WO2005030678A2 - AMINO-SUBSTITUTED ETHYLAMINO β2 ADRENERGIC RECEPTOR AGONISTS - Google Patents
AMINO-SUBSTITUTED ETHYLAMINO β2 ADRENERGIC RECEPTOR AGONISTS Download PDFInfo
- Publication number
- WO2005030678A2 WO2005030678A2 PCT/US2004/030833 US2004030833W WO2005030678A2 WO 2005030678 A2 WO2005030678 A2 WO 2005030678A2 US 2004030833 W US2004030833 W US 2004030833W WO 2005030678 A2 WO2005030678 A2 WO 2005030678A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- phenyl
- ethylamino
- hydroxy
- phenylethylamino
- Prior art date
Links
- 0 Cc1c(*)c(*)c(*)c(C(CNC(*)(*)Cc(cc2)ccc2NCC(*)c2ccc(*)c(*)c2*)O)c1 Chemical compound Cc1c(*)c(*)c(*)c(C(CNC(*)(*)Cc(cc2)ccc2NCC(*)c2ccc(*)c(*)c2*)O)c1 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/34—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by amino groups
- C07C233/42—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by amino groups with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by a carbon atom of a six-membered aromatic ring
- C07C233/43—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by amino groups with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by a carbon atom of a six-membered aromatic ring having the carbon atom of the carboxamide group bound to a hydrogen atom or to a carbon atom of a saturated carbon skeleton
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/08—Bronchodilators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/06—Antiabortive agents; Labour repressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/20—Oxygen atoms
- C07D215/24—Oxygen atoms attached in position 8
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/20—Oxygen atoms
- C07D215/24—Oxygen atoms attached in position 8
- C07D215/26—Alcohols; Ethers thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/38—Nitrogen atoms
Definitions
- the invention is directed to novel ⁇ 2 adrenergic receptor agonists.
- the invention is also directed to pharmaceutical compositions comprising such compounds, methods of using such compounds to treat diseases associated with ⁇ 2 adrenergic receptor activity, and processes and intermediates useful for preparing such compounds.
- Background of the invention ⁇ 2 Adrenergic receptor agonists are recognized as effective drugs for the treatment of pulmonary diseases such as asthma and chronic obstructive pulmonary disease (including chronic bronchitis and emphysema).
- ⁇ 2 Adrenergic receptor agonists are also useful for treating pre-term labor, and are potentially useful for treating neurological disorders and cardiac disorders, i spite of the success that has been achieved with certain ' ⁇ 2 adrenergic receptor agonists, current agents possess less than desirable duration of action, potency, selectivity, and/or onset.
- ⁇ 2 adrenergic receptor agonists having improved properties, such as. improved duration of action, potency, selectivity, and/or onset.
- the invention provides novel compounds that possess ⁇ 2 adrenergic receptor agonist activity.
- compounds of the invention have been found to be potent and selective ⁇ 2 adrenergic receptor agonists.
- compounds of the invention have been found to possess a surprising and unexpectedly long duration of action, which allows for once-daily, or even less frequent, dosing. Accordingly, this invention provides a compound of formula (I):
- the invention also provides pharmaceutical compositions comprising a compound of the invention and a pharmaceutically-acceptable carrier.
- the invention further provides combinations comprising a compound of the invention and one or more other therapeutic agents and pharmaceutical compositions comprising such combinations.
- the invention also provides a method of treating a disease or condition associated with ⁇ 2 adrenergic receptor activity (e.g. a pulmonary disease, such as asthma or chronic obstructive pulmonary disease, pre-term labor, a neurological disorder, a cardiac disorder, or inflammation) in a mammal, comprising administering to the mammal, a therapeutically effective amount of a compound of the invention.
- a disease or condition associated with ⁇ 2 adrenergic receptor activity e.g. a pulmonary disease, such as asthma or chronic obstructive pulmonary disease, pre-term labor, a neurological disorder, a cardiac disorder, or inflammation
- the invention further provides a method of treatment comprising administering a therapeutically effective amount of a combination of a compound of the invention together with one or more other therapeutic agents.
- the invention also provides a method of treating a disease or condition associated with ⁇ 2 adrenergic receptor activity in a mammal, comprising administering to the mammal, a therapeutically effective amount of a pharmaceutical composition of the invention.
- the compounds of the invention can also be used as research tools, i.e. to study biological systems or samples, or to discover new ⁇ 2 adrenergic receptor agonists.
- the invention is directed to a method of agonizing a ⁇ 2 adrenergic receptor in a biological system or sample, the method comprising contacting a biological system or sample comprising a ⁇ 2 adrenergic receptor with ⁇ 2 adrenergic receptor-agonizing amount of a compound of formula (I), or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- the invention also provides synthetic processes and intermediates described herein, which are useful for preparing compounds of the invention.
- the invention also provides a compound of the invention as described herein for use in medical therapy, as well as the use of a compound of the invention in the manufacture of a formulation or medicament for treating a disease or condition associated with ⁇ 2 adrenergic receptor activity, e.g. a pulmonary disease, such as asthma or chronic obstructive pulmonary disease, pre-term labor, a neurological disorder, a cardiac disorder, or inflammation, in a mammal.
- a pulmonary disease such as asthma or chronic obstructive pulmonary disease, pre-term labor, a neurological disorder, a cardiac disorder, or inflammation, in a mammal.
- the invention provides novel amino-substituted ethylamino ⁇ 2 adrenergic receptor agonists of formula (I), or pharmaceutically-acceptable salts or solvates or stereoisomers thereof.
- the following exemplary and preferred values for radicals, substituents, and ranges, are for illustration only; they do not exclude other defined values or other values within defined ranges for the radicals and substituents.
- Examples of particular values of R 1 are halo, -CH 2 OH, and-NHCHO, including chloro, -CH 2 OH, and-NHCHO. Another particular value for R 1 is -CH 2 OH or -NHCHO.
- a particular value for R 2 is hydrogen.
- a particular value for R 1 and R 2 is R 1 and R 2 taken together are
- R 3 is hydroxy, and R 2 and R 4 are each hydrogen.
- Another specific value for R 1 , R 2 , R 3 , and R 4 is R 1 is -CH 2 OH, R 3 is hydroxy, and
- R and R are each hydrogen. Yet another specific value for R 1 , R 2 , R 3 , and R 4 is R 1 and R 4 are chloro, R 3 is amino, and R 2 is hydrogen. Examples of particular values for R 5 and R 6 are R 5 and R 6 are each independently selected from hydrogen, C 1-6 alkyl, and C 3-6 cycloalkyl, wherein each C 1-6 alkyl is optionally substituted with one or more substituents independently selected from heterocyclyl, -OR , and -NR R c .
- R 5 and R 6 are R 5 and R 6 together with the nitrogen atom to which they are attached form a heterocyclic ring having from 5 to 7 ring atoms and containing 1 or 2 heteroatoms independently selected from oxygen, nitrogen, and sulfur.
- R 5 and R 6 are each independently hydrogen or C 1-3 alkyl, wherein each C 1-3 alkyl is optionally substituted with one substituent independently selected from hydroxyl, amino, piperidinyl, and pyrrolidinyl.
- R 5 and R 6 together with the nitrogen atom to which they are attached form a morpholinyl or piperidinyl ring.
- R 5 and R 6 are each independently hydrogen or C 1-3 alkyl.
- a particular value for R 7 is hydrogen.
- a particular value for R 8 is hydrogen.
- Examples of particular values for R 9 are hydrogen, halo and -OR a where R a is hydrogen or C 1-3 alkyl. Another example of particular values for R 9 is hydroxy and methoxy.
- Another particular value for R 9 is hydrogen.
- Examples of particular values for R 10 are hydrogen, halo and -OR a where R a is hydrogen or C 1-3 alkyl. Another example of particular values for R 10 is hydroxy and methoxy.
- Another particular value for R 10 is hydrogen.
- Examples of particular values for R 11 are hydrogen, halo and -OR a where R a is hydrogen or C 1-3 alkyl. Another example of particular values for R 11 is hydroxy and methoxy. Another particular value for R 11 is hydrogen.
- a compound of formula (I) is a compound of formula (11):
- One group of compounds of formula (13) are those in which each of R 5 and R 6 is independently selected from hydrogen, C 1-6 alkyl, and C 3-6 cycloalkyl, wherein each C 1-6 alkyl is optionally substituted with one or more substituents independently selected from heterocyclyl, -OR a , and -NR R c , or R 5 and R 6 together with the nitrogen atom to which they are attached form a heterocyclic ring having from 5 to 7 ring atoms and containing 1 or 2 heteroatoms independently selected from oxygen, nitrogen, and sulfur.
- each of R and R is independently hydrogen or C 1-3 alkyl, wherein each C 1-3 alkyl is optionally substituted with one substituent independently selected from hydroxyl, amino, piperidinyl, and pyrrolidinyl; or R 5 and R 6 together with the nitrogen atom to which they are attached form a morpholinyl or piperidinyl ring, hi yet another group of compounds of formula (13), each of R 5 and R 6 is hydrogen or C 1-3 alkyl.
- the compounds of the invention contain one or more chiral centers. Accordingly, the invention includes racemic mixtures, pure stereoisomers (i.e. individual enantiomers or diastereomers), and stereoisomer-enriched mixtures of such isomers, unless otherwise indicated.
- stereoisomer is shown, it will be understood by those skilled in the art, that minor amounts of other stereoisomers may be present in the compositions of this invention unless otherwise indicated, provided that the utility of the composition as a whole is not eliminated by the presence of such other isomers.
- compounds of the invention contain a chiral center at the alkylene carbon in formulas (I) and (13) to which the hydroxy group is attached.
- the amount of the stereoisomer with the (R) orientation at the chiral center bearing the hydroxy group is advantageous for the amount of the stereoisomer with the (R) orientation at the chiral center bearing the hydroxy group to be greater than the amount of the corresponding (S) stereoisomer.
- the (R) stereoisomer is preferred over the (S) stereoisomer.
- alkyl means a monovalent saturated hydrocarbon group which may be linear or branched or combinations thereof.
- Representative alkyl groups include, by way of example, methyl, ethyl, w-propyl, isopropyi, n-butyl, sec-butyl, isobutyl, tert-butyl, n- pentyl, n-hexyl, w-heptyl, n-octyl, ra-nonyl, «-decyl and the like.
- C 1-6 alkyl means an alkyl group having from 1 to 6 carbon atoms.
- alkenyl means a monovalent unsaturated hydrocarbon group containing at least one carbon-carbon double bond, typically 1 or 2 carbon-carbon double bonds, and which may be linear or branched or combinations thereof.
- alkenyl groups include, by way of example, vinyl, allyl, isopropenyl, but-2-enyl, ra-pent-2- enyl, ra-hex-2-enyl, n-hept-2-enyl, n-oct-2-enyl, n-non-2-enyl, n-dec-4-enyl, w-dec-2,4- dienyl and the like.
- alkynyl means a monovalent unsaturated hydrocarbon group containing at least one carbon-carbon triple bond, typically 1 carbon-carbon triple bond, and which may be linear or branched or combinations thereof.
- Representative alkynyl groups include, by way of example, ethynyl, propargyl, but-2-ynyl and the like.
- cycloalkyl means a monovalent saturated carbocyclic group which may be monocyclic or multicyclic.
- Representative cycloalkyl groups include, by way of example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, and the like.
- aryl means a monovalent aromatic hydrocarbon having a single ring (i.e. phenyl) or fused rings (i.e.napthalene).
- aryl groups typically contain from 6 to 10 carbon ring atoms.
- Representative aryl groups include, by way of example, phenyl, and napthalene-1-yl, napthalene-2-yl and the like.
- heteroaryl means a monovalent aromatic group having a single ring or two fused rings and containing in the ring at least one heteroatom (typically 1 to 3 heteroatoms) selected from nitrogen, oxygen, and sulfur. Unless otherwise defined, such heteroaryl groups typically contain from 5 to 10 atoms total ring atoms.
- heteroaryl groups include, byway of example, pyrroyl, isoxazolyl, isothiazolyl, pyrazolyl, pyridyl (or, equivalently, pyridinyl), oxazolyl, oxadiazolyl, thiadiazolyl, thiazolyl, imidazolyl, triazolyl, tetrazolyl, furanyl, triazinyl, thienyl, pyrimidyl, pyridazinyl, pyrazinyi, benzoxazolyl, benzothiazolyl, benzimidazolyl, benzoftiranyl, benzothiophenyl, quinolyl, indolyl, isoquinolyl and the like, where the point of attachment is at any available carbon or nitrogen ring atom.
- heterocyclyl or “heterocyclic ring” means a monovalent saturated or partially unsaturated cyclic non-aromatic group, which may be monocyclic or multicyclic (i.e., fused or bridged), and which contains at least one heteroatom (typically 1 to 3 heteroatoms) selected from nitrogen, oxygen, and sulfur. Unless otherwise defined, such heterocyclyl groups typically contain from 5 to 10 total ring atoms.
- heterocyclyl groups include, by way of example, pyrrolidinyl, piperidinyl, piperazinyl, imidazolidinyl, morpholinyl, indolin-3-yl, 2-imidazolinyl, 1,2,3,4-tetrahydroisoquinolin- 2-yl, quinuclidinyl, and the like.
- halo means fluoro, chloro, bromo or iodo.
- amino means -NH 2 .
- therapeutically effective amount means an amount sufficient to effect treatment when administered to a patient in need of treatment.
- treatment means the treatment of a disease or medical condition in a patient, such as a mammal (particularly a human) which includes: (a) preventing the disease or medical condition from occurring, i.e., prophylactic treatment of a patient; (b) ameliorating the disease or medical condition, i.e., eliminating or causing regression of the disease or medical condition in a patient; (c) suppressing the disease or medical condition, i.e., slowing or arresting the development of the disease or medical condition in a patient; or (d) alleviating the symptoms of the disease or medical condition in a patient.
- disease or condition associated with ⁇ 2 adrenergic receptor activity includes all disease states and/or conditions that are acknowledged now, or that are found in the future, to be associated with ⁇ 2 adrenergic receptor activity.
- disease states include, but are not limited to, pulmonary diseases, such as asthma and chronic obstructive pulmonary disease (including chronic bronchitis and emphysema), as well as neurological disorders and cardiac disorders.
- pulmonary diseases such as asthma and chronic obstructive pulmonary disease (including chronic bronchitis and emphysema)
- ⁇ 2 adrenergic receptor activity is also known to be associated with pre-term labor (see United States Patent Number 5,872,126) and some types of inflammation (see International Patent Application Publication Number WO 99/30703 and United States Patent Number 5,290,815).
- pharmaceutically-acceptable salt means a salt prepared from a base or acid which is acceptable for administration to a patient, such as a mammal.
- Such salts can be derived from pharmaceutically-acceptable inorganic or organic bases and from pharmaceutically-acceptable inorganic or organic acids.
- Salts derived from pharmaceutically-acceptable acids include, but are not limited to, acetic, benzenesulfonic, benzoic, camphosulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pantothenic, phosphoric, succinic, sulfuric, tartaric, p-toluenesulfonic, xinafoic (l-hydroxy-2-naphthoic acid) and the like.
- Salts derived from fumaric, hydrobromic, hydrochloric, acetic, sulfuric, methanesulfonic, xinafoic, and tartaric acids are particularly preferred.
- Salts derived from pharmaceutically-acceptable inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic, manganous, potassium, sodium, zinc and the like.
- Particularly preferred are ammonium, calcium, magnesium, potassium and sodium salts.
- Salts derived from pharmaceutically- acceptable organic bases include salts of primary, secondary and tertiary amines, including substituted amines, cyclic amines, naturally-occurring amines and the like, such as arginine, betaine, caffeine, choline, N,N'-dibenzylethylenediamine, diethylamine, 2- diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N- ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperadine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine and the like.
- arginine betaine
- caffeine choline
- solvate means a complex or aggregate formed by one or more molecules of a solute, i.e. a compound of the invention or a pharmaceutically-acceptable salt thereof, and one or more molecules of a solvent.
- solvates are typically crystalline solids having a substantially fixed molar ratio of solute and solvent.
- Representative solvents include by way of example, water, methanol, ethanol, isopropanol, acetic acid, and the like. When the solvent is water, the solvate formed is a hydrate.
- a pharmaceutically-acceptable salt or solvate of stereoisomer thereof is intended to include all permutations of salts, solvates and stereoisomers, such as a solvate of a pharmaceutically-acceptable salt of a stereoisomer of a compound of formula (I).
- leaving group means a functional group or atom which can be displaced by another functional group or atom in a substitution reaction, such as a nucleophilic substitution reaction.
- representative leaving groups include chloro, bromo and iodo groups; sulfonic ester groups, such as mesylate, tosylate, brosylate, nosylate and the like; and acyloxy groups, such as acetoxy, trifluoroacetoxy and the like.
- amino-protecting group means a protecting group suitable for preventing undesired reactions at an amino nitrogen.
- Representative amino-protecting groups include, but are not limited to, formyl; acyl groups, for example alkanoyl groups, such as acetyl; alkoxycarbonyl groups, such as tert-butoxycarbonyl (Boc); arylmethoxycarbonyl groups, such as benzyloxycarbonyl (Cbz) and 9-fluorenylmethoxycarbonyl (Fmoc); arylmethyl groups, such as benzyl (Bn), trityl (Tr), and l,l-di-(4'-methoxyphenyl)methyl; silyl groups, such as trimethylsilyl (TMS) and tert- butyldimethylsilyl (TBS); and the like.
- acyl groups for example alkanoyl groups, such as acetyl
- alkoxycarbonyl groups such as tert-butoxycarbonyl (Boc)
- arylmethoxycarbonyl groups
- hydroxy-protecting group means a protecting group suitable for preventing undesired reactions at a hydroxy group.
- Representative hydroxy-protecting groups include, but are not limited to, alkyl groups, such as methyl, ethyl, and tert-butyl; acyl groups, for example alkanoyl groups, such as acetyl; arylmethyl groups, such as benzyl (Bn), -methoxybenzyl (PMB), 9-fluorenylmethyl (Fm), and diphenylmethyl (benzhydryl, DPM); silyl groups, such as trimethylsilyl (TMS) and tert-butyldimethylsilyl (TBS); and the like.
- alkyl groups such as methyl, ethyl, and tert-butyl
- acyl groups for example alkanoyl groups, such as acetyl
- arylmethyl groups such as benzyl (Bn), -methoxybenz
- the protecting group P 2 is typically a benzyl protecting group, which is typically removed from the intermediate of formula 4 by hydrogenation using a palladium on carbon catalyst, to provide the product.
- the compounds of formula 1 employed in the reactions described in this application are readily prepared by procedures known in the art, and described, for example, in U.S. Patent Nos. 6,653,323 B2 and 6,670,376 Bl, which are incorporated herein by reference, and references therein.
- Intermediate 2 is prepared from readily available starting materials, for example, by procedures illustrated in Scheme B.
- P represents an amino-protecting group and P represents an amino- protecting group.
- a protecting group, P 3 is added to the amino nitrogen of 2-(4-nitrophenyl)ethylamine, 5, to provide an intermediate of formula 6.
- Protecting group P 3 is typically a tert-butoxycarbonyl (Boc) group, which is typically added by reaction of di-tert-butyl dicarbonate (Boc 2 O) under basic conditions.
- the intermediate 6 is reduced to provide an intermediate of formula 7. Reduction of the intermediate 6 is typically effected by hydrogenation using a palladium on carbon catalyst.
- the amine of intermediate 7 is coupled with the protected phenyl glycine 8 to provide an intermediate of formula 9.
- the coupling of the intermediate 7 with 8 can be effected using a peptide coupling agent, for example, l-[3-(dimethylamino)propyl]-3-ethylcarbodiimide (EDC), and may employ a catalyst, for example, 1-hydroxybenzotriazole hydrate (HOBT) or l-hydroxy-7-azabenzotriazole hydrate (HO AT).
- EDC l-[3-(dimethylamino)propyl]-3-ethylcarbodiimide
- HOBT 1-hydroxybenzotriazole hydrate
- HO AT l-hydroxy-7-azabenzotriazole hydrate
- Intermediate 9 is deprotected, typically under acidic conditions, to provide an intermediate of formula 10, which is reduced, typically using a borane reductant, to form ( J f )-N -[4-(2-aminoethyl)phenyl]-l-phenyl- ethane- 1,2-diamine (2).
- intermediate 1 is coupled with ( ⁇ S)-2-[4-(2-aminoethyl)- phenylamino]-l-phenylethanol (11) to provide an intermediate of formula 12.
- this reaction is conducted in a polar, aprotic solvent in the presence of base with heating.
- Intermediate 12 is reacted with a reagent such as diphenylphosphoryl azide, which converts the alcohol to a leaving group and provides a nucleophilic azide anion to provide the intermediate of formula 13.
- two-reagent systems can be used to convert intermediate 12 to the azide 13.
- the protecting group P 1 which is typically a silyl protecting group, is removed, typically by use of a fluoride or acid reagent, to provide an intermediate of formula 14.
- the product can be provided by simultaneous hydrogenation of the azide and deprotection of the protecting group P 2 of the intermediate of formula 14, when P 2 is a group, such as benzyl, that is removed by hydrogenation. If the protecting group P 2 is not labile to hydrogenation, an additional deprotection step is required.
- Intermediate 11 is readily prepared by the reaction of 2-(4- aminophenyl)ethylamine with chiral styrene oxide, as described in Example 3, part a, below.
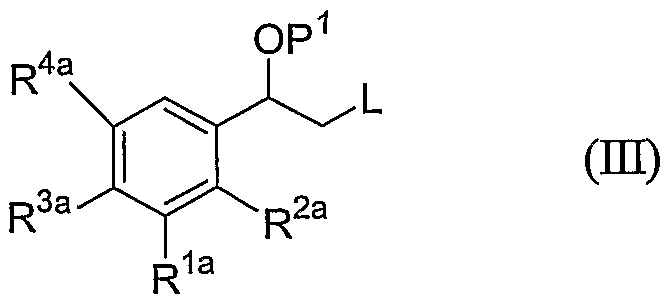
- the invention provides a process for preparing a compound of formula (I), or a salt or stereoisomer or protected derivative thereof, the process comprising: reacting a compound of formula (Hi):
- R la , R 2a , R 3a , and R 4a is independently defined to be the same as R 1 , R 2 , R 3 , and R 4 in formula (I), or -OP 2 , wherein P 2 is a hydroxy-protecting group, with a compound of formula (IV):
- R 5 , R 6 , R 7 , R 8 , R 9 , R 10 , and R 1 * are defined as in formula (I), to provide a compound of formula (V): removing the protecting group P 1 to provide a compound of formula (V3):
- compositions comprising a compound of the invention.
- the compound preferably in the form of a pharmaceutically-acceptable salt, can be formulated for any suitable form of administration, such as oral or parenteral administration, or administration by inhalation.
- the compound can be admixed with conventional pharmaceutical carriers and excipients and used in the form of powders, tablets, capsules, elixirs, suspensions, syrups, wafers, and the like.
- Such pharmaceutical compositions will contain from about 0.05 to about 90% by weight of the active compound, and more generally from about 0.1 to about 30%.
- compositions may contain common carriers and excipients, such as cornstarch or gelatin, lactose, magnesium sulfate, magnesium stearate, sucrose, microcrystalline cellulose, kaolin, mannitol, dicalcium phosphate, sodium chloride, and alginic acid.
- Disintegrators commonly used in the formulations of this invention include croscarmellose, microcrystalline cellulose, cornstarch, sodium starch glycolate and alginic acid.
- a liquid composition will generally consist of a suspension or solution of the compound or pharmaceutically-acceptable salt in a suitable liquid carrier(s), for example ethanol, glycerine, sorbitol, non-aqueous solvent such as polyethylene glycol, oils or water, optionally with a suspending agent, a solubilizing agent (such as a cyclodextrin), preservative, surfactant, wetting agent, flavoring or coloring agent.
- a liquid formulation can be prepared from a reconstitutable powder.
- a powder containing active compound, suspending agent, sucrose and a sweetener can be reconstituted with water to form a suspension; a syrup can be prepared from a powder containing active ingredient, sucrose and a sweetener.
- a composition in the form of a tablet can be prepared using any suitable pharmaceutical carrier(s) routinely used for preparing solid compositions.
- suitable pharmaceutical carrier(s) include magnesium stearate, starch, lactose, sucrose, microcrystalline cellulose and binders, for example polyvinylpyrrolidone.
- the tablet can also be provided with a color film coating, or color included as part of the carrier(s).
- active compound can be formulated in a controlled release dosage form as a tablet comprising a hydrophilic or hydrophobic matrix.
- a composition in the form of a capsule can be prepared using routine encapsulation procedures, for example by incorporation of active compound and excipients into a hard gelatin capsule.
- a semi-solid matrix of active compound and high molecular weight polyethylene glycol can be prepared and filled into a hard gelatin capsule; or a solution of active compound in polyethylene glycol or a suspension in edible oil, for example liquid paraffin or fractionated coconut oil can be prepared and filled into a soft gelatin capsule.
- Tablet binders that can be included are acacia, methylcellulose, sodium carboxymethylcellulose, poly-vinylpyrrolidone (Povidone), hydroxypropyl methylcellulose, sucrose, starch and ethylcellulose.
- Lubricants that can be used include magnesium stearate or other metallic stearates, stearic acid, silicone fluid, talc, waxes, oils and colloidal silica.
- Flavoring agents such as peppennint, oil of wintergreen, cherry flavoring or the like can also be used. Additionally, it may be desirable to add a coloring agent to make the dosage form more attractive in appearance or to help identify the product.
- the compounds of the invention and their pharmaceutically-acceptable salts that are active when given parenterally can be formulated for intramuscular, intrathecal, or intravenous administration.
- a typical composition for infra-muscular or intrathecal administration will consist of a suspension or solution of active ingredient in an oil, for example arachis oil or sesame oil.
- a typical composition for intravenous or intrathecal administration will consist of a sterile isotonic aqueous solution containing, for example active ingredient and dextrose or sodium chloride, or a mixture of dextrose and sodium chloride.
- a sterile isotonic aqueous solution containing, for example active ingredient and dextrose or sodium chloride, or a mixture of dextrose and sodium chloride.
- Other examples are lactated Ringer's injection, lactated Ringer's plus dextrose injection, Normosol-M and dextrose, Isolyte E, acylated Ringer's injection, and the like.
- a co-solvent for example, polyethylene glycol
- a chelating agent for example, ethylenediamine tetraacetic acid
- a solubilizing agent for example, a cyclodextrin
- an anti-oxidant for example, sodium metabisulphite
- the solution can be freeze dried and then reconstituted with a suitable solvent just prior to administration.
- the compounds of this invention and their pharmaceutically-acceptable salts which are active on topical administration can be formulated as transdermal compositions or transdennal delivery devices ("patches").
- Such compositions include, for example, a backing, active compound reservoir, a control membrane, liner and contact adhesive.
- transdermal patches may be used to provide continuous or discontinuous infusion of the compounds of the present invention in controlled amounts.
- the construction and use of transdermal patches for the delivery of pharmaceutical agents is well known in the art. See, for example, U.S. Patent No. 5,023,252.
- Such patches may be constructed for continuous, pulsatile, or on demand delivery of pharmaceutical agents.
- One preferred manner for administering a compound of the invention is inhalation. Inhalation is an effective means for delivering an agent directly to the respiratory tract.
- phannaceutical inhalation devices There are three general types of phannaceutical inhalation devices: nebulizer inhalers, dry powder inhalers (DPI), and metered-dose inhalers (MDI).
- nebulizer devices produce a stream of high velocity air that causes a therapeutic agent to spray as a mist which is carried into the patient's respiratory tract.
- the therapeutic agent is formulated in a liquid form such as a solution or a suspension of micronized particles of respirable size, where micronized is typically defined as having about 90 % or more of the particles with a diameter of less than about 10 ⁇ m.
- a typical formulation for use in a conventional nebulizer device is an isotonic aqueous solution of a pharmaceutical salt of the active agent at a concentration of the active agent of between about 0.05 ⁇ g/mL and about 1 mg/mL.
- Suitable nebulizer devices are provided commercially, for example, by PARI GmbH (Starnberg, Germany).
- nebulizer devices have been disclosed, for example, in U.S. Patent 6,123,068.
- DPI's typically administer a therapeutic agent in the form of a free flowing powder that can be dispersed in a patient's air-stream during inspiration.
- Alternative DPI devices which use an external energy source to disperse the powder are also being developed.
- the therapeutic agent can be formulated with a suitable excipient (e.g., lactose or starch).
- a dry powder formulation can be made, for example, by combining dry lactose particles with micronized particles of a suitable form, typically a pharmaceutically-acceptable salt, of a compound of the invention (i.e. the active agent) and dry blending.
- the agent can be formulated without excipients.
- the formulation is loaded into a dry powder dispenser, or into inhalation cartridges or capsules for use with a dry powder delivery device.
- DPI delivery devices provided commercially include Diskhaler (GlaxoSmithKline, Research Triangle Park, NC) (see, e.g., U.S. Patent No. 5,035,237); Diskus (GlaxoSmithKline) (see, e.g., U.S. Patent No. 6,378,519; Turbuhaler (AstraZeneca, Wilmington, DE) (see, e.g., U.S. Patent No.
- MDI's typically discharge a measured amount of therapeutic agent using compressed propellant gas.
- Formulations for MDI administration include a solution or suspension of active ingredient in a liquefied propellant.
- HFA hydiOfluoroalklanes
- HFA 134a 1,1,1,2-tetrafluoroethane
- HFA 2207 1,1, 1,2,3, 3,3, -heptafluoro-n- propane
- co-solvents such as ethanol or pentane
- surfactants such as sorbitan trioleate, oleic acid, lecithin, and glycerin.
- a suitable formulation for MDI administration can include from about 0.001 % to about 2 % by weight of the present crystalline form, from about 0 % to about 20 % by weight ethanol, and from about 0 % to about 5 % by weight surfactant, with the remainder being the HFA propellant.
- chilled or pressurized hydrofluoroalkane is added to a vial containing the present crystalline form, ethanol (if present) and the surfactant (if present).
- the pharmaceutical salt is provided as micronized particles.
- the formulation is loaded into an aerosol canister, which forms a portion of an MDI device.
- a suspension formulation is prepared by spray drying a coating of surfactant on micronized particles of a pharmaceutical salt of active compound.
- a pharmaceutical salt of active compound See, for example, WO 99/53901 and WO 00/61108.
- processes of preparing respirable particles, and formulations and devices suitable for inhalation dosing see U.S. Patent Nos. 6,268,533, 5,983,956, 5,874,063, and 6,221,398, and WO 99/55319 and WO 00/30614. It will be understood that any form of the compounds of the invention, (i.e.
- the active compounds are useful as ⁇ 2 adrenergic receptor agonists and therefore are useful for treating medical diseases or conditions mediated by ⁇ 2 adrenergic receptors or associated with ⁇ 2 adrenergic receptor activity in a mammal, i.e. medical conditions which are ameliorated by treatment with a ⁇ 2 adrenergic receptor agonist.
- medical conditions include but are not limited to a pulmonary disease, such as asthma or chronic obstructive pulmonary disease, pre-term labor, a neurological disorder, a cardiac disorder, or inflammation.
- the active compounds are effective over a wide dosage range and are generally administered in a therapeutically effective amount. It will be understood, however, that the amount of the compound actually administered will be determined by a physician, in the light of the relevant circumstances, including the condition to be treated, the chosen route of administration, the actual compound administered and its relative activity, the age, weight, and response of the individual patient, the severity of the patient's symptoms, and the like. Suitable doses of the therapeutic agents for inhalation administration are in the general range of from about 0.05 ⁇ g/day to about 1000 ⁇ g/day, preferably from about 0.1 ⁇ g/day to about 500 ⁇ g/day.
- a compound can be administered in a periodic dose: weekly, multiple times per week, daily, or multiple doses per day.
- the treatment regimen may require administration over extended periods of time, for example, for several weeks or months, or the treatment regimen may require chronic admimstration.
- Suitable doses for oral administration are in the general range of from about 0.05 ⁇ g/day to about 100 mg/day, preferably 0.5 to 1000 ⁇ g/day.
- compounds of the invention have been found to be potent and selective agonists of the ⁇ 2 adrenergic receptor.
- compounds of the invention demonstrate excellent selectivity for the ⁇ 2 adrenergic receptor as compared with the ⁇ ! and ⁇ adrenergic receptors. Furthermore, compounds of the invention have been found to possess surprising and unexpected duration of action. As described in the biological assays below, compounds of the invention demonstrated duration of action greater than 24 hours in an animal model of bronchoprotection.
- the invention thus provides a method of treating a disease or condition in a maimnal associated with ⁇ 2 adrenergic receptor activity comprising administering to the mammal a therapeutically effective amount of a compound of the invention or of a pharmaceutical composition comprising a compound of the invention.
- the present active agents can also be co-administered with one or more other therapeutic agents.
- the present agents can be administered in combination with one or more therapeutic agents selected from anti-inflammatory agents (e.g. corticosteroids and non-steroidal anti-inflammatory agents (NSAIDs), antichlolinergic agents (particularly muscarinic receptor antagonists), other ⁇ 2 adrenergic receptor agonists, antiinfective agents (e.g. antibiotics or antivirals) or antihistamines.
- anti-inflammatory agents e.g. corticosteroids and non-steroidal anti-inflammatory agents (NSAIDs)
- antichlolinergic agents particularly muscarinic receptor antagonists
- other ⁇ 2 adrenergic receptor agonists e.g. antibiotics or antivirals
- antiinfective agents e.g. antibiotics or antivirals
- antihistamines e.g. antibiotics or antivirals
- the other therapeutic agents can be used in the form of pharmaceutically- acceptable salts or solvates. As appropriate, the other therapeutic agents can be used as optically pure stereoisomers.
- Suitable anti-inflammatory agents include corticosteroids and NSAIDs.
- Suitable corticosteroids which may be used in combination with the compounds of the invention are those oral and inhaled corticosteroids and their pro-drugs which have anti- inflammatory activity.
- Examples include methyl prednisolone, prednisolone, dexamethasone, fluticasone propionate, 6 ⁇ ,9 ⁇ -difluoro-17 ⁇ -[(2-furanylcarbonyl)oxy]- 1 l ⁇ -hydroxy-16 ⁇ -methyl-3-oxo-androsta-l,4-diene-17 ⁇ -carbothioic acid S-fluoromethyl ester, 6 ,9 ⁇ -difluoro-ll ⁇ -hydroxy-16 -methyl-3-oxo-17 ⁇ -propionyloxy- androsta-1,4- diene-17 ⁇ -carbothioic acid S-(2-oxo-tetrahydro-furan-3S-yl) ester, beclomethasone esters (e.g.
- the 17-propionate ester or the 17,21-di ⁇ ropionate ester the 17-propionate ester or the 17,21-di ⁇ ropionate ester
- budesonide flunisolide
- mometasone esters e.g. the furoate ester
- triamcinolone acetonide e.g. the furoate ester
- rofleponide triamcinolone acetonide
- ciclesonide butixocort propionate
- RPR-106541 the 17-propionate ester or the 17,21-di ⁇ ropionate ester
- ST-126 the 17-propionate ester or the 17,21-di ⁇ ropionate ester
- Preferred corticosteroids include fluticasone propionate, 6 ,9 ⁇ -difluoro-l l ⁇ -hydroxy-16 -methyl-17 ⁇ -[(4-methyl- l,3-thiazole-5-carbonyl)oxy]-3-oxo-androsta-l,4-diene-17 ⁇ -carbothioic acid S- fluoromethyl ester and 6 ⁇ ,9 ⁇ -difluoro-17 ⁇ -[(2-furanylcarbonyl)oxy]-ll ⁇ -hydroxy-16 ⁇ - methyl-3-oxo-androsta-l,4-diene-17 ⁇ -carbothioic acid S-fluoromethyl ester, more preferably 6 ⁇ ,9 ⁇ -difluoro-17 ⁇ -[(2-furanylcarbonyl)oxy]-l 1 ⁇ -hydroxy-16 ⁇ -methyl-3-oxo- androsta- 1 ,4-diene- 17 ⁇ -carbothioic acid iS-fluoromethyl ester.
- Suitable NSAIDs include sodium cromoglycate; nedocromil sodium; phosphodiesterase (PDE) inhibitors (e.g. theophylline, PDE4 inhibitors or mixed PDE3/PDE4 inhibitors); leukotriene antagonists (e.g. monteleukast); inhibitors of leukotriene synthesis; iNOS inliibitors; protease inhibitors, such as tryptase and elastase inhibitors; beta-2 integrin antagonists and adenosine receptor agonists or antagonists (e.g. adenosine 2a agonists); cytokine antagonists (e.g.
- chemokine antagonists such as, an interleukin antibody ( ⁇ lL antibody), specifically, an ⁇ IL-4 therapy, an o L-13 therapy, or a combination thereof); or inhibitors of cytokine synthesis.
- Suitable other ⁇ 2 -adrenoreceptor agonists include salmeterol (e.g. as the xinafoate), salbutamol (e.g. as the sulphate or the free base), fonnoterol (e.g. as the fumarate), fenoterol or terbutaline and salts thereof.
- PDE4 phosphodiesterase 4
- PDE3 mixed PDE3/PDE4 inhibitor.
- Representative phosphodiesterase-4 (PDE4) inhibitors or mixed PDE3/PDE4 inhibitors include, but are not limited to cis 4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)cyclohexan-l-carboxylic acid, 2-carbomethoxy-4-cyano-4-(3 -cyclopropylmethoxy-4- difluoromethoxyphenyl)cyclohexan-l-one; czs-[4-cyano-4-(3-cyclopropylmethoxy-4- difluoromethoxyphenyl)cyclohexan-l-ol]; c ⁇ -4-cyano-4-[3-(cyclopentyloxy)-4- methoxyphenyl]cyclohexane-l-carboxylic acid and the like, or phannaceutically- acceptable salts thereof.
- PDE4 inhibitors or mixed PDE3/PDE4 inhibitors include, but are not limited to cis 4-cyano-4-
- PDE4 or mixed PDE4/PDE3 inhibitors include AWD-12-281 (elbion); NCS-613 (INSERM); D-4418 (Chiroscience and Schering-Plough); CI-1018 or PD-168787 (Pfizer); benzodioxole compounds disclosed in WO99/16766 (Kyowa Hakko); K-34 (Kyowa Hakko); V-l 1294A (Napp); roflumilast (Byk-Gulden); pthalazinone compounds disclosed in WO99/47505 (Byk-Gulden); Pumafentrine (Byk-Gulden, now Altana); arofylline (Almirall-Prodesfarma); VM554/UM565 (Vernalis); T-440 (Tanabe Seiyaku); and T2585 (Tanabe Seiyaku).
- Suitable anticholinergic agents are those compounds that act as antagonists at the muscarinic receptor, in particular those compounds wliich are antagonists of the Mi, M 2 , or M 3 receptors, or of combinations thereof.
- Exemplary compounds include the alkaloids of the belladonna plants as illustrated by the likes of atropine, scopolamine, homatropine, hyoscyamine; these compounds are normally administered as a salt, being tertiary amines.
- drugs are readily available from a number of commercial sources or can be made or prepared from literature data via, to wit: Atropine - CAS-51-55-8 or CAS-51-48-1 (anhydrous form), atropine sulfate - CAS-5908-99-6; atropine oxide - CAS-4438-22-6 or its HCI salt - CAS-4574-60-1 and methylatropine nitrate - CAS-52-88-0. Homatropine - CAS-87-00-3, hydrobromide salt - CAS-51-56-9, methylbromide salt - CAS-80-49-9.
- Preferred anticholinergics include ipratropium (e.g. as the bromide), sold under the name Atrovent, oxitropium (e.g. as the bromide) and tiotropium (e.g. as the bromide) (CAS-139404-48-1).
- methantheline (CAS-53-46-3), propantheline bromide (CAS- 50-34-9), anisotropine methyl bromide or Valpin 50 (CAS- 80-50-2), clidinium bromide (Quarzan, CAS-3485-62-9), copyrrolate (Robinul), isopropamide iodide (CAS-71-81-8), mepenzolate bromide (U.S. patent 2,918,408), tridihexethyl chloride (Pathilone, CAS-4310-35-4), and hexocyclium methylsulfate (Tral, CAS-115-63-
- Suitable antihistamines include any one or more of the numerous antagonists known wliich inhibit Hi -receptors, and are safe for human use.
- first generation antagonists are characterized, based on their core structures, as ethanolamines, ethylenediamines, and alkylamines.
- first generation antihistamines include those which can be characterized as based on piperizine and phenothiazines.
- Second generation antagonists which are non-sedating, have a similar structure-activity relationship in that they retain the core ethylene group (the alkylamines) or mimic a tertiary amine group with piperizine or piperidine.
- Exemplary antagonists are as follows: Ethanolamines: carbinoxamine maleate, clemastine fumarate, diphenylhydramine hydrochloride, and dimenhydrinate. Ethylenediamines: pyrilamine amleate, tripelennamine HCI, and tripelennamine citrate. Alkylamines: chlorpheniramine and its salts such as the maleate salt, and acrivastine. Piperazines: hydroxyzine HCI, hydroxyzine pamoate, cyclizine HCI, cyclizine lactate, meclizine HCI, and cetirizine HCI.
- Piperidines Astemizole, levocabastine HCI, loratadine or its descarboethoxy analogue, and terfenadine and fexofenadine hydrochloride or another pharmaceutically- acceptable salt.
- Azelastine hydrochloride is yet another Hi receptor antagonist which may be used in combination with a compound of the invention.
- preferred anti-histamines include methapyrilene and loratadine.
- the invention thus provides, in a further aspect, a combination comprising a compound of fonnula (I) or a pharmaceutically-acceptable salt or solvate or stereoisomer thereof and a corticosteroid.
- the invention provides a combination wherein the corticosteroid is fluticasone propionate or wherein the corticosteroid is 6 ,9 - difluoro- 17 ⁇ - [(2-furanylcarbonyl)oxy] - 11 ⁇ -hydroxy- 16 ⁇ -methyl-3 -oxo-androsta- 1,4- diene-17 ⁇ -carbothioic acid tf-fluoromethyl ester or 6 ,9 ⁇ -difluoro-ll ⁇ -hydroxy-16 ⁇ - methyl-3-oxo-17 ⁇ -propionyloxy- androsta-l,4-diene-17 ⁇ -carbothioic acid £-(2-0X0- tefrahydro-furan-3S-yl) ester.
- the invention thus provides, in a further aspect, a combination comprising a compound of formula (I) or a pharmaceutically-acceptable salt or solvate or stereoisomer thereof and a PDE4 inhibitor.
- the invention thus provides, in a further aspect, a combination comprising a compound of formula (I) or a pharmaceutically-acceptable salt or solvate or stereoisomer thereof and an anticholinergic agent.
- the invention thus provides, in a further aspect, a combination comprising a compound of formula (I) or a pharmaceutically-acceptable salt or solvate or stereoisomer thereof and an antihistamine.
- the invention thus provides, in a further aspect, a combination comprising a compound of formula (I) or a pharmaceutically-acceptable salt or solvate or stereoisomer thereof together with a PDE4 inhibitor and a corticosteroid.
- the invention thus provides, in a further aspect, a combination comprising a compound of formula (I) or a pharmaceutically-acceptable salt or solvate or stereoisomer thereof together with an anticholinergic agent and a corticosteroid.
- a compound of formula (I) includes a compound of formula (13) and preferred groups thereof, and any individually disclosed compound or compounds.
- compositions of the invention can optionally comprise combinations of a compound of formula (I) or a pharmaceutically-acceptable salt or solvate or stereoisomer thereof with one or more other therapeutic agents, as described above.
- the individual compounds of such combinations may be administered either sequentially or simultaneously in separate or combined pharmaceutical formulations. Appropriate doses of known therapeutic agents will be readily appreciated by those skilled in the art. Methods of treatment of the invention, therefore, include administration of the individual compounds of such combinations either sequentially or simultaneously in separate or combined pharmaceutical formulations.
- the invention provides a method of treating a disease or condition associated with ⁇ 2 adrenergic receptor activity in a mammal, comprising administering to the mammal a therapeutically effective amount of a combination of a compound of formula (I) or a phannaceutically-acceptable salt or solvate or stereoisomer thereof with one or more other therapeutic agents.
- compounds of the invention are ⁇ 2 adrenergic receptor agonists, such compounds are also useful as research tools for investigating or studying biological systems or samples having ⁇ 2 adrenergic receptors, or for discovering new ⁇ 2 adrenergic receptor agonists.
- compounds of the invention exhibit selectivity for ⁇ 2 adrenergic receptors as compared with binding and functional activity at receptors of other ⁇ adrenergic subtypes, such compounds are also useful for studying the effects of selective agonism of ⁇ adrenergic receptors in a biological system or sample.
- Any suitable biological system or sample having ⁇ 2 adrenergic receptors may be employed in such studies which may be conducted either in vitro or in vivo.
- Representative biological systems or samples suitable for such studies include, but are not limited to, cells, cellular extracts, plasma membranes, tissue samples, mammals (such as mice, rats, guinea pigs, rabbits, dogs, pigs, etc.) and the like.
- agonizing the ⁇ 2 adrenergic receptor are determined using conventional procedures and equipment, such as radioligand binding assays and functional assays, for example the assay for ligand-mediated changes in intracellular cyclic adenosine monophosphate (cAMP) described below, or assays of a similar nature.
- a ⁇ 2 adrenergic receptor- agonizing amount of a compound of the invention will typically range from about 1 nanomolar to about 1000 nanomolar.
- the invention also includes, as separate embodiments, both the generation of comparison data (using the appropriate assays) and the analysis of the test data to identify test compounds of interest.
- compositions of the invention illustrate representative pharmaceutical compositions of the invention. Additional suitable carriers for formulations of the active compounds of the present invention can also be found in Remington: The Science and Practice of Pharmacy, 20th Edition, Lippincott Williams & Wilkins, Philadelphia, PA, 2000.
- Formulation Example A This example illustrates the preparation of a representative pharmaceutical composition for oral administration of a compound of this invention: Ingredients Quantity per tablet, (mg) Active Compound 1 Lactose, spray-dried 148 Magnesium stearate 2 The above ingredients are mixed and introduced into a hard-shell gelatin capsule.
- Formulation Example B This example illustrates the preparation of another representative pharmaceutical composition for oral administration of a compound of this invention:
- Fonnulation Example C This example illustrates the preparation of a representative pharmaceutical composition for oral administration of a compound of this invention.
- An oral suspension is prepared having the following composition. Ingredients
- Example E This example illustrates the preparation of a representative pharmaceutical composition for injection of a compound of this invention.
- a reconstituted solution is prepared by adding 20 mL of sterile water to 1 mg of the compound of this invention. Before use, the solution is then diluted with 200 mL of an intravenous fluid that is compatible with the active compound.
- Such fluids are chosen from 5% dextrose solution, 0.9% sodium chloride, or a mixture of 5% dextrose and 0.9%> sodium chloride.
- Other examples are lactated Ringer's injection, lactated Ringer's plus 5% dextrose injection, Normosol-M and 5% dextrose, Isolyte E, and acylated Ringer's injection.
- Formulation Example F This example illustrates the preparation of a representative pharmaceutical composition for topical application of a compound of this invention. Ingredients grams
- An aqueous aerosol formulation for use in a nebulizer is prepared by dissolving 0.1 mg of a pharmaceutical salt of active compound in a 0.9 % sodium chloride solution acidified with citric acid. The mixture is stirred and sonicated until the active salt is dissolved. The pH of the solution is adjusted to a value in the range of from 3 to 8 by the slow addition of NaOH.
- Formulation Example H This example illustrates the preparation of a dry powder formulation containing a compound of the invention for use in inhalation cartridges.
- Gelatin inhalation cartridges are filled with a pharmaceutical composition having the following ingredients: Ingredients mg/cartridge Pharmaceutical salt of active compound 0.2 Lactose 25 The pharmaceutical salt of active compound is micronized prior to blending with lactose. The contents of the cartridges are administered using a powder inhaler.
- Formulation Example I This example illustrates the preparation of a dry powder formulation containing a compound of the invention for use in a dry powder inhalation device.
- a pharmaceutical composition is prepared having a bulk formulation ratio of micronized pharmaceutical salt to lactose of 1 :200.
- the composition is packed into a dry powder inhalation device capable of delivering between about 10 ⁇ g and about 100 ⁇ g of active drug ingredient per dose.
- Formulation Example J This example illustrates the preparation of a formulation containing a compound of the invention for use in a metered dose inhaler.
- a suspension containing 5 % pharmaceutical salt of active compound, 0.5 % lecithin, and 0.5 % trehalose is prepared by dispersing 5 g of active compound as micronized particles with mean size less than 10 ⁇ m in a colloidal solution formed from 0.5 g of trehalose and 0.5 g of lecithin dissolved in 100 mL of demineralized water.
- the suspension is spray dried and the resulting material is micronized to particles having a mean diameter less than 1.5 ⁇ m.
- the particles are loaded into canisters with pressurized 1,1,1,2-tetrafluoroethane.
- Formulation Example K This example illustrates the preparation of a formulation containing a compound of the invention for use in a metered dose inhaler.
- a suspension containing 5 % pharmaceutical salt of active compound and 0.1 % lecithin is prepared by dispersing 10 g of active compound as micronized particles with mean size less than 10 ⁇ m in a solution formed from 0.2 g of lecithin dissolved in 200 mL of demineralized water. The suspension is spray dried and the resulting material is micronized to particles having a mean diameter less than 1.5 ⁇ m. The particles are loaded into canisters with pressurized 1,1,1,2,3,3,3-heptafluoro-n-propane.
- the compounds of this invention exhibit biological activity and are useful for medical treatment.
- the ability of a compound to bind to the ⁇ 2 adrenergic receptor, as well as its selectivity, agonist potency, and intrinsic activity can be demonstrated using Tests A-B below, or can be demonstrated using other tests that are known in the art.
- Binding assays were performed in 96-well microtiter plates in a total assay volume of 100 ⁇ L with 5 ⁇ g membrane protein for membranes containing the human ⁇ 2 adrenergic receptor, or 2.5 ⁇ g membrane protein for membranes containing the human ⁇ i adrenergic receptor in assay buffer (75 mM Tris/HCl pH 7.4 @ 25°C, 12.5 mM MgCl 2 ,
- Filter plates were washed three times with filtration buffer (75 mM Tris/HCl pH 7.4 @ 4°C, 12.5 mM MgCl 2 , 1 mM EDTA) to remove unbound radioactivity. Plates were dried, 50 ⁇ L Microscint-20 liquid scintillation fluid (Packard BioScience Co., Meriden, CT) was added and plates were counted in a Packard Topcount liquid scintillation counter (Packard BioScience Co., Meriden, CT). Binding data were analyzed by nonlinear regression analysis with the GraphPad Prism Software package (GraphPad Software, Inc., San Diego, CA) using the 3-parameter model for one-site competition.
- filtration buffer 75 mM Tris/HCl pH 7.4 @ 4°C, 12.5 mM MgCl 2 , 1 mM EDTA
- the curve minimum was fixed to the value for nonspecific binding, as detennined in the presence of 10 ⁇ M alprenolol.
- K values for compounds were calculated from observed IC 50 values and the K ⁇ value of the radioligand using the Cheng-Prusoff equation (Cheng Y, and Prusoff WH., Biochemical Pharmacology, 1973, 22, 23, 3099-108).
- the receptor subtype selectivity was calculated as the ratio of Kj( ⁇ )/Kj( ⁇ 2 ).
- Compounds of the invention demonstrated greater binding at the ⁇ 2 adrenergic receptor than at the ⁇ i adrenergic receptor, i.e. Kj( ⁇ i) > K( ⁇ 2 ) with selectivity greater than about 100.
- Test B Whole-cell cAMP Flashplate Assays With Cell Lines Heterologously Expressing Human ⁇ i Adrenoceptor, ⁇ 2 Adrenoceptor, and ⁇ 3 Adrenoceptor, Respectively.
- a HEK-293 cell line stably expressing cloned human ⁇ i adrenergic receptor (clone H34.1) was grown to about 70%-90% confluency in medium consisting of DMEM supplemented with 10% FBS and 500 ⁇ g/mL Geneticin.
- a HEK-293 cell line stably expressing cloned human ⁇ 2 -adrenoceptor (clone H24.14) was grown in the same medium to full confluency.
- a CHO-K1 cell line stably expressing cloned human ⁇ 3 -adrenoceptor was grown to about 70%-90% confluency in Ham's F-12 medium supplemented with 10% FBS and with 800 ⁇ g/mL Geneticin added to every fifth passage. The day before the assay, cultures were switched to the same growth-media without antibiotics. cAMP assays were performed in a radioimmunoassay format using the Flashplate
- Adenylyl Cyclase Activation Assay System with 125 I-cAMP (NEN SMP004, PerkinElmer Life Sciences Inc., Boston, MA), according to the manufacturers instructions.
- NNN SMP004, PerkinElmer Life Sciences Inc., Boston, MA the manufacturer instructions.
- cells were rinsed once with PBS, lifted with Versene 1 :5,000 (0.2 g/L EDTA in PBS) and counted.
- Cells were pelleted by centrifugation at 1,000 rpm and resuspended in stimulation buffer prewarmed to 37°C.
- 10 nM ICI 118,551 were added to the stimulation buffer, and cells were incubated for 10 min at 37°C.
- Cells were used at final concentrations of 30,000, 40,000 and 70,000 cells / well for the ⁇ i -adrenoceptor-, the ⁇ -adrenoceptor- and the ⁇ 3 - adrenoceptor expressing cells, respectively.
- Compounds were dissolved to a concentration of 10 mM in DMSO, then diluted to 1 mM in 50 mM Gly-HCl pH 3.0, and from there serially diluted into assay buffer (75 mM Tris/HCl pH 7.4 @ 25°C, 12.5 mM MgCl 2 , 1 mM EDTA, 0.2% BSA).
- test C Whole-cell cAMP Flashplate Assay With a Lung Epithelial Cell Line , Endogenously Expressing Human ⁇ 2 Adrenergic Receptor
- a human lung epithelial cell line (BEAS-2B) was used (ATCC CRL-9609, American Type Culture Collection, Manassas, VA) (January B, et al., British Journal of Pharmacology, 1998, 123, 4, 701- 11).
- Reactions were incubated for 10 min at 37°C and stopped by addition of 100 ⁇ l ice-cold detection buffer. Plates were sealed, incubated over night at 4°C and counted the next morning in a topcount scintillation counter (Packard BioScience Co., Meriden, CT). The amount of cAMP produced per mL of reaction was calculated based on the counts observed for samples and cAMP standards, as described in the manufacturer's user manual. Data were analyzed by nonlinear regression analysis with the GraphPad Prism Software package (GraphPad Software, Inc., San Diego, CA) using the 4-parameter model for sigmoidal dose-response with variable slope.
- Test compounds were administered via inhalation over 10 minutes in a whole-body exposure dosing chamber (R&S Molds, San Carlos, CA). The dosing chambers were arranged so that an aerosol was simultaneously delivered to 6 individual chambers from a central manifold. Following a 60 minute acclimation period and a 10 minute exposure to nebulized water for injection (WFI), guinea pigs were exposed to an aerosol of test compound or vehicle (WFI). These aerosols were generated from aqueous solutions using an LC Star Nebulizer Set (Model 22F51, PARI Respiratory Equipment, ie.
- each guinea pig was anesthetized with an intramuscular injection of ketamine (43.75 mg/kg), xylazine (3.50 mg/kg) and acepromazine (1.05 mg/kg). After the surgical site was shaved and cleaned with 70% alcohol, a 2-5 cm midline incision of the ventral aspect of the neck was made.
- the jugular vein was isolated and cannulated with a saline-filled polyethylene catheter (PE-50, Becton Dickinson, Sparks, MD) to allow for intravenous infusions of a 0.1 mg/mL solution of acetylcholine (Ach), (Sigma- Aldrich, St. Louis, MO) in saline.
- Ach acetylcholine
- the trachea was then dissected free and cannulated with a 14G teflon tube (#NE- 014, Small Parts, Miami Lakes, FL). If required, anesthesia was maintained by additional intramuscular injections of the aforementioned anesthetic cocktail.
- the depth of anesthesia was monitored and adjusted if the animal responded to pinching of its paw or if the respiration rate was greater than 100 breaths/minute.
- the animal was placed into a plethysmograph (#PLY3114, Buxco Electronics, hie, Sharon, CT) and an esophageal pressure cannula was inserted to measure pulmonary driving pressure (pressure).
- the teflon tracheal tube was attached to the opening of the plethysmograph to allow the guinea pig to breathe room air from outside the chamber. The chamber was then sealed.
- a heating lamp was used to maintain body temperature and the guinea pig's lungs were inflated 3 times with 4 mL of air using a 10 mL calibration syringe (#5520 Series, Hans Rudolph, Kansas City, MO) to ensure that the lower airways had not collapsed and that the animal did not suffer from hyperventilation. Once it was determined that baseline values were within the range
- the pulmonary evaluation was initiated.
- a Buxco pulmonary measurement computer progam enabled the collection and derivation of pulmonary values.
- Starting this program initiated the experimental protocol and data collection.
- the changes in volume over time that occured witliin the plethysmograph with each breath were measured via a Buxco pressure transducer. By integrating this signal over time, a measurement of flow was calculated for each breath.
- the guinea pig's lungs were inflated 3 times with 4 mL of air from a 10 mL calibration syringe. Recorded pulmonary parameters included respiration frequency (breaths/minute), compliance (mL/cm H 2 O) and pulmonary resistance (cm H 2 O/ mL per second) (Giles et al, 1971). Once the pulmonary function measurements were completed at minute 35 of this protocol, the guinea pig was removed from the plethysmograph and euthanized by CO 2 asphyxiation.
- the quantity PD which is defined as the amount of Ach needed to cause a doubling of the baseline pulmonary resistance, was calculated using the pulmonary resistance values derived from the flow and the pressure over a range of Ach challenges using the following equation. This was derived from the equation used to calculate PC 0 values in the clinic (Am. Thoracic Soc, 2000).
- Ci Second to last Ach concentration (concentration preceding C 2 )
- C 2 Final concentration of Ach (concentration resulting in a 2-fold increase in pulmonary resistance (R j J)
- R Q Baseline R L value
- R j R L value after C
- R 2 R L value after C 2
- Statistical analysis of the data was performed using a One- Way Analysis of
- Example 1 Synthesis of 5-((R)-2- ⁇ 2-[4-((R)-2-amino-2-phenyIethylamino)- phenyl]ethylamino ⁇ -l-hydroxyethyI)-8-hydroxy-llf ⁇ quinolin-2-one a. Preparation of [2-(4-nitrophenyl)ethyl]carbamic acid tert-butyl ester Di-tert-butyl dicarbonate (20 g, 92 mmol) was suspended in saturated sodium hydrogen carbonate (200 mL) and cooled to 0°C. Dioxane (10 mL) was added.
- Example 2 Synthesis of iV-[5-((R)-2- ⁇ 2-[4-((R)-2-amino-2-phenyl- ethylamino)phenyl]ethylamino ⁇ -l-hydroxyethyl)-2-hydroxyphenyl]- formamide a.
- Example 3 Alternative synthesis of 5-((R)-2- ⁇ 2-[4-((R)-2-amino-2-phenyI- ethylamino)phenyl]ethylamino ⁇ -l-hydroxyethyl)-8-hydroxy-li ⁇ -quinolin-2- one a.
- the reaction mixture was charged with 165 mL (165 mmol) of 1.0 M sodium bis(trimethylsilyl)amide in tetrahydrofuran (the temperature remained below 30°C).
- the sodium bis(trimethylsilyl)amide solution was added in one portion with vigorous stirring.
- the reaction mixture was cooled to -10 °C and (S)-styrene oxide (17 mL, 150 mmol) was added. The rate of addition was controlled to maintain a temperature below -10 °C.
- the reaction was allowed to warm to 20 °C within 15 minutes after the addition of (»S)-styrene oxide, reaching 28 °C within 30 minutes.
- the reaction was cooled to 25 °C, and was quenched by dropwise addition of 90 mL water.
- the reaction mixture was transfened to a separatory funnel, diluted with 100 mL isopropyi acetate and washed with 90 mL saturated aqueous sodium chloride.
- the organic layer was washed three times with a mixture of 90 mL water and 90 mL saturated aqueous sodium chloride and finally with 180 mL saturated aqueous sodium chloride.
- the organic layer was concentrated under vacuum. The residue was twice reconcentrated from isopropanol (100 mL portions) and then redissolved in isopropanol (500 mL) and heated to 70 °C with stining. Concentrated hydrochloric acid (27 mL, 327 mmol) was added over two minutes.
- the mixture was allowed to cool to room temperature and stined for 14 h.
- the precipitated product was isolated by filtration and washed with isopropanol and isopropyi acetate.
- the product was dried under vacuum over a 50 °C water bath for 1 h and then dissolved in 80 mL water and transfened to a separatory funnel.
- Isopropyi acetate (80 mL) and IO N aqueous sodium hydroxide (40 mL, 400 mmol) were added.
- the separatory funnel was shaken and the phases separated.
- the organic layer was washed once with 40 mL saturated NaCl and dried over magnesium sulfate.
- the solids were collected and the filtrate was concentrated.
- Example 4 Alternative synthesis of N-[5-((R)-2- ⁇ 2-[4-((R)-2-amino-2-phenyl- ethylamino)phenyI]ethyIamino ⁇ -l-hydroxyethyl)-2-hydroxyphenyl]- formamide a.
- the mixture was refluxed for 3.5 hours then cooled to room temperature for 16 hours. Additional diphenylphophoryl azide (305 ⁇ L, 1.4 mmol) and l,8-diazabicyclo[5.4.0]undec-7-ene (210 ⁇ L, 1.4 mmol) were added and the mixture refluxed for a further 3 hours, then cooled to room temperature. The mixture was partitioned between ethyl acetate and water. The organics were washed with 0.9 M sodium acetate/acetic acid, saturated sodium hydrogen carbonate and saturated sodium chloride, dried over sodium sulfate and evaporated to dryness.
- the suspension was stined vigorously under hydrogen (atmospheric pressure) for
- Example 5 Alternative Preparation of (R)-N 2 ⁇ [4-(2-aminoethy ⁇ )phenyl]-l- phenylethane-l,2-diamine a.
- Preparation of [2-(4-aminophenyl)ethyl]carbamic acid tez't-butyl ester To a suspension of 4-aminophenethylamine(65.1 g, 1.0 equiv) in dichloromethane (1.5 L) at 0 °C was added di-tert-butyl dicarbonate (99.2 g, 0.95 equiv) in dichloromethane (300 mL) dropwise. The solution was slowly warmed to room temperature and stined for 18 hours.
- the crystals were dried under vacuum to afford the L-malate salt of the title intermediate (38.6 g).
- the L-malate salt was dissolved in water (150 mL) and dichloromethane (175 mL) was added. The mixture was stirred and cooled to 0 °C and 10.0 N sodium hydroxide (50 mL) was added. The layers were separated and the aqueous layer was extracted with dichloromethane (2 x 175 mL). The organic layers were combined and dried over anhydrous sodium sulfate (20 g). The solids were removed by filtration and the filtrate concentrated to yield the title intermediate (24.9 g, e.e. >99 %) as a colorless oil.
- Example 5 (22.4 g, 1.4 equiv), N-[2-benzyloxy-5-((i?)-2-bromo-l-(tert- butyldimethylsilanyloxy)ethyl)phenyl]formamide (29.2 g, 1.0 equiv), potassium carbonate (34.7 g, 4.0 equiv) were combined with dimethyl sulfoxide (35 mL). The resulting slurry was stined at 100 °C for 85 minutes. The mixture was cooled to room temperature and water (200 mL) and isopropyi acetate (200 mL) were added.
- the organic supernatant was decanted and discarded leaving a product-containing gummy solid to which was added isopropyi acetate (200 mL) followed by 1.0 N aqueous sodium hydroxide (200 mL). The mixture was stirred until most of the solid had dissolved. The top layer of the biphasic mixture was decanted and saved. Isopropyi acetate (150 mL) was added to the aqueous layer and stined until all solids dissolved and then the biphasic mixture was combined with the reserved organic layer. The layers were separated and the basic aqueous layer was again extracted with isopropyi acetate (150 mL). The organic layers were combined and dried over anhydrous sodium sulfate.
- the reaction flask was purged with nitrogen and the reaction mixture was filtered tlirough celite (30.0 g) and washed with tetrahydrofuran (100 mL). The solvent was removed under vacuum to yield crude title intermediate (16.0 g) as an oil.
- the crude product was divided into three batches, hi a representative batch, crude product (4.0 g) was solubilized in water (10 mL) and stined for 10 minutes to dissolution.
- Example 7 Synthesis of crystalline iV-[5-((R)-2- ⁇ 2-[4-((R)-2-amino-2- phenylethylamino)phenyl]ethylamino ⁇ -l-hydroxyethyl)-2- hydroxyphenyl] formamide hydrochloride a.
- Example 8 Characterization of crystalline iV-[5-((R)-2- ⁇ 2-[4-((R)-2-amino-2- phenylethylamino)phenyl]ethylamino ⁇ -l-hydroxyethy ⁇ )-2- hy droxyphenyl] formamide hydrochloride
- a sample of the title crystalline hydrochloride salt prepared as in Example 7, part d was characterized as follows: 1H NMR (300MHz): 9.5 (s, IH), 8.2 (s, IH), 8.0 (br s, IH), 7.1-7.4 (m, 6H), 6.7-6.9 (m, 5H), 6.4 (d, 2H), 5.5 (br s, 2H), 4.5 (d, IH), 4.0 (t,
- Example 10 Synthesis of 5-((R)-2- ⁇ 2-[4-((S)-2-amino-2-phenyIethylamino)- phenyl]ethylamino ⁇ -l-hydroxyethyl)-8-hydroxy-lH-quinolin-2-one Using procedures similar to steps d, e, and f of Example 9, substituting 8-benzyloxy-5-[(i?)-2-bromo-l-(tert-butyldimethylsilanyloxy)ethyl]-lH-quinolin-2-one for N-[2-benzyloxy-5-((i?)-2-bromo-l-(tert-butyldimethylsilanyloxy)ethyl)phenyl]- formamide in step d, the trifluoroacetate salt of the title compound was obtained. m/z: [M+ ⁇ + ] calcd for C 27 H 30 N 4 O 3 : 459.2; found 459.4.
- Example 11 Synthesis of 5-((R)-2- ⁇ 2-[4-((R)-2-methylamino-2- phenylethylamino)phenyl]ethylamino ⁇ -l-hydroxyethyl)-8-hydroxy-lH- quinolin-2-one a.
- Example 12 Synthesis of N-[5-((R)-2- ⁇ 2-[4-((S)-2-methylamino-2-phenyl- ethylamino)phenyl]ethylamino ⁇ -l-hydroxyethyl)-2-hydroxyphenyl]- formamide Using procedures similar to steps f, g, and h of Example 11, substituting N-[2- benzyloxy-5-((i?)-2-bromo-l-(tert-butyldimethylsilanyloxy)ethyl)phenyl]formamide for
Abstract
Description














Claims





Priority Applications (16)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002538037A CA2538037A1 (en) | 2003-09-22 | 2004-09-21 | Amino-substituted ethylamino .beta.2 adrenergic receptor agonists |
| AT04784634T ATE428685T1 (en) | 2003-09-22 | 2004-09-21 | AMINO-SUBSTITUTED ETHYLAMINOAGONISTS OF THE BETA2-ADRENERGIC RECEPTOR |
| BRPI0414634-4A BRPI0414634A (en) | 2003-09-22 | 2004-09-21 | b2 receptor agonists, amino substituted ethylamino adrenergic |
| DK04784634T DK1687257T3 (en) | 2003-09-22 | 2004-09-21 | Amino-substituted ethylamino agonists of the beta2-adrenergic receptor |
| NZ545764A NZ545764A (en) | 2003-09-22 | 2004-09-21 | Amino-substituted ethylamino B2 adrenergic receptor agonists |
| JP2006527127A JP2007505931A (en) | 2003-09-22 | 2004-09-21 | Amino-substituted ethylamino β2 adrenergic receptor agonist |
| PL04784634T PL1687257T3 (en) | 2003-09-22 | 2004-09-21 | Amino-substituted ethylamino beta2 adrenergic receptor agonists |
| MXPA06003093A MXPA06003093A (en) | 2003-09-22 | 2004-09-21 | AMINO-SUBSTITUTED ETHYLAMINO beta2 ADRENERGIC RECEPTOR AGONISTS. |
| AU2004276254A AU2004276254A1 (en) | 2003-09-22 | 2004-09-21 | Amino-substituted ethylamino beta2 adrenergic receptor agonists |
| DE602004020655T DE602004020655D1 (en) | 2003-09-22 | 2004-09-21 | AMINO-SUBSTITUTED ETHYLAMINOAGONISTS OF THE BETA2 ADRENEER RECEPTOR |
| EP04784634A EP1687257B1 (en) | 2003-09-22 | 2004-09-21 | Amino-substituted ethylamino beta2 adrenergic receptor agonists |
| SI200431162T SI1687257T1 (en) | 2003-09-22 | 2004-09-21 | Amino-substituted ethylamino beta2 adrenergic receptor agonists |
| IL174026A IL174026A0 (en) | 2003-09-22 | 2006-03-01 | Amino-substituted ethylamino b2 adrenergic receptor agonists |
| IS8346A IS8346A (en) | 2003-09-22 | 2006-03-09 | Amino-substituted ethylamino beta2 adrenergic receptor agonists |
| NO20061737A NO20061737L (en) | 2003-09-22 | 2006-04-20 | Amino-substituted erylamino B2 adrenergic receptor agonists |
| HK07100415.0A HK1093486A1 (en) | 2003-09-22 | 2007-01-11 | Amino-substituted ethylamino beta2 adrenergic receptor agonists |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US50568503P | 2003-09-22 | 2003-09-22 | |
| US60/505,685 | 2003-09-22 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2005030678A2 true WO2005030678A2 (en) | 2005-04-07 |
| WO2005030678A3 WO2005030678A3 (en) | 2005-05-26 |
Family
ID=34393055
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2004/030833 WO2005030678A2 (en) | 2003-09-22 | 2004-09-21 | AMINO-SUBSTITUTED ETHYLAMINO β2 ADRENERGIC RECEPTOR AGONISTS |
Country Status (28)
| Country | Link |
|---|---|
| US (2) | US7399863B2 (en) |
| EP (1) | EP1687257B1 (en) |
| JP (1) | JP2007505931A (en) |
| KR (1) | KR20070015496A (en) |
| CN (1) | CN100465158C (en) |
| AR (1) | AR046085A1 (en) |
| AT (1) | ATE428685T1 (en) |
| AU (1) | AU2004276254A1 (en) |
| BR (1) | BRPI0414634A (en) |
| CA (1) | CA2538037A1 (en) |
| CO (1) | CO5680412A2 (en) |
| DE (1) | DE602004020655D1 (en) |
| DK (1) | DK1687257T3 (en) |
| ES (1) | ES2326172T3 (en) |
| HK (1) | HK1093486A1 (en) |
| IL (1) | IL174026A0 (en) |
| IS (1) | IS8346A (en) |
| MA (1) | MA28089A1 (en) |
| MX (1) | MXPA06003093A (en) |
| NO (1) | NO20061737L (en) |
| NZ (1) | NZ545764A (en) |
| PL (1) | PL1687257T3 (en) |
| PT (1) | PT1687257E (en) |
| RU (1) | RU2385317C2 (en) |
| SI (1) | SI1687257T1 (en) |
| TW (1) | TW200526547A (en) |
| WO (1) | WO2005030678A2 (en) |
| ZA (1) | ZA200602308B (en) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007106016A1 (en) * | 2006-03-14 | 2007-09-20 | Astrazeneca Ab | Bezothiazol derivatives as beta2 adrenoreceptor agonists |
| WO2007124898A1 (en) | 2006-04-27 | 2007-11-08 | Laboratorios, Almirall S.A. | DERIVATIVES OF 4-(2-AMINO-1-HYDROXIETHYL)PHENOL AS AGONISTS OF THE β2 ADRENERGIC RECEPTOR |
| US7700782B2 (en) | 2006-12-20 | 2010-04-20 | Astrazeneca Ab | Compounds 569 |
| US7709511B2 (en) | 2005-08-09 | 2010-05-04 | Astrazeneca Ab | Benzothiazolone derivatives |
| WO2011061527A1 (en) | 2009-11-17 | 2011-05-26 | Astrazeneca Ab | Combinations comprising a glucocorticoid receptor modulator for the treatment of respiratory diseases |
| US7964615B2 (en) | 2005-05-20 | 2011-06-21 | Almirall, S.A. | Derivatives of 4-(2-amino-1-hydroxyethyl)phenol as agonists of the β2 adrenergic receptor |
| US8017602B2 (en) | 2008-06-18 | 2011-09-13 | Astrazeneca Ab | N-(2-(2-(5-hydroxy-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazin-8-yl)ethylamino)ethyl)-3-(phenethoxy)propanamide derivatives, processes for their preparation, pharmaceutical compositions containing them and their use in therapy |
| US8058294B2 (en) | 2007-02-08 | 2011-11-15 | Astrazeneca Ab | Pharmaceutical salts of N-[2-(diethylamino)ethyl]-N-(2-{[2-(4-hydroxy-2-oxo-2,3-dihydro-1,3-benzothiazol-7-yl)ethyl]amino}ethyl)-3-[2-(1-napthyl)ethoxy]propanamide |
| WO2012046050A1 (en) | 2010-10-07 | 2012-04-12 | Astrazeneca Ab | Novel combinations |
| US8524908B2 (en) | 2009-03-12 | 2013-09-03 | Almirall, S.A. | Process for manufacturing 5-(2-{[6-(2,2-difluoro-2-phenylethoxy)hexyl]amino}-1-hydroxyethyl)-8-hydroxyquinolin-2(1H)-one |
| US9108918B2 (en) | 2011-10-07 | 2015-08-18 | Almirall, S.A. | Process for preparing 5-(2-{[6-(2,2-difluoro-2-phenylethoxy)hexyl]amino}-1(R)-hydroxyethyl)-8-hydroxyquinolin-2(1H)-one via a novel intermediate |
| US9346759B2 (en) | 2012-03-20 | 2016-05-24 | Almirall, S.A. | Polymorphic crystal forms of 5-(2-{[6-(2,2-difluoro-2-phenylethoxy)hexyl]amino}-1-(R)-hydroxyethyl)-8-hydroxyquinolin-2(1H)-one, heminapadisytlate as agonist of the β2 adrenergic receptor |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI249515B (en) * | 2001-11-13 | 2006-02-21 | Theravance Inc | Aryl aniline beta2 adrenergic receptor agonists |
| TW200526547A (en) * | 2003-09-22 | 2005-08-16 | Theravance Inc | Amino-substituted ethylamino β2 adrenergic receptor agonists |
| TW200531692A (en) * | 2004-01-12 | 2005-10-01 | Theravance Inc | Aryl aniline derivatives as β2 adrenergic receptor agonists |
| CN1984876A (en) * | 2004-06-03 | 2007-06-20 | 施万制药 | Diamine beta2 adrenergic receptor agonists |
| EP1778638A1 (en) * | 2004-07-21 | 2007-05-02 | Theravance, Inc. | Diaryl ether beta2 adrenergic receptor agonists |
| JP2008512470A (en) * | 2004-09-10 | 2008-04-24 | セラヴァンス, インコーポレーテッド | Amidine-substituted arylaniline compounds |
| BRPI0606712A2 (en) * | 2005-01-11 | 2010-03-16 | Glaxo Group Ltd | compound salt, pharmaceutical formulation, combination, use of a compound, and method for preparing a crystalline compound |
| TWI372749B (en) | 2005-03-10 | 2012-09-21 | Theravance Inc | Crystalline forms of a biphenyl compound |
| ES2302447B1 (en) * | 2006-10-20 | 2009-06-12 | Laboratorios Almirall S.A. | DERIVATIVES OF 4- (2-AMINO-1-HYDROXYETHYL) Phenol as agonists of the BETA2 ADRENERGIC RECEIVER. |
| CN101959887B (en) | 2008-01-08 | 2013-07-31 | 阵列生物制药公司 | Pyrrolopyridines as kinase inhibitors |
| CA2711741A1 (en) * | 2008-01-09 | 2009-07-16 | Array Biopharma Inc. | Pyrazolopyridines as kinase inhibitors |
| EP2096105A1 (en) * | 2008-02-28 | 2009-09-02 | Laboratorios Almirall, S.A. | Derivatives of 4-(2-amino-1-hydroxyethyl)phenol as agonists of the b2 adrenergic receptor |
| CN112645875A (en) * | 2020-12-09 | 2021-04-13 | 深圳海王医药科技研究院有限公司 | Preparation method of procaterol hydrochloride impurity |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003042164A1 (en) * | 2001-11-13 | 2003-05-22 | Theravance, Inc | Aryl aniline beta-2 adrenergic receptor agonists |
| WO2003042160A1 (en) * | 2001-11-13 | 2003-05-22 | Theravance, Inc. | Aryl aniline beta-2 adrenergic receptor agonists |
Family Cites Families (46)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US665323A (en) * | 1897-12-15 | 1901-01-01 | Merrell Soule Co | Method of preparing vegetable-soup powders. |
| BE630296A (en) | 1962-03-31 | |||
| AT310146B (en) | 1971-04-26 | 1973-09-25 | Boehringer Sohn Ingelheim | Process for the production of new N, N'-bis- (β-hydroxyarylethyl) -diaminoalkanes and their acid addition salts |
| CH545764A (en) | 1971-07-27 | 1974-02-15 | ||
| BE794414A (en) | 1972-01-25 | 1973-07-23 | Sandoz Sa | NEW AMINO-ALCOHOLS, THEIR PREPARATION AND THEIR APPLICATION AS A MEDICINAL PRODUCT |
| JPS5913510B2 (en) | 1975-12-26 | 1984-03-30 | オオツカセイヤク カブシキガイシヤ | Calbostyril Yudou Tainoseizohou |
| JPS5283619A (en) | 1976-01-01 | 1977-07-12 | Yamanouchi Pharmaceut Co Ltd | Alpha-substituted aminomethyl benzyl alcohol derivative |
| JPS609713B2 (en) | 1976-10-08 | 1985-03-12 | 大塚製薬株式会社 | carbostyril derivatives |
| ZW6584A1 (en) | 1983-04-18 | 1985-04-17 | Glaxo Group Ltd | Phenethanolamine derivatives |
| GB8334494D0 (en) | 1983-12-24 | 1984-02-01 | Tanabe Seiyaku Co | Carbostyril derivatives |
| GB8507942D0 (en) | 1985-03-27 | 1985-05-01 | Beecham Group Plc | Compounds |
| DK9987A (en) | 1986-01-11 | 1987-07-12 | Beecham Group Plc | ETHANOLAMINE DERIVATIVES |
| US5223614A (en) | 1987-12-19 | 1993-06-29 | Boehringer Ingelheim Gmbh | New quaternary ammonium compounds, their preparation and use |
| US4894219A (en) | 1988-03-29 | 1990-01-16 | University Of Florida | Beta-agonist carbostyril derivatives, assay method and pharmacological composition |
| US5434304A (en) | 1990-09-26 | 1995-07-18 | Aktiebolaget Astra | Process for preparing formoterol and related compounds |
| GB9405019D0 (en) | 1994-03-15 | 1994-04-27 | Smithkline Beecham Plc | Novel compounds |
| US6040344A (en) | 1996-11-11 | 2000-03-21 | Sepracor Inc. | Formoterol process |
| GB9713819D0 (en) | 1997-06-30 | 1997-09-03 | Glaxo Group Ltd | Method of reducing the systemic effects of compounds |
| US6713651B1 (en) | 1999-06-07 | 2004-03-30 | Theravance, Inc. | β2-adrenergic receptor agonists |
| US6362371B1 (en) | 1998-06-08 | 2002-03-26 | Advanced Medicine, Inc. | β2- adrenergic receptor agonists |
| US20020143034A1 (en) | 1998-12-30 | 2002-10-03 | Fujisawa Pharmaceutical Co. Ltd. | Aminoalcohol derivatives and their use as beta 3 adrenergic agonists |
| GB9913083D0 (en) | 1999-06-04 | 1999-08-04 | Novartis Ag | Organic compounds |
| US6683115B2 (en) * | 1999-06-02 | 2004-01-27 | Theravance, Inc. | β2-adrenergic receptor agonists |
| OA11558A (en) | 1999-12-08 | 2004-06-03 | Advanced Medicine Inc | Beta 2-adrenergic receptor agonists. |
| UA73965C2 (en) | 1999-12-08 | 2005-10-17 | Theravance Inc | b2 ADRENERGIC RECEPTOR ANTAGONISTS |
| CA2405745A1 (en) | 2000-04-27 | 2001-11-08 | Boehringer Ingelheim Pharma Kg | New betamimetics having a long-lasting activity, processes for preparingthem and their use as medicaments |
| US6759398B2 (en) | 2000-08-05 | 2004-07-06 | Smithkline Beecham Corporation | Anti-inflammatory androstane derivative |
| EP1341759B1 (en) | 2000-11-10 | 2006-06-14 | Eli Lilly And Company | 3-substituted oxindole beta 3 agonists |
| GB0103630D0 (en) | 2001-02-14 | 2001-03-28 | Glaxo Group Ltd | Chemical compounds |
| US7144908B2 (en) | 2001-03-08 | 2006-12-05 | Glaxo Group Limited | Agonists of beta-adrenoceptors |
| ATE381537T1 (en) | 2001-03-22 | 2008-01-15 | Glaxo Group Ltd | FORMANILIDE DERIVATIVES AS BETA2 ADRENORECEPTOR AGONISTS |
| PT1425001E (en) | 2001-09-14 | 2009-02-18 | Glaxo Group Ltd | Phenethanolamine derivatives for treatment of respiratory diseases |
| US20030229058A1 (en) | 2001-11-13 | 2003-12-11 | Moran Edmund J. | Aryl aniline beta2 adrenergic receptor agonists |
| GB0204719D0 (en) | 2002-02-28 | 2002-04-17 | Glaxo Group Ltd | Medicinal compounds |
| JP2005523920A (en) | 2002-04-25 | 2005-08-11 | グラクソ グループ リミテッド | Phenetanolamine derivative |
| JP2005527618A (en) | 2002-05-28 | 2005-09-15 | セラヴァンス インコーポレーテッド | Alkoxyaryl β2 adrenergic receptor agonist |
| GB0217225D0 (en) | 2002-07-25 | 2002-09-04 | Glaxo Group Ltd | Medicinal compounds |
| TW200409746A (en) | 2002-07-26 | 2004-06-16 | Theravance Inc | Crystalline β2 adrenergic receptor agonist |
| EP1622875B1 (en) | 2003-05-08 | 2009-12-02 | Theravance, Inc. | Crystalline forms of an arylaniline beta-2 adrenergic receptor agonist |
| US20040229058A1 (en) * | 2003-05-12 | 2004-11-18 | Trouilhet Yves M. | Multilayer film |
| TW200510277A (en) | 2003-05-27 | 2005-03-16 | Theravance Inc | Crystalline form of β2-adrenergic receptor agonist |
| TW200526547A (en) * | 2003-09-22 | 2005-08-16 | Theravance Inc | Amino-substituted ethylamino β2 adrenergic receptor agonists |
| TW200531692A (en) | 2004-01-12 | 2005-10-01 | Theravance Inc | Aryl aniline derivatives as β2 adrenergic receptor agonists |
| CN1984876A (en) * | 2004-06-03 | 2007-06-20 | 施万制药 | Diamine beta2 adrenergic receptor agonists |
| EP1778638A1 (en) | 2004-07-21 | 2007-05-02 | Theravance, Inc. | Diaryl ether beta2 adrenergic receptor agonists |
| JP2008512470A (en) | 2004-09-10 | 2008-04-24 | セラヴァンス, インコーポレーテッド | Amidine-substituted arylaniline compounds |
-
2004
- 2004-09-20 TW TW093128456A patent/TW200526547A/en unknown
- 2004-09-21 DK DK04784634T patent/DK1687257T3/en active
- 2004-09-21 NZ NZ545764A patent/NZ545764A/en unknown
- 2004-09-21 AU AU2004276254A patent/AU2004276254A1/en not_active Abandoned
- 2004-09-21 PL PL04784634T patent/PL1687257T3/en unknown
- 2004-09-21 AT AT04784634T patent/ATE428685T1/en not_active IP Right Cessation
- 2004-09-21 CA CA002538037A patent/CA2538037A1/en not_active Abandoned
- 2004-09-21 ES ES04784634T patent/ES2326172T3/en active Active
- 2004-09-21 JP JP2006527127A patent/JP2007505931A/en not_active Withdrawn
- 2004-09-21 SI SI200431162T patent/SI1687257T1/en unknown
- 2004-09-21 BR BRPI0414634-4A patent/BRPI0414634A/en not_active IP Right Cessation
- 2004-09-21 DE DE602004020655T patent/DE602004020655D1/en active Active
- 2004-09-21 RU RU2006113700/04A patent/RU2385317C2/en not_active IP Right Cessation
- 2004-09-21 EP EP04784634A patent/EP1687257B1/en active Active
- 2004-09-21 CN CNB2004800274097A patent/CN100465158C/en not_active Expired - Fee Related
- 2004-09-21 PT PT04784634T patent/PT1687257E/en unknown
- 2004-09-21 MX MXPA06003093A patent/MXPA06003093A/en active IP Right Grant
- 2004-09-21 US US10/946,544 patent/US7399863B2/en active Active
- 2004-09-21 WO PCT/US2004/030833 patent/WO2005030678A2/en active Application Filing
- 2004-09-21 KR KR1020067005604A patent/KR20070015496A/en not_active Application Discontinuation
- 2004-09-22 AR ARP040103406A patent/AR046085A1/en unknown
-
2006
- 2006-03-01 IL IL174026A patent/IL174026A0/en unknown
- 2006-03-09 IS IS8346A patent/IS8346A/en unknown
- 2006-03-17 CO CO06027355A patent/CO5680412A2/en not_active Application Discontinuation
- 2006-03-20 ZA ZA200602308A patent/ZA200602308B/en unknown
- 2006-04-07 MA MA28922A patent/MA28089A1/en unknown
- 2006-04-20 NO NO20061737A patent/NO20061737L/en not_active Application Discontinuation
-
2007
- 2007-01-11 HK HK07100415.0A patent/HK1093486A1/en not_active IP Right Cessation
-
2008
- 2008-04-30 US US12/112,726 patent/US7964730B2/en not_active Expired - Fee Related
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003042164A1 (en) * | 2001-11-13 | 2003-05-22 | Theravance, Inc | Aryl aniline beta-2 adrenergic receptor agonists |
| WO2003042160A1 (en) * | 2001-11-13 | 2003-05-22 | Theravance, Inc. | Aryl aniline beta-2 adrenergic receptor agonists |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7964615B2 (en) | 2005-05-20 | 2011-06-21 | Almirall, S.A. | Derivatives of 4-(2-amino-1-hydroxyethyl)phenol as agonists of the β2 adrenergic receptor |
| US7709511B2 (en) | 2005-08-09 | 2010-05-04 | Astrazeneca Ab | Benzothiazolone derivatives |
| WO2007106016A1 (en) * | 2006-03-14 | 2007-09-20 | Astrazeneca Ab | Bezothiazol derivatives as beta2 adrenoreceptor agonists |
| US7951954B2 (en) | 2006-03-14 | 2011-05-31 | Astrazeneca Ab | Bezothiazol derivatives as Beta2 adrenoreceptor agonists |
| WO2007124898A1 (en) | 2006-04-27 | 2007-11-08 | Laboratorios, Almirall S.A. | DERIVATIVES OF 4-(2-AMINO-1-HYDROXIETHYL)PHENOL AS AGONISTS OF THE β2 ADRENERGIC RECEPTOR |
| US7700782B2 (en) | 2006-12-20 | 2010-04-20 | Astrazeneca Ab | Compounds 569 |
| US8058294B2 (en) | 2007-02-08 | 2011-11-15 | Astrazeneca Ab | Pharmaceutical salts of N-[2-(diethylamino)ethyl]-N-(2-{[2-(4-hydroxy-2-oxo-2,3-dihydro-1,3-benzothiazol-7-yl)ethyl]amino}ethyl)-3-[2-(1-napthyl)ethoxy]propanamide |
| US8017602B2 (en) | 2008-06-18 | 2011-09-13 | Astrazeneca Ab | N-(2-(2-(5-hydroxy-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazin-8-yl)ethylamino)ethyl)-3-(phenethoxy)propanamide derivatives, processes for their preparation, pharmaceutical compositions containing them and their use in therapy |
| US8524908B2 (en) | 2009-03-12 | 2013-09-03 | Almirall, S.A. | Process for manufacturing 5-(2-{[6-(2,2-difluoro-2-phenylethoxy)hexyl]amino}-1-hydroxyethyl)-8-hydroxyquinolin-2(1H)-one |
| WO2011061527A1 (en) | 2009-11-17 | 2011-05-26 | Astrazeneca Ab | Combinations comprising a glucocorticoid receptor modulator for the treatment of respiratory diseases |
| WO2012046050A1 (en) | 2010-10-07 | 2012-04-12 | Astrazeneca Ab | Novel combinations |
| US9108918B2 (en) | 2011-10-07 | 2015-08-18 | Almirall, S.A. | Process for preparing 5-(2-{[6-(2,2-difluoro-2-phenylethoxy)hexyl]amino}-1(R)-hydroxyethyl)-8-hydroxyquinolin-2(1H)-one via a novel intermediate |
| US9346759B2 (en) | 2012-03-20 | 2016-05-24 | Almirall, S.A. | Polymorphic crystal forms of 5-(2-{[6-(2,2-difluoro-2-phenylethoxy)hexyl]amino}-1-(R)-hydroxyethyl)-8-hydroxyquinolin-2(1H)-one, heminapadisytlate as agonist of the β2 adrenergic receptor |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7964730B2 (en) | Amino-substituted ethylamino β2 adrenergic receptor agonists | |
| US7402673B2 (en) | Diamine β2 adrenergic receptor agonists | |
| US7622467B2 (en) | Aryl aniline derivatives as β2 adrenergic receptor agonists | |
| US7566785B2 (en) | Amidine substituted aryl aniline compounds | |
| US7317023B2 (en) | Diaryl ether β2 adrenergic receptor agonists | |
| KR20060136436A (en) | Aryl aniline derivatives as beta2 adrenergic receptor agonists | |
| KR20070022773A (en) | Diamine Beta 2?2 Adrenergic Receptor Agonists | |
| MXPA06007723A (en) | Aryl aniline derivatives as beta2 adrenergic receptor agonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200480027409.7 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 174026 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2538037 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1242/DELNP/2006 Country of ref document: IN Ref document number: 545764 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 06027355 Country of ref document: CO Ref document number: PA/a/2006/003093 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006/02308 Country of ref document: ZA Ref document number: 200602308 Country of ref document: ZA Ref document number: 2006527127 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004276254 Country of ref document: AU Ref document number: 1020067005604 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12006500587 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004784634 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2004276254 Country of ref document: AU Date of ref document: 20040921 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004276254 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1200600639 Country of ref document: VN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006113700 Country of ref document: RU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004784634 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0414634 Country of ref document: BR |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020067005604 Country of ref document: KR |