patient, the method comprising administering to a patient requiring bronchodilation a therapeutically effective amount of a compound of formula I or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- This invention is also directed to a method of treating chronic obstructive pulmonary disease or asthma, the method comprising administering to a patient in need of treatment a therapeutically effective amount of a compound of formula I or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- this invention is directed to a method for using a compound of formula I or a pharmaceutically acceptable salt or solvate or stereoisomer thereof as a research tool for studying a biological system or sample, or for discovering new chemical compounds having both ⁇ 2 adrenergic agonist activity and muscarinic receptor antagonist activity.
- This invention is also directed to processes and novel intermediates useful for preparing compounds of formula I or a pharmaceutically acceptable salt or solvate or stereoisomer thereof. Accordingly, in another of its method aspects, this invention is directed to a process of preparing a compound of formula I, the process comprising:
- the above process further comprises the step of forming a pharmaceutically acceptable salt of a compound of formula I.
- this invention is directed to the other processes described herein; and to the product prepared by any ofthe processes described herein.
- This invention is also directed to a compound of formula I or a pharmaceutically acceptable salt or solvate or stereoisomer thereof, for use in therapy or as a medicament. Additionally, this invention is directed to the use of a compound of formula I or a pharmaceutically acceptable salt or solvate or stereoisomer thereof, for the manufacture of a medicament; especially for the manufacture of a medicament for the treatment of a pulmonary disorder.
- this invention is directed to novel biphenyl derivatives of formula I or pharmaceutically acceptable salts or solvates or stereoisomers thereof. These compounds contain one or more chiral centers and therefore, this invention is directed to racemic mixtures; pure stereoisomers (i.e., enantiomers or diastereomers); stereoisomer-enriched mixtures and the like unless otherwise indicated.
- pure stereoisomers i.e., enantiomers or diastereomers
- stereoisomer-enriched mixtures unless otherwise indicated.
- compounds of formula I contain a chiral center at the carbon atom indicated by the symbol * in the following formula:
- the carbon atom identified by the symbol * has the (R) configuration. In this embodiment, it is preferred for compounds of formula I to have the (R) configuration at the carbon atom identified by the symbol * or to be enriched in a stereoisomeric form having the (R) configuration at this carbon atom. In another embodiment of this invention, the carbon atom identified by the symbol * has the (S) configuration. In this embodiment, it is preferred for compounds of formula I to have the (S) configuration at the carbon atom identified by the symbol * or to be enriched in a stereoisomeric form having the (S) configuration at this carbon atom. In some cases, in order to optimize the ⁇ 2 adrenergic agonist activity ofthe compounds of this invention, it is preferred that the carbon atom identified by the symbol * has the (R) configuration.
- the compounds of formula I also contain several basic groups (e.g., amino groups) and therefore, the compounds of formula I can exist as the free base or in various salt forms. All such salt forms are included within the scope of this invention. Furthermore, solvates of compounds of formula I or salts thereof are included within the scope of this invention.
- each R 1 may be at the 2, 3, 4, 5 or 6-position ofthe phenyl ring to which it is attached. In one embodiment, each R 1 is independently selected from (1-
- R 1 4C)alkyl, halo, -OR Ia and -NR lf R lg ; such as methyl, fluoro, chloro, bromo, hydroxy, methoxy, amino, methylamino, dimethylamino and the like. Particular values for R 1 are fluoro or chloro.
- each R 2 may be at the 3, 4, 5 or 6-position on the phenylene ring to which it is attached (where the carbon atom on the phenylene ring attached to the nitrogen atom is position 1).
- each R 2 is independently selected from (1- 4C)alkyl, halo, -OR 2a and -NR 2f R 2g ; such as methyl, fluoro, chloro, bromo, hydroxy, methoxy, amino, methylamino, dimethylamino and the like. Particular values for R 2 are fluoro or chloro.
- Each R la , R lb , R lc , R ld , R le , R lf and R lg and R 2a , R 2b , R 2c , R 2d , R 2e , R 2f and R 2g as used in R 1 and R 2 , respectively, is independently hydrogen, (l-4C)alkyl or phenyl-(l- 4C)alkyl; such as hydrogen, methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, isobutyl, tert-butyl or benzyl.
- these groups are independently hydrogen or (1- 3C)alkyl.
- these groups are independently hydrogen, methyl or ethyl.
- W is O. In another embodiment, W is NW a . Generally, it has been found that compounds in which W represents O exhibit particularly high affinity for muscarinic and ⁇ 2 adrenergic receptors. Accordingly, in a particular embodiment of this invention, W preferably represents O.
- W When referring to W, particular mention may be made of compounds wherein W is attached to the piperidine ring at the 4-position with respect to the nitrogen atom ofthe piperidine ring.
- W a is hydrogen or (l-4C)alkyl; such as hydrogen, methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, isobutyl and tert-butyl.
- W a is hydrogen or (l-3C)alkyl.
- W a is hydrogen, methyl or ethyl; such as hydrogen or methyl.
- W a is hydrogen and NW a is NH.
- c is 0, 1 or 2; including 0 or 1. In one embodiment, c is 0. In one embodiment, each R 3 is at the 3, 4 or 5-position on the piperidine ring
- each R 3 is independently selected from (l-4C)alkyl; such as methyl, ethyl, n-propyl, isopropyl, n- butyl, sec-butyl, isobutyl and tert-butyl. In another aspect, each R 3 is independently methyl or ethyl.
- R 3 is at the 1 -position ofthe piperidine ring, i.e., on the nitrogen atom ofthe piperidine ring thus forming a quaternary amine salt.
- each R 3 is independently selected from (l-4C)alkyl; such as methyl, ethyl, n-propyl, isopropyl, n-butyl, .fee-butyl, isobutyl and tert-butyl.
- each R 3 is independently methyl or ethyl.
- two R 3 groups are joined to form a (l-3C)alkylene or (2-3C)alkenylene group.
- two R 3 groups at the 2 and 6-positions on the piperidine ring can be joined to form an ethylene bridge (i.e., the piperidine ring and the R 3 groups form an 8-azabicyclo[3.2.1]octane ring); or two R 3 groups at the 1 and 4- ⁇ ositions on the piperidine ring can be joined to form an ethylene bridge (i.e., the piperidine ring and the R 3 groups form an l-azabicyclo[2.2.2]octane ring).
- other R 3 groups as defined herein may also be present.
- two R 3 groups are joined to form a oxiran-2,3-diyl group.
- two R 3 groups at the 2 and 6-positions on the piperidine ring can be joined to form a 3-oxatricyclo[3.3.1.0 2 ' 4 ]nonane ring).
- other R 3 groups as defined herein may also be present.
- Each R 3a , R 3b , R 3c , R 3d , R 3e , R 3f and R 3g as used in R 3 is independently hydrogen or (l-4C)alkyl; such as hydrogen, methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, isobutyl and tert-butyl.
- these groups are independently hydrogen or (l-3C)alkyl.
- these groups are independently hydrogen, methyl or ethyl.
- R 5 is hydrogen or (l-4C)alkyl; such as hydrogen, methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, isobutyl and tert- butyl.
- each R 5 is independently hydrogen, methyl or ethyl.
- R 5 is hydrogen.
- R 6 is -NR 6 CR 6b (O) and R 7 is hydrogen, where each of R 6a and R 6b is independently hydrogen or (l-4C)alkyl, such as hydrogen, methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, isobutyl and tert-butyl.
- these groups are independently hydrogen or (l-3C)alkyl.
- these groups are independently hydrogen, methyl or ethyl.
- a particular value for R 6 in this embodiment is -NHCHO.
- R 7a , R 7b , R 7 °, R 7d , R 7e , R 7f , R 7g , R 7h , R 7i , R 7j , R 7k , R 71 , R 7m , R 7n , R 7 ° and R 7p is independently hydrogen or (l-4C)alkyl; such as hydrogen, methyl, ethyl, n- propyl, isopropyl, n-butyl, sec-butyl, isobutyl and tert-butyl. In one embodiment, these groups are independently hydrogen or (l-3C)alkyl.
- these groups are independently hydrogen, methyl or ethyl.
- R 4 is a divalent group ofthe formula:
- R 4a , A 1 , R 4b , Q, R 4c , A 2 , R 4d , d, e, f, g h and i are as defined herein.
- the values of each ofthe components R 4a , A 1 , R 4b , Q, R 4c , A 2 and R 4d are selected such that the number of contiguous atoms in the shortest chain between the two nitrogen atoms to which R 4 is attached is in the range of from 4 to 16, (specifically, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 or 16); including 8, 9, 10, 11, 12, 13 or 14; such as 8, 9, 10 or 11 ; or 9 or 10.
- values for each variable in R it will be appreciated by those skilled in the art that values should be selected such that a chemically stable group is formed.
- each contiguous atom ofthe chain is counted consecutively starting from the first atom in the R 4 group adjacent to the nitrogen ofthe piperidine ring ending with the last atom in the R 4 group adjacent to the nitrogen of the aminohydroxyethyl group.
- the shortest chain is used to determine the number of contiguous atoms.
- R 4 is -(CH 2 ) 2 -NHC(O)-CH 2 -(phen-l,4-ylene)-CH 2 -
- R 4a is selected from (l-lOC)alkylene, (2-10C)alkenylene and (2-10C)alkynylene wherein the alkylene group is unsubstituted or substituted with 1 or 2 substituents independently selected from (l-4C)alkyl, hydroxy and phenyl.
- R 4a Representative examples of particular values for R 4a are -(CH 2 ) 2 -, -(CH 2 ) 3 -, -(CH 2 ) 4 -, -(CH 2 ) 5 -, -(CH 2 ) 6 -, -(CH 2 ) 7 -, -(CH 2 ) 8 -, -(CH 2 ) 9 -, -(CH 2 ) 10 -, -(CH 2 )CH(CH 3 )-,
- d is 1.
- a 1 is an optionally substituted (3-7C)cycloalkylene group; including a cyclohexylene group, such as cyclohex-l,4-ylene and cyclohex-l,3-ylene; and a cyclopentylene group, such as cyclopent- 1 ,3-ylene.
- a 1 is an optionally substituted (6-10C)arylene group, including a phenylene group, such as phen-l,4-ylene, phen-l,3-ylene and phen-l,2-ylene; and a naphthylene group, such as naphth-l,4-ylene and napth-l,5-ylene.
- a phenylene group such as phen-l,4-ylene, phen-l,3-ylene and phen-l,2-ylene
- a naphthylene group such as naphth-l,4-ylene and napth-l,5-ylene.
- a 1 is an optionally substituted (2-9C)heteroarylene group, including a pyridylene group, such as pyrid-l,4-ylene; a furylene group, such as fur- 2,5-ylene and fur-2,4-ylene; a thienylene group, such as thien-2,5-ylene and thien-2,4- ylene; and a pyrrolylene, such as pyrrol-2,5-ylene and pyrrol-2,4-ylene.
- a pyridylene group such as pyrid-l,4-ylene
- a furylene group such as fur- 2,5-ylene and fur-2,4-ylene
- a thienylene group such as thien-2,5-ylene and thien-2,4- ylene
- a pyrrolylene such as pyrrol-2,5-ylene and pyrrol-2,4-ylene.
- a 1 is an optionally substituted (3-6C)heterocyclene group, including a piperidinylene group, such as piperidin-l,4-ylene; and a pyrrolidinylene group, such as pyrrolidin-2,5-ylene.
- a piperidinylene group such as piperidin-l,4-ylene
- a pyrrolidinylene group such as pyrrolidin-2,5-ylene
- a 1 is an optionally substituted phenylene, thienylene, cyclopentylene, cyclohexylene or piperidinylene.
- e 0.
- R 4b is (l-5C)alkylene.
- Representative examples of particular values for R 4b are -CH 2 -, -(CH 2 ) 2 -, -(CH 2 ) 3 -, -(CH 2 ) 4 -, -(CH 2 ) 5 -; including methylene, ethylene and propylene.
- f 0.
- Q is selected from a bond, -N(Q a )C(O)-, -C(O)N(Q b )-, -N(Q°)S(O) 2 -, -S(O) 2 N(Q d )-, -N(Q e )C(O)N(Q f )-, -OC(O)N(Q , -N(Q )' )C(O)O- or-N(Q k ); such as where Q is a bond, -N(Q a )C(O)- or -C(O)N(Q b )-.
- Q is a bond, O, NH, -C(O)NH-, -C(O)N(CH 3 )-, -NHC(O)-, -N(CH 3 )C(O)-, -S(O) 2 NH-, -S(O) 2 N(CH 3 )-, -NHS(O) 2 -, -N(CH 3 )S(O) 2 - and -NHC(O)NH-.
- Another example of a value for Q, together with R 4c is -C(O)(piperidin- 1,4-ylene).
- Q a , Q b , Q c , Q d , Q e , Q f , Q g , Q h , Q Oj and Q k are each independently selected from hydrogen and (l-6C)alkyl, wherein the alkyl group is unsubstituted or substituted with from 1 to 3 substituents independently selected from fluoro, hydroxy and (l-4C)alkoxy.
- Q 1 and Q k are each independently selected from hydrogen, and (l-3C)alkyl, including hydrogen, methyl, ethyl, n-propyl and isopropyl.
- An example of a value for each of Q a , Q b , Q°, Q d , Q Q f , Q g , Q h , O 0 s and Q k is hydrogen.
- Q a , Q b , Q°, Q d , Q e , Q f , Q g , Q h , Q 1 , Oj and Q k together with the nitrogen atom and the group R b or R 4c to which they are attached form a 4-6 membered azacycloalkylene group.
- Q a and Q b together with the nitrogen atom and the group R 4b or R 4 ° to which they are attached form a piperidin-4-ylene group.
- R 4 is a group of formula:
- R 4 is a group of formula:
- R 4c is (l-5C)alkylene.
- Representative examples of particular values for R 4 ° are -CH 2 -, -(CH 2 ) 2 -, -(CH 2 ) 3 -, -(CH 2 ) 4 -, -(CH 2 ) 5 -; including methylene, ethylene and propylene.
- a 2 is an optionally substituted (3-7C)cycloalkylene group; including a cyclohexylene group, such as cyclohex-l,4-ylene and cyclohex-l,3-ylene; and a cyclopentylene group, such as cyclopent- 1,3 -ylene.
- a 2 is an optionally substituted (6-10C)arylene group, including a phenylene group, such as phen- 1 ,4-ylene, phen- 1 ,3 -ylene and phen- 1 ,2-ylene; and a naphthylene group, such as naphth-l,4-ylene and napth-l,5-ylene.
- a phenylene group such as phen- 1 ,4-ylene, phen- 1 ,3 -ylene and phen- 1 ,2-ylene
- a naphthylene group such as naphth-l,4-ylene and napth-l,5-ylene.
- a 2 is an optionally substituted (2-9C)heteroarylene group, including a pyridylene group, such as ⁇ yrid-l,4-ylene; a furylene group, such as fur- 2,5-ylene and fur-2,4-ylene; a thienylene group, such as thien-2,5-ylene and thien-2,4- ylene; and a pyrrolylene, such as pyrrol-2,5-ylene and pyrrol-2,4-ylene.
- a pyridylene group such as ⁇ yrid-l,4-ylene
- a furylene group such as fur- 2,5-ylene and fur-2,4-ylene
- a thienylene group such as thien-2,5-ylene and thien-2,4- ylene
- a pyrrolylene such as pyrrol-2,5-ylene and pyrrol-2,4-ylene.
- a 2 is an optionally substituted (3-6C)heterocyclene group, including a piperidinylene group, such as piperidin-l,4-ylene; and a pyrrolidinylene group, such as pyrrolidin-2,5-ylene.
- a piperidinylene group such as piperidin-l,4-ylene
- a pyrrolidinylene group such as pyrrolidin-2,5-ylene
- a 2 is optionally substituted phenylene, thienylene, cyclopentylene, cyclohexylene or piperidinylene.
- a 1 or A 2 or both can be phenylene, such as phen- 1,4- ylene or phen-l,3-ylene, where the phenylene group is unsubstituted or substituted with from 1 to 4 substituents independently selected from halo, (l-4C)alkyl, (l-4C)alkoxy, -S-(l-4C)alkyl, -S(O)-(l-4C)alkyl, -S(O) 2 -(l-4C)alkyl, -C(O)O(l-4C)alkyl, carboxy, cyano, hydroxy, nitro, trifluoromethyl and trifluoromethoxy.
- substituents independently selected from halo, (l-4C)alkyl, (l-4C)alkoxy, -S-(l-4C)alkyl, -S(O)-(l-4C)alkyl, -S(O) 2 -(l-4C)alkyl, -
- a 1 or A 2 or both can be cyclopentylene or cyclohexylene; wherein the cyclopentylene or cyclohexylene group is unsubstituted or substituted with (l-4C)alkyl.
- Representative examples include cz's-cyclopent- 1,3 -ylene, tr ns-cyclopent-l,3-ylene, cis- cyclohex-l,4-ylene and tr ⁇ ns-cyclohex-l,4-ylene.
- a 1 or A 2 or both can also be optionally substituted thienylene or piperidinylene, for example, thien-2,5-ylene or piperidin-1,4- ylene.
- R 4d is selected from (l-lOC)alkylene, (2-10C)alkenylene and (2-10C)alkynylene wherein the alkylene is unsubstituted or substituted with 1 or 2 substituents independently selected from (l-4C)alkyl, hydroxy and phenyl.
- R 4d Representative examples of particular values for R 4d are -(CH 2 )-, -(CH 2 ) 2 -, -(CH 2 ) 3 -, -(CH 2 ) 4 -, -(CH 2 ) 5 -, -(CH 2 ) 6 -, -(CH 2 ) 7 -, -(CH 2 ) 8 -, -(CH 2 ) 9 -, -(CH 2 ) 10 - and -(CH 2 )CH(CH 3 )-(CH 2 )-C(CH 3 ) 2 - (CH 2 ) 2 -.
- R 4 is a divalent group ofthe formula: -(R 4a ) d - where R 4a is (4-10C)alkylene. In one aspect of this embodiment, R 4 is a divalent group ofthe formula: -(CH )j- where j is 8, 9 or 10.
- R 4 in this embodiment examples include -(CH 2 ) 4 -, -(CH 2 ) 5 -, -(CH 2 ) 6 -, -(CH 2 ) 7 -, -(CH 2 ) 8 -, -(CH 2 ) 9 , and -(CH 2 ) 10 -; including -(CH 2 ) 8 -, -(CH 2 ) 9 , and -(CH 2 ) 10 -.
- R 4 is a divalent group ofthe formula:
- R 4a is (l-lOC)alkylene, such as -(CH 2 )-, -(CH 2 ) 2 -, -(CH 2 ) 3 -;
- a 2 is (6- 10C)arylene, such as phen-l,4-ylene or phen- 1,3 -ylene, or (2-9C)heteroarylene, such as thien-2,5-ylene or thien-2,4-ylene; and
- R 4d is (l-lOC)alkylene, such as -(CH 2 )-, -(CH 2 ) 2 -, -(CH 2 ) 3 -.
- R 4 in this embodiment examples are -(CH 2 )-(phen-l,4- ylene)-(CH 2 )-; -(CH 2 )-(phen- 1 ,4-ylene)-(CH 2 ) 2 -; -(CH 2 )-(phen- 1 ,4-ylene)-(CH 2 ) 3 -; - (CH 2 ) 2 -(phen-l,4-ylene)-(CH 2 )-; -(CH 2 ) 2 -(phen-l,4-ylene)-(CH 2 ) 2 -; -(CH 2 ) 2 -(phen-l,4- ylene)-(CH 2 ) 3 -; -(CH 2 ) 3 -(phen-l,4-ylene)-(CH 2 )-; -(CH 2 ) 3 -(phen-l,4-ylene)-(CH 2 )-; -(CH 2 ) 3 -(phen-l,4-
- Q is -O- or -N(Q k )-;
- Q k is hydrogen or (l-3C)alkyl, such as methyl or ethyl;
- R 4a is (l-lOC)alkylene, such as -(CH 2 )-, -(CH 2 ) 2 -, -(CH 2 ) 3 -;
- a 2 is (6-10C)arylene, such as phen-l,4-ylene or phen-l,3-ylene, or (2-9C)heteroarylene, such as thien-2,5-ylene or thien- 2,4-ylene;
- R 4d is (l-lOC)alkylene, such as -(CH 2 )-, -(CH 2 ) 2 -, -(CH 2 ) 3 -.
- R 4 in this embodiment examples are -(CH 2 ) 2 -O-(phen-l,4-ylene)-(CH 2 )-; -(CH 2 ) 2 -0-(phen-l,4-ylene)-(CH 2 ) 2 -; -(CH 2 ) 2 -O-(phen-l,4-ylene)-(CH 2 ) 3 -; -(CH 2 ) 3 -O- (phen- 1 ,4-ylene)-(CH 2 )-; -(CH 2 ) 3 -O-(phen- 1 ,4-ylene)-(CH 2 ) 2 -; -(CH 2 ) 3 -O-(phen- 1 ,4- ylene)-(CH 2 ) 3 -; -(CH 2 ) 2 -NH-(phen- 1 ,4-ylene)-(CH 2 )-; -(CH 2 ) 2 -NH-(phen- 1 ,4-ylene)-(
- R 4 in this embodiment is the formula:
- m is an integer from 2 to 10; and n is an integer from 2 to 10; provided that m + n is an integer from 4 to 12.
- d and g are 1 and e, f, h and i are 0; and R 4a is -(CH 2 ) m -, R 4c is -(CH 2 ) disguise- and Q is -C(O)NH-.
- Particular values for m are 2 or 3; and for n, 4, 5 or 6.
- R 4 Another particular value for R 4 is the formula:
- R 4 where o is an integer from 2 to 7; and p is an integer from 1 to 6; provided that o + p is an integer from 3 to 8.
- d, h and i are 1 and e, f and g are 0; and R 4a is -(CH 2 )o-, A 2 is phen- 1 ,4-ylene, R 4d is -(CH 2 ) P - and Q is -C(O)NH-.
- Particular values for o are 2 or 3; and for p, 1 or 2.
- the phen-l,4-ylene group may be optionally substituted as defined herein for A 2 .
- Another particular value for R 4 is the formula:
- R 4 is an integer from 2 to 6; r is an integer from 1 to 5; and s is an integer from 1 to 5; provided that q + r + s is an integer from 4 to 8.
- R 4a is -(CH 2 ) q -
- R 4c is -(CH 2 )
- a 2 is 1 ,4-phenylene
- R 4d is -(CH 2 ) S - and Q is -C(O)NH-.
- Particular values for q are 2 or 3; for r, 1 or 2; and for s, 1 or 2.
- the phen-l,4-ylene group may be optionally substituted as defined herein for A 2 .
- R 4 Another particular value for R 4 is the formula:
- t is an integer from 2 to 10; and u is an integer from 2 to 10; provided that t + u is an integer from 4 to 12.
- d and g are 1 and e, f, h and i are 0; and R 4a is -(CH 2 ) r , R 4c is -(CH 2 ) U - and Q is -NHC(O)-.
- Particular values for t are 2 or 3; and for u, 4, 5 or 6.
- R 4 Another particular value for R 4 is the formula:
- R 4 where v is an integer from 2 to 7; and w is an integer from 1 to 6; provided that v + w is an integer from 3 to 8.
- d, h and i are 1 and e, f and g are 0; and R 4a is -(CH 2 ) V -, A 2 is 1 ,4-phenylene, R 4d is -(CH 2 ) W - and Q is -NHC(O)-.
- Particular values for v are 2 or 3; and for w, 1 or 2.
- the phen- 1 ,4-ylene group may be optionally substituted as defined herein for A 2 .
- Another particular value for R 4 is the formula:
- x is an integer from 2 to 6; y is an integer from 1 to 5; and z is an integer from 1 to 5; provided that x + y + z is an integer from 4 to 8.
- R 4 d, g, h and i are 1 and e and f are 0; and R 4a is -(CH 2 ) X -, R 4c is -(CH 2 ) y -, A 2 is 1,4-phenylene, R 4d is -(CH 2 ) Z - and Q is -NHC(O)-.
- Particular values for x are 2 or 3; for y, 1 or 2; and for z,
- the phen-l,4-ylene group may be optionally substituted as defined herein for A 2 .
- R 4 can be selected from:
- a particular group of compounds of formula I are those disclosed in U.S. Provisional Application No. 60/447,843, filed on February 14, 2003.
- This group includes compounds of formula I; wherein: a is 0 or an integer of from 1 to 3; each R 1 is independently selected from (1 -4C)alkyl, (2-4C)alkenyl, (2-4C)alkynyl,
- each of R la , R lb , R ,c , R Id , R le , R lf and R lg is independently hydrogen or (1- 4C)alkyl;
- b is 0 or an integer of from 1 to 3;
- each R 2 is independently selected from (l-4C)alkyl, (2-4C)alkenyl, (2-4C)alkynyl, (3-6C)cycloalkyl, cyano, halo, -OR 2a , -C(O)OR 2b , SR 2 °, -S(O)R 2d , -S(O) 2 R 2e and -NR 2f R 2g ; each of R 2a
- W is attached to the 3- or 4-position with respect to the nitrogen atom in the piperidine ring, and represents O or NW a ;
- W a is hydrogen or (l-4C)alkyl; c is 0 or an integer of from 1 to 4; each R 3 is a substituent on carbon independently selected from (l-4C)alkyl, (2-
- R 4a , R 4b , R 4c and R 4d are each independently selected from (l-lOC)alkylene, (2- 10C)alkenylene and (2-10C)alkynylene wherein each alkylene, alkenylene or alkynylene group is unsubstituted or substituted with from 1 to 5 substituents independently selected from ( 1 -4C)alkyl, fluoro, hydroxy, phenyl and phenyl( 1 -4C)-alkyl;
- a 1 and A 2 are each independently selected from (3-7C)cycloalkylene, (6- 10C)arylene, (2-9C)heteroarylene and (3-6C)heterocyclene; wherein each cycloalkylene is unsubstituted or substituted with from 1 to 4 substitutents selected independently from (1- 4C)alkyl and each arylene, heteroarylene or heterocyclene group is unsubstituted or substituted with from 1 to 4 substituents independently selected from halo, (l-4C)alkyl and (l-4C)alkoxy;
- Q is selected from a bond, -O-, -C(O)O-, -OC(O)-, -S-, -S(O)-, -S(O) 2 -, -N(Q a )C(O)-, -C(O)N(Q b )-, -N(Q c )S(O) 2 -, -S(O) 2 N(Q d )-, -N(Q e )C(O)N(Q f )-, -N(Q g )S(O) 2 N(Q h )-, -OC(O)N(Q and -N( )C(O)O-; Q a , Q , Q c , Q d , Q e , Q f , Q g , Q h , Q 1 and are each independently selected from hydrogen, (l-6C)alkyl, A 3 and (l-4C)alkylene-A
- each cycloalkyl is unsubstituted or substituted with from 1 to 4 substitutents selected independently from (l-4C)alkyl and each aryl, heteroaryl or heterocyclyl group is unsubstituted or substituted with from 1 to 4 substituents independently selected from halo, (l-4C)alkyl and (l-4C)alkoxy; provided that the number of contiguous atoms in the shortest chain between the two nitrogen atoms to which R 4 is attached is in the range of from 8 to 14;
- R 5 represents hydrogen or (l-4C)alkyl
- Another particular group of compounds of formula I are those disclosed in U.S. Provisional Application No. 60/467,035, filed on May 1, 2003.
- This group of compounds includes compounds of formula I; wherein: a is 0 or an integer of from 1 to 3; each R 1 is independently selected from (l-4C)alkyl, (2-4C)alkenyl, (2-4C)alkynyl, (3-6C)cycloalkyl, cyano, halo, -OR la , -C(O)OR l , SR lc , -S(O)R ld , -S(O) 2 R le , and -NR lf R l ; each of R la , R Ib , R lc , R ld , R l ⁇ , R lf and R l is independently hydrogen or (1- 4C)alkyl; b is 0 or an integer of from 1 to 3; each R 2 is independently selected from (l-4C)alkyl,
- W is attached to the 3- or 4-position with respect to the nitrogen atom in the piperidine ring, and represents O or NW a ;
- W a is hydrogen or (l-4C)alkyl; c is 0 or an integer of from 1 to 4; each R 3 is a substituent on carbon independently selected from (l-4C)alkyl, (2- 4C)alkenyl, (2-4C)alkynyl, (3-6C)cycloalkyl, cyano, halo, -OR 3a , -C(O)OR 3b , SR 3c , -S(O)R 3d , -S(O) 2 R 3e , and -NR 3f R 3g ; each of R 3a , R 3b , R 3 °, R 3d , R 3e , R 3f and R 3g is independently hydrogen or (1- 4C)alkyl;
- R 4 is a divalent group ofthe formula:
- R 4a , R 4b , R 4 ° and R 4d are each independently selected from (l-lOC)alkylene, (2- 10C)alkenylene and (2-10C)alkynylene wherein each alkylene, alkenylene or alkynylene group is unsubstituted or substituted with from 1 to 5 substituents independently selected from (l-4C)alkyl, fluoro, hydroxy, phenyl and phenyl(l-4C)-alkyl;
- a and A are each independently selected from (3-7C)cycloalkylene, (6- 10C)arylene, (2-9C)heteroarylene and (3-6C)heterocyclene; wherein each cycloalkylene is unsubstituted or substituted with from 1 to 4 substitutents selected independently from (1- 4C)alkyl and each arylene, heteroarylene or heterocyclene group is unsubstituted or substituted with from 1 to 4 substituents independently selected from halo, (l-4C)alkyl and (l-4C)alkoxy;
- Q is selected from a bond, -O-, -C(O)O-, -OC(O)-, -S-, -S(O , -S(O) 2 -, -N(Q a )C(O)-, -C(O)N(Q b )-, -N(Q°)S(O) 2 -, -S(O) 2 N(Q d )-, -N(Q e )C(O)N(Q f )-, -N(Q g )S(O) 2 N(Q h )-, -OC(O)N(Q and
- Q a , Q b , Q c , Q d , Q e , Q f , Q g , Q h , Q ; and OJ are each independently selected from hydrogen, (l-6C)alkyl, A 3 and (l-4C)alkylene-A 4 ; wherein the alkyl group is unsubstituted or substituted with from 1 to 3 substituents independently selected from fluoro, hydroxy and ( 1 -4C)alkoxy; or together with the nitrogen atom and the group R 4b or R 4c to which they are attached, form a 4-6 membered azacycloalkylene group;
- a 3 and A 4 are each independently selected from (3-6C)cycloalkyl, (6-10C)aryl, (2-9C)heteroaryl and (3-6C)heterocyclyl; wherein each cycloalkyl is unsubstituted or substituted with from 1 to 4 substitutents selected independently from (l-4C)alkyl and each aryl, heteroaryl or heterocyclyl group is unsubstituted or substituted with from 1 to 4 substituents independently selected from halo, (l-4C)alkyl and (l-4C)alkoxy; provided that the number of contiguous atoms in the shortest chain between the two nitrogen atoms to which R is attached is in the range of from 4 to 14;
- R 5 represents hydrogen or (l-4C)alkyl
- Another particular group of compounds of formula I are those where: a is 0; b is 0; c is 0; W is O; W is attached at the 4-position ofthe piperidinyl ring; R 5 is hydrogen; and R 4 , R 6 and R 7 are as defined herein; or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- Still another particular group of compounds of formula I are those wherein: a is 0; b is 0; c is 0; W is NH; W is attached at the 4-position ofthe piperidinyl ring; R 5 is hydrogen; and R 4 , R and R 7 are as defined herein; or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- Yet another particular group of compounds of formula I are those wherein: a is 0; b is 0; c is 0; W is O; W is attached at the 4-position ofthe piperidinyl ring; R 4 is -(CH 2 )j- where j is 8, 9 or 10; R 5 is hydrogen; and R 6 and R 7 are as defined herein; or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- Another particular group of compounds of formula I are those wherein: a is 0; b is 0; c is 0; W is NH; W is attached at the 4-position ofthe piperidinyl ring; R 4 is -(CH 2 )j- where j is 8, 9 or 10; R 5 is hydrogen; and R 6 and R 7 are as defined herein; or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- Yet another particular group of compounds of fonnula I are those wherein: a is 0; b is 0; c is 0; W is O; W is attached at the 4-position ofthe piperidinyl ring; R 4 is -(CH 2 ) 2 - C(O)NH-(CH ) 5 -; R 5 is hydrogen; and R 6 and R 7 are as defined herein; or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- Another particular group of compounds of formula I are those wherein: a is 0; b is 0; c is 0; W is NH; W is attached at the 4-position ofthe piperidinyl ring; R 4 is -(CH 2 ) 2 - C(O)NH-(CH 2 ) 5 -; R 5 is hydrogen; and R 6 and R 7 are as defined herein; or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- Another particular group of compounds of formula I are those of formula II as defined herein; or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- Another particular group of compounds of formula I are those of formula III as defined herein; or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- Another particular group of compounds of formula I are those of formula IN as defined herein; or a phannaceutically acceptable salt or solvate or stereoisomer thereof.
- Another particular group of compounds of formula I are those of formula II, III or IN as defined herein, wherein the piperidinyl ring is substitued at the 4-position with a methyl group; or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- Another particular group of compounds of formula I are compounds of formula N:
- W, R 4 , R 6 and R 7 are as defined in Table I; or a pharmaceutically acceptable salt or solvate thereof.
- racemic means the compound is racemic at the chiral carbon bearing the hydroxyl group in formula V, VI or NIL
- W, R 4 , R 6 and R 7 are as defined in Table III; or a pharmaceutically acceptable salt or solvate thereof.
- alkyl means a monovalent saturated hydrocarbon group which may be linear or branched. Unless otherwise defined, such alkyl groups typically contain from 1 to 10 carbon atoms. Representative alkyl groups include, by way of example, methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, isobutyl, tert-butyl, n-pentyl, n-hexyl, n-heptyl, n- octyl, n-nonyl, n-decyl and the like.
- alkylene means a divalent saturated hydrocarbon group which may be linear or branched. Unless otherwise defined, such alkylene groups typically contain from
- alkylene groups include, by way of example, methylene, ethane- 1,2-diyl ("ethylene”), propane- 1,2-diyl, propane- 1,3-diyl, butane- 1,4- diyl, pentane-l,5-diyl and the like.
- alkoxy means a monovalent group ofthe formula (alkyl)-O-, where alkyl is as defined herein.
- Representative alkoxy groups include, by way of example, methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, sec-butoxy, isobutoxy, tert-butoxy and the like.
- alkenyl means a monovalent unsaturated hydrocarbon group which may be linear or branched and which has at least one, and typically 1, 2 or 3, carbon-carbon double bonds. Unless otherwise defined, such alkenyl groups typically contain from 2 to 10 carbon atoms. Representative alkenyl groups include, by way of example, ethenyl, n- propenyl, isopropenyl, n-but-2-enyl, n-hex-3-enyl and the like.
- alkenylene means a divalent alkenyl group.
- alkynyl means a monovalent unsaturated hydrocarbon group which may be linear or branched and which has at least one, and typically 1 , 2 or 3, carbon- carbon triple bonds. Unless otherwise defined, such alkynyl groups typically contain from
- alkynyl groups include, by way of example, ethynyl, n-propynyl, n-but-2-ynyl, n-hex-3-ynyl and the like.
- alkynylene means a divalent alkynyl group.
- aryl means a monovalent aromatic hydrocarbon having a single ring
- aryl groups typically contain from 6 to 10 carbon ring atoms.
- Representative aryl groups include, by way of example, phenyl and naphthalene- 1-yl, naphthalene-2-yl, and the like.
- arylene means a divalent aryl group.
- azacycloalkyl means a monovalent heterocyclic ring containing one nitrogen atom, i.e., a cycloalkyl group in which one carbon atom has been replaced with a nitrogen atom. Unless otherwise defined, such azacycloalkyl groups typically contain from 2 to 9 carbon atoms. Representative examples of an azacycloalkyl group are pyrrolidinyl and piperidinyl groups.
- azacycloalkylene means a divalent azacycloakyl group. Representative examples of an azacycloalkylene group are pyrrolidinylene and piperidinylene groups.
- the tenn "cycloalkyl” means a monovalent saturated carbocyclic hydrocarbon group.
- cycloalkyl groups typically contain from 3 to 10 carbon atoms.
- Representative cycloalkyl groups include, by way of example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and the like.
- cycloalkylene means a divalent cycloalkyl group.
- halo means fluoro, chloro, bromo and iodo.
- heteroaryl means a monovalent aromatic group having a single ring or two fused rings and containing in the ring at least one heteroatom (typically 1 to 3 heteroatoms) selected from nitrogen, oxygen or sulfur. Unless otherwise defined, such heteroaryl groups typically contain from 5 to 10 total ring atoms.
- heteroaryl groups include, by way of example, monovalent species of pyrrole, imidazole, thiazole, oxazole, furan, thiophene, triazole, pyrazole, isoxazole, isothiazole, pyridine, pyrazine, pyridazine, pyrimidine, triazine, indole, benzofuran, benzothiophene, benzimidazole, benzthiazole, quinoline, isoquinoline, quinazoline, quinoxaline and the like, where the point of attachment is at any available carbon or nitrogen ring atom.
- heteroarylene means a divalent heteroaryl group.
- heterocyclyl or “heterocyclic” means a monovalent saturated or unsaturated (non-aromatic) group having a single ring or multiple condensed rings and containing in the ring at least one heteroatom (typically 1 to 3 heteroatoms) selected from nitrogen, oxygen or sulfur. Unless otherwise defined, such heterocyclic groups typically contain from 2 to 9 total ring carbon atoms.
- Representative heterocyclic groups include, by way of example, monovalent species of pyrrolidine, imidazolidine, pyrazolidine, piperidine, 1,4-dioxane, morpholine, thiomorpholine, piperazine, 3-pyrroline and the like, where the point of attachment is at any available carbon or nitrogen ring atom.
- heterocyclene means a divalent heterocyclyl or heterocyclic group.
- salt means a salt which is acceptable for administration to a patient, such as a mammal (e.g., salts having acceptable mammalian safety for a given dosage regime). Such salts can be derived from pharmaceutically acceptable inorganic or organic bases and from phannaceutically acceptable inorganic or organic acids.
- Salts derived from pharmaceutically acceptable inorganic bases include ammonium, calcium, copper, ferric, fenous, lithium, magnesium, manganic, manganous, potassium, sodium, zinc and the like. Particularly prefened are ammonium, calcium, magnesium, potassium and sodium salts.
- Salts derived from pharmaceutically acceptable organic bases include salts of primary, secondary and tertiary amines, including substituted amines, cyclic amines, naturally-occurring amines and the like, such as arginine, betaine, caffeine, choline, N,N'-dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2- dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N- ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperadine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine and the like.
- arginine betaine
- caffeine choline
- Salts derived from pharmaceutically acceptable acids include acetic, ascorbic, benzenesulfonic, benzoic, camphosulfonic, citric, ethanesulfonic, edisylic, fumaric, gentisic, gluconic, glucoronic, glutamic, hippuric, hydrobromic, hydrochloric, isethionic, lactic, lactobionic, maleic, malic, mandelic, methanesulfonic, mucic, naphthalenesulfonic, naphthalene- 1,5-disulfonic, naphthalene-2,6-disulfonic, nicotinic, nitric, orotic, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, p-toluenesulfonic, xinafoic and the like. Particularly prefened are citric, hydrobromic, hydrochloric, isethionic,
- salt thereof means a compound formed when the hydrogen of an acid is replaced by a cation, such as a metal cation or an organic cation and the like.
- the salt is a pharmaceutically acceptable salt, although this is not required for salts of intermediate compounds that are not intended for administration to a patient.
- solvate means a complex or aggregate formed by one or more molecules of a solute, i.e. a compound of formula I or a pharmaceutically acceptable salt thereof, and one or more molecules of a solvent.
- Such solvates are typically crystalline solids having a substantially fixed molar ratio of solute and solvent.
- Representative solvents include, by way of example, water, methanol, ethanol, isopropanol, acetic acid and the like. When the solvent is water, the solvate formed is a hydrate.
- a phannaceutically acceptable salt or solvate or stereoisomer thereof is intended to include all permutations of salts, solvates and stereoisomers, such as a solvate of a pharmaceutically acceptable salt of a stereoisomer of a compound of formula I.
- terapéuticaally effective amount means an amount sufficient to effect treatment when administered to a patient in need of treatment.
- treating means the treating or treatment of a disease or medical condition (such as COPD) in a patient, such as a mammal (particularly a human) that includes:
- treating the disease or medical condition i.e., eliminating or causing regression ofthe disease or medical condition in a patient
- suppressing the disease or medical condition i.e., slowing or arresting the development ofthe disease or medical condition in a patient
- alleviating the symptoms ofthe disease or medical condition in a patient means a functional group or atom which can be displaced by another functional group or atom in a substitution reaction, such as a nucleophilic substitution reaction.
- representative leaving groups include chloro, bromo and iodo groups; sulfonic ester groups, such as mesylate, tosylate, brosylate, nosylate and the like; and acyloxy groups, such as acetoxy, trifluoroacetoxy and the like.
- protected derivatives thereof means a derivative ofthe specified compound in which one or more functional groups ofthe compound are protected from undesired reactions with a protecting or blocking group.
- Functional groups which may be protected include, by way of example, carboxylic acid groups, amino groups, hydroxyl groups, thiol groups, carbonyl groups and the like.
- protecting groups for carboxylic acids include esters (such as aj ⁇ -methoxybenzyl ester), amides and hydrazides; for amino groups, carbamates (such as tert-butoxycarbonyl) and amides; for hydroxyl groups, ethers and esters; for thiol groups, thioethers and thioesters; for carbonyl groups, acetals and ketals; and the like.
- Such protecting groups are well-known to those skilled in the art and are described, for example, in T. W. Greene and G. M. Wuts, Protecting Groups in Organic Synthesis, Third Edition, Wiley, New York, 1999, and references cited therein.
- amino-protecting group means a protecting group suitable for preventing undesired reactions at an amino group.
- Representative amino-protecting groups include, but are not limited to, tert-butoxycarbonyl (BOC), trityl (Tr), benzyloxycarbonyl (Cbz), 9-fluorenylmethoxycarbonyl (Fmoc), formyl, trimethylsilyl (TMS), tert- butyldimethylsilyl (TBS), and the like.
- carboxy-protecting group means a protecting group suitable for preventing undesired reactions at a carboxy group.
- Representative carboxy-protecting groups include, but are not limited to, esters, such as methyl, ethyl, tert-butyl, benzyl (Bn), / nethoxybenzyl (PMB), 9-fluroenylmethyl (Fm), trimethylsilyl (TMS), tert- butyldimethylsilyl (TBS), diphenylmethyl (benzhydryl, DPM) and the like.
- hydroxyl-protecting group means a protecting group suitable for preventing undesirable reactions at a hydroxyl group.
- Representative hydroxyl-protecting groups include, but are not limited to, silyl groups including tri(l-6C)alkylsilyl groups, such as trimethylsilyl (TMS), triethylsilyl (TES), tert-butyldimethylsilyl (TBS) and the like; esters (acyl groups) including (l-6C)alkanoyl groups, such as formyl, acetyl and the like; arylmethyl groups, such as benzyl (Bn) 5 j p-methoxybenzyl (PMB), 9-fluorenylmethyl (Fm), diphenylmethyl (benzhydryl, DPM) and the like.
- two hydroxyl groups can also be protected as an alkylidene group, such as prop-2-ylidine, formed, for example, by reaction with a ketone, such as acetone.
- the biphenyl derivatives of this invention can be prepared from readily available starting materials using the following general methods and procedures or by using other information readily available to those of ordinary skill in the art. Although a particular embodiment ofthe present invention may be shown or described herein, those skilled in the art will recognize that all embodiments or aspects ofthe present invention can be prepared using the methods described herein or by using other methods, reagents and starting materials known to those skilled in the art. It will also be appreciated that where typical or prefened process conditions (i.e., reaction temperatures, times, mole ratios of reactants, solvents, pressures, etc.) are given, other process conditions can also be used unless otherwise stated. While the optimum reaction conditions may vary depending on the particular reactants or solvent used, such conditions can be readily determined by one skilled in the art by routine optimization procedures.
- protecting groups may be necessary or desired to prevent certain functional groups from undergoing undesired reactions.
- the choice of a suitable protecting group for a particular functional group as well as suitable conditions for protection and deprotection of such functional groups are well-known in the art.
- Protecting groups other than those illustrated in the procedures described herein may be used, if desired. For example, numerous protecting groups, and their introduction and removal, are described in T. W. Greene and G. M. Wuts, Protecting Groups in Organic Synthesis, Third Edition, Wiley, New York, 1999, and references cited therein.
- biphenyl derivatives of this invention can be prepared by a process comprising:
- X Qa and X Qb each independently represent functional groups that couple to form a group Q
- P 5a represents a hydrogen atom or an amino-protecting group
- P 5b and P 6 each independently represent a hydrogen atom or a hydroxyl-protecting group
- P 8 and P 9 each independently represent a hydrogen atom or a hydroxyl-protecting group
- P 10 represents a hydrogen atom or an amino-protecting group
- R 4 represents a residue that, together with the carbon to which it is attached, affords a group R 4 upon completion ofthe reaction
- R 4 represents a residue that, together with the carbon to which it is attached, affords a group R 4 upon completion ofthe reaction; with a compound of formula 10 in the presence of a reducing agent; and then removing any protecting group P 1 , P 2 , P 3 , P 4 , P 5a , P 5b , P 6 , P 7 , P 8 , P 9 , P 10 , P 11 , P 12 or P to provide a compound of formula I; and optionally, forming a pharmaceutically acceptable salt thereof.
- a salt of one ofthe starting materials such as an acid addition salt
- the salt is typically neutralized before or during the reaction process. This neutralization reaction is typically accomplished by contacting the salt with one molar equivalent of a base for each molar equivalent of acid addition salt.
- the leaving group represented by X can be, for example, halo, such as chloro, bromo or iodo, or a sulfonic ester group, such as mesylate or tosylate.
- the groups P 1 and P 2 can be, for example, trimethylsilyl and benzyl, respectively.
- This reaction is typically conducted in an inert diluent, such as acetonitrile, in the presence of a base.
- this reaction can be conducted in the presence of a tertiary amine, such as diisopropylethylamine.
- this reaction is conducted at a temperature in the range of from 0 °C to 100 °C until the reaction is substantially complete.
- the reaction product is then isolated using conventional procedures, such as extraction, recrystallization, chromatography and the like.
- P 14 represents an amino-protecting group, such as a benzyl group.
- a benzyl group can be readily removed by reduction using, for example, hydrogen or ammonium formate and a group NIII metal catalyst, such as palladium on carbon.
- W represents ⁇ W a
- the hydrogenation reaction is conveniently performed using Pearlman's catalyst (i.e., Pd(OH) 2 ).
- Compounds of formula 12 can be prepared by reacting an isocyanate compound of formula 13:
- hydroxyl group of a compound of formula 23 can be readily converted into a leaving group using well-known reagents and procedures.
- a hydroxyl group can be converted into a halo group using an inorganic acid halide, such as thionyl chloride, phosphorous trichloride, phosphorous tribromide, phosphorous oxychloride and the like, or a halogen acid, such a hydrogen bromide.
- the leaving represented by X 2 can be, for example, halo, such as chloro, bromo or iodo, or a sulfonic ester group, such as mesylate or tosylate.
- the groups P 3 and P 4 can be, for example, tert-butyldimethylsilyl and benzyl, respectively.
- This reaction is typically conducted in the presence of a base, such as sodium bicarbonate, and an alkali metal iodide, such as sodium iodide.
- P 15 and P 16 independently represents a protecting group, such as tert-butoxycarbonyl, and any remainder represents a hydrogen atom.
- a tert-butoxycarbonyl group can be removed by treating the protected compound with trifluoroacetic acid.
- X represents a leaving group such as halo, such as chloro, bromo or iodo, or sulfonic ester group, such as mesylate or tosylate.
- This reaction is typically conducted by contacting a compound of formula 1 with a compound of fonnula 16 in an inert diluent, such as acetonitrile, DMF or mixtures thereof, at a temperature ranging from about 0 °C to about 100 °C until the reaction is substantially complete.
- an inert diluent such as acetonitrile, DMF or mixtures thereof
- compounds of formula 3 can be obtained by reductive amination of a compound of formula 11.
- the reductive amination can be performed by reacting the compound of formula 11 with, for example, benzylamine and hydrogen in the presence of palladium on carbon.
- oxidizing agent such as sulfur trioxide pyridine complex and dimethyl sulfoxide.
- This oxidation reaction is typically conducted in an inert diluent, such as dichloromethane, the presence of a tertiary amine, such as diisopropylethylamine, at a temperature ranging from about -20 °C to about 25 °C.
- Compounds of fonnula 17 can be prepared by reacting a compound of formula 1 with a compound of fonnula 18:
- X 4 represents a leaving group such as halo, such as chloro, bromo or iodo, or a sulfonic ester group, such as mesylate or tosylate.
- compounds of formula 19 can be reduced in the presence of a chiral catalyst formed from (R)-(+)- ⁇ , ⁇ -diphenyl-2-pynolidinemethanol and trimethylboroxine; or alternatively, from (_S)-(-)- ⁇ , ⁇ -diphenyl-2-pyreolidinemethanol and trimethylboroxine.
- a chiral catalyst formed from (R)-(+)- ⁇ , ⁇ -diphenyl-2-pynolidinemethanol and trimethylboroxine; or alternatively, from (_S)-(-)- ⁇ , ⁇ -diphenyl-2-pyreolidinemethanol and trimethylboroxine.
- the resulting hydroxyl group can then be protected with a hydroxyl-protecting group, P , by reaction with, for example, tert-butyldimethylsilyl trifluoromethanesulfonate.
- one of X Qa and X Qb can be an amine group (i.e., -NHQ a or -NHQ b ) and the other can be a carboxyl group (i.e., -COOH) or a reactive derivative thereof (such as acyl halide, such as an acyl chloride or acyl bromide).
- the groups P 5a , P 5b and P 6 can be, for example, benzyl, trimethylsilyl and benzyl, respectively.
- the reaction can be performed under conventional amide coupling conditions.
- Q is a sulfonamide, i.e., -N(Q c )S(O) 2 - or -S(O) 2 N(Q d )-
- one of X Qa and X Qb can be an amine group, -NHQ C or -NHQ d and the other can be a sulfonyl halide group (such as sulfonyl chloride or sulfonyl bromide).
- X 5 represents a leaving group including halo, such as chloro, bromo or iodo, and a sulfonic ester group, such as mesylate or tosylate
- X Qa represents X Qa , such as a carboxyl group or an amino group NHQ a , or a protected derivative thereof, such as a (1- 6C)alkoxycarbonylamino group or a tert-butoxycarbonylamino group.
- X 6 represents a leaving group including halo, such as chloro, bromo or iodo, and a sulfonic ester group, such as mesylate or tosylate
- X Qb represents X Q , such as a carboxyl group or an amino group NHQ b , or a protected derivative thereof, such as a (1- 6C)alkoxycarbonyl group or a tert-butoxycarbonylamino group.
- any suitable reducing agent may be used in this reaction.
- the reducing agent can be hydrogen in the presence of a Group NIII metal catalyst, such as palladium on carbon; or a metal hydride reagent, such as sodium triacetoxyborohydride.
- the group P 7 can be, for example, benzyl.
- This reaction is typically conducted in an inert diluent and a protic solvent, such as a mixture of dichloroethane and methanol, at a temperature in the range of from 0 °C to 100 °C until the reaction is substantially complete.
- Compounds of formula 7 in the form of a hydrate can be prepared by conventional procedures, for example, by dibrominating a compound of formula 19 (where X 2 in this case can also be hydrogen), and then hydrolyzing the resulting dibromide to form a glyoxal or a hydrate thereof.
- a compound of formula 19 can be reacted with hydrogen bromide and then hydrolyzed with water to form the corresponding glyoxal hydrate.
- any suitable reducing agent may be used in this reaction.
- the reducing agent may be hydrogen in the presence of a Group NIII metal catalyst, such as palladium on carbon; or a metal hydride reagent, such as sodium triacetoxyborohydride.
- the groups P 8 , P 9 and P 10 can be, for example, trimethylsilyl, benzyl and benzyl, respectively.
- this reduction reaction is conducted in an inert diluent and a protic solvent, such as dichloroethane and methanol, at a temperature in the range of from 0 °C to 100 °C until the reaction is substantially complete.
- oxidizing agent such as sulfur trioxide pyridine complex and dimethyl sulfoxide.
- This reaction is typically conducted in the presence of a tertiary amine, such as diisopropylethylamine, at a temperature in the range of from about -20 °C to about 25 °C until the oxidation is substantially complete.
- X 7 represents a leaving group including halo, such as chloro, bromo or iodo, and a sulfonic ester group, such as mesylate or tosylate.
- the leaving group represented by X 3 can be, for example, halo, such as chloro, bromo or iodo, or a sulfonic ester group, such as mesylate or tosylate.
- the groups P 11 , P 12 and P 13 can be, for example, trimethylsilyl, benzyl and benzyl, respectively.
- This reaction is typically conducted an inert diluent, such as acetonitrile, in the presence of a suitable base.
- this reaction can be conducted in the presence of a tertiary amine, such as diisopropylethylamine. Generally, this reaction is conducted at a temperature in the range of from 0 °C to 100 °C until the reaction is substantially complete.
- a tertiary amine such as diisopropylethylamine.
- this reaction is conducted at a temperature in the range of from 0 °C to 100 °C until the reaction is substantially complete.
- Compounds of formula 9 can be prepared by steps analogous to those of methods
- compounds of fonnula 10 can be prepared from compounds of formula 4 by reaction with an amine of formula P I3 NH 2 .
- any suitable reducing agent may be used in this reaction.
- the reducing agent may be hydrogen in the presence of a Group NIII metal catalyst, such as palladium on carbon; or a metal hydride reagent, such as sodium triacetoxyborohydride.
- the groups P 11 , P 12 and P 13 can be, for example, tert- butyldimethylsilyl, benzyl and benzyl, respectively.
- this reduction reaction is conducted in an inert diluent and a protic solvent, such as dichloroethane and methanol, at a temperature in the range of from 0 °C to 100 °C until the reaction is substantially complete.
- a protic solvent such as dichloroethane and methanol
- P 17 represents a hydrogen atom or an amino-protecting group
- each of P 18 , P 19 and P 20 independently represent a hydrogen atom or a hydroxyl-protecting group; provided that at least one of P 17 , P 18 , P 19 or P 20 is a protecting group; (i) deprotecting a compound of formula 26:
- P 21 represents a hydrogen atom or an amino-protecting group
- each of P 22 and P 23 independently represent a hydrogen atom or a hydroxyl-protecting group; provided that at least one of P , P or P is a protecting group; or (j) deprotecting a compound of formula 27:
- P 24 represents a hydrogen atom or an amino-protecting group
- each of P 25 and P 26 independently represent a hydrogen atom or a hydroxyl-protecting group
- benzyl protecting groups are conveniently removed by catalytic hydrogenation in the presence of a Group NIII metal catalyst, such as palladium on carbon; a tert-butyldimethylsilyl group is conveniently removed by treatment with hydrogen fluoride, such as triethylamine trihydrofluoride; and a propylidine group is conveniently removed by treatment with an acid, such as trifluoroacetic acid.
- a Group NIII metal catalyst such as palladium on carbon
- hydrogen fluoride such as triethylamine trihydrofluoride
- a propylidine group is conveniently removed by treatment with an acid, such as trifluoroacetic acid.
- R 8 represents -CH 2 OP 19 , -CHO, -COOH or -C(O)O(l-6C)alkoxy, such as carbomethoxy
- R 9 represents -OP 18 and R 10 represents a hydrogen atom
- Any suitable reducing agent may be used in this reaction including, by way of example, metal hydride reducing agents, such as sodium borohydride, lithium aluminum hydride and the like.
- X represents a leaving group, such as a bromo.
- examples of particular values for P , P and P are: for P 21 , hydrogen or benzyl; for P 22 hydrogen or tert-butyldimethylsilyl; and for P 23 hydrogen or benzyl.
- benzyl protecting groups are conveniently removed by catalytic hydrogenation in the presence of a Group NIII metal catalyst, such as palladium on carbon; and a tert-butyldimethylsilyl group is conveniently removed by treatment with hydrogen fluoride, such as triethylamine trihydrofluoride.
- Compounds of formula 26 can be prepared by the methods described herein, such as by processes (a) to (g).
- examples of particular values for P , P and P are: for P , hydrogen or benzyl; for P hydrogen or tert-butyldimethylsilyl; and for P hydrogen or benzyl.
- benzyl protecting groups are conveniently removed by catalytic hydrogenation in the presence of a Group NIII metal catalyst, such as palladium on carbon; and a tert-butyldimethylsilyl group is conveniently removed by treatment with hydrogen fluoride, such as triethylamine trihydrofluoride.
- Compounds of formula 27 can be prepared by the methods described herein, such as by processes (a) to (g).
- the biphenyl derivatives of this invention are typically administered to a patient in the form of a pharmaceutical composition or formulation.
- Such pharmaceutical compositions may be administered to the patient by any acceptable route of administration including, but not limited to, inhaled, oral, nasal, topical (including transdermal) and parenteral modes of administration.
- any form ofthe compounds of this invention, (i.e., free base, pharmaceutically acceptable salt, solvate, etc.) that is suitable for the particular mode of administration can be used in the pharmaceutical compositions discussed herein.
- this invention is directed to a pharmaceutical composition comprising a pharmaceutically acceptable carrier or excipient and a therapeutically effective amount of a compound of formula I, or a pharmaceutically acceptable salt thereof.
- such pharmaceutical compositions may contain other therapeutic and/or formulating agents if desired.
- compositions of this invention typically contain a therapeutically effective amount of a compound ofthe present invention or a pharmaceutically acceptable salt thereof.
- pharmaceutical compositions will contain from about 0.01 to about 95% by weight ofthe active agent; including, from about 0.01 to about 30% by weight; such as from about 0.01 to about 10% by weight ofthe active agent.
- any conventional carrier or excipient may be used in the pharmaceutical compositions of this invention.
- the choice of a particular carrier or excipient, or combinations of carriers or exipients, will depend on the mode of administration being used to treat a particular patient or type of medical condition or disease state.
- the preparation of a suitable pharmaceutical composition for a particular mode of administration is well within the scope of those skilled in the pharmaceutical arts.
- the ingredients for such compositions are commercially available from, for example, Sigma, P.O. Box 14508, St. Louis, MO 63178.
- conventional formulation techniques are described in Remington: The Science and Practice of Pharmacy, 20 th Edition, Lippincott Williams & White, Baltimore, Maryland (2000); and H.C. Ansel et al., Pharmaceutical Dosage Forms and Drug Delivery Systems, 7 l Edition, Lippincott Williams & White, Baltimore, Maryland (1999).
- compositions which can serve as pharmaceutically acceptable carriers include, but are not limited to, the following: (1) sugars, such as lactose, glucose and sucrose; (2) starches, such as corn starch and potato starch; (3) cellulose, and its derivatives, such as sodium carboxymethyl cellulose, ethyl cellulose and cellulose acetate; (4) powdered tragacanth; (5) malt; (6) gelatin; (7) talc; (8) excipients, such as cocoa butter and suppository waxes; (9) oils, such as peanut oil, cottonseed oil, safflower oil, sesame oil, olive oil, corn oil and soybean oil; (10) glycols, such as propylene glycol; (11) polyols, such as glycerin, sorbitol, mannitol and polyethylene glycol; (12) esters, such as ethyl oleate and ethyl laurate; (13) agar; (14) buffering agents, such as magnesium magnesium
- compositions of this invention are typically prepared by thoroughly and intimately mixing or blending a compound ofthe invention with a pharmaceutically acceptable carrier and one or more optional ingredients. If necessary or desired, the resulting uniformly blended mixture can then be shaped or loaded into tablets, capsules, pills, canisters, cartridges, dispensers and the like using conventional procedures and equipment.
- the pharmaceutical compositions of this invention are suitable for inhaled administration. Suitable pharmaceutical compositions for inhaled administration will typically be in the form of an aerosol or a powder.
- Such compositions are generally administered using well-known delivery devices, such as a nebulizer inhaler, a metered-dose inhaler (MDI), a dry powder inhaler (DPI) or a similar delivery device.
- the pharmaceutical composition comprising the active agent is administered by inhalation using a nebulizer inhaler.
- a nebulizer inhaler typically produce a stream of high velocity air that causes the pharmaceutical composition comprising the active agent to spray as a mist that is carried into the patient's respiratory tract.
- the active agent when formulated for use in a nebulizer inhaler, is typically dissolved in a suitable carrier to form a solution.
- the active agent can be micronized and combined with a suitable carrier to form a suspension of micronized particles of respirable size, where micronized is typically defined as having about 90% or more ofthe particles with a diameter of less than about 10 ⁇ m.
- Suitable nebulizer devices are provided commercially, for example, by PARI GmbH (Stamberg, German). Other nebulizer devices include Respimat (Boehringer Ingelheim) and those disclosed, for example, in U.S. Patent No. 6,123,068 and WO 97/12687.
- a representative pharmaceutical composition for use in a nebulizer inhaler comprises an isotonic aqueous solution comprising from about 0.05 ⁇ g/mL to about 10 mg/mL of a compound of formula I or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- the pharmaceutical composition comprising the active agent is administered by inhalation using a dry powder inhaler.
- dry powder inhalers typically administer the active agent as a free-flowing powder that is dispersed in a patient's air-stream during inspiration.
- the active agent is typically formulated with a suitable excipient such as lactose or starch.
- a representative pharmaceutical composition for use in a dry powder inhaler comprises dry lactose having a particle size between about 1 ⁇ m and about 100 ⁇ m and micronized particles of a compound of formula I, or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- Such a dry powder formulation can be made, for example, by combining the lactose with the active agent and then dry blending the components.
- the active agent can be formulated without an excipient.
- the phannaceutical composition is then typically loaded into a dry powder dispenser, or into inhalation cartridges or capsules for use with a dry powder delivery device.
- Examples of dry powder inhaler delivery devices include Diskhaler (GlaxoSmithKline, Research Triangle Park, NC) (see, e.g., U.S. Patent No. 5,035,237); Diskus (GlaxoSmithKline) (see, e.g., U.S. Patent No. 6,378,519; Turbuhaler (AstraZeneca, Wilmington, DE) (see, e.g., U.S. Patent No. 4,524,769); Rotahaler (GlaxoSmithKline) (see, e.g., U.S. Patent No. 4,353,365) and Handihaler (Boehringer Ingelheim). Further examples of suitable DPI devices are described in U.S. Patent Nos.
- the pharmaceutical composition comprising the active agent is administered by inhalation using a metered- dose inhaler.
- metered-dose inhalers typically discharge a measured amount ofthe active agent or a pharmaceutically acceptable salt thereof using compressed propellant gas.
- pharmaceutical compositions administered using a metered-dose inhaler typically comprise a solution or suspension ofthe active agent in a liquefied propellant.
- Any suitable liquefied propellant may be employed including chlorofluorocarbons, such as CC1 3 F, and hydro fluoroalkanes (HFAs), such as 1,1,1,2-tetrafluoroethane (HFA 134a) and 1,1,1,2,3,3,3-heptafluoro-n-propane, (HFA 227).
- chlorofluorocarbons such as CC1 3 F
- HFAs hydro fluoroalkanes
- HFA 134a 1,1,1,2-tetrafluoroethane
- HFA 227 1,1,1,2,3,3,3-heptafluoro-n-propane
- co-solvents such as ethanol or pentane
- surfactants such as sorbitan trioleate, oleic acid, lecithin, and glycerin. See, for example, U.S.
- a representative pharmaceutical composition for use in a metered-dose inhaler comprises from about 0.01 % to about 5 % by weight of a compound of formula I, or a phannaceutically acceptable salt or solvate or stereoisomer thereof; from about 0 % to about 20 % by weight ethanol; and from about 0 % to about 5 % by weight surfactant; with the remainder being an HFA propellant.
- compositions are typically prepared by adding chilled or pressurized hydro fluoroalkane to a suitable container containing the active agent, ethanol (if present) and the surfactant (if present).
- the active agent is micronized and then combined with the propellant.
- the formulation is then loaded into an aerosol canister, which forms a portion of a metered-dose inhaler device. Examples of metered-dose inhaler devices developed specifically for use with HFA propellants are provided in U.S. Patent Nos. 6,006,745 and 6,143,277.
- a suspension formulation can be prepared by spray drying a coating of surfactant on micronized particles ofthe active agent. See, for example, WO 99/53901 and WO 00/61108.
- compositions of this invention are suitable for oral administration.
- suitable pharmaceutical compositions for oral administration may be in the form of capsules, tablets, pills, lozenges, cachets, dragees, powders, granules; or as a solution or a suspension in an aqueous or non-aqueous liquid; or as an oil-in-water or water-in-oil liquid emulsion; or as an elixir or syrup; and the like; each containing a predetermined amount of a compound ofthe present invention as an active ingredient.
- the pharmaceutical compositions of this invention When intended for oral administration in a solid dosage form (i.e., as capsules, tablets, pills and the like), the pharmaceutical compositions of this invention will typically comprise a compound ofthe present invention as the active ingredient and one or more pharmaceutically acceptable earners, such as sodium citrate or dicalcium phosphate.
- a pharmaceutically acceptable earners such as sodium citrate or dicalcium phosphate.
- such solid dosage forms may also comprise: (1) fillers or extenders, such as starches, lactose, sucrose, glucose, mannitol, and/or silicic acid; (2) binders, such as carboxymethylcellulose, alginates, gelatin, polyvinyl pyrrolidone, sucrose and/or acacia; (3) humectants, such as glycerol; (4) disintegrating agents, such as agar- agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates, and/or sodium carbonate; (5) solution retarding agents, such as paraffin; (6) absorption accelerators, such as quaternary ammonium compounds; (7) wetting agents, such as cetyl alcohol and/or glycerol monostearate; (8) absorbents, such as kaolin and/or bentonite clay; (9) lubricants, such as talc, calcium stearate, magnesium stearate, solid poly
- antioxidants include: (1) water-soluble antioxidants, such as ascorbic acid, cysteine hydrochloride, sodium bisulfate, sodium metabisulfate sodium sulfite and the like; (2) oil-soluble antioxidants, such as ascorbyl palmitate, butylated hydroxyanisole (BHA), butylated hydroxytoluene (BHT), lecithin, propyl gallate, alpha-tocopherol, and the like; and (3) metal-chelating agents, such as citric acid, ethylenediamine tetraacetic acid (EDTA), sorbitol, tartaric acid, phosphoric acid, and the like.
- water-soluble antioxidants such as ascorbic acid, cysteine hydrochloride, sodium bisulfate, sodium metabisulfate sodium sulfite and the like
- oil-soluble antioxidants such as ascorbyl palmitate, butylated hydroxyanisole (BHA), butylated hydroxytoluene (BHT), lecithin
- Coating agents for tablets, capsules, pills and like include those used for enteric coatings, such as cellulose acetate phthalate (CAP), polyvinyl acetate phthalate (PNAP), hydroxypropyl methylcellulose phthalate, methacrylic acid-methacrylic acid ester copolymers, cellulose acetate trimellitate (CAT), carboxymethyl ethyl cellulose (CMEC), hydroxypropyl methyl cellulose acetate succinate (HPMCAS), and the like.
- enteric coatings such as cellulose acetate phthalate (CAP), polyvinyl acetate phthalate (PNAP), hydroxypropyl methylcellulose phthalate, methacrylic acid-methacrylic acid ester copolymers, cellulose acetate trimellitate (CAT), carboxymethyl ethyl cellulose (CMEC), hydroxypropyl methyl cellulose acetate succinate (HPMCAS), and the like.
- the pharmaceutical compositions ofthe present invention may also be formulated to provide slow or controlled release ofthe active ingredient using, by way of example, hydroxypropyl methyl cellulose in varying proportions; or other polymer matrices, liposomes and/or microspheres.
- the pharmaceutical compositions of the present invention may optionally contain opacifying agents and may be formulated so that they release the active ingredient only, or preferentially, in a certain portion ofthe gastrointestinal tract, optionally, in a delayed manner. Examples of embedding compositions which can be used include polymeric substances and waxes.
- the active ingredient can also be in micro- encapsulated form, if appropriate, with one or more ofthe above-described excipients.
- Suitable liquid dosage forms for oral administration include, by way of illustration, pharmaceutically acceptable emulsions, microemulsions, solutions, suspensions, syrups and elixirs.
- Such liquid dosage forms typically comprise the active ingredient and an inert diluent, such as, for example, water or other solvents, solubilizing agents and emulsifiers, such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1, 3-butyl ene glycol, oils (esp., cottonseed, groundnut, corn, germ, olive, castor and sesame oils), glycerol, tetrahydrofuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan, and mixtures thereof.
- an inert diluent such as, for example, water or other solvents, solubilizing agents and emul
- Suspensions in addition to the active ingredient, may contain suspending agents such as, for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminium metahydroxide, bentonite, agar-agar and tragacanth, and mixtures thereof.
- the pharmaceutical compositions of this invention are preferably packaged in a unit dosage form.
- the term "unit dosage form" means a physically discrete unit suitable for dosing a patient, i.e., each unit containing a predetermined quantity of active agent calculated to produce the desired therapeutic effect either alone or in combination with one or more additional units.
- such unit dosage forms may be capsules, tablets, pills, and the like.
- the compounds of this invention can also be administered transdermally using known transdermal delivery systems and excipents.
- a compound of this invention can be admixed with permeation enhancers, such as propylene glycol, polyethylene glycolm monolaurate, azacycloalkan-2-ones and the like, and incorporated into a patch or similar delivery system. Additional excipients including gelling agents, emulsifiers and buffers, may be used in such transdermal compositions if desired.
- the pharmaceutical compositions of this invention may also contain other therapeutic agents that are co-administered with a compound of formula I, or pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- the phannaceutical compositions of this invention may further comprise one or more therapeutic agents selected from other bronchodilators (e.g., PDE 3 inhibitors,adenosine 2b modulators and ⁇ 2 adrenergic receptor agonists); anti-inflammatory agents (e.g. steroidal anti-inflammatory agents, such as corticosteroids; non-steroidal anti-inflammatory agents (NSAIDs), and PDE 4 inhibitors); other muscarinic receptor antagonists (i.e., antichlolinergic agents); antiinfective agents (e.g.
- other bronchodilators e.g., PDE 3 inhibitors,adenosine 2b modulators and ⁇ 2 adrenergic receptor agonists
- anti-inflammatory agents e.g. steroidal anti-inflammatory agents, such as corticosteroids; non-steroidal anti-inflammatory agents (NSAIDs), and PDE 4 inhibitors
- other muscarinic receptor antagonists i.e., antichloline
- the other therapeutic agents can be used in the form of pharmaceutically acceptable salts or solvates. Additionally, if appropriate, the other therapeutic agents can be used as optically pure stereoisomers.
- Representative ⁇ adrenergic receptor agonists that can be used in combination with, and in addition to, the compounds of this invention include, but are not limited to, salmeterol, salbutamol, formoterol, salmefamol, fenoterol, terbutaline, albuterol, isoetharine, metaproterenol, bitolterol, pirbuterol, levalbuterol and the like, or pharmaceutically acceptable salts thereof.
- ⁇ 2 adrenergic receptor agonists that can be used in combination with the compounds of this invention include, but are not limited to, 3-(4- ⁇ [6-( ⁇ (2R)-2-hydroxy-2-[4-hydroxy-3-(hydroxymethyl)-phenyl]ethyl ⁇ amino)- hexyl]oxy ⁇ butyl)benzenesulfonamide and 3-(-3- ⁇ [7-( ⁇ (2R)-2-hydroxy-2-[4-hydroxy-3- (hydroxymethyl)phenyl]ethyl ⁇ -amino)heptyl]oxy ⁇ -propyl)benzenesulfonamide and related compounds disclosed in WO 02/066422, published on August 29, 2002; 3-[3-(4- ⁇ [6- ([(2R)-2-hydroxy-2-[4-hydroxy-3-(hydroxymethyl)phenyl]ethyl ⁇ amino)hexyl]oxy ⁇ butyl)- phenyl]imidazolidine-2,4-d
- the ⁇ 2 -adrenoreceptor agonist When employed, the ⁇ 2 -adrenoreceptor agonist will be present in the pharmaceutical composition in a therapeutically effective amount. Typically, the ⁇ 2 -adrenoreceptor agonist will be present in an amount sufficient to provide from about 0.05 ⁇ g to about 500 ⁇ g per dose.
- steroidal anti-inflammatory agents that can be used in combination with the compounds of this invention include, but are not limited to, methyl prednisolone, prednisolone, dexamethasone, fluticasone propionate, 6,9-difluoro-17 -[(2- furanylcarbonyl)oxy] - 11 -hydroxy- 16-methyl-3-oxoandrosta- 1 ,4-diene- 17-carbothioic acid S-fluoromethyl ester, 6,9-difluoro- 11 -hydroxy- 16 -methyl-3-oxo-17 -propionyloxy- androsta-l,4-diene-l 7-carbothioic acid 5 , -(2-oxo-tetrahydrofuran-3S-yl) ester, beclomethasone esters (e.g.
- the steroidal anti- inflammatory agent will be present in the pharmaceutical composition in a therapeutically effective amount. Typically, the steroidal anti-inflammatory agent will be present in an amount sufficient to provide from about 0.05 ⁇ g to about 500 ⁇ g per dose.
- NSAIDs such as sodium cromoglycate; nedocromil sodium; phosphodiesterase (PDE) inhibitors (e.g. theophylline, PDE4 inhibitors or mixed PDE3/PDE4 inhibitors); leukotriene antagonists (e.g. monteleukast); inhibitors of leukotriene synthesis; iNOS inhibitors; protease inhibitors, such as tryptase and elastase inhibitors; beta-2 integrin antagonists and adenosine receptor agonists or antagonists (e.g. adenosine 2a agonists); cytokine antagonists (e.g. chemokine antagonists such as, an interleukin antibody ( IL antibody), specifically, an IL-4 therapy, an IL-13 therapy, or a combination thereof); or inhibitors of cytokine synthesis.
- IL antibody interleukin antibody
- IL-4 therapy an interleukin antibody
- IL-13 therapy an IL-13 therapy
- representative phosphodiesterase-4 (PDE4) inhibitors or mixed PDE3/PDE4 inhibitors that can be used in combination with the compounds of this invention include, but are not limited to cis 4-cyano-4-(3-cyclopentyloxy-4- methoxyphenyl)cyclohexan- 1 -carboxylic acid, 2-carbomethoxy-4-eyano-4-(3- cyclopropylmethoxy-4-difluoromethoxyphenyl)cyclohexan- 1 -one; c/s-[4-cyano-4-(3- cyclopropylmethoxy-4-difluoromethoxyphenyl)cyclohexan- 1 -ol] ; czs-4-cyano-4-[3- (cyclopentyloxy)-4-methoxyphenyl]cyclohexane-l -carboxylic acid and the like, or pharmaceutically acceptable salts thereof.
- PDE4 or mixed PDE4/PDE3 inhibitors include AWD-12-281 (elbion); NCS-613 (LNSERM); D-4418 (Chiroscience and Schering-Plough); CI-1018 or PD-168787 (Pfizer); benzodioxole compounds disclosed in WO99/16766 (Kyowa Hakko); K-34 (Kyowa Hakko); V-l 1294A (Napp); roflumilast (Byk-Gulden); pthalazinone compounds disclosed in WO99/47505 (Byk-Gulden); Pumafentrine (Byk-Gulden, now Altana); arofylline (Almirall- Prodesfarma); NM554/UM565 (Nernalis); T-440 (Tanabe Seiyaku); and T2585 (Tanabe Seiyaku).
- muscarinic antagonists i.e., anticholinergic agents
- muscarinic antagonists include, but are not limited to, atropine, atropine sulfate, atropine oxide, methylatropine nitrate, homatropine hydrobromide, hyoscyamine (d, l) hydrobromide, scopolamine hydrobromide, ipratropium bromide, oxitropium bromide, tiotropium bromide, methantheline, propantheline bromide, anisotropine methyl bromide, clidinium bromide, copynolate (Robinul), isopropamide iodide, mepenzolate bromide, tridihexethyl chloride (Pathilone), hexocyclium methylsulfate, cyclopentolate hydrochloride, tropicamide, trihexyphenidyl hydrochloride
- antihistamines i.e., Hi-receptor antagonists
- ethanolamines such as carbinoxamine maleate, clemastine fumarate, diphenylhydramine hydrochloride and dimenhydrinate
- ethylenediamines such as pyrilamine amleate, tripelennamine hydrochloride and tripelennamine citrate
- alkylamines such as chlorpheniramine and acrivastine
- piperazines such as hydroxyzine hydrochloride, hydroxyzine pamoate, cyclizine hydrochloride, cyclizine lactate, meclizine hydrochloride and cetirizine hydrochloride
- piperidines such as astemizole, levocabastine hydrochloride, loratadine or its descarboethoxy analogue, terfenadine and fexofenadine hydrochloride
- ethanolamines such as carbinoxamine maleate, clemastine fumarate
- Formulation Example A A dry powder for administration by inhalation is prepared as follows: Ingredients Amount
- the compound ofthe invention is micronized and then blended with lactose. This blended mixture is then loaded into a gelatin inhalation cartridge. The contents ofthe cartridge are administered using a powder inhaler.
- Formulation Example B A dry powder formulation for use in a dry powder inhalation device is prepared as follows:
- a pharmaceutical composition is prepared having a bulk formulation ratio of micronized compound ofthe invention to lactose of 1 :200.
- the composition is packed into a dry powder inhalation device capable of delivering between about 10 ⁇ g and about 100 ⁇ g ofthe compound ofthe invention per dose.
- Formulation Example C A dry powder for administration by inhalation in a metered dose inhaler is prepared as follows:
- a suspension containing 5 wt. % of a compound ofthe invention and 0.1 wt. % lecithin is prepared by dispersing 10 g ofthe compound ofthe invention as micronized particles with mean size less than 10 ⁇ m in a solution formed from 0.2 g of lecithin dissolved in 200 mL of demineralized water. The suspension is spray dried and the resulting material is micronized to particles having a mean diameter less than 1.5 ⁇ m. The particles are loaded into cartridges with pressurized 1,1,1,2-tetrafluoroethane.
- a pharmaceutical composition for use in a metered dose inhaler is prepared as follows: Representative Procedure: A suspension containing 5 % compound ofthe invention, 0.5 % lecithin, and 0.5 % trehalose is prepared by dispersing 5 g of active ingredient as micronized particles with mean size less than 10 m in a colloidal solution formed from 0.5 g of trehalose and 0.5 g of lecithin dissolved in 100 mL of demineralized water. The suspension is spray dried and the resulting material is micronized to particles having a mean diameter less than 1.5 ⁇ m. The particles are loaded into canisters with pressurized 1,1,1 ,2-tetrafluoroethane.
- a pharmaceutical composition for use in a nebulizer inhaler is prepared as follows: Representative Procedure: An aqueous aerosol formulation for use in a nebulizer is prepared by dissolving 0.1 mg ofthe compound ofthe invention in 1 mL of a 0.9 % sodium chloride solution acidified with citric acid. The mixture is stined and sonicated until the active ingredient is dissolved. The pH ofthe solution is adjusted to a value in the range of from 3 to 8 by the slow addition of NaOH.
- Hard gelatin capsules for oral administration are prepared as follows:
- Neegum k (Nanderbilt Co.) 1.0 g
- An injectable formulation is prepared as follows: Ingredients Amount
- the biphenyl derivatives of this invention possess both ⁇ 2 adrenergic receptor agonist and muscarinic receptor antagonist activity and therefore, such compounds are useful for treating medical conditions mediated by ⁇ 2 adrenergic receptors or muscarinic receptors, i.e., medical conditions that are ameliorated by treatment with a ⁇ 2 adrenergic receptor agonist or a muscarinic receptor antagonist.
- medical conditions include, by way of example, pulmonary disorders or diseases associated with reversible airway obstruction, such as chronic obstructive pulmonary disease (e.g., chronic and whez bronchitis and emphysema), asthma, pulmonary fibrosis and the like.
- Other conditions which may be treated include premature labor, depression, congestive heart failure, skin diseases (e.g., inflammatory, allergic, psoriatic and proliferative skin diseases, conditions where lowering peptic acidity is desirable (e.g., peptic and gastric ulceration) and muscle wasting disease.
- skin diseases e.g., inflammatory, allergic, psoriatic and proliferative skin diseases, conditions where lowering peptic acidity is desirable (e.g., peptic and gastric ulceration) and muscle wasting disease.
- this invention is directed to a method for treating a pulmonary disorder, the method comprising administering to a patient in need of treatment a therapeutically effective amount of a compound of formula I or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- the compounds of this invention When used to treat a pulmonary disorder, the compounds of this invention will typically be administered by inhalation in multiple doses per day, in a single daily dose or a single weekly dose.
- the dose for treating a pulmonary disorder will range from about 10 ⁇ g/day to about 200 ⁇ g/day.
- this invention is directed to a method of providing bronchodilation in a patient, the method comprising administering to a patient requiring bronchodilation a therapeutically effective amount of a compound of formula I or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- the dose for providing bronchodilation will range from about 10 ⁇ g/day to about 200 ⁇ g/day.
- this invention is directed to a method of treating chronic obstructive pulmonary disease or asthma, the method comprising administering to a patient in need of treatment a therapeutically effective amount of a compound of formula I or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- the compounds of this invention When used to treat a COPD or asthma, the compounds of this invention will typically be administered by inhalation in multiple doses per day or in a single daily dose. Generally, the dose for treating COPD or asthma will range from about 10 ⁇ g/day to about 200 ⁇ g/day.
- COPD includes chronic obstructive bronchitis and emphysema (see, for example, Barnes, Chronic Obstructive Pulmonary Disease, NEngl JMed 2000: 343:269-78).
- the compounds of this invention are optionally administered in combination with other therapeutic agents.
- a steroidal anti-inflammatory agent e.g. a corticosteroid
- the pharmaceutical compositions of this invention can provide triple therapy, i.e., ⁇ 2 adrenergic receptor agonist, muscarinic receptor antagonist and anti- inflammatory activity, using only two active components. Since pharmaceutical compositions containing two active components are typically easier to formulate compared to compositions containing three active components, such two component compositions provide a significant advantage over compositions containing three active components. Accordingly, in a particular embodiment, the pharmaceutical compositions and methods of this invention further comprise a therapeutically effective amount of a steroidal anti- inflammatory agent.
- compounds of this invention exhibit both muscarinic receptor antagonist and ⁇ 2 adrenergic receptor agonist activity. Accordingly, among other properties, compounds of particular interest are those that demonstrate an inhibitory constant K; value for binding at the M 3 muscarinic receptor and an EC 50 value for ⁇ 2 adrenergic receptor agonist activity of less than about 100 nM; particularly less than 10 nM.
- compounds of special interest include those having muscarinic activity, expressed in terms ofthe inhibitory constant Kj for binding at the M 3 muscarinic receptor, that is about equal to the compound's ⁇ 2 adrenergic agonist activity, expressed in terms ofthe half maximal effective concentration EC 50 , as determined in the in vitro assays described herein, or in similar assays.
- compounds of particular interest are those having a ratio of the inhibitory constant Kj for the M 3 muscarinic receptor to the EC 50 for the ⁇ 2 adrenergic receptor ranging from about 30:1 to about 1:30; including about 20:1 to about 1:20; such as about 10:1 to about 1:10.
- the present invention also provides a method for treating a pulmonary disorder, the method comprising administering to a patient in need of treatment a therapeutically effective amount of a compound having both muscarinic receptor antagonist and ⁇ 2 adrenergic receptor agonist activity.
- the compound administered has an inhibitory constant K; for the M 3 muscarinic receptor that is less than about 100 nM and a half maximal effective concentration EC 50 for agonism at the ⁇ 2 adrenergic receptor that is less than about 100 nM.
- the method for treating a pulmonary disorder comprises administering a therapeutically effective amount of a compound for which the ratio ofthe inhibitory constant K; for the M 3 muscarinic receptor to the EC 50 for agonism ofthe ⁇ 2 adrenergic receptor is between about 30: 1 and about 1 :30.
- compounds of this invention possess both ⁇ 2 adrenergic agonist activity and muscarinic receptor antagonist activity, such compounds are also useful as research tools for investigating or studying biological systems or samples having ⁇ 2 adrenergic receptors or muscarinic receptors, or for discovering new compounds having both ⁇ 2 adrenergic agonist activity and muscarinic receptor antagonist activity.
- biological systems or samples may comprise ⁇ 2 adrenergic receptors and/or muscarinic receptors. Any suitable biological system or sample having ⁇ 2 adrenergic and/or muscarinic receptors may be employed in such studies which may be conducted either in vitro or in vivo.
- Representative biological systems or samples suitable for such studies include, but are not limited to, cells, cellular extracts, plasma membranes, tissue samples, mammals (such as mice, rats, guinea pigs, rabbits, dogs, pigs, etc.), and the like.
- a biological system or sample comprising a ⁇ 2 adrenergic receptor or a muscarinic receptor is contacted with a ⁇ 2 adrenergic receptor-agonizing or muscarinic receptor-antagonizing amount of a compound of this invention.
- the effects are then determined using conventional procedures and equipment, such as radioligand binding assays and functional assays.
- Such functional assays include ligand-mediated changes in intracellular cyclic adenosine monophosphate (cAMP), ligand-mediated changes in activity ofthe enzyme adenylyl cyclase (which synthesizes cAMP), ligand- mediated changes in incorporation of guanosine 5'-O-( -thio)triphosphate ([ 35 S]GTP S) into isolated membranes via receptor catalyzed exchange of [ 35 S]GTP S for GDP, ligand- mediated changes in free intracellular calcium ions (measured, for example, with a fluorescence-linked imaging plate reader or FLIPR ® from Molecular Devices, Inc.).
- cAMP cyclic adenosine monophosphate
- adenylyl cyclase which synthesizes cAMP
- ligand- mediated changes in incorporation of guanosine 5'-O-( -thio)triphosphate [ 35 S]GTP S
- a compound of this invention will agonize or cause activation of a ⁇ 2 adrenergic receptor and antagonize or decrease the activation of muscarinic receptors in any ofthe functional assays listed above, or assays of a similar nature.
- the amount of compound used in these studies will typically range from about 0.1 nanomolar to about 100 nanomolar.
- the compounds of this invention can be used as research tools for discovering new compounds that have both a ⁇ 2 adrenergic receptor agonist and muscarinic receptor antagonist activity.
- a ⁇ 2 adrenergic receptor and muscarinic receptor binding data for example, as determined by in vitro radioligand displacement assays
- a compound of this invention is compared to the ⁇ 2 adrenergic receptor and muscarinic receptor binding data for a compound of this invention to identify those test compounds that have about equal or superior ⁇ 2 adrenergic receptor and/or muscarinic receptor binding, if any.
- This aspect of the invention includes, as separate embodiments, both the generation of comparison data (using the appropriate assays) and the analysis ofthe test data to identify test compounds of interest.
- compounds of this invention may possess either weak muscarinic receptor antagonist activity or weak ⁇ 2 adrenergic receptor agonist activity. In these cases, those of ordinary skill in the art will recognize that such compounds still have utility as primarily either a ⁇ 2 adrenergic receptor agonist or a muscarinic receptor antagonist, respectively.
- HPLC 10-70 data was obtained with a flow rate of 0.5 mL/minute of 10%-70% B over 6 minutes.
- Mobile phase A was 2 % - 98 % - 0.1% AC ⁇ -H 2 O-TFA; and mobile phase B was 90 % - 10 % - 0.1 % ACN-H 2 O-TFA.
- HPLC 5-35 data and HPLC 10-90 data were obtained with a 5 minute gradient.
- LCMS Liquid chromatography mass spectrometry
- N-1,1 '-biphenyl-2-yl-iV-4-piperidinylurea was synthesized by heating together biphenyl-2-isocyanate (50 g, 256 mmol) and 4-amino-N-benzylpiperidine (51.1 g, 269 mmol) at 70 °C for 12 h (the reaction was monitored by LCMS). The reaction mixture was cooled to 50 °C and ethanol (500 mL) added, followed by slow addition of 6M hydrochloric acid (95 mL). The reaction mixture was cooled to room temperature.
- step (b) 5-Acetyl-8-hydroxy-lH-quinolin-2-one
- a sluny of aluminum chloride (85.7 g, 640 mmol) in 1 ,2-dichloroethane (280 mL) was cooled in ice, and the product of step (a) (56.8 g, 280 mmol) was added.
- the mixture was warmed to room temperature and then heated at 85°C. After 30 min, acetyl chloride (1.5 mL, 21 mmol) was added and the mixture was heated an additional 60 min.
- the reaction mixture was then cooled and added to IN hydrochloric acid (3 L) at 0 °C with good stirring.
- step (b) To the product of step (b) (37.7 g, 186 mmol) was added N,N-dimethylformamide (200 mL) and potassium carbonate (34.5 g, 250 mmol) followed by benzyl bromide (31.8 g, 186 mmol). The mixture was stined at room temperature for 2.25 hour and then poured into saturated sodium chloride (3.5 L) at 0 °C and stined for 1 hour. The product was collected and dried on a Buchner funnel for 1 hour, and the resulting solids were dissolved in dichloromethane (2 L) and this mixture was dried over sodium sulfate.
- N,N-dimethylformamide 200 mL
- potassium carbonate 34.5 g, 250 mmol
- benzyl bromide 31.8 g, 186 mmol
- step (d) 8-Benzyloxy-5-(2,2-dihydroxyacetyl)-lH-quinolin-2-one
- DMSO 60 mL
- a 48% w/w hydrobromic acid solution 11.8 mL, 102.3 mmol
- the mixture was warmed to 60 °C for 16 h then allowed to cool to room temperature.
- Water (100 mL) was added and the resulting slurry stirred at room temperature for 0.5 h before being cooled to 0°C.
- the product was collected on a Buchner funnel then dried under reduced pressure to give 8-benzyloxy-5-(2,2-dihydroxyacetyl)-lH-quinolin-2-one (12.2 g) as a solid.
- Biphenyl-2-isocyanate 97.5 g, 521 mmol
- 4-hydroxy-l-benzylpiperidine 105 g, 549 mmol
- the reaction mixture was then cooled to 50 °C and ethanol (1 L) was added, and then 6M hydrochloric acid (191 mL) was added slowly.
- reaction mixture was then cooled to ambient temperature and ammonium formate (98.5 g, 1.56 mol) was added and nitrogen gas was bubbled through the solution vigorously for 20 min. Palladium (10 wt. % (dry basis) on activated carbon) (20 g) was then added. The reaction mixture was heated at 40 °C for 12 h and then filtered through a pad of Celite. The solvent was then removed under reduced pressure and IM hydrochloric acid (40 mL) was added to the crude residue. Sodium hydroxide (ION) was then added to adjust the pH to 12.
- Trifluoroacetic acid (11 mL) was added to a solution ofthe product of Preparation 10 (7.2 g, 11.3 mmol) in dichloromethane (56 mL). After 2 h, LCMS analysis showed that the reaction was completed. The reaction mixture was then concentrated to dryness and diluted with ethyl acetate (75 mL). Sodium hydroxide (IN) was then added until the pH of the mixture reached 14. The organic phase was then collected and washed with saturated sodium bicarbonate (2 x 50 mL) and brine (50 mL). The organic phase was then dried over magnesium sulfate and concentrated to provide the title compound (5.5 g). MS m/z: [M + H + ] calcd for C 27 H 39 N 3 O 2 438.30; found 439.
- step (b) To the product of step (b) (70.2 g, 189 mmol) was added NN-dimethylformamide (260 mL) and this mixture was cooled in an ice bath under nitrogen. 2,6-Lutidine (40.3 g, 376 mmol) was added over 5 min and then tert-butyldimethylsilyl trifluoromethanesulfonate (99.8 g, 378 mmol) was added slowly while maintaining the temperature below 20°C. The mixture was allowed to Trust to room temperature for 45 min.
- the product of Preparation 13 (3.9 g, 8.17 mmol) was added to a solution ofthe product of Preparation 11 (5.0 g, 11.4 mmol) in THF (20 mL), followed by sodium bicarbonate (2.0 g, 24.5 mmol) and sodium iodide (1.8 g, 12.2 mmol).
- the reaction mixture was heated to 80 °C for 72 h.
- the reaction mixture was then cooled, diluted with dichloromethane (20 mL) and the organic phase was washed with saturated sodium bicarbonate (2 x 50 mL) and brine (50 mL).
- the organic phase was then dried (magnesium sulfate) and concentrated to give 6.5 g of a crude product.
- the crude product was purified by chromatography on silica gel eluting with 3% methanol in dichloromethane to provide the title compound (1.4 g, 21% yield).
- Triethylamine hydrogen fluoride (376 ⁇ L, 2.3 mmol) was added to a solution ofthe product of Preparation 14 (1.3 g, 1.5 mmol) in THF (8 mL) and the reaction mixture was stirred at ambient temperature. After 5 h, the reaction was complete as determined by LCMS analysis. The reaction mixture was then quenched with IN NaOH until the pH was 14 and then diluted with ethyl acetate (20 mL) and washed with IN NaOH (20 mL) and brine (20 mL). The organic phase was then separated, dried over magnesium sulfate, and concentrated to yield the title compound (1.1 g).
- TEMPO 2,2,6,6-Tetramethyl-l-piperidinyloxy free radical
- the residue was purified by silica gel chromatography (15g silica/1 g crude) using 5% MeOH in DCM/0.5% NH 4 OH (10 x 150 mL), 8% MeOH in DCM/0.5% NH 4 OH (10 x 150 mL) and 10% MeOH in DCM/0.5% NH 4 OH (10 x 150 mL). The appropriate fractions were combined and the solvent was removed under reduced pressure while maintaining the temperature ⁇ 35 °C to give the title intermediate (4.05 g , 97% purity).
- the reaction mixture was then diluted with a mixture of isopropyl acetate (53 mL) and hexanes (27 mL) and transfened to a separatory funnel.
- the organic layer was washed twice with a mixture of water (27 mL) and saturated aqueous sodium chloride (27 mL) followed by a final wash with saturated aqueous sodium chloride (27 mL).
- the organic layer was dried over sodium sulfate.
- Silica gel (23.6 g) and hexanes (27 mL) were added and the suspension was stirred for 10 min. The solids were removed by filtration and the filtrate concentrated under vacuum.
- Methyl 3-bromopropionate (553 ⁇ L, 5.07 mmol) was added to a stined solution of the product of Preparation 8 (1.00 g, 3.38 mmol) and DIPEA (1.76 mL, 10.1 mmol) in acetonitrile (34 mL) at 50 °C and the reaction mixture was heated at 50 °C overnight. The solvent was then removed under reduced pressure and the residue was dissolved in dichloromethane (30 mL). The resulting solution was washed with saturated aqueous sodium bicarbonate solution (10 mL), dried (magnesium sulfate) and the solvent was removed under reduced pressure. The crude residue was purified by column chromatography (5-10% MeOH/DCM) to give the title compound (905 mg, 70%).
- palladium (10 wt. % (dry basis) on activated carbon) 81 mg
- the title compound can be prepared as follows: (a) 5-ChloropentanaI
- TEMPO 2,2,6,6-Tetramethyl-l-piperidinyloxy free radical
- the layers ofthe mixture were then separated, and the aqueous layer was extracted with dichloromethane (1 x 50 mL). The combined dichloromethane layers were then washed with water (1 x 50 mL), dried (MgSO 4 ), filtered and concentrated under reduced pressure to afford the title compound (53 g). The product was distilled at 65°C/8 ton to afford the title compound (31.16 g) as an orange oil (GC purity was 70 to 80 %). The product was further purified by adding the crude material (4 g) to a mixture of ethanol (920 mL), ethyl acetate (12 mL) and water (4 mL).
- Aqueous saturated sodium bicarbonate solution (200 mL) was then added slowly (gas evolution) and stirring was continued for 15 min. The pH of the solution was then adjusted with solid sodium carbonate to a pH of about 9 and the layers were separated. The organic layer was washed with aqueous 5% sodium chloride solution (200 mL), dried (MgSO 4 ), filtered and concentrated under reduced pressure to afford the title compound (53 g).
- step (c) 5-[(R)-2-[(5-N ⁇ V-Diformylaminopentyl)benzylamino]-l-(tert- butyldimethyl-silanyloxy)ethyl]-8-benzyloxy-lH-quinolin-2-one
- sodium diformylamide 6.1 g, 64.2 mmol
- sodium iodide 2.13 g, 14.3 mmol
- step (e) A mixture ofthe product of step (e) (3.68 g, 10 mmol) and N,N-dimethylformamide (50 mL) was heated at 60 °C until the solid completely dissolved and then cooled to room temperature.
- the product of step (d) (6 g, 10 mmol) and diisopropylethylamine (3.5 mL) was added and the reaction mixture was cooled to 0°C.
- PyBOP (6.25 g, 12 mmol) was added in one portion and the reaction mixture was stined at 0 °C to room temperature for 2 hours.
- the reaction mixture was then poured into cold water (500 mL) with stirring and the pH ofthe resulting mixture was adjusted to about 2 using aqueous 1 M hydrochloric acid. This mixture was stined for 15 min and then filtered to collect the solid, which was washed with water (100 mL) and dried to afford the title compound (8.7 g, HPLC purity >95%) as an
- step (f) can be deprotected using essentially the same procedures as those described in Preparation 24 and Example 6 to afford the title compound.
- DMSO 71 mL,l mol
- DIPEA 87.1 mL, 0.5 mol
- the reaction mixture was stined at -10 °C for 15 min and then sulfur trioxide pyridine complex (79.6 g, 0.5 mol) was added and the resulting mixture was stined for 1 hour.
- the reaction mixture was quenched with addition of IM hydrochloric acid (200 mL).
- the organic layer was separated and washed with saturated aqueous sodium bicarbonate (100 mL), brine (100 mL), dried (potassium carbonate) and solvent removed under reduced pressure to give the title compound (20.7 g, ⁇ 100% yield).
- N-tert-butoxycarbonyl-l,5-diaminopentane (1.04 g, 5.12 mmol) was added to a solution ofthe product of Preparation 13 (1.00 g, 2.05 mmol) in dimethyl sulfoxide (2 mL).
- the solution was stined at 75 °C for 12 hours, at which time LCMS analysis showed that the reaction was complete.
- the reaction mixture was then concentrated under vacuum to dryness. To the residue was added dichloromethane (2 mL) and trifluoroacetic acid (1 mL) was then added. The solution was stined at room temperature for about 3 hours, at which time MS analysis showed that the reaction was complete.
- Triethylamine trihydrofluoride (0.16 mL, 1.02 mmol) was added to a solution of the product of Preparation 41 (597 mg, 0.68 mmol) in tetrahydrofuran (3.4 mL) and this mixture was stined at room temperature for about 12 hours, at which time the reaction was determined to be completed by MS analysis.
- pH 7.0
- the diethyl ether phase was washed once with brine, dried over Na 2 SO 4 , filtered and concentrated under reduced pressure to give a light orange oil.
- the oil was dissolved in a minimum volume of ethyl acetate, diluted with hexanes, and to give the title compound as a crystalline solid.
- step (b) To the product of step (b) (23.4 g, 0.113 mol) in 600 mL of THF at -78 °C was added 135 mL of 1.0 M sodium hexamethyldisilazane in THF (Sigma- Aldrich). After 1 hour, trimethylsilyl chloride (15.8 mL, 0.124 mol) was added. After another 30 minutes, bromine (5.82 mL, 0.113 mol) was added. After 10 minutes, the reaction was quenched by diluting the reaction mixture with diethyl ether and pouring it onto 500 mL of 5% aqueous Na 2 SO 3 premixed with 500 mL of 5% aqueous NaHCO 3 . The phases were separated and the organic phase was washed with brine, dried over Na 2 SO 4 , filtered and concentrated under reduced pressure to give the title compound as an oil that solidified upon storage in the freezer.
- step (d) (R)-2-Bromo-l-(2,2-dimethyl-4H-benzo[l,3]dioxin-6-yI)ethanol
- step (c)(1) (0.97 g, 3.5 mmol).
- the solution was cooled to between -20 °C and -10 °C and BH 3 -THF (35 mL, 35 mmol) diluted with 50 mL THF was added dropwise via a dropping funnel. After the addition was complete, the reaction mixture was allowed to warm to ambient temperature.
- reaction mixture was quenched by slow addition of 50 mL of methanol and then concentrated to a thick oil.
- the oil was purified by silica gel chromatography eluted with 1 :2 ethyl acetate/hexanes. The fractions were combined and concentrated to give the title compound as an off-white solid.
- step (d) To the product of step (d) (10 g, 34.8 mmol) and imidazole (4.7 g, 69.7 mmol) dissolved in 100 mL DMF was added tert-butyldimethylsilyl chloride (5.78 g, 38.3 mmol). The reaction mixture was stined for 18 hours. The reaction mixture was then partitioned between 200 mL of saturated sodium chloride and 200 mL of diethyl ether. The aqueous layer was extracted with 200 mL of diethyl ether. The organic layers were then combined, washed with saturated sodium chloride (3 x 100 mL), dried over MgSO and concentrated. The product was purified by silica gel chromatography, eluting with hexanes followed by 5% ethyl acetate in hexanes. The desired fractions were combined and concentrated to give the title compound as an oil.
- Triethylamine trihydrofluoride (342 ⁇ L, 2.10 mmol) was added to a stirred solution ofthe Product of Preparation 44 (798 mg, 1.05 mmol) in dichloromethane (10.5 mL) at ambient temperature. The reaction mixture was stined for 24 h and it was then diluted with dichloromethane (20 mL) and washed with saturated aqueous sodium bicarbonate (15 mL). The organic layer was dried (magnesium sulfate) and the solvent was removed under reduced pressure. The crude title compound was isolated as an oil (659 mg, 1.02 mmol), which was used in the next step without further purification. MS m/z: [M + H + ] calcd for C 39 H 53 N 3 O 5 644.4; found 644.8.
- step (a) The product of step (a) (10.0 g, 35.2 mmol) was dissolved in chloroform (250 mL) in a 500 mL flask under a nitrogen atmosphere. Bromine (1.63 mL, 31.7 mmol) dissolved in chloroform (50 mL) was added using a dropping funnel over 30 min. The reaction mixture was stined for 2.5 h and then concentrated to give a solid. The solid was dissolved in toluene (150 mL) with some gentle heat, followed by the addition of ethyl ether (150 mL) to yield the title compound as a crystalline solid (55% yield).
- the product of Preparation 47 (371 mg, 1.00 mmol) was added to a solution ofthe product of Preparation 46 (448 mg, 0.85 mmol) in dimethyl sulfoxide (4.5 mL) followed by the addition of potassium carbonate (234 mg, 1.7 mmol).
- the reaction mixture was stined at 40 °C for 6 h, at which time the product of Preparation 46 was no longer observed by HPLC analysis.
- the reaction mixture was cooled to ambient temperature and filtered, and then diluted with ethanol (4 mL). Sodium borohydride (63 mg, 1.7 mmol) was added to the reaction mixture and the reaction was stined at ambient temperature for 24 h.
- reaction mixture was quenched with 0.5 M ammonium chloride (5 mL) and extracted into ethyl acetate (2 x 10 mL). The combined organic layers were washed with saturated sodium bicarbonate (10 mL) and then with brine (5 mL). The organic layer was dried over magnesium sulfate and the solvent was removed under reduced pressure. The crude residue was purified by chromatography on silica gel (3% methanol in chloroform) to give the title compound.
- the crude product was purified by flash chromatography (5-10% MeOH/DCM) and then dissolved in a trifluoroacetic acid/DCM mixture (1 mL/5 mL) and stirred at room temperature for lh. The solvent was removed under reduced pressure. The residue was dissolved in dichloromethane (20 mL) and washed with IM sodium hydroxide (10 mL), dried (magnesium sulfate) and the solvent reduced to yield the title compound (167 mg, 39% yield).
- step (a) To the product of step (a) (1.71 g, 7.2 mmol) was added a solution of 4 M hydrochloric acid in dioxane (9 mL, 36 mmol) and the resulting mixture was stined at room temperature for 1 h. The reaction mixture was then concentrated and the residue was diluted with diethyl ether (50 mL) and filtered to provide the title compound as a white solid (1.09 g).
- step (a) To the product from step (a) was added acetonitrile (20 mL), diisopropylethylamine (0.5 mL, 2.8 mmol) and the product of Preparation 8 (626 mg, 2.11 mmol). The reaction mixture was heated to 50 °C for 20 h and then cooled to room temperature and the solvent was removed under reduced pressure. The residue was purified by silica gel chromatography (5% MeOH/DCM with 0.6% NH 3 (aq)) to afford the title compound (450 mg, 44% yield). MS m/z (MH + ) 479.6; R 4.15 min (10-70% ACN: H 2 O, reverse phase HPLC).
Abstract
Description




































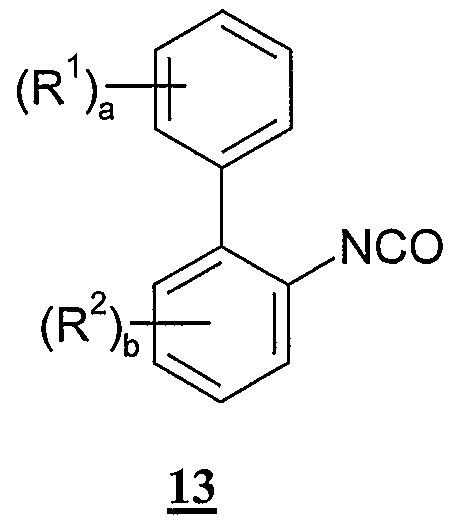





























Claims

























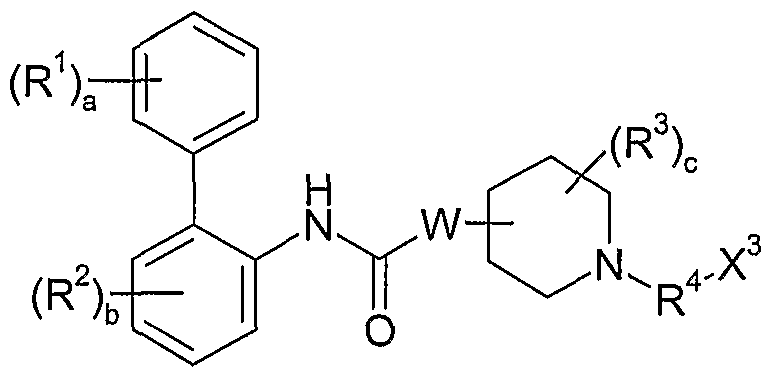


Priority Applications (18)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006503604A JP4555283B2 (en) | 2003-02-14 | 2004-02-13 | Biphenyl derivatives having β2 adrenergic receptor agonist activity and muscarinic receptor antagonist activity |
| DK04711253.7T DK1615889T3 (en) | 2003-02-14 | 2004-02-13 | Biphenyl derivatives with beta2-adrenergic receptor agonist and muscarinic receptor antagonist activity |
| AU2004213411A AU2004213411B2 (en) | 2003-02-14 | 2004-02-13 | Biphenyl derivatives having beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
| KR1020057014929A KR101223991B1 (en) | 2003-02-14 | 2004-02-13 | Biphenyl derivatives having beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
| MXPA05008528A MXPA05008528A (en) | 2003-02-14 | 2004-02-13 | Biphenyl derivatives. |
| BRPI0407508A BRPI0407508B8 (en) | 2003-02-14 | 2004-02-13 | biphenyl derivative compounds, pharmaceutical composition and use thereof |
| AT04711253T ATE482934T1 (en) | 2003-02-14 | 2004-02-13 | BIPHENYL DERIVATIVES WITH BETA2 ADRENERG RECEPTOR AGONISTIC AND MUSCARIN RECEPTOR ANTAGONISTIC ACTIVITY |
| CA2515777A CA2515777C (en) | 2003-02-14 | 2004-02-13 | Biphenyl derivatives |
| DE602004029347T DE602004029347D1 (en) | 2003-02-14 | 2004-02-13 | BIPHENYL DERIVATIVES WITH BETA2 ADRENERG RECEPTOR AGONISTIC AND MUSCARIN RECEPTOR OF ANTAGONISTIC ACTIVITY |
| SI200431566T SI1615889T1 (en) | 2003-02-14 | 2004-02-13 | Biphenyl derivatives having beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
| NZ541579A NZ541579A (en) | 2003-02-14 | 2004-02-13 | Biphenyl derivatives having beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
| EP04711253A EP1615889B1 (en) | 2003-02-14 | 2004-02-13 | Biphenyl derivatives having beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
| KR1020117010078A KR101224497B1 (en) | 2003-02-14 | 2004-02-13 | Biphenyl derivatives having beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
| IL169922A IL169922A (en) | 2003-02-14 | 2005-07-27 | Biphenyl derivatives having beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
| IS7964A IS2847B (en) | 2003-02-14 | 2005-07-28 | Biphenyl derivatives with beta2 adrenergic receptor antagonist and muscarinic receptor antagonist activity |
| NO20054206A NO331947B1 (en) | 2003-02-14 | 2005-09-09 | Biphenyl derivative, its use and pharmaceutical composition comprising the compound. |
| HK06106230.1A HK1086266A1 (en) | 2003-02-14 | 2006-05-29 | Biphenyl derivatives having beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
| AU2010219338A AU2010219338B2 (en) | 2003-02-14 | 2010-09-08 | Biphenyl derivatives having beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US44784303P | 2003-02-14 | 2003-02-14 | |
| US60/447,843 | 2003-02-14 | ||
| US46703503P | 2003-05-01 | 2003-05-01 | |
| US60/467,035 | 2003-05-01 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2004074246A2 true WO2004074246A2 (en) | 2004-09-02 |
| WO2004074246A3 WO2004074246A3 (en) | 2004-11-18 |
Family
ID=32912274
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2004/004449 WO2004074246A2 (en) | 2003-02-14 | 2004-02-13 | Biphenyl derivatives having beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
| PCT/US2004/004224 WO2004074276A1 (en) | 2003-02-14 | 2004-02-13 | BIPHENYL DERIVATIVES HAVING β2 ADRENERGIC RECEPTOR AGONIST AND MUSCARINIC RECEPTOR ANTAGONIST ACTIVITY |
| PCT/US2004/004273 WO2004074812A2 (en) | 2003-02-14 | 2004-02-13 | Library of biphenyl derivatives useful for identifying compounds having both beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2004/004224 WO2004074276A1 (en) | 2003-02-14 | 2004-02-13 | BIPHENYL DERIVATIVES HAVING β2 ADRENERGIC RECEPTOR AGONIST AND MUSCARINIC RECEPTOR ANTAGONIST ACTIVITY |
| PCT/US2004/004273 WO2004074812A2 (en) | 2003-02-14 | 2004-02-13 | Library of biphenyl derivatives useful for identifying compounds having both beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
Country Status (28)
| Country | Link |
|---|---|
| US (15) | US20040209915A1 (en) |
| EP (5) | EP1615889B1 (en) |
| JP (6) | JP4555283B2 (en) |
| KR (3) | KR101223991B1 (en) |
| AR (1) | AR043176A1 (en) |
| AT (1) | ATE482934T1 (en) |
| AU (2) | AU2004213411B2 (en) |
| BR (1) | BRPI0407508B8 (en) |
| CA (1) | CA2515777C (en) |
| CY (2) | CY1111276T1 (en) |
| DE (1) | DE602004029347D1 (en) |
| DK (2) | DK1615889T3 (en) |
| ES (2) | ES2566155T3 (en) |
| HK (3) | HK1086266A1 (en) |
| HU (1) | HUE027380T2 (en) |
| IL (1) | IL169922A (en) |
| IS (1) | IS2847B (en) |
| MX (1) | MXPA05008528A (en) |
| MY (1) | MY148487A (en) |
| NO (2) | NO331947B1 (en) |
| NZ (1) | NZ541579A (en) |
| PE (1) | PE20040950A1 (en) |
| PL (1) | PL216397B1 (en) |
| PT (1) | PT1615889E (en) |
| RU (1) | RU2330841C2 (en) |
| SI (2) | SI2246345T1 (en) |
| TW (1) | TWI331995B (en) |
| WO (3) | WO2004074246A2 (en) |
Cited By (89)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006023460A3 (en) * | 2004-08-16 | 2006-04-06 | Theravance Inc | COMPOUNDS HAVING β2 ADRENERGIC RECEPTOR AGONIST AND MUSCARINIC RECEPTOR ANTAGONIST ACTIVITY |
| WO2006056471A1 (en) | 2004-11-29 | 2006-06-01 | Novartis Ag | 5-hydroxy-benzothiazole derivatives having beta-2-adrenorecptor agonist activity |
| WO2006050239A3 (en) * | 2004-10-29 | 2006-10-12 | Glaxo Group Ltd | Muscarinic acetylcholine receptor antagonists |
| WO2006108643A2 (en) | 2005-04-14 | 2006-10-19 | Novartis Ag | Organic compounds |
| WO2007090859A1 (en) * | 2006-02-10 | 2007-08-16 | Glaxo Group Limited | Succinic acid salt of biphenyl-2-ylcarbamic acid 1-[2-(2-chloro-4-{ [ (r)-2-hydroxy-2- (8-hydroxy-2-oxo-1, 2-dihydroquinolin-5-yl) eth ylaminoimethyl] } -5-methoxyphenylcarbamoyl) ethyl] piperidin-4-yl ester and its use for the treatment of pulmonary disorders |
| WO2007121920A2 (en) | 2006-04-21 | 2007-11-01 | Novartis Ag | Purine derivatives for use as adenosin a2a receptor agonists |
| WO2007127297A2 (en) * | 2006-04-25 | 2007-11-08 | Theravance, Inc. | Crystalline forms of a dimethylphenyl compound |
| JP2008510014A (en) * | 2004-08-16 | 2008-04-03 | セラヴァンス, インコーポレーテッド | Compounds having β2 adrenergic receptor agonist activity and muscarinic receptor antagonist activity |
| WO2008041095A1 (en) * | 2006-10-04 | 2008-04-10 | Pfizer Limited | Sulfonamide derivatives as adrenergic agonists and muscarinic antagonists |
| WO2009087224A1 (en) | 2008-01-11 | 2009-07-16 | Novartis Ag | Pyrimidines as kinase inhibitors |
| US7612084B2 (en) | 2006-03-20 | 2009-11-03 | Pfizer Inc | Amine derivatives for the treatment of asthma and COPD |
| JP2009541209A (en) * | 2006-04-25 | 2009-11-26 | セラヴァンス, インコーポレーテッド | Dialkylphenyl compounds having β2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
| WO2009150137A2 (en) | 2008-06-10 | 2009-12-17 | Novartis Ag | Organic compounds |
| WO2010004517A1 (en) * | 2008-07-11 | 2010-01-14 | Pfizer Limited | Triazole derivatives useful for the treatment of diseases |
| WO2010119064A1 (en) | 2009-04-14 | 2010-10-21 | Glaxo Group Limited | Process for the preparation of a biphenyl-2-ylcarbamic acid ester |
| EP2279777A2 (en) | 2007-01-10 | 2011-02-02 | Irm Llc | Compounds and compositions as channel activating protease inhibitors |
| EP2280006A1 (en) | 2005-08-08 | 2011-02-02 | Pulmagen Therapeutics (Synergy) Limited | Pharmaceutical composition for inhalation comprising an oxazole or thiazole m3 muscarinic receptor antagonist |
| EP2281813A1 (en) | 2005-08-08 | 2011-02-09 | Pulmagen Therapeutics (Synergy) Limited | Bicyclo[2.2.1]hept-7-ylamine derivatives and their uses |
| EP2286813A2 (en) | 2006-01-31 | 2011-02-23 | Novartis AG | Use of naphthyridine derivatives as medicaments |
| EP2292619A1 (en) | 2004-10-22 | 2011-03-09 | Novartis AG | Purine derivatives for use as adenonsin A-2A receptor agonists |
| WO2011050325A1 (en) | 2009-10-22 | 2011-04-28 | Vertex Pharmaceuticals Incorporated | Compositions for treatment of cystic fibrosis and other chronic diseases |
| EP2332933A1 (en) | 2007-05-07 | 2011-06-15 | Novartis AG | Epithelial sodium channel (ENaC) inhibitors |
| WO2011081937A1 (en) | 2009-12-15 | 2011-07-07 | Gilead Sciences, Inc. | Corticosteroid-beta-agonist-muscarinic antagonist compounds for use in therapy |
| WO2011113894A1 (en) | 2010-03-19 | 2011-09-22 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of cf |
| WO2012007539A1 (en) | 2010-07-14 | 2012-01-19 | Novartis Ag | Ip receptor agonist heterocyclic compounds |
| WO2012034095A1 (en) | 2010-09-09 | 2012-03-15 | Irm Llc | Compounds and compositions as trk inhibitors |
| WO2012034091A1 (en) | 2010-09-09 | 2012-03-15 | Irm Llc | Imidazo [1, 2] pyridazin compounds and compositions as trk inhibitors |
| WO2012035158A1 (en) | 2010-09-17 | 2012-03-22 | Novartis Ag | Pyrazine derivatives as enac blockers |
| EP2444120A1 (en) | 2007-12-10 | 2012-04-25 | Novartis AG | Spirocyclic amiloride analogues as ENac blockers |
| WO2012032546A3 (en) * | 2010-09-08 | 2012-06-28 | Cadila Healthcare Limited | Process for the preparation of salmeterol and its intermediates |
| WO2012116217A1 (en) | 2011-02-25 | 2012-08-30 | Irm Llc | Compounds and compositions as trk inhibitors |
| EP2532679A1 (en) | 2005-10-21 | 2012-12-12 | Novartis AG | Human antibodies against il13 and therapeutic uses |
| WO2012168359A1 (en) | 2011-06-10 | 2012-12-13 | Chiesi Farmaceutici S.P.A. | Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
| WO2012168349A1 (en) | 2011-06-10 | 2012-12-13 | Chiesi Farmaceutici S.P.A. | Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
| WO2013030802A1 (en) | 2011-09-01 | 2013-03-07 | Novartis Ag | Bicyclic heterocycle derivatives for the treatment of pulmonary arterial hypertension |
| WO2013038386A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Heterocyclic compounds for the treatment of cystic fibrosis |
| WO2013038390A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | N-substituted heterocyclyl carboxamides |
| WO2013038378A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Pyridine amide derivatives |
| WO2013038381A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Pyridine/pyrazine amide derivatives |
| WO2013038373A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Pyridine amide derivatives |
| WO2013105061A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | Fused dihydropyrido [2,3 -b] pyrazines as ip receptor agonists for the treatment of pulmonary arterial hypertension (pah) and related disorders |
| WO2013105058A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | 7,8- dihydropyrido [3, 4 - b] pyrazines as ip receptor agonists for the treatment of pulmonary arterial hypertension (pah) and related disorders |
| WO2013105065A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | Fused piperidines as ip receptor agonists for the treatment of pah and related disorders |
| WO2013105063A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | Fused piperidines as ip receptor agonists for the treatment of pulmonary arterial hypertension (pah) and related disorders |
| WO2013105057A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | Fused pyrroles as ip receptor agonists for the treatment of pulmonary arterial hypertension (pah) and related disorders |
| WO2013105066A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | Salts of an ip receptor agonist |
| WO2013140319A1 (en) | 2012-03-19 | 2013-09-26 | Novartis Ag | Crystalline form of a succinate salt |
| WO2014086927A1 (en) | 2012-12-06 | 2014-06-12 | Chiesi Farmaceutici S.P.A. | Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
| WO2014086924A1 (en) | 2012-12-06 | 2014-06-12 | Chiesi Farmaceutici S.P.A. | Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
| WO2014125413A1 (en) | 2013-02-13 | 2014-08-21 | Novartis Ag | Ip receptor agonist heterocyclic compounds |
| WO2014132220A1 (en) | 2013-03-01 | 2014-09-04 | Novartis Ag | Solid forms of bicyclic heterocyclic derivatives as pdgf receptor mediators |
| US8883800B2 (en) | 2011-07-15 | 2014-11-11 | Boehringer Ingelheim International Gmbh | Substituted quinazolines, the preparation thereof and the use thereof in pharmaceutical compositions |
| US8883805B2 (en) | 2004-11-05 | 2014-11-11 | Boehringer Ingelheim International Gmbh | Process for the preparation of chiral 8-(3-aminopiperidin-1-yl)-xanthines |
| WO2015008230A1 (en) | 2013-07-18 | 2015-01-22 | Novartis Ag | Autotaxin inhibitors comprising a heteroaromatic ring-benzyl-amide-cycle core |
| WO2015008229A1 (en) | 2013-07-18 | 2015-01-22 | Novartis Ag | Autotaxin inhibitors |
| US9034883B2 (en) | 2010-11-15 | 2015-05-19 | Boehringer Ingelheim International Gmbh | Vasoprotective and cardioprotective antidiabetic therapy |
| US9108964B2 (en) | 2002-08-21 | 2015-08-18 | Boehringer Ingelheim International Gmbh | 8-[3-amino-piperidin-1-yl]-xanthines, the preparation thereof and their use as pharmaceutical compositions |
| WO2015162456A1 (en) | 2014-04-24 | 2015-10-29 | Novartis Ag | Amino pyridine derivatives as phosphatidylinositol 3-kinase inhibitors |
| WO2015162459A1 (en) | 2014-04-24 | 2015-10-29 | Novartis Ag | Amino pyrazine derivatives as phosphatidylinositol 3-kinase inhibitors |
| WO2015162461A1 (en) | 2014-04-24 | 2015-10-29 | Novartis Ag | Pyrazine derivatives as phosphatidylinositol 3-kinase inhibitors |
| US9173859B2 (en) | 2006-05-04 | 2015-11-03 | Boehringer Ingelheim International Gmbh | Uses of DPP IV inhibitors |
| US9212183B2 (en) | 2008-12-23 | 2015-12-15 | Boehringer Ingelheim International Gmbh | Salt forms of 1-[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1-yl)-8-(3-(R)-amino-piperidin-1-yl)-xanthine |
| US9233108B2 (en) | 2011-11-11 | 2016-01-12 | Almirall, S.A. | Cyclohexylamine derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activities |
| US9266888B2 (en) | 2006-05-04 | 2016-02-23 | Boehringer Ingelheim International Gmbh | Polymorphs |
| US9315463B2 (en) | 2010-05-13 | 2016-04-19 | Almirall, S.A. | Cyclohexylamine derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activities |
| US9415016B2 (en) | 2008-04-03 | 2016-08-16 | Boehringer Ingelheim International Gmbh | DPP-IV inhibitor combined with a further antidiabetic agent, tablets comprising such formulations, their use and process for their preparation |
| WO2016128456A1 (en) | 2015-02-12 | 2016-08-18 | Chiesi Farmaceutici S.P.A. | Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
| US9457029B2 (en) | 2009-11-27 | 2016-10-04 | Boehringer Ingelheim International Gmbh | Treatment of genotyped diabetic patients with DPP-IV inhibitors such as linagliptin |
| US9486526B2 (en) | 2008-08-06 | 2016-11-08 | Boehringer Ingelheim International Gmbh | Treatment for diabetes in patients inappropriate for metformin therapy |
| WO2016193241A1 (en) | 2015-06-01 | 2016-12-08 | Chiesi Farmaceutici S.P.A. | Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
| US9518050B2 (en) | 2012-12-18 | 2016-12-13 | Almirall, S.A. | Cyclohexyl and quinuclidinyl carbamate derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activity |
| US9526730B2 (en) | 2012-05-14 | 2016-12-27 | Boehringer Ingelheim International Gmbh | Use of a DPP-4 inhibitor in podocytes related disorders and/or nephrotic syndrome |
| US9526728B2 (en) | 2014-02-28 | 2016-12-27 | Boehringer Ingelheim International Gmbh | Medical use of a DPP-4 inhibitor |
| US9555001B2 (en) | 2012-03-07 | 2017-01-31 | Boehringer Ingelheim International Gmbh | Pharmaceutical composition and uses thereof |
| US9562039B2 (en) | 2013-02-27 | 2017-02-07 | Almirall, S.A. | Salts of 2-amino-1-hydroxyethyl-8-hydroxyquinolin-2(1H)-one derivatives having both β2 adrenergic receptor agonist and M3 muscarinic receptor antagonist activities |
| US9603851B2 (en) | 2010-05-05 | 2017-03-28 | Boehringer Ingelheim International Gmbh | Combination therapy |
| US9643961B2 (en) | 2010-05-13 | 2017-05-09 | Almirall, S.A. | Cyclohexylamine derivatives having β2 adrenergic antagonist and M3 muscarinic antagonist activities |
| WO2017093208A1 (en) | 2015-12-03 | 2017-06-08 | Chiesi Farmaceutici S.P.A. | Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
| US9713618B2 (en) | 2012-05-24 | 2017-07-25 | Boehringer Ingelheim International Gmbh | Method for modifying food intake and regulating food preference with a DPP-4 inhibitor |
| WO2018011090A1 (en) | 2016-07-13 | 2018-01-18 | Chiesi Farmaceutici S.P.A. | Hydroxyquinolinone compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
| WO2018108089A1 (en) | 2016-12-14 | 2018-06-21 | 北京硕佰医药科技有限责任公司 | Class of bifunctional compounds with quaternary ammonium salt structure |
| US10005771B2 (en) | 2014-09-26 | 2018-06-26 | Almirall, S.A. | Bicyclic derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activities |
| US10040765B2 (en) | 2012-09-21 | 2018-08-07 | Crystal Pharma S.A.U. | Methods for the preparation of indacaterol and pharmaceutically acceptable salts thereof |
| US10155000B2 (en) | 2016-06-10 | 2018-12-18 | Boehringer Ingelheim International Gmbh | Medical use of pharmaceutical combination or composition |
| US10456390B2 (en) | 2013-07-25 | 2019-10-29 | Almirall, S.A. | Combinations comprising MABA compounds and corticosteroids |
| WO2020250116A1 (en) | 2019-06-10 | 2020-12-17 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of cf, copd, and bronchiectasis |
| WO2021038426A1 (en) | 2019-08-28 | 2021-03-04 | Novartis Ag | Substituted 1,3-phenyl heteroaryl derivatives and their use in the treatment of disease |
| US11033552B2 (en) | 2006-05-04 | 2021-06-15 | Boehringer Ingelheim International Gmbh | DPP IV inhibitor formulations |
| US11911388B2 (en) | 2008-10-16 | 2024-02-27 | Boehringer Ingelheim International Gmbh | Treatment for diabetes in patients with insufficient glycemic control despite therapy with an oral or non-oral antidiabetic drug |
Families Citing this family (74)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6693202B1 (en) * | 1999-02-16 | 2004-02-17 | Theravance, Inc. | Muscarinic receptor antagonists |
| ES2165768B1 (en) | 1999-07-14 | 2003-04-01 | Almirall Prodesfarma Sa | NEW DERIVATIVES OF QUINUCLIDINE AND PHARMACEUTICAL COMPOSITIONS THAT CONTAIN THEM. |
| PE20050130A1 (en) * | 2002-08-09 | 2005-03-29 | Novartis Ag | ORGANIC COMPOUNDS |
| PE20040950A1 (en) | 2003-02-14 | 2005-01-01 | Theravance Inc | BIPHENYL DERIVATIVES AS AGONISTS OF ß2-ADRENERGIC RECEPTORS AND AS ANTAGONISTS OF MUSCARINAL RECEPTORS |
| US7317102B2 (en) | 2003-04-01 | 2008-01-08 | Theravance, Inc. | Diarylmethyl and related compounds |
| WO2004106333A1 (en) * | 2003-05-28 | 2004-12-09 | Theravance, Inc. | Azabicycloalkane compounds as muscarinic receptor antagonists |
| PT1631295E (en) * | 2003-06-06 | 2010-05-24 | Arexis Ab | Use of condensed heterocyclic compounds as scce inhibitors for the treatment of skin diseases |
| US20050026948A1 (en) * | 2003-07-29 | 2005-02-03 | Boehringer Ingelheim International Gmbh | Medicaments for inhalation comprising an anticholinergic and a betamimetic |
| ES2329586T3 (en) * | 2003-11-21 | 2009-11-27 | Theravance, Inc. | COMPOUNDS THAT HAVE AGONIST ACTIVITY OF THE BETA2 ADRENERGIC RECEIVER AND ANTAGONIST OF THE MUSCARINE RECEIVER. |
| TW200531692A (en) | 2004-01-12 | 2005-10-01 | Theravance Inc | Aryl aniline derivatives as β2 adrenergic receptor agonists |
| WO2005080375A1 (en) * | 2004-02-13 | 2005-09-01 | Theravance, Inc. | Crystalline form of a biphenyl compound |
| EP1723115A1 (en) * | 2004-03-11 | 2006-11-22 | Theravance, Inc. | Biphenyl compounds useful as muscarinic receptor antagonists |
| JP2007528412A (en) * | 2004-03-11 | 2007-10-11 | セラヴァンス, インコーポレーテッド | Biphenyl compounds useful as muscarinic receptor antagonists |
| TW200538095A (en) * | 2004-03-11 | 2005-12-01 | Theravance Inc | Biphenyl compounds useful as muscarinic receptor antagonists |
| WO2005087735A1 (en) * | 2004-03-11 | 2005-09-22 | Theravance, Inc. | Biphenyl compounds useful as muscarinic receptor antagonists |
| TWI341836B (en) | 2004-03-11 | 2011-05-11 | Theravance Inc | Biphenyl compounds useful as muscarinic receptor antagonists |
| JP2007528408A (en) * | 2004-03-11 | 2007-10-11 | セラヴァンス, インコーポレーテッド | Useful biphenyl compounds as muscarinic receptor antagonists |
| EP1723113A1 (en) * | 2004-03-11 | 2006-11-22 | Theravance, Inc. | Biphenyl compounds useful as muscarinic receptor antagonists |
| US7501442B2 (en) * | 2004-03-11 | 2009-03-10 | Theravance, Inc. | Biphenyl compounds useful as muscarinic receptor antagonists |
| ES2317245T3 (en) * | 2004-05-31 | 2009-04-16 | Laboratorios Almirall, S.A. | COMBINATIONS THAT INCLUDE ANTIMUSCARINIC AGENTS AND BETA-ADRENERGIC AGONISTS. |
| ES2257152B1 (en) * | 2004-05-31 | 2007-07-01 | Laboratorios Almirall S.A. | COMBINATIONS THAT INCLUDE ANTIMUSCARINIC AGENTS AND BETA-ADRENERGIC AGONISTS. |
| TWI374883B (en) * | 2004-08-16 | 2012-10-21 | Theravance Inc | Crystalline form of a biphenyl compound |
| TW200714587A (en) * | 2005-03-10 | 2007-04-16 | Theravance Inc | Biphenyl compounds useful as muscarinic receptor antagonists |
| US7642355B2 (en) | 2005-03-10 | 2010-01-05 | Theravance, Inc. | Biphenyl compounds useful as muscarinic receptor antagonists |
| WO2006099031A1 (en) * | 2005-03-10 | 2006-09-21 | Theravance, Inc. | Biphenyl compounds useful as muscarinic receptor antagonists |
| US7659403B2 (en) * | 2005-03-10 | 2010-02-09 | Theravance, Inc. | Biphenyl compounds useful as muscarinic receptor antagonists |
| JP2008533028A (en) * | 2005-03-10 | 2008-08-21 | セラヴァンス, インコーポレーテッド | Biphenyl compounds useful as muscarinic receptor antagonists |
| ES2265276B1 (en) | 2005-05-20 | 2008-02-01 | Laboratorios Almirall S.A. | DERIVATIVES OF 4- (2-AMINO-1-HYDROXYETHYL) Phenol as agonists of the BETA2 ADRENERGIC RECEIVER. |
| JP2008546695A (en) | 2005-06-13 | 2008-12-25 | セラヴァンス, インコーポレーテッド | Biphenyl compounds useful as muscarinic receptor antagonists |
| US8429052B2 (en) * | 2005-07-19 | 2013-04-23 | Lincoln National Life Insurance Company | Method and system for providing employer-sponsored retirement plan |
| TW200738658A (en) | 2005-08-09 | 2007-10-16 | Astrazeneca Ab | Novel compounds |
| US7973055B2 (en) * | 2006-03-09 | 2011-07-05 | Theravance, Inc. | Crystalline forms of a biphenyl compound |
| TW200745067A (en) | 2006-03-14 | 2007-12-16 | Astrazeneca Ab | Novel compounds |
| ES2296516B1 (en) * | 2006-04-27 | 2009-04-01 | Laboratorios Almirall S.A. | DERIVATIVES OF 4- (2-AMINO-1-HYDROXYETHYL) Phenol as agonists of the BETA2 ADRENERGIC RECEIVER. |
| GB0613154D0 (en) * | 2006-06-30 | 2006-08-09 | Novartis Ag | Organic Compounds |
| ES2302447B1 (en) * | 2006-10-20 | 2009-06-12 | Laboratorios Almirall S.A. | DERIVATIVES OF 4- (2-AMINO-1-HYDROXYETHYL) Phenol as agonists of the BETA2 ADRENERGIC RECEIVER. |
| TW200833670A (en) | 2006-12-20 | 2008-08-16 | Astrazeneca Ab | Novel compounds 569 |
| PT2119700E (en) * | 2007-01-31 | 2012-10-02 | Toray Industries | Benzylamine derivative or pharmaceutically acceptable acid addition salt thereof, and use thereof for medical purposes |
| GB0702458D0 (en) | 2007-02-08 | 2007-03-21 | Astrazeneca Ab | Salts 668 |
| ES2306595B1 (en) | 2007-02-09 | 2009-09-11 | Laboratorios Almirall S.A. | NAPADISYLATE SALT OF 5- (2 - ((6- (2,2-DIFLUORO-2-PHENYLETOXI) HEXIL) AMINO) -1-HYDROXYETHYL) -8-HYDROXYCHINOLIN-2 (1H) -ONE AS ADRENERGIC RECEIVER AGONIST BETA2 . |
| US8682466B2 (en) * | 2007-05-04 | 2014-03-25 | Taiwan Semiconductor Manufacturing Company, Ltd. | Automatic virtual metrology for semiconductor wafer result prediction |
| ES2320961B1 (en) * | 2007-11-28 | 2010-03-17 | Laboratorios Almirall, S.A. | DERIVATIVES OF 4- (2-AMINO-1-HYDROXYETHYL) PHENOL AS BETA2 ADRENERGIC RECEIVER AGONISTS. |
| DK2242759T3 (en) * | 2008-02-06 | 2012-12-17 | Astrazeneca Ab | compounds |
| EP2096105A1 (en) * | 2008-02-28 | 2009-09-02 | Laboratorios Almirall, S.A. | Derivatives of 4-(2-amino-1-hydroxyethyl)phenol as agonists of the b2 adrenergic receptor |
| EP2100599A1 (en) | 2008-03-13 | 2009-09-16 | Laboratorios Almirall, S.A. | Inhalation composition containing aclidinium for treatment of asthma and chronic obstructive pulmonary disease |
| EP2100598A1 (en) | 2008-03-13 | 2009-09-16 | Laboratorios Almirall, S.A. | Inhalation composition containing aclidinium for treatment of asthma and chronic obstructive pulmonary disease |
| AU2009260899B2 (en) | 2008-06-18 | 2012-02-23 | Astrazeneca Ab | Benzoxazinone derivatives acting as beta2-adrenoreceptor agonist for the treatment of respiratory disorders |
| WO2010007561A1 (en) * | 2008-07-15 | 2010-01-21 | Pfizer Limited | Novel compounds active as muscarinic receptor antagonists |
| US20100016366A1 (en) * | 2008-07-15 | 2010-01-21 | Paul Alan Glossop | Novel Compounds Active as Muscarinic Receptor Antagonists |
| UY32297A (en) | 2008-12-22 | 2010-05-31 | Almirall Sa | MESILATE SALT OF 5- (2 - {[6- (2,2-DIFLUORO-2-PHENYLITOXI) HEXIL] AMINO} -1-HYDROXYETHYL) -8-HYDROXYCHINOLIN-2 (1H) -ONA AS A RECEIVER AGONIST B (BETA ) 2 ACRENERGIC |
| TW201036957A (en) | 2009-02-20 | 2010-10-16 | Astrazeneca Ab | Novel salt 628 |
| EP2228368A1 (en) | 2009-03-12 | 2010-09-15 | Almirall, S.A. | Process for manufacturing 5-(2-{[6-(2,2-difluoro-2-phenylethoxy) hexyl]amino}-1-hydroxyethyl)-8-hydroxyquinolin-2(1H)-one |
| EP2409977A4 (en) | 2009-03-17 | 2012-09-26 | Daiichi Sankyo Co Ltd | Amide derivative |
| ES2715965T3 (en) | 2009-04-23 | 2019-06-07 | Theravance Respiratory Co Llc | Diamine compounds that have muscarinic receptor antagonist activity and beta 2 adrenergic receptor agonist |
| AU2015234331B2 (en) * | 2009-04-23 | 2017-08-03 | Theravance Biopharma R&D Ip, Llc | Diamide compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
| EP2426121A4 (en) * | 2009-04-30 | 2012-10-31 | Teijin Pharma Ltd | Quaternary ammonium salt compound |
| WO2011061527A1 (en) | 2009-11-17 | 2011-05-26 | Astrazeneca Ab | Combinations comprising a glucocorticoid receptor modulator for the treatment of respiratory diseases |
| KR101742253B1 (en) * | 2010-07-13 | 2017-05-31 | 세라밴스 바이오파마 알앤디 아이피, 엘엘씨 | Process for preparing a biphenyl-2-ylcarbamic acid |
| AR083115A1 (en) * | 2010-09-30 | 2013-01-30 | Theravance Inc | CRYSTAL OXALATE SALTS OF A DIAMID COMPOUND |
| GB201016912D0 (en) | 2010-10-07 | 2010-11-24 | Astrazeneca Ab | Novel combination |
| JP6060242B2 (en) * | 2010-11-30 | 2017-01-11 | 株式会社日立国際電気 | Substrate processing apparatus, semiconductor device manufacturing method, and baffle structure |
| EP2510928A1 (en) | 2011-04-15 | 2012-10-17 | Almirall, S.A. | Aclidinium for use in improving the quality of sleep in respiratory patients |
| EP2578570A1 (en) | 2011-10-07 | 2013-04-10 | Almirall, S.A. | Novel process for preparing 5-(2-{[6-(2,2-difluoro-2-phenylethoxy)hexyl]amino}-1(r)-hydroxyethyl)-8-hydroxyquinolin-2(1h)-one via novel intermediates of synthesis. |
| EP2641900A1 (en) | 2012-03-20 | 2013-09-25 | Almirall, S.A. | Novel polymorphic Crystal forms of 5-(2-{[6-(2,2-difluoro-2-phenylethoxy) hexyl]amino}-1-(R)-hydroxyethyl)-8-hydroxyquinolin-2(1h)-one, heminapadisylate as agonist of the ß2 adrenergic receptor. |
| CZ306252B6 (en) | 2013-03-15 | 2016-10-26 | Zentiva, K.S. | Process for preparing 5-[(R)-2-(5,6-diethylindan-2-ylamino)-1-hydroxyethyl]-8-hydroxy-(1H)-quinolin-2-one (indacaterol) |
| TWI641373B (en) | 2013-07-25 | 2018-11-21 | 阿爾米雷爾有限公司 | SALTS OF 2-AMINO-1-HYDROXYETHYL-8-HYDROXYQUINOLIN-2(1H)-ONE DERIVATIVES HAVING BOTH MUSCARINIC RECEPTOR ANTAGONIST AND β2 ADRENERGIC RECEPTOR AGONIST ACTIVITIES |
| WO2017001907A1 (en) * | 2015-06-29 | 2017-01-05 | Teva Pharmaceuticals International Gmbh | Biocatalytic processes for the preparation of vilanterol |
| MX2018008799A (en) * | 2016-01-22 | 2019-01-14 | Sichuan Haisco Pharmaceutical Co Ltd | Nitrogenous heterocyclic amide derivative, preparation method thereof, and pharmaceutical application. |
| CN106706868B (en) * | 2016-11-15 | 2018-12-04 | 河南省农业科学院 | Application of the artificial expression HSP27 albumen in terms of detecting beta-2-adrenoreceptor agonists class medicament residue |
| CN106632257B (en) * | 2016-12-15 | 2019-02-12 | 上海市奉贤区中心医院 | The synthetic method of GSK961081 and its intermediate |
| US11866482B2 (en) | 2018-02-07 | 2024-01-09 | Duke University | System and method for homogenous GPCR phosphorylation and identification of beta-2 adrenergic receptor positive allosteric modulators |
| US11484531B2 (en) | 2018-08-30 | 2022-11-01 | Theravance Biopharma R&D Ip, Llc | Methods for treating chronic obstructive pulmonary disease |
| WO2022175982A1 (en) * | 2021-02-19 | 2022-08-25 | Gbr Laboratories Private Limited | A process for preparing batefenterol and intermediates thereof |
| CN114409890A (en) * | 2022-02-28 | 2022-04-29 | 中国科学院长春应用化学研究所 | Amino-functionalized polyethylene glycol derivative and preparation method thereof |
Family Cites Families (83)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4026897A (en) * | 1974-01-31 | 1977-05-31 | Otsuka Pharmaceutical Company | 5-[1-Hydroxy-2-(substituted-amino)]alkyl-8-hydroxycarbostyril derivatives |
| BR8007911A (en) | 1979-12-06 | 1981-06-16 | Glaxo Group Ltd | PERFECTED INHALER |
| CY1492A (en) | 1981-07-08 | 1990-02-16 | Draco Ab | Powder inhalator |
| DE3134590A1 (en) | 1981-09-01 | 1983-03-10 | Boehringer Ingelheim KG, 6507 Ingelheim | NEW BENZO HETEROCYCLES |
| US4460581A (en) * | 1982-10-12 | 1984-07-17 | Boehringer Ingelheim Kg | (1-Hydroxy-2-amino-alkyl)-substituted benzoxazinones and benzoxazolinones |
| ZW6584A1 (en) * | 1983-04-18 | 1985-04-17 | Glaxo Group Ltd | Phenethanolamine derivatives |
| PT83094B (en) | 1985-07-30 | 1993-07-30 | Glaxo Group Ltd | DEVICES PROPER FOR THE ADMINISTRATION OF MEDICINES TO PATIENTS |
| US4894219A (en) | 1988-03-29 | 1990-01-16 | University Of Florida | Beta-agonist carbostyril derivatives, assay method and pharmacological composition |
| US5225183A (en) | 1988-12-06 | 1993-07-06 | Riker Laboratories, Inc. | Medicinal aerosol formulations |
| DK443489D0 (en) | 1989-09-08 | 1989-09-08 | Ferrosan As | SUBSTITUTED URINARY COMPOUNDS, THEIR PREPARATION AND USE |
| SK280967B6 (en) | 1990-03-02 | 2000-10-09 | Glaxo Group Limited | Inhalation device |
| US6006745A (en) | 1990-12-21 | 1999-12-28 | Minnesota Mining And Manufacturing Company | Device for delivering an aerosol |
| US5874063A (en) | 1991-04-11 | 1999-02-23 | Astra Aktiebolag | Pharmaceutical formulation |
| AU2178392A (en) | 1991-06-12 | 1993-01-12 | Minnesota Mining And Manufacturing Company | Albuterol sulfate suspension aerosol formulations |
| US5337740A (en) | 1991-08-01 | 1994-08-16 | New England Pharmaceuticals, Inc. | Inhalation devices |
| ES2216800T3 (en) | 1991-12-18 | 2004-11-01 | Minnesota Mining And Manufacturing Company | AEROSOL FORMULATIONS IN SUSPENSION. |
| HUT72321A (en) | 1992-03-31 | 1996-04-29 | Glaxo Group Ltd | Substituted phenylcarbamate and phenyl-urea derivatives, pharmaceutical compositions containing them and process for their preparation |
| US5239993A (en) | 1992-08-26 | 1993-08-31 | Glaxo Inc. | Dosage inhalator providing optimized compound inhalation trajectory |
| US5497763A (en) | 1993-05-21 | 1996-03-12 | Aradigm Corporation | Disposable package for intrapulmonary delivery of aerosolized formulations |
| CA2123728A1 (en) * | 1993-05-21 | 1994-11-22 | Noriyoshi Sueda | Urea derivatives and their use as acat inhibitors |
| WO1995006635A1 (en) | 1993-09-02 | 1995-03-09 | Yamanouchi Pharmaceutical Co., Ltd. | Carbamate derivative and medicine containing the same |
| US5415162A (en) | 1994-01-18 | 1995-05-16 | Glaxo Inc. | Multi-dose dry powder inhalation device |
| WO1995021820A1 (en) | 1994-02-10 | 1995-08-17 | Yamanouchi Pharmaceutical Co., Ltd. | Novel carbamate derivative and medicinal composition containing the same |
| US5495054A (en) | 1994-05-31 | 1996-02-27 | Sepracor, Inc. | Tetrahydroindeno[1,2-D][1,3,2]oxazaboroles and their use as enantioselective catalysts |
| US5983956A (en) | 1994-10-03 | 1999-11-16 | Astra Aktiebolag | Formulation for inhalation |
| SE9501384D0 (en) | 1995-04-13 | 1995-04-13 | Astra Ab | Process for the preparation of respirable particles |
| SK284447B6 (en) | 1995-04-14 | 2005-04-01 | Glaxo Wellcome Inc. | Metered dose inhaler |
| DE19536902A1 (en) | 1995-10-04 | 1997-04-10 | Boehringer Ingelheim Int | Miniature fluid pressure generating device |
| EP0863141B1 (en) | 1995-10-13 | 2001-09-12 | Banyu Pharmaceutical Co., Ltd. | Substituted heteroaromatic derivatives |
| PE92198A1 (en) | 1996-08-01 | 1999-01-09 | Banyu Pharma Co Ltd | DERIVATIVES OF FLUORINE-CONTAINED 1,4-PIPERIDINE |
| US6040344A (en) | 1996-11-11 | 2000-03-21 | Sepracor Inc. | Formoterol process |
| AU9281298A (en) | 1997-10-01 | 1999-04-23 | Kyowa Hakko Kogyo Co. Ltd. | Benzodioxole derivatives |
| JP2002508366A (en) | 1997-12-12 | 2002-03-19 | スミスクライン・ビーチャム・パブリック・リミテッド・カンパニー | Quinoline piperazine and quinoline piperidine derivatives, methods for their preparation, and their use as complex 5-HT1A, 5-HT1B and 5-HT1D receptor antagonists |
| SI1070056T1 (en) | 1998-03-14 | 2004-12-31 | Altana Pharma Ag | Phthalazinone pde iii/iv inhibitors |
| PT1073417E (en) | 1998-04-18 | 2003-04-30 | Glaxo Group Ltd | PHARMACEUTICAL FORMULA IN AEROSOL |
| GB9808802D0 (en) | 1998-04-24 | 1998-06-24 | Glaxo Group Ltd | Pharmaceutical formulations |
| GB9812088D0 (en) * | 1998-06-05 | 1998-08-05 | Celltech Therapeutics Ltd | Chemical compounds |
| US6713651B1 (en) * | 1999-06-07 | 2004-03-30 | Theravance, Inc. | β2-adrenergic receptor agonists |
| AU763638B2 (en) | 1998-06-08 | 2003-07-31 | Theravance Biopharma R&D Ip, Llc | Muscarinic receptor antagonists |
| US6541669B1 (en) | 1998-06-08 | 2003-04-01 | Theravance, Inc. | β2-adrenergic receptor agonists |
| SE9804000D0 (en) | 1998-11-23 | 1998-11-23 | Astra Ab | New composition of matter |
| WO2000035877A1 (en) * | 1998-12-18 | 2000-06-22 | Du Pont Pharmaceuticals Company | Heterocyclic piperidines as modulators of chemokine receptor activity |
| US6693202B1 (en) | 1999-02-16 | 2004-02-17 | Theravance, Inc. | Muscarinic receptor antagonists |
| DE19921693A1 (en) | 1999-05-12 | 2000-11-16 | Boehringer Ingelheim Pharma | Pharmaceutical composition for treating respiratory disorders, e.g. asthma, comprises combination of anticholinergic and beta-mimetic agents having synergistic bronchospasmolytic activity and reduced side-effects |
| GB9913083D0 (en) | 1999-06-04 | 1999-08-04 | Novartis Ag | Organic compounds |
| DK1169019T3 (en) | 1999-04-14 | 2003-06-02 | Glaxo Group Ltd | Pharmaceutical aerosol formulation |
| US6593497B1 (en) * | 1999-06-02 | 2003-07-15 | Theravance, Inc. | β2-adrenergic receptor agonists |
| MXPA02005596A (en) | 1999-12-07 | 2004-09-10 | Theravance Inc | Carbamate derivatives having muscarinic receptor antagonist activity. |
| UA73543C2 (en) * | 1999-12-07 | 2005-08-15 | Тераванс, Інк. | Urea derivatives, a pharmaceutical composition and use of derivative in the preparation of medicament for the treatment of disease being mediated by muscarine receptor |
| UA73965C2 (en) * | 1999-12-08 | 2005-10-17 | Theravance Inc | b2 ADRENERGIC RECEPTOR ANTAGONISTS |
| US6933296B2 (en) | 2000-06-05 | 2005-08-23 | Altana Pharma B.V. | Compounds effective as β2-adrenoreceptor agonists as well as PDE4-inhibitors |
| IL156499A0 (en) | 2000-12-22 | 2004-01-04 | Almirall Prodesfarma Ag | Quinuclidine carbamate derivatives and their use as m3 antagonists |
| GB0103630D0 (en) | 2001-02-14 | 2001-03-28 | Glaxo Group Ltd | Chemical compounds |
| DE60220887T2 (en) | 2001-03-08 | 2008-02-28 | Glaxo Group Ltd., Greenford | AGONISTS OF BETA ADRENORE RECEPTORS |
| JP4143413B2 (en) | 2001-03-22 | 2008-09-03 | グラクソ グループ リミテッド | Formanilide derivatives as β2-adrenergic receptor agonists |
| US20030018019A1 (en) | 2001-06-23 | 2003-01-23 | Boehringer Ingelheim Pharma Kg | Pharmaceutical compositions based on anticholinergics, corticosteroids and betamimetics |
| US7361787B2 (en) | 2001-09-14 | 2008-04-22 | Glaxo Group Limited | Phenethanolamine derivatives for treatment of respiratory diseases |
| TWI249515B (en) * | 2001-11-13 | 2006-02-21 | Theravance Inc | Aryl aniline beta2 adrenergic receptor agonists |
| US6653323B2 (en) | 2001-11-13 | 2003-11-25 | Theravance, Inc. | Aryl aniline β2 adrenergic receptor agonists |
| BR0309185A (en) | 2002-04-12 | 2005-02-15 | Boehringer Ingelheim Pharma | Beta-mimetic drug and a new anticholinergic |
| WO2003099764A1 (en) * | 2002-05-28 | 2003-12-04 | Theravance, Inc. | ALKOXY ARYL β2 ADRENERGIC RECEPTOR AGONISTS |
| US6711451B2 (en) * | 2002-07-02 | 2004-03-23 | 3D Systems, Inc. | Power management in selective deposition modeling |
| GB0217225D0 (en) | 2002-07-25 | 2002-09-04 | Glaxo Group Ltd | Medicinal compounds |
| TW200410951A (en) | 2002-08-06 | 2004-07-01 | Glaxo Group Ltd | M3 muscarinic acetylcholine receptor antagonists |
| PE20040950A1 (en) * | 2003-02-14 | 2005-01-01 | Theravance Inc | BIPHENYL DERIVATIVES AS AGONISTS OF ß2-ADRENERGIC RECEPTORS AND AS ANTAGONISTS OF MUSCARINAL RECEPTORS |
| US7317102B2 (en) * | 2003-04-01 | 2008-01-08 | Theravance, Inc. | Diarylmethyl and related compounds |
| WO2004106333A1 (en) * | 2003-05-28 | 2004-12-09 | Theravance, Inc. | Azabicycloalkane compounds as muscarinic receptor antagonists |
| TW200510298A (en) * | 2003-06-13 | 2005-03-16 | Theravance Inc | Substituted pyrrolidine and related compounds |
| JP2007524641A (en) * | 2003-07-11 | 2007-08-30 | セラヴァンス, インコーポレーテッド | Substituted 4-amino-1-benzylpiperidine compounds |
| ES2329586T3 (en) | 2003-11-21 | 2009-11-27 | Theravance, Inc. | COMPOUNDS THAT HAVE AGONIST ACTIVITY OF THE BETA2 ADRENERGIC RECEIVER AND ANTAGONIST OF THE MUSCARINE RECEIVER. |
| WO2005080375A1 (en) * | 2004-02-13 | 2005-09-01 | Theravance, Inc. | Crystalline form of a biphenyl compound |
| US7501442B2 (en) * | 2004-03-11 | 2009-03-10 | Theravance, Inc. | Biphenyl compounds useful as muscarinic receptor antagonists |
| JP2007528408A (en) * | 2004-03-11 | 2007-10-11 | セラヴァンス, インコーポレーテッド | Useful biphenyl compounds as muscarinic receptor antagonists |
| WO2005087735A1 (en) * | 2004-03-11 | 2005-09-22 | Theravance, Inc. | Biphenyl compounds useful as muscarinic receptor antagonists |
| JP2007528412A (en) * | 2004-03-11 | 2007-10-11 | セラヴァンス, インコーポレーテッド | Biphenyl compounds useful as muscarinic receptor antagonists |
| EP1723113A1 (en) * | 2004-03-11 | 2006-11-22 | Theravance, Inc. | Biphenyl compounds useful as muscarinic receptor antagonists |
| US7071224B2 (en) * | 2004-03-11 | 2006-07-04 | Theravance, Inc. | Diphenylmethyl compounds useful as muscarinic receptor antagonists |
| TW200538095A (en) * | 2004-03-11 | 2005-12-01 | Theravance Inc | Biphenyl compounds useful as muscarinic receptor antagonists |
| WO2006023460A2 (en) * | 2004-08-16 | 2006-03-02 | Theravance, Inc. | COMPOUNDS HAVING β2 ADRENERGIC RECEPTOR AGONIST AND MUSCARINIC RECEPTOR ANTAGONIST ACTIVITY |
| TWI374883B (en) * | 2004-08-16 | 2012-10-21 | Theravance Inc | Crystalline form of a biphenyl compound |
| JP2008510014A (en) * | 2004-08-16 | 2008-04-03 | セラヴァンス, インコーポレーテッド | Compounds having β2 adrenergic receptor agonist activity and muscarinic receptor antagonist activity |
| TW200811105A (en) * | 2006-04-25 | 2008-03-01 | Theravance Inc | Dialkylphenyl compounds having beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
| TW200811104A (en) * | 2006-04-25 | 2008-03-01 | Theravance Inc | Crystalline forms of a dimethylphenyl compound |
-
2004
- 2004-02-11 PE PE2004000152A patent/PE20040950A1/en active IP Right Grant
- 2004-02-13 BR BRPI0407508A patent/BRPI0407508B8/en not_active IP Right Cessation
- 2004-02-13 ES ES10173127.1T patent/ES2566155T3/en not_active Expired - Lifetime
- 2004-02-13 ES ES15196303.0T patent/ES2636480T3/en not_active Expired - Lifetime
- 2004-02-13 AT AT04711253T patent/ATE482934T1/en active
- 2004-02-13 HU HUE10173127A patent/HUE027380T2/en unknown
- 2004-02-13 WO PCT/US2004/004449 patent/WO2004074246A2/en active Application Filing
- 2004-02-13 JP JP2006503604A patent/JP4555283B2/en not_active Expired - Fee Related
- 2004-02-13 PL PL378025A patent/PL216397B1/en unknown
- 2004-02-13 DK DK04711253.7T patent/DK1615889T3/en active
- 2004-02-13 NZ NZ541579A patent/NZ541579A/en not_active IP Right Cessation
- 2004-02-13 AU AU2004213411A patent/AU2004213411B2/en not_active Ceased
- 2004-02-13 PT PT04711253T patent/PT1615889E/en unknown
- 2004-02-13 JP JP2006503544A patent/JP2006517971A/en not_active Withdrawn
- 2004-02-13 WO PCT/US2004/004224 patent/WO2004074276A1/en active Application Filing
- 2004-02-13 EP EP04711253A patent/EP1615889B1/en not_active Expired - Lifetime
- 2004-02-13 EP EP04711117A patent/EP1594860A2/en not_active Withdrawn
- 2004-02-13 WO PCT/US2004/004273 patent/WO2004074812A2/en active Application Filing
- 2004-02-13 US US10/778,290 patent/US20040209915A1/en not_active Abandoned
- 2004-02-13 MY MYPI20040480A patent/MY148487A/en unknown
- 2004-02-13 DK DK10173127.1T patent/DK2246345T3/en active
- 2004-02-13 CA CA2515777A patent/CA2515777C/en not_active Expired - Lifetime
- 2004-02-13 DE DE602004029347T patent/DE602004029347D1/en not_active Expired - Lifetime
- 2004-02-13 KR KR1020057014929A patent/KR101223991B1/en active IP Right Grant
- 2004-02-13 MX MXPA05008528A patent/MXPA05008528A/en active IP Right Grant
- 2004-02-13 AR ARP040100450A patent/AR043176A1/en active IP Right Grant
- 2004-02-13 SI SI200432320A patent/SI2246345T1/en unknown
- 2004-02-13 SI SI200431566T patent/SI1615889T1/en unknown
- 2004-02-13 EP EP10173127.1A patent/EP2246345B1/en not_active Expired - Lifetime
- 2004-02-13 EP EP04711137A patent/EP1592685A1/en not_active Withdrawn
- 2004-02-13 JP JP2006503553A patent/JP2006518739A/en active Pending
- 2004-02-13 US US10/778,649 patent/US20040209860A1/en not_active Abandoned
- 2004-02-13 RU RU2005128557/04A patent/RU2330841C2/en active
- 2004-02-13 US US10/779,157 patent/US7141671B2/en active Active
- 2004-02-13 EP EP15196303.0A patent/EP3012254B1/en not_active Expired - Lifetime
- 2004-02-13 KR KR1020117027619A patent/KR20110132634A/en not_active Application Discontinuation
- 2004-02-13 KR KR1020117010078A patent/KR101224497B1/en active IP Right Grant
- 2004-02-13 TW TW093103510A patent/TWI331995B/en active
-
2005
- 2005-07-27 IL IL169922A patent/IL169922A/en active IP Right Grant
- 2005-07-28 IS IS7964A patent/IS2847B/en unknown
- 2005-09-09 NO NO20054206A patent/NO331947B1/en not_active IP Right Cessation
-
2006
- 2006-05-29 HK HK06106230.1A patent/HK1086266A1/en not_active IP Right Cessation
- 2006-06-07 US US11/448,317 patent/US7879879B2/en active Active
- 2006-06-07 US US11/448,294 patent/US7355046B2/en not_active Expired - Lifetime
- 2006-06-07 US US11/448,293 patent/US7345175B2/en not_active Expired - Lifetime
- 2006-06-07 US US11/449,004 patent/US7521561B2/en active Active
- 2006-10-18 US US11/582,885 patent/US7524959B2/en active Active
- 2006-11-27 US US11/604,607 patent/US7514558B2/en active Active
-
2007
- 2007-02-09 JP JP2007031325A patent/JP2007119496A/en not_active Withdrawn
- 2007-04-19 US US11/788,343 patent/US20070208176A1/en not_active Abandoned
- 2007-07-13 US US11/879,004 patent/US7829583B2/en active Active
- 2007-08-01 US US11/888,526 patent/US7507751B2/en not_active Expired - Lifetime
-
2010
- 2010-04-14 JP JP2010093582A patent/JP5247757B2/en not_active Expired - Fee Related
- 2010-09-08 AU AU2010219338A patent/AU2010219338B2/en not_active Ceased
- 2010-11-30 CY CY20101101092T patent/CY1111276T1/en unknown
-
2011
- 2011-01-21 US US13/011,100 patent/US8242135B2/en not_active Expired - Lifetime
- 2011-04-06 HK HK16104933.4A patent/HK1216892A1/en unknown
- 2011-04-06 HK HK11103503.1A patent/HK1149548A1/en not_active IP Right Cessation
- 2011-08-05 JP JP2011172162A patent/JP5436503B2/en not_active Expired - Lifetime
-
2012
- 2012-02-15 NO NO20120150A patent/NO333394B1/en not_active IP Right Cessation
- 2012-07-10 US US13/545,523 patent/US8618131B2/en not_active Expired - Lifetime
-
2013
- 2013-11-22 US US14/087,404 patent/US8969571B2/en not_active Expired - Lifetime
-
2016
- 2016-04-28 CY CY20161100361T patent/CY1117459T1/en unknown
Non-Patent Citations (1)
| Title |
|---|
| None |
Cited By (137)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9108964B2 (en) | 2002-08-21 | 2015-08-18 | Boehringer Ingelheim International Gmbh | 8-[3-amino-piperidin-1-yl]-xanthines, the preparation thereof and their use as pharmaceutical compositions |
| US9556175B2 (en) | 2002-08-21 | 2017-01-31 | Boehringer Ingelheim International Gmbh | 8-[3-amino-piperidin-1-yl]-xanthines, the preparation thereof and thier use as pharmaceutical compositions |
| US9321791B2 (en) | 2002-08-21 | 2016-04-26 | Boehringer Ingelheim International Gmbh | 8-[3-amino-piperidin-1-yl]-xanthines, the preparation thereof and their use as pharmaceutical compositions |
| US10202383B2 (en) | 2002-08-21 | 2019-02-12 | Boehringer Ingelheim International Gmbh | 8-[3-amino-piperidin-1-yl]-xanthines, the preparation thereof and their use as pharmaceutical compositions |
| US10023574B2 (en) | 2002-08-21 | 2018-07-17 | Boehringer Ingelheim International Gmbh | 8-[3-amino-piperidin-1-yl]-xanthines, the preparation thereof and their use as pharmaceutical compositions |
| JP2008510014A (en) * | 2004-08-16 | 2008-04-03 | セラヴァンス, インコーポレーテッド | Compounds having β2 adrenergic receptor agonist activity and muscarinic receptor antagonist activity |
| WO2006023460A3 (en) * | 2004-08-16 | 2006-04-06 | Theravance Inc | COMPOUNDS HAVING β2 ADRENERGIC RECEPTOR AGONIST AND MUSCARINIC RECEPTOR ANTAGONIST ACTIVITY |
| US7569586B2 (en) | 2004-08-16 | 2009-08-04 | Theravance, Inc. | Compounds having β2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
| US8143277B2 (en) | 2004-08-16 | 2012-03-27 | Theravance, Inc. | Method of using compounds having β2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
| EP2292619A1 (en) | 2004-10-22 | 2011-03-09 | Novartis AG | Purine derivatives for use as adenonsin A-2A receptor agonists |
| WO2006050239A3 (en) * | 2004-10-29 | 2006-10-12 | Glaxo Group Ltd | Muscarinic acetylcholine receptor antagonists |
| US8883805B2 (en) | 2004-11-05 | 2014-11-11 | Boehringer Ingelheim International Gmbh | Process for the preparation of chiral 8-(3-aminopiperidin-1-yl)-xanthines |
| US9499546B2 (en) | 2004-11-05 | 2016-11-22 | Boehringer Ingelheim International Gmbh | Process for the preparation of chiral 8-(3-aminopiperidin-1-yl)-xanthines |
| US9751855B2 (en) | 2004-11-05 | 2017-09-05 | Boehringer Ingelheim International Gmbh | Process for the preparation of chiral 8-(3-aminopiperidin-1-yl)-xanthines |
| EP2305659A1 (en) | 2004-11-29 | 2011-04-06 | Novartis AG | 5-hydroxy-benzothiazole derivatives having beta-2-adrenoreceptor agonist activity |
| WO2006056471A1 (en) | 2004-11-29 | 2006-06-01 | Novartis Ag | 5-hydroxy-benzothiazole derivatives having beta-2-adrenorecptor agonist activity |
| EP2253612A1 (en) | 2005-04-14 | 2010-11-24 | Novartis AG | Organic compounds |
| WO2006108643A2 (en) | 2005-04-14 | 2006-10-19 | Novartis Ag | Organic compounds |
| US7994211B2 (en) | 2005-08-08 | 2011-08-09 | Argenta Discovery Limited | Bicyclo[2.2.1]hept-7-ylamine derivatives and their uses |
| EP2280006A1 (en) | 2005-08-08 | 2011-02-02 | Pulmagen Therapeutics (Synergy) Limited | Pharmaceutical composition for inhalation comprising an oxazole or thiazole m3 muscarinic receptor antagonist |
| EP2281813A1 (en) | 2005-08-08 | 2011-02-09 | Pulmagen Therapeutics (Synergy) Limited | Bicyclo[2.2.1]hept-7-ylamine derivatives and their uses |
| EP2532679A1 (en) | 2005-10-21 | 2012-12-12 | Novartis AG | Human antibodies against il13 and therapeutic uses |
| EP2532677A1 (en) | 2005-10-21 | 2012-12-12 | Novartis AG | Human antibodies against il13 and therapeutic uses |
| EP2286813A2 (en) | 2006-01-31 | 2011-02-23 | Novartis AG | Use of naphthyridine derivatives as medicaments |
| WO2007090859A1 (en) * | 2006-02-10 | 2007-08-16 | Glaxo Group Limited | Succinic acid salt of biphenyl-2-ylcarbamic acid 1-[2-(2-chloro-4-{ [ (r)-2-hydroxy-2- (8-hydroxy-2-oxo-1, 2-dihydroquinolin-5-yl) eth ylaminoimethyl] } -5-methoxyphenylcarbamoyl) ethyl] piperidin-4-yl ester and its use for the treatment of pulmonary disorders |
| AU2007213767B2 (en) * | 2006-02-10 | 2012-04-26 | Theravance Biopharma R&D Ip, Llc | Succinic acid salt of biphenyl-2-ylcarbamic acid 1-[2-(2-chloro-4-{ [ (R)-2-hydroxy-2- (8-hydroxy-2-oxo-1, 2-dihydroquinolin-5-yl) ethylaminoimethyl] } -5-methoxyphenylcarbamoyl) ethyl] piperidin-4-yl ester and its use for the treatment of pulmonary disorders |
| US7960551B2 (en) | 2006-02-10 | 2011-06-14 | Glaxo Group Limited | Compound |
| NO341343B1 (en) * | 2006-02-10 | 2017-10-16 | Glaxo Group Ltd | Succinic acid salt of biphenyl-2-ylcarbamic acid 1- [2- (2-chloro-4 - {[(R) -2-hydroxy-2- (8-hydroxy-2-oxo-1,2-dihydroquinolin-5-yl) ethylamino] methyl] - 5-methoxyphenylcarbamoyl) ethyl] piperidin-4-yl ester, a pharmaceutical composition comprising the succinic salt, its use, method of preparation thereof and combination thereof with an anti-inflammatory agent |
| EA016580B1 (en) * | 2006-02-10 | 2012-06-29 | Глаксо Груп Лимитед | Succinic acid salt 1-[2-(2-chloro-4-{[(r)-2-hydroxy-2-(8-hydroxy-2-oxo-1,2-dihydroquinolin-5-yl)ethylamino]methyl]}-5-methoxyphenylcarbamoyl)ethyl]piperidin-4-yl ester biphenyl-2-ylcarbamic acid and its use for the treatment of pulmonary |
| KR101488691B1 (en) * | 2006-02-10 | 2015-02-02 | 글락소 그룹 리미티드 | Succinic acid salt of biphenyl-2-ylcarbamic acid 1-〔2-(2-chloro-4-{[(r)-2-hydroxy-2-(8-hydroxy-2-oxo-1,2-dihydroquinolin-5-yl)ethylamino]methyl}-5-methoxyphenylcarbamoyl)ethyl〕piperidin-4-yl ester and its use for the treatment of pulmonary disorders |
| US7612084B2 (en) | 2006-03-20 | 2009-11-03 | Pfizer Inc | Amine derivatives for the treatment of asthma and COPD |
| EP2322525A1 (en) | 2006-04-21 | 2011-05-18 | Novartis AG | Purine derivatives for use as adenosin A2A receptor agonists |
| WO2007121920A2 (en) | 2006-04-21 | 2007-11-01 | Novartis Ag | Purine derivatives for use as adenosin a2a receptor agonists |
| JP2009535340A (en) * | 2006-04-25 | 2009-10-01 | セラヴァンス, インコーポレーテッド | Crystalline form of dimethylphenyl compounds |
| US7880010B2 (en) | 2006-04-25 | 2011-02-01 | Theravance, Inc. | Crystalline forms of a dimethylphenyl compound |
| JP2009541209A (en) * | 2006-04-25 | 2009-11-26 | セラヴァンス, インコーポレーテッド | Dialkylphenyl compounds having β2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
| WO2007127297A3 (en) * | 2006-04-25 | 2007-12-21 | Theravance Inc | Crystalline forms of a dimethylphenyl compound |
| WO2007127297A2 (en) * | 2006-04-25 | 2007-11-08 | Theravance, Inc. | Crystalline forms of a dimethylphenyl compound |
| US9815837B2 (en) | 2006-05-04 | 2017-11-14 | Boehringer Ingelheim International Gmbh | Polymorphs |
| US11919903B2 (en) | 2006-05-04 | 2024-03-05 | Boehringer Ingelheim International Gmbh | Polymorphs |
| US9266888B2 (en) | 2006-05-04 | 2016-02-23 | Boehringer Ingelheim International Gmbh | Polymorphs |
| US9173859B2 (en) | 2006-05-04 | 2015-11-03 | Boehringer Ingelheim International Gmbh | Uses of DPP IV inhibitors |
| US9493462B2 (en) | 2006-05-04 | 2016-11-15 | Boehringer Ingelheim International Gmbh | Polymorphs |
| US10080754B2 (en) | 2006-05-04 | 2018-09-25 | Boehringer Ingelheim International Gmbh | Uses of DPP IV inhibitors |
| US10301313B2 (en) | 2006-05-04 | 2019-05-28 | Boehringer Ingelheim International Gmbh | Polymorphs |
| US11033552B2 (en) | 2006-05-04 | 2021-06-15 | Boehringer Ingelheim International Gmbh | DPP IV inhibitor formulations |
| US11084819B2 (en) | 2006-05-04 | 2021-08-10 | Boehringer Ingelheim International Gmbh | Polymorphs |
| US11291668B2 (en) | 2006-05-04 | 2022-04-05 | Boehringer Ingelheim International Gmbh | Uses of DPP IV inhibitors |
| WO2008041095A1 (en) * | 2006-10-04 | 2008-04-10 | Pfizer Limited | Sulfonamide derivatives as adrenergic agonists and muscarinic antagonists |
| EP2279777A2 (en) | 2007-01-10 | 2011-02-02 | Irm Llc | Compounds and compositions as channel activating protease inhibitors |
| EP2332933A1 (en) | 2007-05-07 | 2011-06-15 | Novartis AG | Epithelial sodium channel (ENaC) inhibitors |
| EP2444120A1 (en) | 2007-12-10 | 2012-04-25 | Novartis AG | Spirocyclic amiloride analogues as ENac blockers |
| EP2520574A1 (en) | 2007-12-10 | 2012-11-07 | Novartis AG | Amiloride analogues substituted on the cyclic guanidine moiety as ENaC blockers for treating respiratory diseases |
| WO2009087224A1 (en) | 2008-01-11 | 2009-07-16 | Novartis Ag | Pyrimidines as kinase inhibitors |
| US9415016B2 (en) | 2008-04-03 | 2016-08-16 | Boehringer Ingelheim International Gmbh | DPP-IV inhibitor combined with a further antidiabetic agent, tablets comprising such formulations, their use and process for their preparation |
| US10973827B2 (en) | 2008-04-03 | 2021-04-13 | Boehringer Ingelheim International Gmbh | DPP-IV inhibitor combined with a further antidiabetic agent, tablets comprising such formulations, their use and process for their preparation |
| US10022379B2 (en) | 2008-04-03 | 2018-07-17 | Boehringer Ingelheim International Gmbh | DPP-IV inhibitor combined with a further antidiabetic agent, tablets comprising such formulations, their use and process for their preparation |
| WO2009150137A2 (en) | 2008-06-10 | 2009-12-17 | Novartis Ag | Organic compounds |
| KR101235962B1 (en) * | 2008-07-11 | 2013-02-21 | 화이자 리미티드 | Triazole derivatives useful for the treatment of diseases |
| WO2010004517A1 (en) * | 2008-07-11 | 2010-01-14 | Pfizer Limited | Triazole derivatives useful for the treatment of diseases |
| JP2011527672A (en) * | 2008-07-11 | 2011-11-04 | ファイザー・リミテッド | Triazole derivatives useful for treating diseases |
| US8263623B2 (en) | 2008-07-11 | 2012-09-11 | Pfizer Inc. | Triazol derivatives useful for the treatment of diseases |
| US10034877B2 (en) | 2008-08-06 | 2018-07-31 | Boehringer Ingelheim International Gmbh | Treatment for diabetes in patients inappropriate for metformin therapy |
| US9486526B2 (en) | 2008-08-06 | 2016-11-08 | Boehringer Ingelheim International Gmbh | Treatment for diabetes in patients inappropriate for metformin therapy |
| US11911388B2 (en) | 2008-10-16 | 2024-02-27 | Boehringer Ingelheim International Gmbh | Treatment for diabetes in patients with insufficient glycemic control despite therapy with an oral or non-oral antidiabetic drug |
| US9212183B2 (en) | 2008-12-23 | 2015-12-15 | Boehringer Ingelheim International Gmbh | Salt forms of 1-[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1-yl)-8-(3-(R)-amino-piperidin-1-yl)-xanthine |
| WO2010119064A1 (en) | 2009-04-14 | 2010-10-21 | Glaxo Group Limited | Process for the preparation of a biphenyl-2-ylcarbamic acid ester |
| WO2011050325A1 (en) | 2009-10-22 | 2011-04-28 | Vertex Pharmaceuticals Incorporated | Compositions for treatment of cystic fibrosis and other chronic diseases |
| EP2813227A1 (en) | 2009-10-22 | 2014-12-17 | Vertex Pharmaceuticals Incorporated | Compositions for treatment of cystic fibrosis and other chronic diseases |
| US10092571B2 (en) | 2009-11-27 | 2018-10-09 | Boehringer Ingelheim International Gmbh | Treatment of genotyped diabetic patients with DPP-IV inhibitors such as linagliptin |
| US9457029B2 (en) | 2009-11-27 | 2016-10-04 | Boehringer Ingelheim International Gmbh | Treatment of genotyped diabetic patients with DPP-IV inhibitors such as linagliptin |
| WO2011081937A1 (en) | 2009-12-15 | 2011-07-07 | Gilead Sciences, Inc. | Corticosteroid-beta-agonist-muscarinic antagonist compounds for use in therapy |
| EP2845593A1 (en) | 2010-03-19 | 2015-03-11 | Novartis AG | Pyridine and pyrazine derivative for the treatment of chronic obstructive pulmonary disease |
| WO2011113894A1 (en) | 2010-03-19 | 2011-09-22 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of cf |
| US10004747B2 (en) | 2010-05-05 | 2018-06-26 | Boehringer Ingelheim International Gmbh | Combination therapy |
| US9603851B2 (en) | 2010-05-05 | 2017-03-28 | Boehringer Ingelheim International Gmbh | Combination therapy |
| US9643961B2 (en) | 2010-05-13 | 2017-05-09 | Almirall, S.A. | Cyclohexylamine derivatives having β2 adrenergic antagonist and M3 muscarinic antagonist activities |
| US9315463B2 (en) | 2010-05-13 | 2016-04-19 | Almirall, S.A. | Cyclohexylamine derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activities |
| WO2012007539A1 (en) | 2010-07-14 | 2012-01-19 | Novartis Ag | Ip receptor agonist heterocyclic compounds |
| WO2012032546A3 (en) * | 2010-09-08 | 2012-06-28 | Cadila Healthcare Limited | Process for the preparation of salmeterol and its intermediates |
| WO2012034095A1 (en) | 2010-09-09 | 2012-03-15 | Irm Llc | Compounds and compositions as trk inhibitors |
| WO2012034091A1 (en) | 2010-09-09 | 2012-03-15 | Irm Llc | Imidazo [1, 2] pyridazin compounds and compositions as trk inhibitors |
| WO2012035158A1 (en) | 2010-09-17 | 2012-03-22 | Novartis Ag | Pyrazine derivatives as enac blockers |
| US9034883B2 (en) | 2010-11-15 | 2015-05-19 | Boehringer Ingelheim International Gmbh | Vasoprotective and cardioprotective antidiabetic therapy |
| US11911387B2 (en) | 2010-11-15 | 2024-02-27 | Boehringer Ingelheim International Gmbh | Vasoprotective and cardioprotective antidiabetic therapy |
| WO2012116217A1 (en) | 2011-02-25 | 2012-08-30 | Irm Llc | Compounds and compositions as trk inhibitors |
| WO2012168349A1 (en) | 2011-06-10 | 2012-12-13 | Chiesi Farmaceutici S.P.A. | Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
| WO2012168359A1 (en) | 2011-06-10 | 2012-12-13 | Chiesi Farmaceutici S.P.A. | Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
| US9012644B2 (en) | 2011-06-10 | 2015-04-21 | Chiesi Farmaceutici S.P.A. | Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
| US8883800B2 (en) | 2011-07-15 | 2014-11-11 | Boehringer Ingelheim International Gmbh | Substituted quinazolines, the preparation thereof and the use thereof in pharmaceutical compositions |
| US8962636B2 (en) | 2011-07-15 | 2015-02-24 | Boehringer Ingelheim International Gmbh | Substituted quinazolines, the preparation thereof and the use thereof in pharmaceutical compositions |
| US9199998B2 (en) | 2011-07-15 | 2015-12-01 | Boehringer Ingelheim Internatioal Gmbh | Substituted quinazolines, the preparation thereof and the use thereof in pharmaceutical compositions |
| WO2013030802A1 (en) | 2011-09-01 | 2013-03-07 | Novartis Ag | Bicyclic heterocycle derivatives for the treatment of pulmonary arterial hypertension |
| WO2013038386A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Heterocyclic compounds for the treatment of cystic fibrosis |
| WO2013038390A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | N-substituted heterocyclyl carboxamides |
| WO2013038378A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Pyridine amide derivatives |
| WO2013038381A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Pyridine/pyrazine amide derivatives |
| WO2013038373A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Pyridine amide derivatives |
| US9233108B2 (en) | 2011-11-11 | 2016-01-12 | Almirall, S.A. | Cyclohexylamine derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activities |
| US9549934B2 (en) | 2011-11-11 | 2017-01-24 | Almirall, S.A. | Cyclohexylamine derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activities |
| US9757383B2 (en) | 2011-11-11 | 2017-09-12 | Almirall, S.A. | Cyclohexylamine derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activities |
| US10300072B2 (en) | 2011-11-11 | 2019-05-28 | Almirall, S.A. | Cyclohexylamine derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activities |
| WO2013105061A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | Fused dihydropyrido [2,3 -b] pyrazines as ip receptor agonists for the treatment of pulmonary arterial hypertension (pah) and related disorders |
| WO2013105058A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | 7,8- dihydropyrido [3, 4 - b] pyrazines as ip receptor agonists for the treatment of pulmonary arterial hypertension (pah) and related disorders |
| WO2013105065A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | Fused piperidines as ip receptor agonists for the treatment of pah and related disorders |
| WO2013105063A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | Fused piperidines as ip receptor agonists for the treatment of pulmonary arterial hypertension (pah) and related disorders |
| WO2013105057A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | Fused pyrroles as ip receptor agonists for the treatment of pulmonary arterial hypertension (pah) and related disorders |
| WO2013105066A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | Salts of an ip receptor agonist |
| US9555001B2 (en) | 2012-03-07 | 2017-01-31 | Boehringer Ingelheim International Gmbh | Pharmaceutical composition and uses thereof |
| WO2013140319A1 (en) | 2012-03-19 | 2013-09-26 | Novartis Ag | Crystalline form of a succinate salt |
| US10195203B2 (en) | 2012-05-14 | 2019-02-05 | Boehringr Ingelheim International GmbH | Use of a DPP-4 inhibitor in podocytes related disorders and/or nephrotic syndrome |
| US9526730B2 (en) | 2012-05-14 | 2016-12-27 | Boehringer Ingelheim International Gmbh | Use of a DPP-4 inhibitor in podocytes related disorders and/or nephrotic syndrome |
| US9713618B2 (en) | 2012-05-24 | 2017-07-25 | Boehringer Ingelheim International Gmbh | Method for modifying food intake and regulating food preference with a DPP-4 inhibitor |
| US10040765B2 (en) | 2012-09-21 | 2018-08-07 | Crystal Pharma S.A.U. | Methods for the preparation of indacaterol and pharmaceutically acceptable salts thereof |
| EP3345904A1 (en) | 2012-12-06 | 2018-07-11 | Chiesi Farmaceutici S.p.a. | Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
| WO2014086927A1 (en) | 2012-12-06 | 2014-06-12 | Chiesi Farmaceutici S.P.A. | Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
| WO2014086924A1 (en) | 2012-12-06 | 2014-06-12 | Chiesi Farmaceutici S.P.A. | Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
| US9518050B2 (en) | 2012-12-18 | 2016-12-13 | Almirall, S.A. | Cyclohexyl and quinuclidinyl carbamate derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activity |
| WO2014125413A1 (en) | 2013-02-13 | 2014-08-21 | Novartis Ag | Ip receptor agonist heterocyclic compounds |
| US9562039B2 (en) | 2013-02-27 | 2017-02-07 | Almirall, S.A. | Salts of 2-amino-1-hydroxyethyl-8-hydroxyquinolin-2(1H)-one derivatives having both β2 adrenergic receptor agonist and M3 muscarinic receptor antagonist activities |
| WO2014132220A1 (en) | 2013-03-01 | 2014-09-04 | Novartis Ag | Solid forms of bicyclic heterocyclic derivatives as pdgf receptor mediators |
| WO2015008229A1 (en) | 2013-07-18 | 2015-01-22 | Novartis Ag | Autotaxin inhibitors |
| WO2015008230A1 (en) | 2013-07-18 | 2015-01-22 | Novartis Ag | Autotaxin inhibitors comprising a heteroaromatic ring-benzyl-amide-cycle core |
| US10456390B2 (en) | 2013-07-25 | 2019-10-29 | Almirall, S.A. | Combinations comprising MABA compounds and corticosteroids |
| US9526728B2 (en) | 2014-02-28 | 2016-12-27 | Boehringer Ingelheim International Gmbh | Medical use of a DPP-4 inhibitor |
| WO2015162461A1 (en) | 2014-04-24 | 2015-10-29 | Novartis Ag | Pyrazine derivatives as phosphatidylinositol 3-kinase inhibitors |
| WO2015162456A1 (en) | 2014-04-24 | 2015-10-29 | Novartis Ag | Amino pyridine derivatives as phosphatidylinositol 3-kinase inhibitors |
| WO2015162459A1 (en) | 2014-04-24 | 2015-10-29 | Novartis Ag | Amino pyrazine derivatives as phosphatidylinositol 3-kinase inhibitors |
| US10005771B2 (en) | 2014-09-26 | 2018-06-26 | Almirall, S.A. | Bicyclic derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activities |
| WO2016128456A1 (en) | 2015-02-12 | 2016-08-18 | Chiesi Farmaceutici S.P.A. | Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
| WO2016193241A1 (en) | 2015-06-01 | 2016-12-08 | Chiesi Farmaceutici S.P.A. | Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
| WO2017093208A1 (en) | 2015-12-03 | 2017-06-08 | Chiesi Farmaceutici S.P.A. | Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
| US10155000B2 (en) | 2016-06-10 | 2018-12-18 | Boehringer Ingelheim International Gmbh | Medical use of pharmaceutical combination or composition |
| WO2018011090A1 (en) | 2016-07-13 | 2018-01-18 | Chiesi Farmaceutici S.P.A. | Hydroxyquinolinone compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity |
| WO2018108089A1 (en) | 2016-12-14 | 2018-06-21 | 北京硕佰医药科技有限责任公司 | Class of bifunctional compounds with quaternary ammonium salt structure |
| WO2020250116A1 (en) | 2019-06-10 | 2020-12-17 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of cf, copd, and bronchiectasis |
| WO2021038426A1 (en) | 2019-08-28 | 2021-03-04 | Novartis Ag | Substituted 1,3-phenyl heteroaryl derivatives and their use in the treatment of disease |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2246345B1 (en) | Biphenyl derivatives having beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity | |
| EP1778626A1 (en) | Compounds having beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 169922 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004213411 Country of ref document: AU Ref document number: 3375/DELNP/2005 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 541579 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005/06215 Country of ref document: ZA Ref document number: 200506215 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 05079299 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2515777 Country of ref document: CA Ref document number: PA/a/2005/008528 Country of ref document: MX Ref document number: 378025 Country of ref document: PL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006503604 Country of ref document: JP Ref document number: 1020057014929 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 2004213411 Country of ref document: AU Date of ref document: 20040213 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004213411 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004711253 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20048065284 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005128557 Country of ref document: RU |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020057014929 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1-2005-501486 Country of ref document: PH |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004711253 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0407508 Country of ref document: BR |