WO2006116156A2 - Toxin peptides with extended blood halflife - Google Patents
Toxin peptides with extended blood halflife Download PDFInfo
- Publication number
- WO2006116156A2 WO2006116156A2 PCT/US2006/015199 US2006015199W WO2006116156A2 WO 2006116156 A2 WO2006116156 A2 WO 2006116156A2 US 2006015199 W US2006015199 W US 2006015199W WO 2006116156 A2 WO2006116156 A2 WO 2006116156A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- peptide
- composition
- matter
- seq
- amino acid
- Prior art date
Links
- 108090000765 processed proteins & peptides Proteins 0.000 title claims abstract description 641
- 239000003053 toxin Substances 0.000 title claims abstract description 185
- 231100000765 toxin Toxicity 0.000 title claims abstract description 184
- 108700012359 toxins Proteins 0.000 title claims abstract description 184
- 102000004196 processed proteins & peptides Human genes 0.000 title description 137
- 210000004369 blood Anatomy 0.000 title description 11
- 239000008280 blood Substances 0.000 title description 11
- 239000000203 mixture Substances 0.000 claims abstract description 253
- 238000000034 method Methods 0.000 claims abstract description 70
- 125000000539 amino acid group Chemical group 0.000 claims abstract description 68
- 201000006417 multiple sclerosis Diseases 0.000 claims abstract description 30
- 208000024891 symptom Diseases 0.000 claims abstract description 22
- 208000023275 Autoimmune disease Diseases 0.000 claims abstract description 20
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 20
- 239000013604 expression vector Substances 0.000 claims abstract description 17
- 230000001404 mediated effect Effects 0.000 claims abstract description 12
- 239000003937 drug carrier Substances 0.000 claims abstract description 11
- 208000022559 Inflammatory bowel disease Diseases 0.000 claims abstract description 10
- 201000004681 Psoriasis Diseases 0.000 claims abstract description 10
- 206010025135 lupus erythematosus Diseases 0.000 claims abstract description 10
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 claims abstract description 9
- 206010039073 rheumatoid arthritis Diseases 0.000 claims abstract description 9
- 206010052779 Transplant rejections Diseases 0.000 claims abstract description 8
- 201000004624 Dermatitis Diseases 0.000 claims abstract description 7
- 208000009329 Graft vs Host Disease Diseases 0.000 claims abstract description 7
- 201000001263 Psoriatic Arthritis Diseases 0.000 claims abstract description 7
- 208000036824 Psoriatic arthropathy Diseases 0.000 claims abstract description 7
- 208000024908 graft versus host disease Diseases 0.000 claims abstract description 7
- 230000000116 mitigating effect Effects 0.000 claims abstract description 7
- 125000003275 alpha amino acid group Chemical group 0.000 claims description 129
- 210000004027 cell Anatomy 0.000 claims description 115
- 229920001223 polyethylene glycol Polymers 0.000 claims description 102
- 101000994669 Homo sapiens Potassium voltage-gated channel subfamily A member 3 Proteins 0.000 claims description 73
- 239000003112 inhibitor Substances 0.000 claims description 61
- 102100034355 Potassium voltage-gated channel subfamily A member 3 Human genes 0.000 claims description 47
- 239000005557 antagonist Substances 0.000 claims description 47
- 108091006146 Channels Proteins 0.000 claims description 44
- 150000001413 amino acids Chemical class 0.000 claims description 38
- 239000002202 Polyethylene glycol Substances 0.000 claims description 32
- 230000000694 effects Effects 0.000 claims description 31
- 229920000642 polymer Polymers 0.000 claims description 31
- 101001049892 Scorpio palmatus Potassium channel toxin alpha-KTx 6.2 Proteins 0.000 claims description 22
- 229940125400 channel inhibitor Drugs 0.000 claims description 22
- 239000011575 calcium Substances 0.000 claims description 20
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 20
- 229910052791 calcium Inorganic materials 0.000 claims description 17
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 claims description 16
- 229920002307 Dextran Polymers 0.000 claims description 15
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 claims description 15
- 239000003446 ligand Substances 0.000 claims description 14
- 208000035475 disorder Diseases 0.000 claims description 13
- 150000003384 small molecules Chemical class 0.000 claims description 13
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 claims description 12
- 101001077188 Leiurus hebraeus Potassium channel toxin alpha-KTx 3.2 Proteins 0.000 claims description 12
- 101100242312 Oryza sativa subsp. japonica OSK1 gene Proteins 0.000 claims description 12
- 101100533605 Oryza sativa subsp. japonica SKP1 gene Proteins 0.000 claims description 12
- 101710126338 Apamin Proteins 0.000 claims description 11
- 108060003951 Immunoglobulin Proteins 0.000 claims description 11
- 102000018358 immunoglobulin Human genes 0.000 claims description 11
- 108091006905 Human Serum Albumin Proteins 0.000 claims description 10
- 102000008100 Human Serum Albumin Human genes 0.000 claims description 10
- YVIIHEKJCKCXOB-STYWVVQQSA-N molport-023-276-178 Chemical compound C([C@H](NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H]1CSSC[C@H]2C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N3CCC[C@H]3C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@H](C(=O)N[C@@H](C)C(=O)N[C@H](C(N[C@@H](CSSC[C@H](N)C(=O)N[C@@H](CC(N)=O)C(=O)N2)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1)=O)CC(C)C)[C@@H](C)O)C(N)=O)C1=CNC=N1 YVIIHEKJCKCXOB-STYWVVQQSA-N 0.000 claims description 10
- 208000006673 asthma Diseases 0.000 claims description 9
- FQKNZJLPBMOCKU-ZDGDVGGMSA-N 3-[(1R,4S,4aS,7R,12R,15S,18S,21S,24S,30S,33S,36S,42S,45S,48R,53R,56S,59S,65S,68S,71S,74R,81S,87S,90S,95S,98S)-53-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S,3S)-2-[[(2S,3S)-2-[[(2S,3R)-2-amino-3-hydroxybutanoyl]amino]-3-methylpentanoyl]amino]-3-methylpentanoyl]amino]-4-oxobutanoyl]amino]-3-methylbutanoyl]amino]hexanoyl]amino]-4,15,45,68,81,98-hexakis(4-aminobutyl)-7-[[(2S)-1-[[(2S)-4-amino-1-[[(2S)-1,4-diamino-1,4-dioxobutan-2-yl]amino]-1,4-dioxobutan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]carbamoyl]-87-(2-amino-2-oxoethyl)-71-(3-amino-3-oxopropyl)-56-[(1R)-1-hydroxyethyl]-30,33,59,95-tetrakis(hydroxymethyl)-24-[(4-hydroxyphenyl)methyl]-36,42-dimethyl-21-(2-methylpropyl)-90-(2-methylsulfanylethyl)-2,5,5a,13,16,19,22,25,28,31,34,37,40,43,46,54,57,60,66,69,72,80,83,86,89,92,93,96,99-nonacosaoxo-9,10,50,51,76,77-hexathia-a,3,6,6a,14,17,20,23,26,29,32,35,38,41,44,47,55,58,61,67,70,73,79,82,85,88,91,94,97-nonacosazapentacyclo[46.30.14.1412,74.061,65.0100,104]hexahectan-18-yl]propanoic acid Chemical compound CC[C@H](C)[C@H](NC(=O)[C@@H](N)[C@@H](C)O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@H]1CSSC[C@@H]2NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](Cc3ccc(O)cc3)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@@H]3CSSC[C@H](NC(=O)[C@H](CCCCN)NC(=O)[C@H](CSSC[C@H](NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@@H]4CCCN4C(=O)[C@H](CO)NC(=O)[C@@H](NC1=O)[C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N1CCC[C@H]1C(=O)N3)NC(=O)[C@H](CCCCN)NC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCSC)NC2=O)C(=O)N[C@@H](Cc1ccc(O)cc1)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(N)=O)C(N)=O FQKNZJLPBMOCKU-ZDGDVGGMSA-N 0.000 claims description 8
- 102000009027 Albumins Human genes 0.000 claims description 8
- 108010088751 Albumins Proteins 0.000 claims description 8
- 101001049882 Anuroctonus phaiodactylus Potassium channel toxin alpha-KTx 6.12 Proteins 0.000 claims description 8
- 101000997262 Centruroides noxius Potassium channel toxin alpha-KTx 2.1 Proteins 0.000 claims description 8
- 101001049894 Leiurus hebraeus Potassium channel toxin alpha-KTx 5.1 Proteins 0.000 claims description 8
- 210000004978 chinese hamster ovary cell Anatomy 0.000 claims description 8
- 230000002757 inflammatory effect Effects 0.000 claims description 8
- 230000008685 targeting Effects 0.000 claims description 8
- 208000006386 Bone Resorption Diseases 0.000 claims description 7
- 241000588724 Escherichia coli Species 0.000 claims description 7
- 206010020751 Hypersensitivity Diseases 0.000 claims description 7
- 208000026935 allergic disease Diseases 0.000 claims description 7
- 230000007815 allergy Effects 0.000 claims description 7
- 230000024279 bone resorption Effects 0.000 claims description 7
- 229920001577 copolymer Polymers 0.000 claims description 7
- MXWDLLUGULWYIQ-BFRWRHKQSA-N scyllatoxin Chemical compound C([C@@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CS)C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CS)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1N=CNC=1)C(N)=O)NC(=O)[C@H](C)N)C1=CC=CC=C1 MXWDLLUGULWYIQ-BFRWRHKQSA-N 0.000 claims description 7
- 101000975483 Anemonia sulcata Kappa-actitoxin-Avd6a Proteins 0.000 claims description 6
- 229920002134 Carboxymethyl cellulose Polymers 0.000 claims description 6
- 101001050258 Mesobuthus martensii Potassium channel toxin alpha-KTx 16.2 Proteins 0.000 claims description 6
- 108010071690 Prealbumin Proteins 0.000 claims description 6
- 239000001768 carboxy methyl cellulose Substances 0.000 claims description 6
- 230000002209 hydrophobic effect Effects 0.000 claims description 6
- 229920000036 polyvinylpyrrolidone Polymers 0.000 claims description 6
- 239000001267 polyvinylpyrrolidone Substances 0.000 claims description 6
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 claims description 6
- 101001049893 Androctonus mauritanicus mauritanicus Potassium channel toxin alpha-KTx 5.2 Proteins 0.000 claims description 5
- 101000994677 Androctonus mauritanicus mauritanicus Potassium channel toxin alpha-KTx 8.1 Proteins 0.000 claims description 5
- 102000004506 Blood Proteins Human genes 0.000 claims description 5
- 108010017384 Blood Proteins Proteins 0.000 claims description 5
- 101000997271 Centruroides noxius Potassium channel toxin alpha-KTx 1.11 Proteins 0.000 claims description 5
- 206010016654 Fibrosis Diseases 0.000 claims description 5
- 206010018364 Glomerulonephritis Diseases 0.000 claims description 5
- 101001049895 Hottentotta tamulus Potassium channel toxin alpha-KTx 5.4 Proteins 0.000 claims description 5
- 101001046987 Mesobuthus martensii Potassium channel toxin alpha-KTx 3.6 Proteins 0.000 claims description 5
- 108090000699 N-Type Calcium Channels Proteins 0.000 claims description 5
- 102000004129 N-Type Calcium Channels Human genes 0.000 claims description 5
- 102100033184 Potassium voltage-gated channel subfamily D member 3 Human genes 0.000 claims description 5
- 206010039710 Scleroderma Diseases 0.000 claims description 5
- 208000021386 Sjogren Syndrome Diseases 0.000 claims description 5
- 201000009594 Systemic Scleroderma Diseases 0.000 claims description 5
- 206010042953 Systemic sclerosis Diseases 0.000 claims description 5
- 102000002248 Thyroxine-Binding Globulin Human genes 0.000 claims description 5
- 108010000259 Thyroxine-Binding Globulin Proteins 0.000 claims description 5
- 230000001270 agonistic effect Effects 0.000 claims description 5
- 235000010948 carboxy methyl cellulose Nutrition 0.000 claims description 5
- 239000008112 carboxymethyl-cellulose Substances 0.000 claims description 5
- 230000004761 fibrosis Effects 0.000 claims description 5
- 210000004962 mammalian cell Anatomy 0.000 claims description 5
- IYKSHBADGOZWIF-UTPLJIOFSA-N slotoxin Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CS)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CS)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)NCC(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCSC)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CS)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](C(C)C)C(O)=O)NC(=O)[C@@H](N)[C@@H](C)O)C1=CC=CC=C1 IYKSHBADGOZWIF-UTPLJIOFSA-N 0.000 claims description 5
- XUIIKFGFIJCVMT-UHFFFAOYSA-N thyroxine-binding globulin Natural products IC1=CC(CC([NH3+])C([O-])=O)=CC(I)=C1OC1=CC(I)=C(O)C(I)=C1 XUIIKFGFIJCVMT-UHFFFAOYSA-N 0.000 claims description 5
- 102000011045 Chloride Channels Human genes 0.000 claims description 4
- 108010062745 Chloride Channels Proteins 0.000 claims description 4
- 102100033170 Potassium voltage-gated channel subfamily D member 2 Human genes 0.000 claims description 4
- 230000003042 antagnostic effect Effects 0.000 claims description 4
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 claims description 3
- 101100454808 Caenorhabditis elegans lgg-2 gene Proteins 0.000 claims description 3
- 102000014914 Carrier Proteins Human genes 0.000 claims description 3
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 claims description 3
- 239000004372 Polyvinyl alcohol Substances 0.000 claims description 3
- 101001047017 Tityus serrulatus Potassium channel toxin alpha-KTx 4.2 Proteins 0.000 claims description 3
- 229920002451 polyvinyl alcohol Polymers 0.000 claims description 3
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 claims description 2
- 229930182556 Polyacetal Natural products 0.000 claims description 2
- GOOHAUXETOMSMM-UHFFFAOYSA-N Propylene oxide Chemical compound CC1CO1 GOOHAUXETOMSMM-UHFFFAOYSA-N 0.000 claims description 2
- 125000001931 aliphatic group Chemical group 0.000 claims description 2
- 108091008324 binding proteins Proteins 0.000 claims description 2
- 210000003527 eukaryotic cell Anatomy 0.000 claims description 2
- 229920001519 homopolymer Polymers 0.000 claims description 2
- 150000004668 long chain fatty acids Chemical class 0.000 claims description 2
- 229920000191 poly(N-vinyl pyrrolidone) Polymers 0.000 claims description 2
- 229920001308 poly(aminoacid) Polymers 0.000 claims description 2
- 229920001583 poly(oxyethylated polyols) Polymers 0.000 claims description 2
- 229920006324 polyoxymethylene Polymers 0.000 claims description 2
- 229920005606 polypropylene copolymer Polymers 0.000 claims description 2
- 229920001451 polypropylene glycol Polymers 0.000 claims description 2
- 210000001236 prokaryotic cell Anatomy 0.000 claims description 2
- 101000588067 Homo sapiens Metaxin-1 Proteins 0.000 claims 4
- 101001050257 Mesobuthus martensii Potassium channel toxin alpha-KTx 16.3 Proteins 0.000 claims 4
- 101000997277 Hottentotta tamulus Potassium channel toxin alpha-KTx 1.3 Proteins 0.000 claims 2
- 102000007584 Prealbumin Human genes 0.000 claims 2
- 108020001580 protein domains Proteins 0.000 claims 2
- 229940126121 sodium channel inhibitor Drugs 0.000 claims 2
- 238000001727 in vivo Methods 0.000 abstract description 12
- 208000008589 Obesity Diseases 0.000 abstract description 4
- 235000020824 obesity Nutrition 0.000 abstract description 4
- 239000000047 product Substances 0.000 description 157
- 108090000623 proteins and genes Proteins 0.000 description 113
- 235000018102 proteins Nutrition 0.000 description 111
- 102000004169 proteins and genes Human genes 0.000 description 110
- 230000002829 reductive effect Effects 0.000 description 101
- 235000001014 amino acid Nutrition 0.000 description 59
- 150000001875 compounds Chemical class 0.000 description 57
- 125000005647 linker group Chemical group 0.000 description 50
- -1 Ca2+ cation Chemical class 0.000 description 49
- 229940024606 amino acid Drugs 0.000 description 45
- 125000000151 cysteine group Chemical class N[C@@H](CS)C(=O)* 0.000 description 34
- 102000004310 Ion Channels Human genes 0.000 description 33
- 108090000862 Ion Channels Proteins 0.000 description 33
- 210000001744 T-lymphocyte Anatomy 0.000 description 33
- 238000009472 formulation Methods 0.000 description 33
- 235000018417 cysteine Nutrition 0.000 description 30
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 28
- 239000000463 material Substances 0.000 description 28
- 230000001225 therapeutic effect Effects 0.000 description 28
- 239000000872 buffer Substances 0.000 description 26
- 108020004414 DNA Proteins 0.000 description 25
- 229910052700 potassium Inorganic materials 0.000 description 25
- 239000000523 sample Substances 0.000 description 25
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 25
- AXAVXPMQTGXXJZ-UHFFFAOYSA-N 2-aminoacetic acid;2-amino-2-(hydroxymethyl)propane-1,3-diol Chemical compound NCC(O)=O.OCC(N)(CO)CO AXAVXPMQTGXXJZ-UHFFFAOYSA-N 0.000 description 24
- 239000003814 drug Substances 0.000 description 24
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 23
- 238000003776 cleavage reaction Methods 0.000 description 23
- 239000002953 phosphate buffered saline Substances 0.000 description 23
- 230000007017 scission Effects 0.000 description 23
- 102000035195 Peptidases Human genes 0.000 description 22
- 108091005804 Peptidases Proteins 0.000 description 22
- 239000011591 potassium Substances 0.000 description 22
- 238000011282 treatment Methods 0.000 description 22
- 230000003595 spectral effect Effects 0.000 description 21
- 208000002193 Pain Diseases 0.000 description 20
- NKLPQNGYXWVELD-UHFFFAOYSA-M coomassie brilliant blue Chemical compound [Na+].C1=CC(OCC)=CC=C1NC1=CC=C(C(=C2C=CC(C=C2)=[N+](CC)CC=2C=C(C=CC=2)S([O-])(=O)=O)C=2C=CC(=CC=2)N(CC)CC=2C=C(C=CC=2)S([O-])(=O)=O)C=C1 NKLPQNGYXWVELD-UHFFFAOYSA-M 0.000 description 20
- 239000000539 dimer Substances 0.000 description 20
- 230000014509 gene expression Effects 0.000 description 20
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 19
- 239000004365 Protease Substances 0.000 description 18
- 238000002835 absorbance Methods 0.000 description 18
- 102000005962 receptors Human genes 0.000 description 18
- 108020003175 receptors Proteins 0.000 description 18
- 238000001542 size-exclusion chromatography Methods 0.000 description 18
- 230000001419 dependent effect Effects 0.000 description 16
- 150000002500 ions Chemical class 0.000 description 16
- 238000004519 manufacturing process Methods 0.000 description 16
- 230000036407 pain Effects 0.000 description 16
- 239000010453 quartz Substances 0.000 description 16
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N silicon dioxide Inorganic materials O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 16
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 16
- 229910000162 sodium phosphate Inorganic materials 0.000 description 16
- 238000010183 spectrum analysis Methods 0.000 description 16
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 15
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 15
- 239000000499 gel Substances 0.000 description 15
- 235000018977 lysine Nutrition 0.000 description 15
- 229910052757 nitrogen Inorganic materials 0.000 description 15
- 230000004044 response Effects 0.000 description 15
- 238000001228 spectrum Methods 0.000 description 15
- 239000004472 Lysine Substances 0.000 description 14
- 230000006334 disulfide bridging Effects 0.000 description 14
- 229920001184 polypeptide Polymers 0.000 description 14
- 239000000843 powder Substances 0.000 description 14
- 239000000126 substance Substances 0.000 description 14
- BHPQYMZQTOCNFJ-UHFFFAOYSA-N Calcium cation Chemical compound [Ca+2] BHPQYMZQTOCNFJ-UHFFFAOYSA-N 0.000 description 13
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 13
- 241000282326 Felis catus Species 0.000 description 13
- 102220624568 Kelch repeat and BTB domain-containing protein 3_E16K_mutation Human genes 0.000 description 13
- 102220624589 Kelch repeat and BTB domain-containing protein 3_K20D_mutation Human genes 0.000 description 13
- 230000004913 activation Effects 0.000 description 13
- 210000003719 b-lymphocyte Anatomy 0.000 description 13
- 210000004899 c-terminal region Anatomy 0.000 description 13
- 230000006320 pegylation Effects 0.000 description 13
- 239000000243 solution Substances 0.000 description 13
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 12
- 108010036949 Cyclosporine Proteins 0.000 description 12
- 101001046988 Orthochirus scrobiculosus Potassium channel toxin alpha-KTx 3.7 Proteins 0.000 description 12
- 101001121312 Oryza sativa subsp. japonica Serine/threonine protein kinase OSK1 Proteins 0.000 description 12
- 239000003636 conditioned culture medium Substances 0.000 description 12
- 229940079593 drug Drugs 0.000 description 12
- 108010068927 iberiotoxin Proteins 0.000 description 12
- 230000005764 inhibitory process Effects 0.000 description 12
- 238000000816 matrix-assisted laser desorption--ionisation Methods 0.000 description 12
- 239000002609 medium Substances 0.000 description 12
- 108020001213 potassium channel Proteins 0.000 description 12
- 238000006467 substitution reaction Methods 0.000 description 12
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 12
- 238000001262 western blot Methods 0.000 description 12
- 102000004257 Potassium Channel Human genes 0.000 description 11
- 230000002378 acidificating effect Effects 0.000 description 11
- 230000004927 fusion Effects 0.000 description 11
- VDNVVLOBNHIMQA-UHFFFAOYSA-N iberiotoxin Chemical compound C1SSCC(C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(O)=O)NC(=O)C(CCCNC(N)=N)NC(=O)C1NC(=O)C(CCCCN)NC(=O)C(CCCCN)NC(=O)CNC(=O)C(CCSC)NC(=O)C(NC(=O)C(CCCCN)NC(=O)CNC(=O)C(CCCNC(N)=N)NC(=O)C(CC(O)=O)NC(=O)C(C(C)C)NC(=O)CNC(=O)C(CC=1C=CC=CC=1)NC(=O)C(CC(C)C)NC(=O)C(CC(O)=O)NC(=O)C(CCCCN)NC1=O)CSSCC1NC(=O)C(C(C)C)NC(=O)C(CO)NC(=O)C(CC=1C2=CC=CC=C2NC=1)NC(=O)C(NC(=O)C(CCC(O)=O)NC(=O)C(CCCCN)NC(=O)C(CO)NC(=O)C(C(C)C)NC(=O)C(CO)NC1=O)CSSCC1NC(=O)C(CC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(O)=O)NC(=O)C(C(C)O)NC(=O)C(NC(=O)C1NC(=O)CC1)CC1=CC=CC=C1 VDNVVLOBNHIMQA-UHFFFAOYSA-N 0.000 description 11
- 238000000746 purification Methods 0.000 description 11
- UQDJGEHQDNVPGU-UHFFFAOYSA-N serine phosphoethanolamine Chemical compound [NH3+]CCOP([O-])(=O)OCC([NH3+])C([O-])=O UQDJGEHQDNVPGU-UHFFFAOYSA-N 0.000 description 11
- 239000013598 vector Substances 0.000 description 11
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 10
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 10
- 238000004458 analytical method Methods 0.000 description 10
- 230000001580 bacterial effect Effects 0.000 description 10
- 230000008901 benefit Effects 0.000 description 10
- 239000008103 glucose Substances 0.000 description 10
- 230000003834 intracellular effect Effects 0.000 description 10
- 230000003389 potentiating effect Effects 0.000 description 10
- 239000004094 surface-active agent Substances 0.000 description 10
- 229910001868 water Inorganic materials 0.000 description 10
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 9
- 239000004471 Glycine Substances 0.000 description 9
- 108010002350 Interleukin-2 Proteins 0.000 description 9
- 102000000588 Interleukin-2 Human genes 0.000 description 9
- 241000700159 Rattus Species 0.000 description 9
- 230000015572 biosynthetic process Effects 0.000 description 9
- 238000006243 chemical reaction Methods 0.000 description 9
- 230000021615 conjugation Effects 0.000 description 9
- 239000012528 membrane Substances 0.000 description 9
- 210000004379 membrane Anatomy 0.000 description 9
- 150000007523 nucleic acids Chemical class 0.000 description 9
- 239000002773 nucleotide Substances 0.000 description 9
- 125000003729 nucleotide group Chemical group 0.000 description 9
- 230000037361 pathway Effects 0.000 description 9
- 230000008569 process Effects 0.000 description 9
- 239000003826 tablet Substances 0.000 description 9
- 229940124597 therapeutic agent Drugs 0.000 description 9
- 231100000611 venom Toxicity 0.000 description 9
- 229930105110 Cyclosporin A Natural products 0.000 description 8
- 229920002472 Starch Polymers 0.000 description 8
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 8
- 208000026278 immune system disease Diseases 0.000 description 8
- 230000004048 modification Effects 0.000 description 8
- 238000012986 modification Methods 0.000 description 8
- 102000039446 nucleic acids Human genes 0.000 description 8
- 108020004707 nucleic acids Proteins 0.000 description 8
- 239000013612 plasmid Substances 0.000 description 8
- 235000019419 proteases Nutrition 0.000 description 8
- 108091008146 restriction endonucleases Proteins 0.000 description 8
- 150000003839 salts Chemical class 0.000 description 8
- 239000011734 sodium Substances 0.000 description 8
- 235000019698 starch Nutrition 0.000 description 8
- 229940032147 starch Drugs 0.000 description 8
- 239000008107 starch Substances 0.000 description 8
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 7
- 102000005702 Calcium-Activated Potassium Channels Human genes 0.000 description 7
- 108010045489 Calcium-Activated Potassium Channels Proteins 0.000 description 7
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 7
- 241000239226 Scorpiones Species 0.000 description 7
- 150000001720 carbohydrates Chemical class 0.000 description 7
- 235000014633 carbohydrates Nutrition 0.000 description 7
- 229910052799 carbon Inorganic materials 0.000 description 7
- 239000003795 chemical substances by application Substances 0.000 description 7
- 238000004132 cross linking Methods 0.000 description 7
- 239000003085 diluting agent Substances 0.000 description 7
- 201000010099 disease Diseases 0.000 description 7
- 239000007884 disintegrant Substances 0.000 description 7
- 239000012636 effector Substances 0.000 description 7
- 229940088598 enzyme Drugs 0.000 description 7
- 239000012091 fetal bovine serum Substances 0.000 description 7
- 239000012634 fragment Substances 0.000 description 7
- 239000011159 matrix material Substances 0.000 description 7
- 230000007246 mechanism Effects 0.000 description 7
- 230000009145 protein modification Effects 0.000 description 7
- 239000000758 substrate Substances 0.000 description 7
- 238000003786 synthesis reaction Methods 0.000 description 7
- 239000002435 venom Substances 0.000 description 7
- 210000001048 venom Anatomy 0.000 description 7
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 6
- 108010029697 CD40 Ligand Proteins 0.000 description 6
- 102100032937 CD40 ligand Human genes 0.000 description 6
- 102000003922 Calcium Channels Human genes 0.000 description 6
- 108090000312 Calcium Channels Proteins 0.000 description 6
- 102220614563 Calmodulin-3_K22A_mutation Human genes 0.000 description 6
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 6
- 102000053602 DNA Human genes 0.000 description 6
- 102000004190 Enzymes Human genes 0.000 description 6
- 108090000790 Enzymes Proteins 0.000 description 6
- 101000932804 Homo sapiens Voltage-dependent T-type calcium channel subunit alpha-1H Proteins 0.000 description 6
- 206010020772 Hypertension Diseases 0.000 description 6
- 108010074328 Interferon-gamma Proteins 0.000 description 6
- 102000008070 Interferon-gamma Human genes 0.000 description 6
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 6
- 229930195725 Mannitol Natural products 0.000 description 6
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 6
- 102000003729 Neprilysin Human genes 0.000 description 6
- 108090000028 Neprilysin Proteins 0.000 description 6
- 108091028043 Nucleic acid sequence Proteins 0.000 description 6
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 6
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 6
- HATRDXDCPOXQJX-UHFFFAOYSA-N Thapsigargin Natural products CCCCCCCC(=O)OC1C(OC(O)C(=C/C)C)C(=C2C3OC(=O)C(C)(O)C3(O)C(CC(C)(OC(=O)C)C12)OC(=O)CCC)C HATRDXDCPOXQJX-UHFFFAOYSA-N 0.000 description 6
- 102100025482 Voltage-dependent T-type calcium channel subunit alpha-1H Human genes 0.000 description 6
- 235000004279 alanine Nutrition 0.000 description 6
- 150000001299 aldehydes Chemical class 0.000 description 6
- 230000004071 biological effect Effects 0.000 description 6
- 230000028956 calcium-mediated signaling Effects 0.000 description 6
- 238000007385 chemical modification Methods 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- 239000003599 detergent Substances 0.000 description 6
- 206010012601 diabetes mellitus Diseases 0.000 description 6
- 238000012377 drug delivery Methods 0.000 description 6
- 230000006870 function Effects 0.000 description 6
- 239000001963 growth medium Substances 0.000 description 6
- 230000001965 increasing effect Effects 0.000 description 6
- 230000003993 interaction Effects 0.000 description 6
- 229960003130 interferon gamma Drugs 0.000 description 6
- 239000010410 layer Substances 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- 239000000594 mannitol Substances 0.000 description 6
- 235000010355 mannitol Nutrition 0.000 description 6
- 210000002569 neuron Anatomy 0.000 description 6
- 150000002482 oligosaccharides Chemical class 0.000 description 6
- 238000010647 peptide synthesis reaction Methods 0.000 description 6
- 238000002823 phage display Methods 0.000 description 6
- 230000035755 proliferation Effects 0.000 description 6
- 235000013930 proline Nutrition 0.000 description 6
- 230000002459 sustained effect Effects 0.000 description 6
- IXFPJGBNCFXKPI-FSIHEZPISA-N thapsigargin Chemical compound CCCC(=O)O[C@H]1C[C@](C)(OC(C)=O)[C@H]2[C@H](OC(=O)CCCCCCC)[C@@H](OC(=O)C(\C)=C/C)C(C)=C2[C@@H]2OC(=O)[C@@](C)(O)[C@]21O IXFPJGBNCFXKPI-FSIHEZPISA-N 0.000 description 6
- 241000242759 Actiniaria Species 0.000 description 5
- 241000894006 Bacteria Species 0.000 description 5
- 102000004631 Calcineurin Human genes 0.000 description 5
- 108010042955 Calcineurin Proteins 0.000 description 5
- 108020004705 Codon Proteins 0.000 description 5
- 241000699802 Cricetulus griseus Species 0.000 description 5
- 102000004127 Cytokines Human genes 0.000 description 5
- 108090000695 Cytokines Proteins 0.000 description 5
- 108010091135 Immunoglobulin Fc Fragments Proteins 0.000 description 5
- 102000018071 Immunoglobulin Fc Fragments Human genes 0.000 description 5
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 5
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 5
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 5
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 5
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 5
- 206010028980 Neoplasm Diseases 0.000 description 5
- 102000019315 Nicotinic acetylcholine receptors Human genes 0.000 description 5
- 108050006807 Nicotinic acetylcholine receptors Proteins 0.000 description 5
- 241000239299 Orthochirus scrobiculosus Species 0.000 description 5
- 229930006000 Sucrose Natural products 0.000 description 5
- 102000003691 T-Type Calcium Channels Human genes 0.000 description 5
- 108090000030 T-Type Calcium Channels Proteins 0.000 description 5
- 108090000631 Trypsin Proteins 0.000 description 5
- 102000004142 Trypsin Human genes 0.000 description 5
- 239000011543 agarose gel Substances 0.000 description 5
- 125000000217 alkyl group Chemical group 0.000 description 5
- 230000010056 antibody-dependent cellular cytotoxicity Effects 0.000 description 5
- 230000000903 blocking effect Effects 0.000 description 5
- 230000037396 body weight Effects 0.000 description 5
- 125000001314 canonical amino-acid group Chemical group 0.000 description 5
- 210000000170 cell membrane Anatomy 0.000 description 5
- 229920002678 cellulose Polymers 0.000 description 5
- 239000001913 cellulose Substances 0.000 description 5
- 235000010980 cellulose Nutrition 0.000 description 5
- 230000000295 complement effect Effects 0.000 description 5
- 239000000306 component Substances 0.000 description 5
- 238000007906 compression Methods 0.000 description 5
- 230000006835 compression Effects 0.000 description 5
- 238000001514 detection method Methods 0.000 description 5
- 238000010586 diagram Methods 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 239000000194 fatty acid Substances 0.000 description 5
- 239000000796 flavoring agent Substances 0.000 description 5
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 5
- 230000002401 inhibitory effect Effects 0.000 description 5
- 239000000543 intermediate Substances 0.000 description 5
- 238000001990 intravenous administration Methods 0.000 description 5
- 229960001375 lactose Drugs 0.000 description 5
- 239000008101 lactose Substances 0.000 description 5
- 210000004698 lymphocyte Anatomy 0.000 description 5
- 229920000609 methyl cellulose Polymers 0.000 description 5
- 235000010981 methylcellulose Nutrition 0.000 description 5
- 239000001923 methylcellulose Substances 0.000 description 5
- 229920001542 oligosaccharide Polymers 0.000 description 5
- 125000001151 peptidyl group Chemical group 0.000 description 5
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 5
- 235000019833 protease Nutrition 0.000 description 5
- 230000002685 pulmonary effect Effects 0.000 description 5
- 239000002795 scorpion venom Substances 0.000 description 5
- 210000002966 serum Anatomy 0.000 description 5
- 229910052708 sodium Inorganic materials 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- 239000007790 solid phase Substances 0.000 description 5
- 239000005720 sucrose Substances 0.000 description 5
- 229960004793 sucrose Drugs 0.000 description 5
- 229910052717 sulfur Inorganic materials 0.000 description 5
- 238000013268 sustained release Methods 0.000 description 5
- 239000012730 sustained-release form Substances 0.000 description 5
- 238000002560 therapeutic procedure Methods 0.000 description 5
- 210000001519 tissue Anatomy 0.000 description 5
- 238000012546 transfer Methods 0.000 description 5
- 239000012588 trypsin Substances 0.000 description 5
- 229910052727 yttrium Inorganic materials 0.000 description 5
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 4
- 229940122739 Calcineurin inhibitor Drugs 0.000 description 4
- 101710192106 Calcineurin-binding protein cabin-1 Proteins 0.000 description 4
- 102100024123 Calcineurin-binding protein cabin-1 Human genes 0.000 description 4
- 108090000317 Chymotrypsin Proteins 0.000 description 4
- 239000001856 Ethyl cellulose Substances 0.000 description 4
- 108010010803 Gelatin Proteins 0.000 description 4
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 4
- 206010061218 Inflammation Diseases 0.000 description 4
- RHGKLRLOHDJJDR-BYPYZUCNSA-N L-citrulline Chemical compound NC(=O)NCCC[C@H]([NH3+])C([O-])=O RHGKLRLOHDJJDR-BYPYZUCNSA-N 0.000 description 4
- QUOGESRFPZDMMT-YFKPBYRVSA-N L-homoarginine Chemical compound OC(=O)[C@@H](N)CCCCNC(N)=N QUOGESRFPZDMMT-YFKPBYRVSA-N 0.000 description 4
- LRQKBLKVPFOOQJ-YFKPBYRVSA-N L-norleucine Chemical compound CCCC[C@H]([NH3+])C([O-])=O LRQKBLKVPFOOQJ-YFKPBYRVSA-N 0.000 description 4
- 102000004086 Ligand-Gated Ion Channels Human genes 0.000 description 4
- 108090000543 Ligand-Gated Ion Channels Proteins 0.000 description 4
- 102000004868 N-Methyl-D-Aspartate Receptors Human genes 0.000 description 4
- 108090001041 N-Methyl-D-Aspartate Receptors Proteins 0.000 description 4
- 239000012124 Opti-MEM Substances 0.000 description 4
- FKNQFGJONOIPTF-UHFFFAOYSA-N Sodium cation Chemical compound [Na+] FKNQFGJONOIPTF-UHFFFAOYSA-N 0.000 description 4
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 4
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 4
- 102000009190 Transthyretin Human genes 0.000 description 4
- 102000003734 Voltage-Gated Potassium Channels Human genes 0.000 description 4
- 108090000013 Voltage-Gated Potassium Channels Proteins 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 239000004480 active ingredient Substances 0.000 description 4
- 239000013543 active substance Substances 0.000 description 4
- 239000000654 additive Substances 0.000 description 4
- 150000001408 amides Chemical group 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 4
- 125000003118 aryl group Chemical group 0.000 description 4
- 235000009582 asparagine Nutrition 0.000 description 4
- 229960001230 asparagine Drugs 0.000 description 4
- 239000011230 binding agent Substances 0.000 description 4
- 230000000975 bioactive effect Effects 0.000 description 4
- 229940046731 calcineurin inhibitors Drugs 0.000 description 4
- 201000011510 cancer Diseases 0.000 description 4
- 229920002301 cellulose acetate Polymers 0.000 description 4
- 238000001311 chemical methods and process Methods 0.000 description 4
- 229960002376 chymotrypsin Drugs 0.000 description 4
- 238000000576 coating method Methods 0.000 description 4
- 210000004443 dendritic cell Anatomy 0.000 description 4
- 108010091894 dendrotoxin K Proteins 0.000 description 4
- 238000001212 derivatisation Methods 0.000 description 4
- 235000014113 dietary fatty acids Nutrition 0.000 description 4
- 238000010790 dilution Methods 0.000 description 4
- 239000012895 dilution Substances 0.000 description 4
- 208000033068 episodic angioedema with eosinophilia Diseases 0.000 description 4
- 210000003743 erythrocyte Anatomy 0.000 description 4
- 229920001249 ethyl cellulose Polymers 0.000 description 4
- 238000002474 experimental method Methods 0.000 description 4
- 229930195729 fatty acid Natural products 0.000 description 4
- 239000000945 filler Substances 0.000 description 4
- 235000013355 food flavoring agent Nutrition 0.000 description 4
- 125000000524 functional group Chemical group 0.000 description 4
- 108020001507 fusion proteins Proteins 0.000 description 4
- 102000037865 fusion proteins Human genes 0.000 description 4
- 239000008273 gelatin Substances 0.000 description 4
- 229920000159 gelatin Polymers 0.000 description 4
- 229940014259 gelatin Drugs 0.000 description 4
- 235000019322 gelatine Nutrition 0.000 description 4
- 235000011852 gelatine desserts Nutrition 0.000 description 4
- 229930195712 glutamate Natural products 0.000 description 4
- 125000000291 glutamic acid group Chemical group N[C@@H](CCC(O)=O)C(=O)* 0.000 description 4
- 102000046611 human KCNA3 Human genes 0.000 description 4
- 239000000017 hydrogel Substances 0.000 description 4
- 229910052739 hydrogen Inorganic materials 0.000 description 4
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 4
- 230000005847 immunogenicity Effects 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 239000002502 liposome Substances 0.000 description 4
- 210000004072 lung Anatomy 0.000 description 4
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 4
- 210000003071 memory t lymphocyte Anatomy 0.000 description 4
- 230000005012 migration Effects 0.000 description 4
- 238000013508 migration Methods 0.000 description 4
- 108091005601 modified peptides Proteins 0.000 description 4
- 239000006199 nebulizer Substances 0.000 description 4
- 230000002611 ovarian Effects 0.000 description 4
- 239000003961 penetration enhancing agent Substances 0.000 description 4
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 4
- 229920001282 polysaccharide Polymers 0.000 description 4
- 239000005017 polysaccharide Substances 0.000 description 4
- 150000004804 polysaccharides Chemical class 0.000 description 4
- 229920000053 polysorbate 80 Polymers 0.000 description 4
- 230000017854 proteolysis Effects 0.000 description 4
- 208000037803 restenosis Diseases 0.000 description 4
- 230000000717 retained effect Effects 0.000 description 4
- 238000004007 reversed phase HPLC Methods 0.000 description 4
- 238000012552 review Methods 0.000 description 4
- 238000012216 screening Methods 0.000 description 4
- 230000028327 secretion Effects 0.000 description 4
- 230000035945 sensitivity Effects 0.000 description 4
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 4
- 208000007056 sickle cell anemia Diseases 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- 238000010532 solid phase synthesis reaction Methods 0.000 description 4
- 235000000346 sugar Nutrition 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 4
- 230000001988 toxicity Effects 0.000 description 4
- 231100000419 toxicity Toxicity 0.000 description 4
- 238000001890 transfection Methods 0.000 description 4
- 229920003169 water-soluble polymer Polymers 0.000 description 4
- OGNSCSPNOLGXSM-UHFFFAOYSA-N 2,4-diaminobutyric acid Chemical group NCCC(N)C(O)=O OGNSCSPNOLGXSM-UHFFFAOYSA-N 0.000 description 3
- FUOOLUPWFVMBKG-UHFFFAOYSA-N 2-Aminoisobutyric acid Chemical compound CC(C)(N)C(O)=O FUOOLUPWFVMBKG-UHFFFAOYSA-N 0.000 description 3
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- 239000004475 Arginine Substances 0.000 description 3
- 101150013553 CD40 gene Proteins 0.000 description 3
- 208000000094 Chronic Pain Diseases 0.000 description 3
- 241001638933 Cochlicella barbara Species 0.000 description 3
- 102100024746 Dihydrofolate reductase Human genes 0.000 description 3
- 102000005593 Endopeptidases Human genes 0.000 description 3
- 108010059378 Endopeptidases Proteins 0.000 description 3
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 3
- 108010049003 Fibrinogen Proteins 0.000 description 3
- 102000008946 Fibrinogen Human genes 0.000 description 3
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 3
- 206010021639 Incontinence Diseases 0.000 description 3
- QUOGESRFPZDMMT-UHFFFAOYSA-N L-Homoarginine Natural products OC(=O)C(N)CCCCNC(N)=N QUOGESRFPZDMMT-UHFFFAOYSA-N 0.000 description 3
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 3
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 3
- 101000994385 Mesobuthus martensii Potassium channel toxin alpha-KTx 15.2 Proteins 0.000 description 3
- 206010027926 Monoplegia Diseases 0.000 description 3
- 239000002033 PVDF binder Substances 0.000 description 3
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 3
- 108090000526 Papain Proteins 0.000 description 3
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 3
- 102000012479 Serine Proteases Human genes 0.000 description 3
- 108010022999 Serine Proteases Proteins 0.000 description 3
- 230000006044 T cell activation Effects 0.000 description 3
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 3
- 241000239292 Theraphosidae Species 0.000 description 3
- 230000002159 abnormal effect Effects 0.000 description 3
- 238000010521 absorption reaction Methods 0.000 description 3
- 239000002671 adjuvant Substances 0.000 description 3
- 239000000556 agonist Substances 0.000 description 3
- 125000003295 alanine group Chemical group N[C@@H](C)C(=O)* 0.000 description 3
- 235000010443 alginic acid Nutrition 0.000 description 3
- 229920000615 alginic acid Polymers 0.000 description 3
- 210000000612 antigen-presenting cell Anatomy 0.000 description 3
- 238000013459 approach Methods 0.000 description 3
- 235000009697 arginine Nutrition 0.000 description 3
- 229960003121 arginine Drugs 0.000 description 3
- 235000003704 aspartic acid Nutrition 0.000 description 3
- SQVRNKJHWKZAKO-UHFFFAOYSA-N beta-N-Acetyl-D-neuraminic acid Natural products CC(=O)NC1C(O)CC(O)(C(O)=O)OC1C(O)C(O)CO SQVRNKJHWKZAKO-UHFFFAOYSA-N 0.000 description 3
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 3
- 230000008033 biological extinction Effects 0.000 description 3
- 239000012472 biological sample Substances 0.000 description 3
- 238000006664 bond formation reaction Methods 0.000 description 3
- 210000004556 brain Anatomy 0.000 description 3
- 230000009460 calcium influx Effects 0.000 description 3
- 230000001964 calcium overload Effects 0.000 description 3
- 150000001718 carbodiimides Chemical class 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- 230000004663 cell proliferation Effects 0.000 description 3
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 3
- 239000003086 colorant Substances 0.000 description 3
- 238000013270 controlled release Methods 0.000 description 3
- 230000001276 controlling effect Effects 0.000 description 3
- 230000008878 coupling Effects 0.000 description 3
- 238000010168 coupling process Methods 0.000 description 3
- 238000005859 coupling reaction Methods 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 235000005911 diet Nutrition 0.000 description 3
- 230000037213 diet Effects 0.000 description 3
- 230000004069 differentiation Effects 0.000 description 3
- 230000029087 digestion Effects 0.000 description 3
- 108020001096 dihydrofolate reductase Proteins 0.000 description 3
- AFOSIXZFDONLBT-UHFFFAOYSA-N divinyl sulfone Chemical group C=CS(=O)(=O)C=C AFOSIXZFDONLBT-UHFFFAOYSA-N 0.000 description 3
- 231100000673 dose–response relationship Toxicity 0.000 description 3
- 238000005538 encapsulation Methods 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 239000003623 enhancer Substances 0.000 description 3
- 206010015037 epilepsy Diseases 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- 235000019325 ethyl cellulose Nutrition 0.000 description 3
- 150000004665 fatty acids Chemical class 0.000 description 3
- 229940012952 fibrinogen Drugs 0.000 description 3
- 239000012467 final product Substances 0.000 description 3
- BRZYSWJRSDMWLG-CAXSIQPQSA-N geneticin Chemical compound O1C[C@@](O)(C)[C@H](NC)[C@@H](O)[C@H]1O[C@@H]1[C@@H](O)[C@H](O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](C(C)O)O2)N)[C@@H](N)C[C@H]1N BRZYSWJRSDMWLG-CAXSIQPQSA-N 0.000 description 3
- 235000013922 glutamic acid Nutrition 0.000 description 3
- 239000004220 glutamic acid Substances 0.000 description 3
- 125000003630 glycyl group Chemical group [H]N([H])C([H])([H])C(*)=O 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 210000002216 heart Anatomy 0.000 description 3
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 3
- 239000001257 hydrogen Substances 0.000 description 3
- 230000016784 immunoglobulin production Effects 0.000 description 3
- 229940072221 immunoglobulins Drugs 0.000 description 3
- 230000004054 inflammatory process Effects 0.000 description 3
- 230000004941 influx Effects 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 210000003141 lower extremity Anatomy 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 3
- 239000004005 microsphere Substances 0.000 description 3
- 239000000178 monomer Substances 0.000 description 3
- 235000019799 monosodium phosphate Nutrition 0.000 description 3
- 210000000822 natural killer cell Anatomy 0.000 description 3
- 230000007935 neutral effect Effects 0.000 description 3
- 230000000269 nucleophilic effect Effects 0.000 description 3
- AHLPHDHHMVZTML-BYPYZUCNSA-N ornithyl group Chemical group N[C@@H](CCCN)C(=O)O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 3
- 201000002528 pancreatic cancer Diseases 0.000 description 3
- 229940055729 papain Drugs 0.000 description 3
- 235000019834 papain Nutrition 0.000 description 3
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 3
- 230000000144 pharmacologic effect Effects 0.000 description 3
- 229920001515 polyalkylene glycol Polymers 0.000 description 3
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 3
- 229940068968 polysorbate 80 Drugs 0.000 description 3
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 3
- 239000011148 porous material Substances 0.000 description 3
- 230000003823 potassium efflux Effects 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 239000003380 propellant Substances 0.000 description 3
- 125000006239 protecting group Chemical group 0.000 description 3
- 230000004224 protection Effects 0.000 description 3
- 230000027425 release of sequestered calcium ion into cytosol Effects 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 239000012679 serum free medium Substances 0.000 description 3
- 230000011664 signaling Effects 0.000 description 3
- 210000003491 skin Anatomy 0.000 description 3
- 210000000329 smooth muscle myocyte Anatomy 0.000 description 3
- 239000007909 solid dosage form Substances 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical group O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 3
- 239000002562 thickening agent Substances 0.000 description 3
- 150000003573 thiols Chemical class 0.000 description 3
- 230000009466 transformation Effects 0.000 description 3
- 239000003656 tris buffered saline Substances 0.000 description 3
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 3
- 238000000108 ultra-filtration Methods 0.000 description 3
- 230000003827 upregulation Effects 0.000 description 3
- 230000002792 vascular Effects 0.000 description 3
- 210000004509 vascular smooth muscle cell Anatomy 0.000 description 3
- 239000003981 vehicle Substances 0.000 description 3
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 2
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 2
- DQLHSFUMICQIMB-VIFPVBQESA-N (2s)-2-amino-3-(4-methylphenyl)propanoic acid Chemical compound CC1=CC=C(C[C@H](N)C(O)=O)C=C1 DQLHSFUMICQIMB-VIFPVBQESA-N 0.000 description 2
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 2
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 2
- UUDAMDVQRQNNHZ-UHFFFAOYSA-N (S)-AMPA Chemical compound CC=1ONC(=O)C=1CC(N)C(O)=O UUDAMDVQRQNNHZ-UHFFFAOYSA-N 0.000 description 2
- UWYZHKAOTLEWKK-UHFFFAOYSA-N 1,2,3,4-tetrahydroisoquinoline Chemical compound C1=CC=C2CNCCC2=C1 UWYZHKAOTLEWKK-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- OYIFNHCXNCRBQI-UHFFFAOYSA-N 2-aminoadipic acid Chemical compound OC(=O)C(N)CCCC(O)=O OYIFNHCXNCRBQI-UHFFFAOYSA-N 0.000 description 2
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 2
- BRMWTNUJHUMWMS-UHFFFAOYSA-N 3-Methylhistidine Chemical group CN1C=NC(CC(N)C(O)=O)=C1 BRMWTNUJHUMWMS-UHFFFAOYSA-N 0.000 description 2
- PECYZEOJVXMISF-UHFFFAOYSA-N 3-aminoalanine Chemical group [NH3+]CC(N)C([O-])=O PECYZEOJVXMISF-UHFFFAOYSA-N 0.000 description 2
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 2
- 208000024827 Alzheimer disease Diseases 0.000 description 2
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 2
- 208000020925 Bipolar disease Diseases 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- 108010078791 Carrier Proteins Proteins 0.000 description 2
- 108010076667 Caspases Proteins 0.000 description 2
- 102000011727 Caspases Human genes 0.000 description 2
- 108010023798 Charybdotoxin Proteins 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 108091026890 Coding region Proteins 0.000 description 2
- 108091035707 Consensus sequence Proteins 0.000 description 2
- 102000005927 Cysteine Proteases Human genes 0.000 description 2
- 108010005843 Cysteine Proteases Proteins 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- 150000008574 D-amino acids Chemical class 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- QSJXEFYPDANLFS-UHFFFAOYSA-N Diacetyl Chemical compound CC(=O)C(C)=O QSJXEFYPDANLFS-UHFFFAOYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 2
- 239000012591 Dulbecco’s Phosphate Buffered Saline Substances 0.000 description 2
- 108010008165 Etanercept Proteins 0.000 description 2
- 108010087819 Fc receptors Proteins 0.000 description 2
- 102000009109 Fc receptors Human genes 0.000 description 2
- 241000237858 Gastropoda Species 0.000 description 2
- 208000010412 Glaucoma Diseases 0.000 description 2
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 2
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 description 2
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 2
- 101000760570 Homo sapiens ATP-binding cassette sub-family C member 8 Proteins 0.000 description 2
- 101000944272 Homo sapiens ATP-sensitive inward rectifier potassium channel 1 Proteins 0.000 description 2
- 101000944277 Homo sapiens Inward rectifier potassium channel 2 Proteins 0.000 description 2
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 2
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 2
- PMMYEEVYMWASQN-DMTCNVIQSA-N Hydroxyproline Chemical compound O[C@H]1CN[C@H](C(O)=O)C1 PMMYEEVYMWASQN-DMTCNVIQSA-N 0.000 description 2
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 2
- 102100026120 IgG receptor FcRn large subunit p51 Human genes 0.000 description 2
- 101710177940 IgG receptor FcRn large subunit p51 Proteins 0.000 description 2
- 108010038453 Interleukin-2 Receptors Proteins 0.000 description 2
- 102000010789 Interleukin-2 Receptors Human genes 0.000 description 2
- 102100033114 Inward rectifier potassium channel 2 Human genes 0.000 description 2
- ODKSFYDXXFIFQN-BYPYZUCNSA-N L-arginine Chemical compound OC(=O)[C@@H](N)CCCN=C(N)N ODKSFYDXXFIFQN-BYPYZUCNSA-N 0.000 description 2
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 2
- SNDPXSYFESPGGJ-UHFFFAOYSA-N L-norVal-OH Natural products CCCC(N)C(O)=O SNDPXSYFESPGGJ-UHFFFAOYSA-N 0.000 description 2
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 2
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 2
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 2
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 2
- 101001050250 Mesobuthus martensii Potassium channel toxin alpha-KTx 17.1 Proteins 0.000 description 2
- 101000631734 Mesobuthus martensii Sodium channel neurotoxin BmK NT2 Proteins 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- 102000006386 Myelin Proteins Human genes 0.000 description 2
- 108010083674 Myelin Proteins Proteins 0.000 description 2
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 2
- JDHILDINMRGULE-LURJTMIESA-N N(pros)-methyl-L-histidine Chemical group CN1C=NC=C1C[C@H](N)C(O)=O JDHILDINMRGULE-LURJTMIESA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- RHGKLRLOHDJJDR-UHFFFAOYSA-N Ndelta-carbamoyl-DL-ornithine Natural products OC(=O)C(N)CCCNC(N)=O RHGKLRLOHDJJDR-UHFFFAOYSA-N 0.000 description 2
- 208000028389 Nerve injury Diseases 0.000 description 2
- 208000012902 Nervous system disease Diseases 0.000 description 2
- 208000025966 Neurological disease Diseases 0.000 description 2
- 102000007399 Nuclear hormone receptor Human genes 0.000 description 2
- 239000005642 Oleic acid Substances 0.000 description 2
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 2
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 2
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 2
- 229920001734 PEG propionaldehyde Polymers 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 206010033885 Paraparesis Diseases 0.000 description 2
- 108010067902 Peptide Library Proteins 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 108010039918 Polylysine Proteins 0.000 description 2
- 241001415846 Procellariidae Species 0.000 description 2
- 108010076504 Protein Sorting Signals Proteins 0.000 description 2
- 101710137312 Protein orai Proteins 0.000 description 2
- RADKZDMFGJYCBB-UHFFFAOYSA-N Pyridoxal Chemical compound CC1=NC=C(CO)C(C=O)=C1O RADKZDMFGJYCBB-UHFFFAOYSA-N 0.000 description 2
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 2
- 108020004511 Recombinant DNA Proteins 0.000 description 2
- 108091060738 Sea anemone family Proteins 0.000 description 2
- 241000270295 Serpentes Species 0.000 description 2
- 102000018674 Sodium Channels Human genes 0.000 description 2
- 108010052164 Sodium Channels Proteins 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- 241000242730 Stichodactyla helianthus Species 0.000 description 2
- 230000006052 T cell proliferation Effects 0.000 description 2
- 108091008874 T cell receptors Proteins 0.000 description 2
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 2
- 239000006180 TBST buffer Substances 0.000 description 2
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 description 2
- NYTOUQBROMCLBJ-UHFFFAOYSA-N Tetranitromethane Chemical compound [O-][N+](=O)C([N+]([O-])=O)([N+]([O-])=O)[N+]([O-])=O NYTOUQBROMCLBJ-UHFFFAOYSA-N 0.000 description 2
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 2
- 239000004473 Threonine Substances 0.000 description 2
- 108090000190 Thrombin Proteins 0.000 description 2
- 208000030886 Traumatic Brain injury Diseases 0.000 description 2
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 239000007983 Tris buffer Substances 0.000 description 2
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 206010046543 Urinary incontinence Diseases 0.000 description 2
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 2
- 108010053752 Voltage-Gated Sodium Channels Proteins 0.000 description 2
- 102000016913 Voltage-Gated Sodium Channels Human genes 0.000 description 2
- YRKCREAYFQTBPV-UHFFFAOYSA-N acetylacetone Chemical compound CC(=O)CC(C)=O YRKCREAYFQTBPV-UHFFFAOYSA-N 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- 239000000488 activin Substances 0.000 description 2
- 239000000853 adhesive Substances 0.000 description 2
- 230000001070 adhesive effect Effects 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 2
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 2
- 238000002399 angioplasty Methods 0.000 description 2
- 229960004977 anhydrous lactose Drugs 0.000 description 2
- 239000003831 antifriction material Substances 0.000 description 2
- 239000000427 antigen Substances 0.000 description 2
- 108091007433 antigens Proteins 0.000 description 2
- 102000036639 antigens Human genes 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- 206010003246 arthritis Diseases 0.000 description 2
- 235000010323 ascorbic acid Nutrition 0.000 description 2
- 239000011668 ascorbic acid Substances 0.000 description 2
- 229960005070 ascorbic acid Drugs 0.000 description 2
- 125000000613 asparagine group Chemical group N[C@@H](CC(N)=O)C(=O)* 0.000 description 2
- CKLJMWTZIZZHCS-REOHCLBHSA-L aspartate group Chemical group N[C@@H](CC(=O)[O-])C(=O)[O-] CKLJMWTZIZZHCS-REOHCLBHSA-L 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 230000006399 behavior Effects 0.000 description 2
- 235000019445 benzyl alcohol Nutrition 0.000 description 2
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 2
- 230000001588 bifunctional effect Effects 0.000 description 2
- 239000003833 bile salt Substances 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 208000029028 brain injury Diseases 0.000 description 2
- 229910000394 calcium triphosphate Inorganic materials 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 125000002091 cationic group Chemical group 0.000 description 2
- 150000001768 cations Chemical class 0.000 description 2
- 210000005056 cell body Anatomy 0.000 description 2
- 230000024245 cell differentiation Effects 0.000 description 2
- 230000012292 cell migration Effects 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- 229960001265 ciclosporin Drugs 0.000 description 2
- 229960002173 citrulline Drugs 0.000 description 2
- 235000013477 citrulline Nutrition 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 238000004891 communication Methods 0.000 description 2
- 239000002131 composite material Substances 0.000 description 2
- 230000008602 contraction Effects 0.000 description 2
- 230000000875 corresponding effect Effects 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 230000002950 deficient Effects 0.000 description 2
- 230000008021 deposition Effects 0.000 description 2
- 238000009792 diffusion process Methods 0.000 description 2
- 238000007865 diluting Methods 0.000 description 2
- 125000002228 disulfide group Chemical group 0.000 description 2
- PMMYEEVYMWASQN-UHFFFAOYSA-N dl-hydroxyproline Natural products OC1C[NH2+]C(C([O-])=O)C1 PMMYEEVYMWASQN-UHFFFAOYSA-N 0.000 description 2
- 238000001647 drug administration Methods 0.000 description 2
- 238000009510 drug design Methods 0.000 description 2
- 235000013601 eggs Nutrition 0.000 description 2
- 230000007831 electrophysiology Effects 0.000 description 2
- 238000002001 electrophysiology Methods 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 239000002158 endotoxin Substances 0.000 description 2
- 238000001976 enzyme digestion Methods 0.000 description 2
- 125000004185 ester group Chemical group 0.000 description 2
- 229940011871 estrogen Drugs 0.000 description 2
- 239000000262 estrogen Substances 0.000 description 2
- 238000000855 fermentation Methods 0.000 description 2
- 230000004151 fermentation Effects 0.000 description 2
- 239000007888 film coating Substances 0.000 description 2
- 238000009501 film coating Methods 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 230000005714 functional activity Effects 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-L glutamate group Chemical group N[C@@H](CCC(=O)[O-])C(=O)[O-] WHUUTDBJXJRKMK-VKHMYHEASA-L 0.000 description 2
- 125000000404 glutamine group Chemical group N[C@@H](CCC(N)=O)C(=O)* 0.000 description 2
- 150000002334 glycols Chemical class 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 210000003630 histaminocyte Anatomy 0.000 description 2
- 125000001165 hydrophobic group Chemical group 0.000 description 2
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 2
- 229960002591 hydroxyproline Drugs 0.000 description 2
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 2
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 2
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 2
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 2
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 2
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 2
- 150000002463 imidates Chemical class 0.000 description 2
- 210000002865 immune cell Anatomy 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 208000015181 infectious disease Diseases 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 229910001410 inorganic ion Inorganic materials 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 210000000936 intestine Anatomy 0.000 description 2
- 230000004410 intraocular pressure Effects 0.000 description 2
- 238000005040 ion trap Methods 0.000 description 2
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 2
- 230000013016 learning Effects 0.000 description 2
- 239000007937 lozenge Substances 0.000 description 2
- 229920001427 mPEG Polymers 0.000 description 2
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 2
- 239000001095 magnesium carbonate Substances 0.000 description 2
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 230000015654 memory Effects 0.000 description 2
- 229940071648 metered dose inhaler Drugs 0.000 description 2
- 125000001360 methionine group Chemical group N[C@@H](CCSC)C(=O)* 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 239000011859 microparticle Substances 0.000 description 2
- 235000013336 milk Nutrition 0.000 description 2
- 239000008267 milk Substances 0.000 description 2
- 210000004080 milk Anatomy 0.000 description 2
- 230000003278 mimic effect Effects 0.000 description 2
- 238000000302 molecular modelling Methods 0.000 description 2
- 210000004877 mucosa Anatomy 0.000 description 2
- 210000003205 muscle Anatomy 0.000 description 2
- 229940035363 muscle relaxants Drugs 0.000 description 2
- 238000002703 mutagenesis Methods 0.000 description 2
- 231100000350 mutagenesis Toxicity 0.000 description 2
- 210000005012 myelin Anatomy 0.000 description 2
- 239000003158 myorelaxant agent Substances 0.000 description 2
- 239000006225 natural substrate Substances 0.000 description 2
- 229940063121 neoral Drugs 0.000 description 2
- 230000008764 nerve damage Effects 0.000 description 2
- 230000000926 neurological effect Effects 0.000 description 2
- 230000003040 nociceptive effect Effects 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 2
- 235000021313 oleic acid Nutrition 0.000 description 2
- 239000006186 oral dosage form Substances 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 229960003104 ornithine Drugs 0.000 description 2
- JMANVNJQNLATNU-UHFFFAOYSA-N oxalonitrile Chemical compound N#CC#N JMANVNJQNLATNU-UHFFFAOYSA-N 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 108010044644 pegfilgrastim Proteins 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- RFWLACFDYFIVMC-UHFFFAOYSA-D pentacalcium;[oxido(phosphonatooxy)phosphoryl] phosphate Chemical compound [Ca+2].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[O-]P([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O.[O-]P([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O RFWLACFDYFIVMC-UHFFFAOYSA-D 0.000 description 2
- 239000000816 peptidomimetic Substances 0.000 description 2
- 208000028169 periodontal disease Diseases 0.000 description 2
- 239000012466 permeate Substances 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- OJUGVDODNPJEEC-UHFFFAOYSA-N phenylglyoxal Chemical compound O=CC(=O)C1=CC=CC=C1 OJUGVDODNPJEEC-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 230000026731 phosphorylation Effects 0.000 description 2
- 238000006366 phosphorylation reaction Methods 0.000 description 2
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 2
- 229920000656 polylysine Polymers 0.000 description 2
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 2
- 239000004810 polytetrafluoroethylene Substances 0.000 description 2
- 239000003450 potassium channel blocker Substances 0.000 description 2
- BPKIMPVREBSLAJ-UHFFFAOYSA-N prialt Chemical compound N1C(=O)C(CCSC)NC(=O)C(CC(C)C)NC(=O)C(CCCNC(N)=N)NC(=O)C(CO)NC(=O)C(NC(=O)C(CCCCN)NC(=O)C(C)NC(=O)CNC(=O)C(CCCCN)NC(=O)CNC(=O)C(CCCCN)NC(=O)C(N)CSSC2)CSSCC(C(NC(CCCNC(N)=N)C(=O)NC(CO)C(=O)NCC(=O)NC(CCCCN)C(=O)NC(CSSC3)C(N)=O)=O)NC(=O)C(CO)NC(=O)CNC(=O)C(C(C)O)NC(=O)C2NC(=O)C3NC(=O)C(CC(O)=O)NC(=O)C1CC1=CC=C(O)C=C1 BPKIMPVREBSLAJ-UHFFFAOYSA-N 0.000 description 2
- XOJVVFBFDXDTEG-UHFFFAOYSA-N pristane Chemical compound CC(C)CCCC(C)CCCC(C)CCCC(C)C XOJVVFBFDXDTEG-UHFFFAOYSA-N 0.000 description 2
- 239000000583 progesterone congener Substances 0.000 description 2
- 125000001500 prolyl group Chemical group [H]N1C([H])(C(=O)[*])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 2
- NGVDGCNFYWLIFO-UHFFFAOYSA-N pyridoxal 5'-phosphate Chemical compound CC1=NC=C(COP(O)(O)=O)C(C=O)=C1O NGVDGCNFYWLIFO-UHFFFAOYSA-N 0.000 description 2
- 238000011552 rat model Methods 0.000 description 2
- 238000005932 reductive alkylation reaction Methods 0.000 description 2
- 238000006268 reductive amination reaction Methods 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 239000012465 retentate Substances 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- FSYKKLYZXJSNPZ-UHFFFAOYSA-N sarcosine Chemical compound C[NH2+]CC([O-])=O FSYKKLYZXJSNPZ-UHFFFAOYSA-N 0.000 description 2
- 238000012163 sequencing technique Methods 0.000 description 2
- 125000003607 serino group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(O[H])([H])[H] 0.000 description 2
- SQVRNKJHWKZAKO-OQPLDHBCSA-N sialic acid Chemical compound CC(=O)N[C@@H]1[C@@H](O)C[C@@](O)(C(O)=O)OC1[C@H](O)[C@H](O)CO SQVRNKJHWKZAKO-OQPLDHBCSA-N 0.000 description 2
- PCMORTLOPMLEFB-ONEGZZNKSA-N sinapic acid Chemical compound COC1=CC(\C=C\C(O)=O)=CC(OC)=C1O PCMORTLOPMLEFB-ONEGZZNKSA-N 0.000 description 2
- PCMORTLOPMLEFB-UHFFFAOYSA-N sinapinic acid Natural products COC1=CC(C=CC(O)=O)=CC(OC)=C1O PCMORTLOPMLEFB-UHFFFAOYSA-N 0.000 description 2
- HRZFUMHJMZEROT-UHFFFAOYSA-L sodium disulfite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])(=O)=O HRZFUMHJMZEROT-UHFFFAOYSA-L 0.000 description 2
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 2
- 229940001584 sodium metabisulfite Drugs 0.000 description 2
- 235000010262 sodium metabisulphite Nutrition 0.000 description 2
- DAEPDZWVDSPTHF-UHFFFAOYSA-M sodium pyruvate Chemical compound [Na+].CC(=O)C([O-])=O DAEPDZWVDSPTHF-UHFFFAOYSA-M 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 238000012421 spiking Methods 0.000 description 2
- 239000008174 sterile solution Substances 0.000 description 2
- 150000003431 steroids Chemical class 0.000 description 2
- 210000002784 stomach Anatomy 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 230000005062 synaptic transmission Effects 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- 125000000341 threoninyl group Chemical group [H]OC([H])(C([H])([H])[H])C([H])(N([H])[H])C(*)=O 0.000 description 2
- 229960004072 thrombin Drugs 0.000 description 2
- 229940104230 thymidine Drugs 0.000 description 2
- 238000013271 transdermal drug delivery Methods 0.000 description 2
- 230000009529 traumatic brain injury Effects 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- 230000005747 tumor angiogenesis Effects 0.000 description 2
- 210000004881 tumor cell Anatomy 0.000 description 2
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 2
- 239000004474 valine Substances 0.000 description 2
- 102000038650 voltage-gated calcium channel activity Human genes 0.000 description 2
- 108091023044 voltage-gated calcium channel activity Proteins 0.000 description 2
- 230000029663 wound healing Effects 0.000 description 2
- 229930195727 α-lactose Natural products 0.000 description 2
- 108091058538 ω-conotoxin MVIIA Proteins 0.000 description 2
- SFLSHLFXELFNJZ-QMMMGPOBSA-N (-)-norepinephrine Chemical compound NC[C@H](O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-QMMMGPOBSA-N 0.000 description 1
- PFWOJTLVNOZSGQ-RIMJLNMDSA-N (1R,2aS,4S,7S,10S,13S,19S,22S,25S,28S,31R,36R,39S,45R,48S,51S,54S,57R,60S,63S,66S,69S,72S,75S,78S,81S,84S,87S,90R,93S,96S,99S)-10,51,87-tris(4-aminobutyl)-31-[[(2S)-2-[[(2S)-2-amino-5-carbamimidamidopentanoyl]amino]-3-hydroxypropanoyl]amino]-75-(aminomethyl)-93-(3-amino-3-oxopropyl)-60,96-dibenzyl-19,28-bis[(2S)-butan-2-yl]-4,54,69-tris(3-carbamimidamidopropyl)-25-(carboxymethyl)-2a,22,39,48-tetrakis[(1R)-1-hydroxyethyl]-7,63,81-tris(hydroxymethyl)-72-[(4-hydroxyphenyl)methyl]-84-(1H-imidazol-5-ylmethyl)-99-methyl-66-(2-methylpropyl)-78-(2-methylsulfanylethyl)-1a,3,4a,6,9,12,18,21,24,27,30,38,41,44,47,50,53,56,59,62,65,68,71,74,77,80,83,86,89,92,95,98-dotriacontaoxo-6a,7a,10a,11a,33,34-hexathia-a,2,3a,5,8,11,17,20,23,26,29,37,40,43,46,49,52,55,58,61,64,67,70,73,76,79,82,85,88,91,94,97-dotriacontazatetracyclo[55.47.4.445,90.013,17]dodecahectane-36-carboxylic acid Chemical compound CC[C@H](C)[C@@H]1NC(=O)[C@H](CSSC[C@H](NC(=O)[C@@H](NC(=O)CNC(=O)[C@@H]2CSSC[C@@H]3NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](Cc4ccccc4)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CSSC[C@H](NC(=O)[C@H](Cc4ccccc4)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](Cc4ccc(O)cc4)NC(=O)[C@H](CN)NC(=O)[C@H](CCSC)NC(=O)[C@H](CO)NC(=O)[C@H](Cc4cnc[nH]4)NC(=O)[C@H](CCCCN)NC3=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)O)C(=O)N2)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CO)NC(=O)[C@H](CCCCN)NC(=O)[C@@H]2CCCN2C(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC1=O)[C@@H](C)O)[C@@H](C)CC)[C@@H](C)O)[C@@H](C)O)C(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CCCNC(N)=N PFWOJTLVNOZSGQ-RIMJLNMDSA-N 0.000 description 1
- FXYPGCIGRDZWNR-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 3-[[3-(2,5-dioxopyrrolidin-1-yl)oxy-3-oxopropyl]disulfanyl]propanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCSSCCC(=O)ON1C(=O)CCC1=O FXYPGCIGRDZWNR-UHFFFAOYSA-N 0.000 description 1
- IZUAHLHTQJCCLJ-UHFFFAOYSA-N (2-chloro-1,1,2,2-tetrafluoroethyl) hypochlorite Chemical compound FC(F)(Cl)C(F)(F)OCl IZUAHLHTQJCCLJ-UHFFFAOYSA-N 0.000 description 1
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 description 1
- NMDDZEVVQDPECF-LURJTMIESA-N (2s)-2,7-diaminoheptanoic acid Chemical compound NCCCCC[C@H](N)C(O)=O NMDDZEVVQDPECF-LURJTMIESA-N 0.000 description 1
- BVAUMRCGVHUWOZ-ZETCQYMHSA-N (2s)-2-(cyclohexylazaniumyl)propanoate Chemical compound OC(=O)[C@H](C)NC1CCCCC1 BVAUMRCGVHUWOZ-ZETCQYMHSA-N 0.000 description 1
- OPOTYPXOKUZEKY-QMMMGPOBSA-N (2s)-2-nitramido-3-phenylpropanoic acid Chemical compound [O-][N+](=O)N[C@H](C(=O)O)CC1=CC=CC=C1 OPOTYPXOKUZEKY-QMMMGPOBSA-N 0.000 description 1
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- UKAUYVFTDYCKQA-UHFFFAOYSA-N -2-Amino-4-hydroxybutanoic acid Natural products OC(=O)C(N)CCO UKAUYVFTDYCKQA-UHFFFAOYSA-N 0.000 description 1
- LVGUZGTVOIAKKC-UHFFFAOYSA-N 1,1,1,2-tetrafluoroethane Chemical compound FCC(F)(F)F LVGUZGTVOIAKKC-UHFFFAOYSA-N 0.000 description 1
- KILNVBDSWZSGLL-KXQOOQHDSA-N 1,2-dihexadecanoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCCCC KILNVBDSWZSGLL-KXQOOQHDSA-N 0.000 description 1
- SNKAWJBJQDLSFF-NVKMUCNASA-N 1,2-dioleoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCC\C=C/CCCCCCCC SNKAWJBJQDLSFF-NVKMUCNASA-N 0.000 description 1
- NRJAVPSFFCBXDT-HUESYALOSA-N 1,2-distearoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCCCCCC NRJAVPSFFCBXDT-HUESYALOSA-N 0.000 description 1
- WDQFELCEOPFLCZ-UHFFFAOYSA-N 1-(2-hydroxyethyl)pyrrolidin-2-one Chemical compound OCCN1CCCC1=O WDQFELCEOPFLCZ-UHFFFAOYSA-N 0.000 description 1
- 108010073488 1-(N(2)-(3,4-dibromo-N-((4-(3,4-dihydro-2(1H)-oxoquinazolin-3-yl)-1-piperidinyl)carbonyl)tyrosyl)lysyl)-4-(4-pyridinyl)piperazine Proteins 0.000 description 1
- LUTLAXLNPLZCOF-UHFFFAOYSA-N 1-Methylhistidine Chemical group OC(=O)C(N)(C)CC1=NC=CN1 LUTLAXLNPLZCOF-UHFFFAOYSA-N 0.000 description 1
- WQUWKZJWBCOHQH-UHFFFAOYSA-N 1-[2-[2-(2-methoxyethoxy)ethoxy]ethyl]pyrrole-2,5-dione Chemical compound COCCOCCOCCN1C(=O)C=CC1=O WQUWKZJWBCOHQH-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- VOXZDWNPVJITMN-ZBRFXRBCSA-N 17β-estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 VOXZDWNPVJITMN-ZBRFXRBCSA-N 0.000 description 1
- 125000003287 1H-imidazol-4-ylmethyl group Chemical group [H]N1C([H])=NC(C([H])([H])[*])=C1[H] 0.000 description 1
- OHMHBGPWCHTMQE-UHFFFAOYSA-N 2,2-dichloro-1,1,1-trifluoroethane Chemical compound FC(F)(F)C(Cl)Cl OHMHBGPWCHTMQE-UHFFFAOYSA-N 0.000 description 1
- NHJVRSWLHSJWIN-UHFFFAOYSA-N 2,4,6-trinitrobenzenesulfonic acid Chemical compound OS(=O)(=O)C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O NHJVRSWLHSJWIN-UHFFFAOYSA-N 0.000 description 1
- 150000003923 2,5-pyrrolediones Chemical class 0.000 description 1
- JECYNCQXXKQDJN-UHFFFAOYSA-N 2-(2-methylhexan-2-yloxymethyl)oxirane Chemical compound CCCCC(C)(C)OCC1CO1 JECYNCQXXKQDJN-UHFFFAOYSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- JQPFYXFVUKHERX-UHFFFAOYSA-N 2-hydroxy-2-cyclohexen-1-one Natural products OC1=CCCCC1=O JQPFYXFVUKHERX-UHFFFAOYSA-N 0.000 description 1
- 102100027324 2-hydroxyacyl-CoA lyase 1 Human genes 0.000 description 1
- 125000003816 2-hydroxybenzoyl group Chemical group OC1=C(C(=O)*)C=CC=C1 0.000 description 1
- ZOOGRGPOEVQQDX-UUOKFMHZSA-N 3',5'-cyclic GMP Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=C(NC2=O)N)=C2N=C1 ZOOGRGPOEVQQDX-UUOKFMHZSA-N 0.000 description 1
- BIGBDMFRWJRLGJ-UHFFFAOYSA-N 3-benzyl-1,5-didiazoniopenta-1,4-diene-2,4-diolate Chemical compound [N-]=[N+]=CC(=O)C(C(=O)C=[N+]=[N-])CC1=CC=CC=C1 BIGBDMFRWJRLGJ-UHFFFAOYSA-N 0.000 description 1
- CDOUZKKFHVEKRI-UHFFFAOYSA-N 3-bromo-n-[(prop-2-enoylamino)methyl]propanamide Chemical compound BrCCC(=O)NCNC(=O)C=C CDOUZKKFHVEKRI-UHFFFAOYSA-N 0.000 description 1
- ZHQJIJUMPYNVAZ-UHFFFAOYSA-N 3-hydroxy-1-methylpyrrolidin-2-one Chemical compound CN1CCC(O)C1=O ZHQJIJUMPYNVAZ-UHFFFAOYSA-N 0.000 description 1
- 238000010600 3H thymidine incorporation assay Methods 0.000 description 1
- YXDGRBPZVQPESQ-QMMMGPOBSA-N 4-[(2s)-2-amino-2-carboxyethyl]benzoic acid Chemical compound OC(=O)[C@@H](N)CC1=CC=C(C(O)=O)C=C1 YXDGRBPZVQPESQ-QMMMGPOBSA-N 0.000 description 1
- OQEMUGXECXBKDP-UHFFFAOYSA-N 4-[[1-[[6-amino-1-[[15,30-bis(4-aminobutyl)-36,81-bis(2-amino-2-oxoethyl)-65-(3-amino-3-oxopropyl)-24,75,89-tribenzyl-21,42-bis(3-carbamimidamidopropyl)-56-(2-carboxyethyl)-4-(carboxymethyl)-7-[(1-carboxy-2-phenylethyl)carbamoyl]-33-(1-hydroxyethyl)-18,53,59-tris(hydroxymethyl)-62-[(4-hydroxyphenyl)methyl]-2,5,13,16,19,22,25,28,31,34,37,40,43,51,54,57,60,63,66,74,77,80,83,86,87,90,96,99-octacosaoxo-84,98-di(propan-2-yl)-9,10,47,48,70,71-hexathia-a,3,6,14,17,20,23,26,29,32,35,38,41,44,52,55,58,61,64,67,73,76,79,82,85,88,91,97-octacosazatetracyclo[43.27.14.1412,68.091,95]hectan-50-yl]amino]-1-oxohexan-2-yl]amino]-3-methyl-1-oxopentan-2-yl]amino]-3-[[2-[[1-(2-amino-5-carbamimidamidopentanoyl)pyrrolidine-2-carbonyl]amino]-3-hydroxybutanoyl]amino]-4-oxobutanoic acid Chemical compound CCC(C)C(NC(=O)C(CC(O)=O)NC(=O)C(NC(=O)C1CCCN1C(=O)C(N)CCCNC(N)=N)C(C)O)C(=O)NC(CCCCN)C(=O)NC1CSSCC2NC(=O)C(CCCNC(N)=N)NC(=O)CNC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(CCCCN)NC(=O)CNC(=O)C(Cc3ccccc3)NC(=O)C(CCCNC(N)=N)NC(=O)C(CO)NC(=O)C(CCCCN)NC(=O)C3CSSCC(NC(=O)C(CC(O)=O)NC(=O)C(CSSCC(NC(=O)C(CCC(N)=O)NC(=O)C(Cc4ccc(O)cc4)NC(=O)C(CO)NC(=O)C(CCC(O)=O)NC(=O)C(CO)NC1=O)C(=O)NC(Cc1ccccc1)C(=O)N1CCCC1C(=O)NC(C(C)C)C(=O)N3)NC(=O)C(Cc1ccccc1)NC(=O)CNC(=O)C(CC(N)=O)NC(=O)C(NC2=O)C(C)C)C(=O)NC(Cc1ccccc1)C(O)=O)C(C)O OQEMUGXECXBKDP-UHFFFAOYSA-N 0.000 description 1
- CMUHFUGDYMFHEI-QMMMGPOBSA-N 4-amino-L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N)C=C1 CMUHFUGDYMFHEI-QMMMGPOBSA-N 0.000 description 1
- NLPWSMKACWGINL-UHFFFAOYSA-N 4-azido-2-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(N=[N+]=[N-])C=C1O NLPWSMKACWGINL-UHFFFAOYSA-N 0.000 description 1
- 102000035037 5-HT3 receptors Human genes 0.000 description 1
- 108091005477 5-HT3 receptors Proteins 0.000 description 1
- 229940117976 5-hydroxylysine Drugs 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- 102100024645 ATP-binding cassette sub-family C member 8 Human genes 0.000 description 1
- 102100024642 ATP-binding cassette sub-family C member 9 Human genes 0.000 description 1
- 102100033056 ATP-sensitive inward rectifier potassium channel 1 Human genes 0.000 description 1
- 102100030913 Acetylcholine receptor subunit alpha Human genes 0.000 description 1
- 102100022725 Acetylcholine receptor subunit beta Human genes 0.000 description 1
- 241000242758 Actinia Species 0.000 description 1
- 102000005606 Activins Human genes 0.000 description 1
- 108010059616 Activins Proteins 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 108010079677 Agatoxins Proteins 0.000 description 1
- 241000238898 Agelenopsis aperta Species 0.000 description 1
- 102100037242 Amiloride-sensitive sodium channel subunit alpha Human genes 0.000 description 1
- 208000007195 Andersen Syndrome Diseases 0.000 description 1
- 201000006060 Andersen-Tawil syndrome Diseases 0.000 description 1
- 241000242762 Anemonia sulcata Species 0.000 description 1
- 101001023095 Anemonia sulcata Delta-actitoxin-Avd1a Proteins 0.000 description 1
- 108010064733 Angiotensins Proteins 0.000 description 1
- 102000015427 Angiotensins Human genes 0.000 description 1
- 102000037829 Anion channels Human genes 0.000 description 1
- 108091006515 Anion channels Proteins 0.000 description 1
- 208000019901 Anxiety disease Diseases 0.000 description 1
- 241000239290 Araneae Species 0.000 description 1
- 101000641989 Araneus ventricosus Kunitz-type U1-aranetoxin-Av1a Proteins 0.000 description 1
- 102000004580 Aspartic Acid Proteases Human genes 0.000 description 1
- 108010017640 Aspartic Acid Proteases Proteins 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 206010003591 Ataxia Diseases 0.000 description 1
- 208000032116 Autoimmune Experimental Encephalomyelitis Diseases 0.000 description 1
- 108010071023 Bacterial Outer Membrane Proteins Proteins 0.000 description 1
- 208000012904 Bartter disease Diseases 0.000 description 1
- 208000010062 Bartter syndrome Diseases 0.000 description 1
- 201000004569 Blindness Diseases 0.000 description 1
- 108010039209 Blood Coagulation Factors Proteins 0.000 description 1
- 102000015081 Blood Coagulation Factors Human genes 0.000 description 1
- 101800004538 Bradykinin Proteins 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 241001441936 Bunodosoma Species 0.000 description 1
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 1
- 102000014817 CACNA1A Human genes 0.000 description 1
- 229940127597 CGRP antagonist Drugs 0.000 description 1
- 108091022898 Calcium release-activated calcium channel Proteins 0.000 description 1
- 102000020167 Calcium release-activated calcium channel Human genes 0.000 description 1
- 102000004612 Calcium-Transporting ATPases Human genes 0.000 description 1
- 108010017954 Calcium-Transporting ATPases Proteins 0.000 description 1
- 102000000584 Calmodulin Human genes 0.000 description 1
- 108010041952 Calmodulin Proteins 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 101001028691 Carybdea rastonii Toxin CrTX-A Proteins 0.000 description 1
- 102000000844 Cell Surface Receptors Human genes 0.000 description 1
- 108010001857 Cell Surface Receptors Proteins 0.000 description 1
- 208000015374 Central core disease Diseases 0.000 description 1
- 101000685083 Centruroides infamatus Beta-toxin Cii1 Proteins 0.000 description 1
- 241000239328 Centruroides noxius Species 0.000 description 1
- 101001028462 Centruroides noxius Potassium channel toxin alpha-KTx 10.2 Proteins 0.000 description 1
- 101000685085 Centruroides noxius Toxin Cn1 Proteins 0.000 description 1
- 241000239282 Centruroides suffusus Species 0.000 description 1
- 208000031976 Channelopathies Diseases 0.000 description 1
- 101710091342 Chemotactic peptide Proteins 0.000 description 1
- 101001028688 Chironex fleckeri Toxin CfTX-1 Proteins 0.000 description 1
- 108010035563 Chloramphenicol O-acetyltransferase Proteins 0.000 description 1
- 101710164760 Chlorotoxin Proteins 0.000 description 1
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 206010056370 Congestive cardiomyopathy Diseases 0.000 description 1
- 241000237970 Conus <genus> Species 0.000 description 1
- 101000644407 Cyriopagopus schmidti U6-theraphotoxin-Hs1a Proteins 0.000 description 1
- 201000003883 Cystic fibrosis Diseases 0.000 description 1
- ODKSFYDXXFIFQN-SCSAIBSYSA-N D-arginine Chemical compound OC(=O)[C@H](N)CCCNC(N)=N ODKSFYDXXFIFQN-SCSAIBSYSA-N 0.000 description 1
- 229930028154 D-arginine Natural products 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- 238000001712 DNA sequencing Methods 0.000 description 1
- 206010011878 Deafness Diseases 0.000 description 1
- 208000024940 Dent disease Diseases 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 238000009007 Diagnostic Kit Methods 0.000 description 1
- 240000006497 Dianthus caryophyllus Species 0.000 description 1
- 235000009355 Dianthus caryophyllus Nutrition 0.000 description 1
- 201000010046 Dilated cardiomyopathy Diseases 0.000 description 1
- 101000761020 Dinoponera quadriceps Poneritoxin Proteins 0.000 description 1
- 102100033991 E3 ubiquitin-protein ligase DTX1 Human genes 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 102400000686 Endothelin-1 Human genes 0.000 description 1
- 101800004490 Endothelin-1 Proteins 0.000 description 1
- 241000283073 Equus caballus Species 0.000 description 1
- 108010091443 Exopeptidases Proteins 0.000 description 1
- 102000018389 Exopeptidases Human genes 0.000 description 1
- 208000010541 Familial or sporadic hemiplegic migraine Diseases 0.000 description 1
- 239000004606 Fillers/Extenders Substances 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 108091006027 G proteins Proteins 0.000 description 1
- 102000030782 GTP binding Human genes 0.000 description 1
- 108091000058 GTP-Binding Proteins 0.000 description 1
- 208000034619 Gingival inflammation Diseases 0.000 description 1
- 102000058061 Glucose Transporter Type 4 Human genes 0.000 description 1
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 description 1
- 229920002683 Glycosaminoglycan Polymers 0.000 description 1
- VPZXBVLAVMBEQI-VKHMYHEASA-N Glycyl-alanine Chemical compound OC(=O)[C@H](C)NC(=O)CN VPZXBVLAVMBEQI-VKHMYHEASA-N 0.000 description 1
- 102100034471 H(+)/Cl(-) exchange transporter 5 Human genes 0.000 description 1
- 102100028685 H(+)/Cl(-) exchange transporter 7 Human genes 0.000 description 1
- QXZGBUJJYSLZLT-UHFFFAOYSA-N H-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg-OH Natural products NC(N)=NCCCC(N)C(=O)N1CCCC1C(=O)N1C(C(=O)NCC(=O)NC(CC=2C=CC=CC=2)C(=O)NC(CO)C(=O)N2C(CCC2)C(=O)NC(CC=2C=CC=CC=2)C(=O)NC(CCCN=C(N)N)C(O)=O)CCC1 QXZGBUJJYSLZLT-UHFFFAOYSA-N 0.000 description 1
- 206010018910 Haemolysis Diseases 0.000 description 1
- 102000014702 Haptoglobin Human genes 0.000 description 1
- 108050005077 Haptoglobin Proteins 0.000 description 1
- 206010019476 Hemiplegic migraine Diseases 0.000 description 1
- 102000001554 Hemoglobins Human genes 0.000 description 1
- 108010054147 Hemoglobins Proteins 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 241001517046 Heteractis Species 0.000 description 1
- 241001517044 Heteractis magnifica Species 0.000 description 1
- 241000250994 Heterometrus Species 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101001009252 Homo sapiens 2-hydroxyacyl-CoA lyase 1 Proteins 0.000 description 1
- 101000760581 Homo sapiens ATP-binding cassette sub-family C member 9 Proteins 0.000 description 1
- 101000614701 Homo sapiens ATP-sensitive inward rectifier potassium channel 11 Proteins 0.000 description 1
- 101000726895 Homo sapiens Acetylcholine receptor subunit alpha Proteins 0.000 description 1
- 101000678746 Homo sapiens Acetylcholine receptor subunit beta Proteins 0.000 description 1
- 101000740448 Homo sapiens Amiloride-sensitive sodium channel subunit alpha Proteins 0.000 description 1
- 101001017463 Homo sapiens E3 ubiquitin-protein ligase DTX1 Proteins 0.000 description 1
- 101000710225 Homo sapiens H(+)/Cl(-) exchange transporter 5 Proteins 0.000 description 1
- 101000766971 Homo sapiens H(+)/Cl(-) exchange transporter 7 Proteins 0.000 description 1
- 101001019117 Homo sapiens Mediator of RNA polymerase II transcription subunit 23 Proteins 0.000 description 1
- 101001123834 Homo sapiens Neprilysin Proteins 0.000 description 1
- 101000997283 Homo sapiens Potassium voltage-gated channel subfamily C member 1 Proteins 0.000 description 1
- 101000994648 Homo sapiens Potassium voltage-gated channel subfamily KQT member 4 Proteins 0.000 description 1
- 101000753197 Homo sapiens Sodium/potassium-transporting ATPase subunit alpha-2 Proteins 0.000 description 1
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 1
- 101000867811 Homo sapiens Voltage-dependent L-type calcium channel subunit alpha-1C Proteins 0.000 description 1
- 101000935117 Homo sapiens Voltage-dependent P/Q-type calcium channel subunit alpha-1A Proteins 0.000 description 1
- 101000867850 Homo sapiens Voltage-dependent T-type calcium channel subunit alpha-1G Proteins 0.000 description 1
- 101000932785 Homo sapiens Voltage-dependent T-type calcium channel subunit alpha-1I Proteins 0.000 description 1
- YZJSUQQZGCHHNQ-UHFFFAOYSA-N Homoglutamine Chemical compound OC(=O)C(N)CCCC(N)=O YZJSUQQZGCHHNQ-UHFFFAOYSA-N 0.000 description 1
- 102000002265 Human Growth Hormone Human genes 0.000 description 1
- 108010000521 Human Growth Hormone Proteins 0.000 description 1
- 239000000854 Human Growth Hormone Substances 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 241000257303 Hymenoptera Species 0.000 description 1
- 208000004454 Hyperalgesia Diseases 0.000 description 1
- 206010020590 Hypercalciuria Diseases 0.000 description 1
- 206010060378 Hyperinsulinaemia Diseases 0.000 description 1
- 208000007599 Hyperkalemic periodic paralysis Diseases 0.000 description 1
- 206010020853 Hypertonic bladder Diseases 0.000 description 1
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 1
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 1
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 206010022489 Insulin Resistance Diseases 0.000 description 1
- 108010002386 Interleukin-3 Proteins 0.000 description 1
- 208000004404 Intractable Pain Diseases 0.000 description 1
- 102000017786 KCNJ1 Human genes 0.000 description 1
- 102000017792 KCNJ11 Human genes 0.000 description 1
- 108060005987 Kallikrein Proteins 0.000 description 1
- 102000001399 Kallikrein Human genes 0.000 description 1
- 102100035792 Kininogen-1 Human genes 0.000 description 1
- SNDPXSYFESPGGJ-BYPYZUCNSA-N L-2-aminopentanoic acid Chemical compound CCC[C@H](N)C(O)=O SNDPXSYFESPGGJ-BYPYZUCNSA-N 0.000 description 1
- 108090000420 L-Type Calcium Channels Proteins 0.000 description 1
- 102000004016 L-Type Calcium Channels Human genes 0.000 description 1
- 150000008575 L-amino acids Chemical class 0.000 description 1
- 229930064664 L-arginine Natural products 0.000 description 1
- 235000014852 L-arginine Nutrition 0.000 description 1
- XIGSAGMEBXLVJJ-YFKPBYRVSA-N L-homocitrulline Chemical compound NC(=O)NCCCC[C@H]([NH3+])C([O-])=O XIGSAGMEBXLVJJ-YFKPBYRVSA-N 0.000 description 1
- FFFHZYDWPBMWHY-VKHMYHEASA-N L-homocysteine Chemical compound OC(=O)[C@@H](N)CCS FFFHZYDWPBMWHY-VKHMYHEASA-N 0.000 description 1
- UKAUYVFTDYCKQA-VKHMYHEASA-N L-homoserine Chemical compound OC(=O)[C@@H](N)CCO UKAUYVFTDYCKQA-VKHMYHEASA-N 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- 229940122282 L-type calcium channel antagonist Drugs 0.000 description 1
- 101001050253 Leiurus hebraeus Potassium channel toxin alpha-KTx 16.5 Proteins 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 108010000817 Leuprolide Proteins 0.000 description 1
- 208000026709 Liddle syndrome Diseases 0.000 description 1
- OYHQOLUKZRVURQ-HZJYTTRNSA-N Linoleic acid Chemical compound CCCCC\C=C/C\C=C/CCCCCCCC(O)=O OYHQOLUKZRVURQ-HZJYTTRNSA-N 0.000 description 1
- 108010046938 Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102000007651 Macrophage Colony-Stimulating Factor Human genes 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical group O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 101000944157 Mesobuthus martensii Potassium channel toxin alpha-KTx 9.1 Proteins 0.000 description 1
- 102000005741 Metalloproteases Human genes 0.000 description 1
- 108010006035 Metalloproteases Proteins 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- HBNPJJILLOYFJU-VMPREFPWSA-N Mibefradil Chemical compound C1CC2=CC(F)=CC=C2[C@H](C(C)C)[C@@]1(OC(=O)COC)CCN(C)CCCC1=NC2=CC=CC=C2N1 HBNPJJILLOYFJU-VMPREFPWSA-N 0.000 description 1
- 208000019695 Migraine disease Diseases 0.000 description 1
- 208000019022 Mood disease Diseases 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- 101000846105 Mus musculus Short transient receptor potential channel 6 Proteins 0.000 description 1
- 208000029578 Muscle disease Diseases 0.000 description 1
- 208000021642 Muscular disease Diseases 0.000 description 1
- 206010028424 Myasthenic syndrome Diseases 0.000 description 1
- 201000009623 Myopathy Diseases 0.000 description 1
- 206010061533 Myotonia Diseases 0.000 description 1
- ONXPDKGXOOORHB-BYPYZUCNSA-N N(5)-methyl-L-glutamine Chemical compound CNC(=O)CC[C@H](N)C(O)=O ONXPDKGXOOORHB-BYPYZUCNSA-N 0.000 description 1
- NTNWOCRCBQPEKQ-YFKPBYRVSA-N N(omega)-methyl-L-arginine Chemical group CN=C(N)NCCC[C@H](N)C(O)=O NTNWOCRCBQPEKQ-YFKPBYRVSA-N 0.000 description 1
- BRMWTNUJHUMWMS-LURJTMIESA-N N(tele)-methyl-L-histidine Chemical group CN1C=NC(C[C@H](N)C(O)=O)=C1 BRMWTNUJHUMWMS-LURJTMIESA-N 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical class ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- HOKKHZGPKSLGJE-GSVOUGTGSA-N N-Methyl-D-aspartic acid Chemical compound CN[C@@H](C(O)=O)CC(O)=O HOKKHZGPKSLGJE-GSVOUGTGSA-N 0.000 description 1
- NTWVQPHTOUKMDI-YFKPBYRVSA-N N-Methyl-arginine Chemical group CN[C@H](C(O)=O)CCCN=C(N)N NTWVQPHTOUKMDI-YFKPBYRVSA-N 0.000 description 1
- SQVRNKJHWKZAKO-PFQGKNLYSA-N N-acetyl-beta-neuraminic acid Chemical compound CC(=O)N[C@@H]1[C@@H](O)C[C@@](O)(C(O)=O)O[C@H]1[C@H](O)[C@H](O)CO SQVRNKJHWKZAKO-PFQGKNLYSA-N 0.000 description 1
- 108010079364 N-glycylalanine Proteins 0.000 description 1
- 125000000729 N-terminal amino-acid group Chemical group 0.000 description 1
- 229940123579 N-type calcium channel antagonist Drugs 0.000 description 1
- 102000003945 NF-kappa B Human genes 0.000 description 1
- 108010057466 NF-kappa B Proteins 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- 208000034827 Neointima Diseases 0.000 description 1
- 102100028782 Neprilysin Human genes 0.000 description 1
- 108090000189 Neuropeptides Proteins 0.000 description 1
- 102000017922 Neurotensin receptor Human genes 0.000 description 1
- 108060003370 Neurotensin receptor Proteins 0.000 description 1
- NCHSYZVVWKVWFQ-BYPYZUCNSA-N Ng-amino-l-arginine Chemical group NNC(N)=NCCC[C@H](N)C(O)=O NCHSYZVVWKVWFQ-BYPYZUCNSA-N 0.000 description 1
- 206010057852 Nicotine dependence Diseases 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- 108010058846 Ovalbumin Proteins 0.000 description 1
- 208000009722 Overactive Urinary Bladder Diseases 0.000 description 1
- 108091006006 PEGylated Proteins Proteins 0.000 description 1
- 208000000114 Pain Threshold Diseases 0.000 description 1
- 108010067372 Pancreatic elastase Proteins 0.000 description 1
- 102000016387 Pancreatic elastase Human genes 0.000 description 1
- 101000997272 Parabuthus transvaalicus Potassium channel toxin alpha-KTx 1.10 Proteins 0.000 description 1
- 101000658306 Paraphysa scrofa Kappa-theraphotoxin-Ps1b Proteins 0.000 description 1
- 239000004264 Petrolatum Substances 0.000 description 1
- 101000679608 Phaeosphaeria nodorum (strain SN15 / ATCC MYA-4574 / FGSC 10173) Cysteine rich necrotrophic effector Tox1 Proteins 0.000 description 1
- 101710124951 Phospholipase C Proteins 0.000 description 1
- 241001315609 Pittosporum crassifolium Species 0.000 description 1
- 229920001363 Polidocanol Polymers 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 229920000954 Polyglycolide Polymers 0.000 description 1
- 229920002701 Polyoxyl 40 Stearate Polymers 0.000 description 1
- 229920000037 Polyproline Polymers 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 229920001219 Polysorbate 40 Polymers 0.000 description 1
- 229920001214 Polysorbate 60 Polymers 0.000 description 1
- 229920002642 Polysorbate 65 Polymers 0.000 description 1
- 102100023205 Potassium channel subfamily K member 4 Human genes 0.000 description 1
- 101710185483 Potassium channel subfamily K member 4 Proteins 0.000 description 1
- 102100034308 Potassium voltage-gated channel subfamily C member 1 Human genes 0.000 description 1
- 102100034363 Potassium voltage-gated channel subfamily KQT member 4 Human genes 0.000 description 1
- RJKFOVLPORLFTN-LEKSSAKUSA-N Progesterone Chemical class C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)C)[C@@]1(C)CC2 RJKFOVLPORLFTN-LEKSSAKUSA-N 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 108010001267 Protein Subunits Proteins 0.000 description 1
- 102000002067 Protein Subunits Human genes 0.000 description 1
- 206010037714 Quadriplegia Diseases 0.000 description 1
- 102000004913 RYR1 Human genes 0.000 description 1
- 108060007240 RYR1 Proteins 0.000 description 1
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 1
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 1
- 206010039085 Rhinitis allergic Diseases 0.000 description 1
- 108010012219 Ryanodine Receptor Calcium Release Channel Proteins 0.000 description 1
- 102000019027 Ryanodine Receptor Calcium Release Channel Human genes 0.000 description 1
- 108091006300 SLC2A4 Proteins 0.000 description 1
- 108010077895 Sarcosine Proteins 0.000 description 1
- 229940124639 Selective inhibitor Drugs 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- 102000007562 Serum Albumin Human genes 0.000 description 1
- 108010071390 Serum Albumin Proteins 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 102100021955 Sodium/potassium-transporting ATPase subunit alpha-2 Human genes 0.000 description 1
- 239000004147 Sorbitan trioleate Substances 0.000 description 1
- PRXRUNOAOLTIEF-ADSICKODSA-N Sorbitan trioleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCC\C=C/CCCCCCCC)[C@H]1OC[C@H](O)[C@H]1OC(=O)CCCCCCC\C=C/CCCCCCCC PRXRUNOAOLTIEF-ADSICKODSA-N 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 240000001058 Sterculia urens Species 0.000 description 1
- 235000015125 Sterculia urens Nutrition 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 230000005867 T cell response Effects 0.000 description 1
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 1
- 229940123939 T-type calcium channel antagonist Drugs 0.000 description 1
- 241000906446 Theraps Species 0.000 description 1
- 241001572088 Thrixopelma pruriens Species 0.000 description 1
- 101000994422 Tityus discrepans Potassium channel toxin alpha-KTx 15.6 Proteins 0.000 description 1
- 101001028442 Tityus serrulatus Potassium channel toxin alpha-KTx 12.1 Proteins 0.000 description 1
- 101001028429 Tityus stigmurus Potassium channel toxin alpha-KTx 12.4 Proteins 0.000 description 1
- 101001028439 Tityus trivittatus Potassium channel toxin alpha-KTx 12.2 Proteins 0.000 description 1
- 208000025569 Tobacco Use disease Diseases 0.000 description 1
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 102000004338 Transferrin Human genes 0.000 description 1
- 108090000901 Transferrin Proteins 0.000 description 1
- 102000011753 Transient Receptor Potential Channels Human genes 0.000 description 1
- 229940123445 Tricyclic antidepressant Drugs 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 1
- 208000002495 Uterine Neoplasms Diseases 0.000 description 1
- 102100032574 Voltage-dependent L-type calcium channel subunit alpha-1C Human genes 0.000 description 1
- 102100025330 Voltage-dependent P/Q-type calcium channel subunit alpha-1A Human genes 0.000 description 1
- 102100033024 Voltage-dependent T-type calcium channel subunit alpha-1G Human genes 0.000 description 1
- 102100025484 Voltage-dependent T-type calcium channel subunit alpha-1I Human genes 0.000 description 1
- TVXBFESIOXBWNM-UHFFFAOYSA-N Xylitol Natural products OCCC(O)C(O)C(O)CCO TVXBFESIOXBWNM-UHFFFAOYSA-N 0.000 description 1
- 108010084455 Zeocin Proteins 0.000 description 1
- MMWCIQZXVOZEGG-HOZKJCLWSA-N [(1S,2R,3S,4S,5R,6S)-2,3,5-trihydroxy-4,6-diphosphonooxycyclohexyl] dihydrogen phosphate Chemical compound O[C@H]1[C@@H](O)[C@H](OP(O)(O)=O)[C@@H](OP(O)(O)=O)[C@H](O)[C@H]1OP(O)(O)=O MMWCIQZXVOZEGG-HOZKJCLWSA-N 0.000 description 1
- GELXFVQAWNTGPQ-UHFFFAOYSA-N [N].C1=CNC=N1 Chemical compound [N].C1=CNC=N1 GELXFVQAWNTGPQ-UHFFFAOYSA-N 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- SNEIUMQYRCDYCH-UHFFFAOYSA-N acetylarginine Chemical compound CC(=O)NC(C(O)=O)CCCN=C(N)N SNEIUMQYRCDYCH-UHFFFAOYSA-N 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 239000011149 active material Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 230000033289 adaptive immune response Effects 0.000 description 1
- 230000004721 adaptive immunity Effects 0.000 description 1
- 229960005305 adenosine Drugs 0.000 description 1
- 239000012790 adhesive layer Substances 0.000 description 1
- 238000012382 advanced drug delivery Methods 0.000 description 1
- 238000012387 aerosolization Methods 0.000 description 1
- 238000003314 affinity selection Methods 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 238000012867 alanine scanning Methods 0.000 description 1
- BNPSSFBOAGDEEL-UHFFFAOYSA-N albuterol sulfate Chemical compound OS(O)(=O)=O.CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1.CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 BNPSSFBOAGDEEL-UHFFFAOYSA-N 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- 201000010105 allergic rhinitis Diseases 0.000 description 1
- 102000015395 alpha 1-Antitrypsin Human genes 0.000 description 1
- 108010050122 alpha 1-Antitrypsin Proteins 0.000 description 1
- 229940024142 alpha 1-antitrypsin Drugs 0.000 description 1
- 150000001370 alpha-amino acid derivatives Chemical class 0.000 description 1
- QWCKQJZIFLGMSD-UHFFFAOYSA-N alpha-aminobutyric acid Chemical compound CCC(N)C(O)=O QWCKQJZIFLGMSD-UHFFFAOYSA-N 0.000 description 1
- DTOSIQBPPRVQHS-PDBXOOCHSA-N alpha-linolenic acid Chemical compound CC\C=C/C\C=C/C\C=C/CCCCCCCC(O)=O DTOSIQBPPRVQHS-PDBXOOCHSA-N 0.000 description 1
- 235000020661 alpha-linolenic acid Nutrition 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 230000001668 ameliorated effect Effects 0.000 description 1
- 238000010640 amide synthesis reaction Methods 0.000 description 1
- 230000006229 amino acid addition Effects 0.000 description 1
- 150000003862 amino acid derivatives Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 229940124277 aminobutyric acid Drugs 0.000 description 1
- 125000002344 aminooxy group Chemical group [H]N([H])O[*] 0.000 description 1
- 230000000202 analgesic effect Effects 0.000 description 1
- 229940035676 analgesics Drugs 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 239000000730 antalgic agent Substances 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000001430 anti-depressive effect Effects 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 239000000935 antidepressant agent Substances 0.000 description 1
- 230000030741 antigen processing and presentation Effects 0.000 description 1
- 230000036506 anxiety Effects 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 210000001742 aqueous humor Anatomy 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 125000000637 arginyl group Chemical group N[C@@H](CCCNC(N)=N)C(=O)* 0.000 description 1
- 206010003119 arrhythmia Diseases 0.000 description 1
- 229960003272 asparaginase Drugs 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 238000000889 atomisation Methods 0.000 description 1
- 210000000227 basophil cell of anterior lobe of hypophysis Anatomy 0.000 description 1
- 238000010009 beating Methods 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- UREZNYTWGJKWBI-UHFFFAOYSA-M benzethonium chloride Chemical compound [Cl-].C1=CC(C(C)(C)CC(C)(C)C)=CC=C1OCCOCC[N+](C)(C)CC1=CC=CC=C1 UREZNYTWGJKWBI-UHFFFAOYSA-M 0.000 description 1
- 229960001950 benzethonium chloride Drugs 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- 150000001576 beta-amino acids Chemical class 0.000 description 1
- 235000013361 beverage Nutrition 0.000 description 1
- 229940093761 bile salts Drugs 0.000 description 1
- 230000008512 biological response Effects 0.000 description 1
- 150000001615 biotins Chemical class 0.000 description 1
- JCZLABDVDPYLRZ-AWEZNQCLSA-N biphenylalanine Chemical compound C1=CC(C[C@H](N)C(O)=O)=CC=C1C1=CC=CC=C1 JCZLABDVDPYLRZ-AWEZNQCLSA-N 0.000 description 1
- 239000003114 blood coagulation factor Substances 0.000 description 1
- 229940019700 blood coagulation factors Drugs 0.000 description 1
- 239000012503 blood component Substances 0.000 description 1
- 238000004820 blood count Methods 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- QXZGBUJJYSLZLT-FDISYFBBSA-N bradykinin Chemical compound NC(=N)NCCC[C@H](N)C(=O)N1CCC[C@H]1C(=O)N1[C@H](C(=O)NCC(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CO)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)CCC1 QXZGBUJJYSLZLT-FDISYFBBSA-N 0.000 description 1
- 230000006931 brain damage Effects 0.000 description 1
- 231100000874 brain damage Toxicity 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 239000007975 buffered saline Substances 0.000 description 1
- 239000004067 bulking agent Substances 0.000 description 1
- 239000003735 calcitonin gene related peptide receptor antagonist Substances 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 1
- 150000001244 carboxylic acid anhydrides Chemical class 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 238000005341 cation exchange Methods 0.000 description 1
- 230000020411 cell activation Effects 0.000 description 1
- 230000036755 cellular response Effects 0.000 description 1
- 201000007303 central core myopathy Diseases 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- PRQROPMIIGLWRP-BZSNNMDCSA-N chemotactic peptide Chemical compound CSCC[C@H](NC=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 PRQROPMIIGLWRP-BZSNNMDCSA-N 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- KYKAJFCTULSVSH-UHFFFAOYSA-N chloro(fluoro)methane Chemical compound F[C]Cl KYKAJFCTULSVSH-UHFFFAOYSA-N 0.000 description 1
- 125000002668 chloroacetyl group Chemical group ClCC(=O)* 0.000 description 1
- QPAKKWCQMHUHNI-GQIQPHNSSA-N chlorotoxin Chemical compound C([C@H]1C(=O)NCC(=O)N2CCC[C@H]2C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H]2CSSC[C@H]3C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@H]4CSSC[C@@H](C(N[C@@H](CCSC)C(=O)N5CCC[C@H]5C(=O)N[C@@H](CSSC[C@@H](C(=O)N1)NC(=O)[C@H](CCCCN)NC(=O)CNC(=O)[C@H](CCCNC(N)=N)NC(=O)CNC(=O)[C@H](CCCCN)NC(=O)CNC(=O)CNC(=O)[C@H](CSSC[C@H](NC(=O)[C@H](CC(C)C)NC2=O)C(=O)N[C@@H](CCCNC(N)=N)C(N)=O)NC4=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N3)=O)NC(=O)[C@@H](N)CCSC)C1=CC=C(O)C=C1 QPAKKWCQMHUHNI-GQIQPHNSSA-N 0.000 description 1
- 229960005534 chlorotoxin Drugs 0.000 description 1
- 210000002825 class switched memory b cell Anatomy 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 230000019771 cognition Effects 0.000 description 1
- 230000001143 conditioned effect Effects 0.000 description 1
- 108050003126 conotoxin Proteins 0.000 description 1
- 230000037020 contractile activity Effects 0.000 description 1
- 239000007822 coupling agent Substances 0.000 description 1
- 239000003431 cross linking reagent Substances 0.000 description 1
- CNVQLPPZGABUCM-LIGYZCPXSA-N ctx toxin Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CC(N)=O)C(=O)N[C@H](C(=O)N[C@@H](CO)C(=O)N[C@H]1CSSC[C@H]2C(=O)N[C@H](C(N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@H]3CSSC[C@@H](C(N[C@@H](CC=4C5=CC=CC=C5NC=4)C(=O)N[C@@H](CO)C(=O)N[C@H](C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CCCNC(N)=N)NC3=O)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CO)C(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=3NC=NC=3)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N2)C(C)C)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H]([C@@H](C)O)NC1=O)=O)CCSC)C(C)C)[C@@H](C)O)NC(=O)[C@H]1NC(=O)CC1)C1=CC=CC=C1 CNVQLPPZGABUCM-LIGYZCPXSA-N 0.000 description 1
- OILAIQUEIWYQPH-UHFFFAOYSA-N cyclohexane-1,2-dione Chemical compound O=C1CCCCC1=O OILAIQUEIWYQPH-UHFFFAOYSA-N 0.000 description 1
- 229930182912 cyclosporin Natural products 0.000 description 1
- 230000016396 cytokine production Effects 0.000 description 1
- 230000001461 cytolytic effect Effects 0.000 description 1
- 230000001120 cytoprotective effect Effects 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000007123 defense Effects 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- YSMODUONRAFBET-UHFFFAOYSA-N delta-DL-hydroxylysine Natural products NCC(O)CCC(N)C(O)=O YSMODUONRAFBET-UHFFFAOYSA-N 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 239000000032 diagnostic agent Substances 0.000 description 1
- 229940039227 diagnostic agent Drugs 0.000 description 1
- 102000038379 digestive enzymes Human genes 0.000 description 1
- 108091007734 digestive enzymes Proteins 0.000 description 1
- 230000003292 diminished effect Effects 0.000 description 1
- CLBIEZBAENPDFY-HNXGFDTJSA-N dinophysistoxin 1 Chemical compound C([C@H](O1)[C@H](C)/C=C/[C@H]2CC[C@@]3(CC[C@H]4O[C@@H](C([C@@H](O)[C@@H]4O3)=C)[C@@H](O)C[C@H](C)[C@@H]3[C@@H](CC[C@@]4(O3)[C@@H](CCCO4)C)C)O2)C(C)=C[C@]21O[C@H](C[C@@](C)(O)C(O)=O)CC[C@H]2O CLBIEZBAENPDFY-HNXGFDTJSA-N 0.000 description 1
- 235000019329 dioctyl sodium sulphosuccinate Nutrition 0.000 description 1
- 150000002009 diols Chemical class 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 150000002019 disulfides Chemical class 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 239000013583 drug formulation Substances 0.000 description 1
- 239000003596 drug target Substances 0.000 description 1
- 229940112141 dry powder inhaler Drugs 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 229940073621 enbrel Drugs 0.000 description 1
- 229940066758 endopeptidases Drugs 0.000 description 1
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 230000019439 energy homeostasis Effects 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 239000002532 enzyme inhibitor Substances 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 235000020776 essential amino acid Nutrition 0.000 description 1
- 239000003797 essential amino acid Substances 0.000 description 1
- 229930182833 estradiol Natural products 0.000 description 1
- 229960005309 estradiol Drugs 0.000 description 1
- 238000009164 estrogen replacement therapy Methods 0.000 description 1
- 229960000403 etanercept Drugs 0.000 description 1
- AZHSSKPUVBVXLK-UHFFFAOYSA-N ethane-1,1-diol Chemical compound CC(O)O AZHSSKPUVBVXLK-UHFFFAOYSA-N 0.000 description 1
- HAPOVYFOVVWLRS-UHFFFAOYSA-N ethosuximide Chemical compound CCC1(C)CC(=O)NC1=O HAPOVYFOVVWLRS-UHFFFAOYSA-N 0.000 description 1
- 229960002767 ethosuximide Drugs 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- SFNALCNOMXIBKG-UHFFFAOYSA-N ethylene glycol monododecyl ether Chemical compound CCCCCCCCCCCCOCCO SFNALCNOMXIBKG-UHFFFAOYSA-N 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 208000012997 experimental autoimmune encephalomyelitis Diseases 0.000 description 1
- 238000013401 experimental design Methods 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 239000007941 film coated tablet Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 230000004907 flux Effects 0.000 description 1
- 239000012537 formulation buffer Substances 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 229910021485 fumed silica Inorganic materials 0.000 description 1
- 238000007306 functionalization reaction Methods 0.000 description 1
- 229940110710 fusidate Drugs 0.000 description 1
- IECPWNUMDGFDKC-MZJAQBGESA-N fusidic acid Chemical compound O[C@@H]([C@@H]12)C[C@H]3\C(=C(/CCC=C(C)C)C(O)=O)[C@@H](OC(C)=O)C[C@]3(C)[C@@]2(C)CC[C@@H]2[C@]1(C)CC[C@@H](O)[C@H]2C IECPWNUMDGFDKC-MZJAQBGESA-N 0.000 description 1
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 1
- UHBYWPGGCSDKFX-VKHMYHEASA-N gamma-carboxy-L-glutamic acid Chemical compound OC(=O)[C@@H](N)CC(C(O)=O)C(O)=O UHBYWPGGCSDKFX-VKHMYHEASA-N 0.000 description 1
- 230000030135 gastric motility Effects 0.000 description 1
- 230000012178 germinal center formation Effects 0.000 description 1
- 230000002518 glial effect Effects 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 230000004153 glucose metabolism Effects 0.000 description 1
- 125000002791 glucosyl group Chemical group C1([C@H](O)[C@@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- VPZXBVLAVMBEQI-UHFFFAOYSA-N glycyl-DL-alpha-alanine Natural products OC(=O)C(C)NC(=O)CN VPZXBVLAVMBEQI-UHFFFAOYSA-N 0.000 description 1
- HHLFWLYXYJOTON-UHFFFAOYSA-N glyoxylic acid Chemical compound OC(=O)C=O HHLFWLYXYJOTON-UHFFFAOYSA-N 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- ZRALSGWEFCBTJO-UHFFFAOYSA-N guanidine group Chemical group NC(=N)N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 230000010370 hearing loss Effects 0.000 description 1
- 231100000888 hearing loss Toxicity 0.000 description 1
- 208000016354 hearing loss disease Diseases 0.000 description 1
- 210000002443 helper t lymphocyte Anatomy 0.000 description 1
- 230000008588 hemolysis Effects 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 235000009200 high fat diet Nutrition 0.000 description 1
- 229960001340 histamine Drugs 0.000 description 1
- 229920002674 hyaluronan Polymers 0.000 description 1
- 229960003160 hyaluronic acid Drugs 0.000 description 1
- 229940042795 hydrazides for tuberculosis treatment Drugs 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 150000005828 hydrofluoroalkanes Chemical class 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 229920001477 hydrophilic polymer Polymers 0.000 description 1
- 230000033444 hydroxylation Effects 0.000 description 1
- 238000005805 hydroxylation reaction Methods 0.000 description 1
- 230000003451 hyperinsulinaemic effect Effects 0.000 description 1
- 201000008980 hyperinsulinism Diseases 0.000 description 1
- 230000002519 immonomodulatory effect Effects 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 238000003364 immunohistochemistry Methods 0.000 description 1
- 230000001506 immunosuppresive effect Effects 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 229940125721 immunosuppressive agent Drugs 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 230000001976 improved effect Effects 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 229940060367 inert ingredients Drugs 0.000 description 1
- 230000006749 inflammatory damage Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 230000014828 interferon-gamma production Effects 0.000 description 1
- 230000004073 interleukin-2 production Effects 0.000 description 1
- 108040006849 interleukin-2 receptor activity proteins Proteins 0.000 description 1
- 102000027411 intracellular receptors Human genes 0.000 description 1
- 108091008582 intracellular receptors Proteins 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- PGLTVOMIXTUURA-UHFFFAOYSA-N iodoacetamide Chemical group NC(=O)CI PGLTVOMIXTUURA-UHFFFAOYSA-N 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 208000028867 ischemia Diseases 0.000 description 1
- 210000004153 islets of langerhan Anatomy 0.000 description 1
- 230000000155 isotopic effect Effects 0.000 description 1
- 210000002510 keratinocyte Anatomy 0.000 description 1
- 125000000468 ketone group Chemical group 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 238000004989 laser desorption mass spectroscopy Methods 0.000 description 1
- 229950006462 lauromacrogol 400 Drugs 0.000 description 1
- 238000002386 leaching Methods 0.000 description 1
- 125000001909 leucine group Chemical group [H]N(*)C(C(*)=O)C([H])([H])C(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 description 1
- 229960004338 leuprorelin Drugs 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 235000020778 linoleic acid Nutrition 0.000 description 1
- OYHQOLUKZRVURQ-IXWMQOLASA-N linoleic acid Natural products CCCCC\C=C/C\C=C\CCCCCCCC(O)=O OYHQOLUKZRVURQ-IXWMQOLASA-N 0.000 description 1
- KQQKGWQCNNTQJW-UHFFFAOYSA-N linolenic acid Natural products CC=CCCC=CCC=CCCCCCCCC(O)=O KQQKGWQCNNTQJW-UHFFFAOYSA-N 0.000 description 1
- 229960004488 linolenic acid Drugs 0.000 description 1
- 230000037356 lipid metabolism Effects 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 239000012160 loading buffer Substances 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 239000003589 local anesthetic agent Substances 0.000 description 1
- 229960005015 local anesthetics Drugs 0.000 description 1
- 208000004731 long QT syndrome Diseases 0.000 description 1
- 210000003563 lymphoid tissue Anatomy 0.000 description 1
- 239000008176 lyophilized powder Substances 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 229940037627 magnesium lauryl sulfate Drugs 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- HBNDBUATLJAUQM-UHFFFAOYSA-L magnesium;dodecyl sulfate Chemical compound [Mg+2].CCCCCCCCCCCCOS([O-])(=O)=O.CCCCCCCCCCCCOS([O-])(=O)=O HBNDBUATLJAUQM-UHFFFAOYSA-L 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 210000000412 mechanoreceptor Anatomy 0.000 description 1
- 108091008704 mechanoreceptors Proteins 0.000 description 1
- 229940127554 medical product Drugs 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000028161 membrane depolarization Effects 0.000 description 1
- 230000034153 membrane organization Effects 0.000 description 1
- HEBKCHPVOIAQTA-UHFFFAOYSA-N meso ribitol Natural products OCC(O)C(O)C(O)CO HEBKCHPVOIAQTA-UHFFFAOYSA-N 0.000 description 1
- 208000030159 metabolic disease Diseases 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000037323 metabolic rate Effects 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- YCXSYMVGMXQYNT-UHFFFAOYSA-N methyl 3-[(4-azidophenyl)disulfanyl]propanimidate Chemical compound COC(=N)CCSSC1=CC=C(N=[N+]=[N-])C=C1 YCXSYMVGMXQYNT-UHFFFAOYSA-N 0.000 description 1
- RMAHPRNLQIRHIJ-UHFFFAOYSA-N methyl carbamimidate Chemical compound COC(N)=N RMAHPRNLQIRHIJ-UHFFFAOYSA-N 0.000 description 1
- NEGQCMNHXHSFGU-UHFFFAOYSA-N methyl pyridine-2-carboximidate Chemical compound COC(=N)C1=CC=CC=N1 NEGQCMNHXHSFGU-UHFFFAOYSA-N 0.000 description 1
- 230000011987 methylation Effects 0.000 description 1
- 238000007069 methylation reaction Methods 0.000 description 1
- 229960004438 mibefradil Drugs 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 206010027599 migraine Diseases 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 102000035118 modified proteins Human genes 0.000 description 1
- 108091005573 modified proteins Proteins 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 210000000214 mouth Anatomy 0.000 description 1
- 210000002200 mouth mucosa Anatomy 0.000 description 1
- 210000004400 mucous membrane Anatomy 0.000 description 1
- 208000031225 myocardial ischemia Diseases 0.000 description 1
- OFYAYGJCPXRNBL-LBPRGKRZSA-N naphthalen-2-yl-3-alanine Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CC=CC2=C1 OFYAYGJCPXRNBL-LBPRGKRZSA-N 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 230000023837 negative regulation of proteolysis Effects 0.000 description 1
- 230000008692 neointimal formation Effects 0.000 description 1
- 231100000417 nephrotoxicity Toxicity 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- 229940071846 neulasta Drugs 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 230000002232 neuromuscular Effects 0.000 description 1
- 208000018360 neuromuscular disease Diseases 0.000 description 1
- 230000004112 neuroprotection Effects 0.000 description 1
- 239000002858 neurotransmitter agent Substances 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- FEMOMIGRRWSMCU-UHFFFAOYSA-N ninhydrin Chemical compound C1=CC=C2C(=O)C(O)(O)C(=O)C2=C1 FEMOMIGRRWSMCU-UHFFFAOYSA-N 0.000 description 1
- 210000000929 nociceptor Anatomy 0.000 description 1
- 108091008700 nociceptors Proteins 0.000 description 1
- 239000013631 noncovalent dimer Substances 0.000 description 1
- 229960002748 norepinephrine Drugs 0.000 description 1
- SFLSHLFXELFNJZ-UHFFFAOYSA-N norepinephrine Natural products NCC(O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-UHFFFAOYSA-N 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-M octanoate Chemical compound CCCCCCCC([O-])=O WWZKQHOCKIZLMA-UHFFFAOYSA-M 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 210000001328 optic nerve Anatomy 0.000 description 1
- 229940126701 oral medication Drugs 0.000 description 1
- 239000008184 oral solid dosage form Substances 0.000 description 1
- 230000008816 organ damage Effects 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 208000002865 osteopetrosis Diseases 0.000 description 1
- 229940092253 ovalbumin Drugs 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 208000020629 overactive bladder Diseases 0.000 description 1
- 150000003891 oxalate salts Chemical class 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 150000002923 oximes Chemical class 0.000 description 1
- 239000005022 packaging material Substances 0.000 description 1
- 230000037040 pain threshold Effects 0.000 description 1
- 208000008443 pancreatic carcinoma Diseases 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 230000001991 pathophysiological effect Effects 0.000 description 1
- BKMHDYJRAAJTAD-VPDXYFDSSA-N pbtx-3 Chemical compound OC([C@]1(C)OC2CC3OC4C5)CC(CC(=C)CO)OC1CC2OC3\C=C/C[C@@]4(C)OC1[C@@]5(C)O[C@]2(C)CCC3OC4C[C@]5(C)OC6C(C)=CC(=O)OC6CC5OC4C[C@@H](C)C3OC2C1 BKMHDYJRAAJTAD-VPDXYFDSSA-N 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- HQQSBEDKMRHYME-UHFFFAOYSA-N pefloxacin mesylate Chemical compound [H+].CS([O-])(=O)=O.C1=C2N(CC)C=C(C(O)=O)C(=O)C2=CC(F)=C1N1CCN(C)CC1 HQQSBEDKMRHYME-UHFFFAOYSA-N 0.000 description 1
- 229960001373 pegfilgrastim Drugs 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 235000019271 petrolatum Nutrition 0.000 description 1
- 229940066842 petrolatum Drugs 0.000 description 1
- 239000002831 pharmacologic agent Substances 0.000 description 1
- 239000012660 pharmacological inhibitor Substances 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- 229960005190 phenylalanine Drugs 0.000 description 1
- CWCMIVBLVUHDHK-ZSNHEYEWSA-N phleomycin D1 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC[C@@H](N=1)C=1SC=C(N=1)C(=O)NCCCCNC(N)=N)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C CWCMIVBLVUHDHK-ZSNHEYEWSA-N 0.000 description 1
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 1
- PTMHPRAIXMAOOB-UHFFFAOYSA-L phosphoramidate Chemical compound NP([O-])([O-])=O PTMHPRAIXMAOOB-UHFFFAOYSA-L 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 210000002381 plasma Anatomy 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920002492 poly(sulfone) Polymers 0.000 description 1
- 230000008488 polyadenylation Effects 0.000 description 1
- 229920000232 polyglycine polymer Polymers 0.000 description 1
- 239000004633 polyglycolic acid Substances 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010483 polyoxyethylene sorbitan monopalmitate Nutrition 0.000 description 1
- 239000000249 polyoxyethylene sorbitan monopalmitate Substances 0.000 description 1
- 235000010989 polyoxyethylene sorbitan monostearate Nutrition 0.000 description 1
- 239000001818 polyoxyethylene sorbitan monostearate Substances 0.000 description 1
- 235000010988 polyoxyethylene sorbitan tristearate Nutrition 0.000 description 1
- 239000001816 polyoxyethylene sorbitan tristearate Substances 0.000 description 1
- 229940099429 polyoxyl 40 stearate Drugs 0.000 description 1
- 108010026466 polyproline Proteins 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 229940101027 polysorbate 40 Drugs 0.000 description 1
- 229940113124 polysorbate 60 Drugs 0.000 description 1
- 229940099511 polysorbate 65 Drugs 0.000 description 1
- 229920006316 polyvinylpyrrolidine Polymers 0.000 description 1
- 230000031339 positive regulation of inflammatory response Effects 0.000 description 1
- 230000004481 post-translational protein modification Effects 0.000 description 1
- 150000003109 potassium Chemical class 0.000 description 1
- 229940125422 potassium channel blocker Drugs 0.000 description 1
- 229910001414 potassium ion Inorganic materials 0.000 description 1
- FCTRVTQZOUKUIV-MCDZGGTQSA-M potassium;[[[(2r,3s,4r,5r)-5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl] hydrogen phosphate Chemical compound [K+].C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)([O-])=O)[C@@H](O)[C@H]1O FCTRVTQZOUKUIV-MCDZGGTQSA-M 0.000 description 1
- 238000011533 pre-incubation Methods 0.000 description 1
- 238000011045 prefiltration Methods 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 108010017378 prolyl aminopeptidase Proteins 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 230000022558 protein metabolic process Effects 0.000 description 1
- 238000001742 protein purification Methods 0.000 description 1
- 230000029983 protein stabilization Effects 0.000 description 1
- 230000012743 protein tagging Effects 0.000 description 1
- 201000001474 proteinuria Diseases 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- 229940024999 proteolytic enzymes for treatment of wounds and ulcers Drugs 0.000 description 1
- 208000005069 pulmonary fibrosis Diseases 0.000 description 1
- 239000013014 purified material Substances 0.000 description 1
- 230000001696 purinergic effect Effects 0.000 description 1
- 229960003581 pyridoxal Drugs 0.000 description 1
- 235000008164 pyridoxal Nutrition 0.000 description 1
- 239000011674 pyridoxal Substances 0.000 description 1
- 235000007682 pyridoxal 5'-phosphate Nutrition 0.000 description 1
- 239000011589 pyridoxal 5'-phosphate Substances 0.000 description 1
- 229960001327 pyridoxal phosphate Drugs 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 229920013730 reactive polymer Polymers 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 230000013878 renal filtration Effects 0.000 description 1
- 230000003252 repetitive effect Effects 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000000979 retarding effect Effects 0.000 description 1
- 210000003660 reticulum Anatomy 0.000 description 1
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 1
- 210000003705 ribosome Anatomy 0.000 description 1
- 238000002702 ribosome display Methods 0.000 description 1
- 238000007151 ring opening polymerisation reaction Methods 0.000 description 1
- XMVJITFPVVRMHC-UHFFFAOYSA-N roxarsone Chemical group OC1=CC=C([As](O)(O)=O)C=C1[N+]([O-])=O XMVJITFPVVRMHC-UHFFFAOYSA-N 0.000 description 1
- 102200115048 rs1445404 Human genes 0.000 description 1
- 102220253478 rs1553261891 Human genes 0.000 description 1
- 102200010892 rs1805192 Human genes 0.000 description 1
- 229940063122 sandimmune Drugs 0.000 description 1
- 229940043230 sarcosine Drugs 0.000 description 1
- 201000000980 schizophrenia Diseases 0.000 description 1
- COSNHKQSJCRYKN-BFRWRHKQSA-N scyllotoxin Chemical group C([C@@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CS)C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CS)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1NC=NC=1)C(O)=O)NC(=O)[C@H](C)N)C1=CC=CC=C1 COSNHKQSJCRYKN-BFRWRHKQSA-N 0.000 description 1
- 125000001554 selenocysteine group Chemical group [H][Se]C([H])([H])C(N([H])[H])C(=O)O* 0.000 description 1
- 229940076279 serotonin Drugs 0.000 description 1
- 230000009450 sialylation Effects 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 235000021309 simple sugar Nutrition 0.000 description 1
- 201000002859 sleep apnea Diseases 0.000 description 1
- 210000002460 smooth muscle Anatomy 0.000 description 1
- 230000016160 smooth muscle contraction Effects 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- APSBXTVYXVQYAB-UHFFFAOYSA-M sodium docusate Chemical group [Na+].CCCCC(CC)COC(=O)CC(S([O-])(=O)=O)C(=O)OCC(CC)CCCC APSBXTVYXVQYAB-UHFFFAOYSA-M 0.000 description 1
- 229940054269 sodium pyruvate Drugs 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 235000019337 sorbitan trioleate Nutrition 0.000 description 1
- 229960000391 sorbitan trioleate Drugs 0.000 description 1
- 239000008347 soybean phospholipid Substances 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 210000000278 spinal cord Anatomy 0.000 description 1
- 208000020431 spinal cord injury Diseases 0.000 description 1
- 210000003594 spinal ganglia Anatomy 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 230000010473 stable expression Effects 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 238000012916 structural analysis Methods 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 229960002317 succinimide Drugs 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 230000008093 supporting effect Effects 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 210000001258 synovial membrane Anatomy 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 238000011191 terminal modification Methods 0.000 description 1
- 229960003604 testosterone Drugs 0.000 description 1
- 229960000814 tetanus toxoid Drugs 0.000 description 1
- 229940126585 therapeutic drug Drugs 0.000 description 1
- 230000008719 thickening Effects 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 208000037816 tissue injury Diseases 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- FGMPLJWBKKVCDB-UHFFFAOYSA-N trans-L-hydroxy-proline Natural products ON1CCCC1C(O)=O FGMPLJWBKKVCDB-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000037317 transdermal delivery Effects 0.000 description 1
- 239000012581 transferrin Substances 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 108091053409 transient receptor (TC 1.A.4) family Proteins 0.000 description 1
- 238000003146 transient transfection Methods 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 230000005945 translocation Effects 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 description 1
- 229940029284 trichlorofluoromethane Drugs 0.000 description 1
- 239000003029 tricyclic antidepressant agent Substances 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- 239000013638 trimer Substances 0.000 description 1
- 208000027930 type IV hypersensitivity disease Diseases 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 206010046766 uterine cancer Diseases 0.000 description 1
- 229940005605 valeric acid Drugs 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 239000012178 vegetable wax Substances 0.000 description 1
- 229940070384 ventolin Drugs 0.000 description 1
- 108010078375 voltage-dependent calcium channel (P-Q type) Proteins 0.000 description 1
- 230000007279 water homeostasis Effects 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
- 239000000230 xanthan gum Substances 0.000 description 1
- 235000010493 xanthan gum Nutrition 0.000 description 1
- 229940082509 xanthan gum Drugs 0.000 description 1
- 239000000811 xylitol Substances 0.000 description 1
- HEBKCHPVOIAQTA-SCDXWVJYSA-N xylitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)CO HEBKCHPVOIAQTA-SCDXWVJYSA-N 0.000 description 1
- 235000010447 xylitol Nutrition 0.000 description 1
- 229960002675 xylitol Drugs 0.000 description 1
- BPKIMPVREBSLAJ-QTBYCLKRSA-N ziconotide Chemical compound C([C@H]1C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]2C(=O)N[C@@H]3C(=O)N[C@H](C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@H](C(N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CSSC2)C(N)=O)=O)CSSC[C@H](NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)CNC(=O)[C@H](CCCCN)NC(=O)CNC(=O)[C@H](CCCCN)NC(=O)[C@@H](N)CSSC3)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C(N1)=O)CCSC)[C@@H](C)O)C1=CC=C(O)C=C1 BPKIMPVREBSLAJ-QTBYCLKRSA-N 0.000 description 1
- 229960002811 ziconotide Drugs 0.000 description 1
- JPZXHKDZASGCLU-LBPRGKRZSA-N β-(2-naphthyl)-alanine Chemical compound C1=CC=CC2=CC(C[C@H](N)C(O)=O)=CC=C21 JPZXHKDZASGCLU-LBPRGKRZSA-N 0.000 description 1
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 1
- 108091058550 ω-conotoxin Proteins 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
- A61K47/60—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes the organic macromolecular compound being a polyoxyalkylene oligomer, polymer or dendrimer, e.g. PEG, PPG, PEO or polyglycerol
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/43504—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from invertebrates
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/62—DNA sequences coding for fusion proteins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/30—Non-immunoglobulin-derived peptide or protein having an immunoglobulin constant or Fc region, or a fragment thereof, attached thereto
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/55—Fusion polypeptide containing a fusion with a toxin, e.g. diphteria toxin
Definitions
- the present invention is related to the biochemical arts, in particular to therapeutic peptides and conjugates.
- Ion channels are a diverse group of molecules that permit the exchange of small inorganic ions across membranes. All cells require ion channels for function, but this is especially so for excitable cells such as those present in the nervous system and the heart.
- the electrical signals orchestrated by ion channels control the thinking brain, the beating heart and the contracting muscle. Ion channels play a role in regulating cell volume, and they control a wide variety of signaling processes.
- the ion channel family includes Na + , K + , and Ca 2+ cation and Cl- anion channels. Collectively, ion channels are distinguished as either ligand-gated or voltage-gated. Ligand-gated channels include both extracellular and intracellular ligand-gated channels.
- the extracellular ligand-gated channels include the nicotinic acetylcholine receptor (nAChR), the serotonin (5- hdroxytryptamine, 5-HT) receptors, the glycine and ⁇ -butyric acid receptors (GABA) and the glutamate-activated channels including kanate, ⁇ -amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) and N-methyl-D-aspartate receptors (NMDA) receptors.
- nAChR nicotinic acetylcholine receptor
- 5-HT serotonin (5- hdroxytryptamine
- GABA glycine and ⁇ -butyric acid receptors
- glutamate-activated channels including kanate, ⁇ -amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) and N-methyl-D-aspartate receptors (NMDA) receptors.
- cAMP, cGMP Ca 2+ and G-proteins.
- the voltage-gated ion channels are categorized by their selectivity for inorganic ion species, including sodium, potassium, calcium and chloride ion channels. (Harte and Ouzounis (2002), FEBS Lett. 514: 129-34).
- the K + channels constitute the largest and best characterized family of ion channels described to date.
- Potassium channels are subdivided into three general groups: the 6 transmembrane (6TM) K + channels, the 2TM-2TM/leak K + channels and the 2TM/Kir inward rectifying channels. (Tang et al. (2004), Ann. Rev. Physiol. 66, 131-159). These three groups are further subdivided into families based on sequence similarity.
- the voltage-gated K + channels including (Kv1-6, Kv8-9), EAG, KQT, and SIo (BKCa), are family members of the 6TM group.
- the 2TM-2TM group comprises TWIK, TREK, TASK, TRAAK, and THIK, whereas the 2TM/Kir group consists of Kir1-7.
- Two additional classes of ion channels include the inward rectifier potassium (IRK) and ATP-gated purinergic (P2X) channels. (Harte and Ouzounis (2002), FEBS Lett. 514: 129-34).
- Toxin peptides produced by a variety of organisms have evolved to target ion channels.
- Snakes, scorpions, spiders, bees, snails and sea anemone are a few examples of organisms that produce venom that can serve as a rich source of small bioactive toxin peptides or "toxins" that potently and selectively target ion channels and receptors.
- these toxin peptides have evolved as potent antagonists or inhibitors of ion channels, by binding to the channel pore and physically blocking the ion conduction pathway.
- the peptide is found to antagonize channel function by binding to a region outside the pore (e.g., the voltage sensor domain).
- the toxin peptides are usually between about 20 and about 80 amino acids in length, contain 2-5 disulfide linkages and form a very compact structure (see, e.g., Figure 10), Toxin peptides (e.g., from the venom of scorpions, sea anemones and cone snails) have been isolated and characterized for their impact on ion channels.
- Such peptides appear to have evolved from a relatively small number of structural frameworks that are particularly well suited to addressing the critical issues of potency and stability.
- scorpion and Conus toxin peptides contain 10-40 amino acids and up to five disulfide bonds, forming extremely compact and constrained structure (microproteins) often resistant to proteolysis.
- the conotoxin and scorpion toxin peptides can be divided into a number of superfamilies based on their disulfide connections and peptide folds.
- the solution structure of many of these has been determined by NMR spectroscopy, illustrating their compact structure and verifying conservation of their family fold. (E.g., Jerusalem et al., lonisation behaviour and solution properties of the potassium-channel blocker ShK toxin, Eur. J. Biochem.
- ⁇ -conotoxins have well-defined fourcysteine/two disulfide loop structures (Loughnan, 2004) and inhibit nicotinic acetylcholine receptors.
- ⁇ -conotoxins have six cysteine/three disulfide loop consensus structures (Nielsen, 2000) and block calcium channels. Structural subsets of toxins have evolved to inhibit either voltage-gated or calcium-activated potassium channels.
- Figure 9 shows that a limited number of conserved disulfide architectures shared by a variety of venomous animals from bee to snail and scorpion to snake target ion channels.
- Figure 7 shows alignment of alpha-scorpion toxin family and illustrates that a conserved structural framework is used to derive toxins targeting a vast array of potassium channels.
- toxin peptides Due to their potent and selective blockade of specific ion channels, toxin peptides have been used for many years as tools to investigate the pharmacology of ion channels. Other than excitable cells and tissues such as those present in heart, muscle and brain, ion channels are also important to non-excitable cells such as immune cells. Accordingly, the potential therapeutic utility of toxin peptides has been considered for treating various immune disorders, in particular by inhibition of potassium channels such as Kv1.3 and IKCaI since these channels indirectly control calcium signaling pathway in lymphocytes, [e.g., Kern et al., ShK toxin compositions and methods of use, US Patent No.
- IP3 is the natural second messenger which activates the calcium signaling pathway. IP3 is produced following ligand-induced activation of the T cell receptor (TCR) and upon binding to its intracellular receptor (a channel) causes unloading of intracellular calcium stores.
- TCR T cell receptor
- a channel intracellular receptor
- the endoplasmic reticulum provides one key calcium store.
- Thapsigargin an inhibitor of the sarcoplasmic-endoplasmic reticulum calcium ATPase (SERCA), also causes unloading of intracellular stores and activation of the calcium signaling pathway in lymphocytes. Therefore, thapsigargin can be used as a specific stimulus of the calcium signaling pathway in T cells.
- SERCA sarcoplasmic-endoplasmic reticulum calcium ATPase
- CRAC calcium release activated channel
- potassium efflux is accomplished by the voltage-gated potassium channel Kv1.3 and the calcium-activated potassium channel IKCaI [K.G. Chandy et al. (2004) TIPS 25, 280]. These potassium channels therefore indirectly control the calcium signaling pathway, by allowing for the necessary potassium efflux that allows for a sustained influx of calcium through CRAC,
- the calcium-calmodulin dependent phosphatase calcineurin is activated upon sustained increases in intracellular calcium and dephosphorylates cytosolic NFAT. Dephosphorylated NFAT quickly translocates to the nucleus and is widely accepted as a critical transcription factor for T cell activation (F. Macian (2005) Nat. Rev. Immunol. 5, 472 and N. Venkatesh etal. (2004) PNAS 101, 8969).
- Inhibitors of calcineurin such as cyclosporin A (Neoral, Sandimmune) and FK506 (Tacrolimus) are a main stay for treatment of severe immune disorders such as those resulting in rejection following solid organ transplant (I.M. Gonzalez-Pinto et al. (2005) Transplant.
- Lupus represents another disorder that may benefit from agents blocking activation of helper T cells.
- calcineurin is also expressed in other tissues (e.g. kidney) and cyclosporine A & FK506 have a narrow safety margin due to mechanism based toxicity. Renal toxicity and hypertension are common side effects that have limited the promise of cyclosporine & FK506. Due to concerns regarding toxicity, calcineurin inhibitors are used mostly to treat only severe immune disease (Bissonnette-R et al. (2006) J. Am. Acad. Dermatol. 54, 472).
- Kv1.3 inhibitors offer a safer alternative to calcineurin inhibitors for the treatment of immune disorders. This is because Kv1.3 also operates to control the calcium signaling pathway in T cells, but does so through a distinct mechanism to that of calcineurin inhibitors, and evidence on Kv1.3 expression and function show that Kv1.3 has a more restricted role in T cell biology relative to calcineurin, which functions also in a variety of non- lymphoid cells and tissues.
- IL-2 interleukin 2
- IFNg interferon gamma
- T cells represent the principle source of IFNg production in mediating adaptive immune responses, whereas natural killer (NK) cells & APCs are likely an important source during host defense against infection [K. Schroder et al. (2004) J. Leukoc. Biol. 75, 163], IFNg, originally called macrophage-activating factor, upregulates antigen processing and presentation by monocytes, macrophages and dendritic cells. IFNg mediates a diverse array of biological activities in many cell types [U. Boehm et al. (1997) Annu. Rev. Immunol. 15, 749] including growth & differentiation, enhancement of NK cell activity and regulation of B cell immunoglobulin production and class switching.
- CD40L is another cytokine expressed on activated T cells following calcium mobilization and upon binding to its receptor on B cells provides critical help allowing for B cell germinal center formation, B cell differentiation and antibody isotype switching.
- CD40L-mediated activation of CD40 on B cells can induce profound differentiation and clonal expansion of immunoglobulin (Ig) producing B cells [S. Quezada et al. (2004) Annu. Rev. Immunol. 22, 307],
- the CD40 receptor can also be found on dendritic cells and CD40L signaling can mediate dendritic cell activation and differentiation as well.
- the antigen presenting capacity of B cells and dendritic cells is promoted by CD40L binding, further illustrating the broad role of this cytokine in adaptive immunity.
- toxin peptides are complex processes in venomous organisms, and is an even more complex process synthetically. Due to their conserved disulfide structures and need for efficient oxidative refolding, toxin peptides present challenges to synthesis. Although toxin peptides have been used for years as highly selective pharmacological inhibitors of ion channels, the high cost of synthesis and refolding of the toxin peptides and their short half-life in vivo have impeded the pursuit of these peptides as a therapeutic modality. Far more effort has been expended to identify small molecule inhibitors as therapeutic antagonists of ion channels, than has been given the toxin peptides themselves.
- ZiconotideTM small ⁇ - conotoxin MVIIA peptide
- a cost-effective process for producing therapeutics is a desideratum provided by the present invention, which involves toxin peptides fused, or otherwise covalently conjugated to a vehicle.
- the present invention relates to a composition of matter of the formula:
- F 1 and F 2 are half-life extending moieties, and d and e are each independently O or 1 , provided that at least one of d and e is 1 ;
- X 1 , X 2 , and X 3 are each independently -(L) ⁇ P-(L) 9 -, and f and g are each independently O or 1 ;
- P is a toxin peptide of no more than about 80 amino acid residues in length, comprising at least two intrapeptide disulfide bonds;
- the present invention thus concerns molecules having variations on Formula 1 , such as the formulae:
- the toxin peptide (P), if more than one is present can be independently the same or different from any other toxin peptide(s) also present in the inventive composition
- the linker moiety ((L) ⁇ and/or (L) 3 ), if present, can be independently the same or different from any other linker, or linkers, that may be present in the inventive composition.
- Conjugation of the toxin peptide(s) to the half-life extending moiety, or moieties can be via the N- terminal and/or C-terminal of the toxin peptide, or can be intercalary as to its primary amino acid sequence, F 1 being linked closer to the toxin peptide's N-terminus than is linked F 2 .
- useful half-life extending moieties include immunoglobulin Fc domain, human serum albumin (HSA), or polyethylene glycol) (PEG).
- the present invention also relates to a composition of matter, which includes, conjugated or unconjugated, a toxin peptide analog of ShK, 0SK1 , ChTx, or Maurotoxin modified from the native sequences at one or more amino acid residues, having greater Kv1.3 or IKCaiantagonist activity, and/or target selectivity, compared to a ShK, 0SK1 , or Maurotoxin (MTX) peptides having a native sequence.
- the toxin peptide analogs comprise an amino acid sequence selected from any of the following: SEQ ID NOS: 88, 89, 92, 148 through 200, 548 through 561 , 884 through 949, or 1295 through
- the present invention also relates to other toxin peptide analogs that comprise an amino acid sequence selected from any of the following:
- the present invention is also directed to a pharmaceutical composition that includes the inventive composition of matter and a pharmaceutically acceptable carrier.
- the compositions of this invention can be prepared by conventional synthetic methods, recombinant DNA techniques, or any other methods of preparing peptides and fusion proteins well known in the art.
- Compositions of this invention that have non-peptide portions can be synthesized by conventional organic chemistry reactions, in addition to conventional peptide chemistry reactions when applicable. 5
- the primary use contemplated is as therapeutic and/or prophylactic agents.
- the inventive compositions incorporating the toxin peptide can have activity and/or ion channel target selectivity comparable to— or even greater than— the unconjugated peptide.
- the present invention includes a method of treating an autoimmune disorder, which involves administering to a patient who has been diagnosed with an autoimmune disorder, O such as multiple sclerosis, type 1 diabetes, psoriasis, inflammatory bowel disease, contact- mediated dermatitis, rheumatoid arthritis, psoriatic arthritis, asthma, allergy, restinosis, systemic sclerosis, fibrosis, scleroderma, glomerulonephritis, Sjogren syndrome, inflammatory bone resorption, transplant rejection, graft-versus-host disease, or lupus, a therapeutically effective amount of the inventive composition of matter (preferably comprising a Kv1.3 antagonist peptide or 5 IKCaI antagonist peptide), whereby at least one symptom of the disorder is alleviated in the patient.
- an autoimmune disorder O such as multiple sclerosis, type 1 diabetes, psoriasis, inflammatory bowel disease, contact- mediated dermatitis, rheumatoid arthritis
- the present invention is further directed to a method of preventing or mitigating a relapse of a symptom of multiple sclerosis, which method involves administering to a patient, who has previously experienced at least one symptom of multiple sclerosis, a prophylactically effective O amount of the inventive composition of matter (preferably comprising a Kv1.3 antagonist peptide or
- compositions of this invention can also be useful in screening for therapeutic or diagnostic agents.
- an Fc- peptide in an assay employing anti-Fc coated plates.
- the half-life extending moiety, such as Fc can make insoluble peptides soluble and thus useful in a number of assays.
- FIG. 1 shows schematic structures of some exemplary Fc dimers that can be derived from an IgG1 antibody.
- Fc in the figure represents any of the Fc variants within the meaning of "Fc domain” herein.
- X1 and “X2” represent peptides or linker-peptide combinations as defined 5 hereinafter.
- the specific dimers are as follows:
- FIG. 1A and Figure 1D Single disulfide-bonded dimers
- FIG. 1B and Figure 1E Doubly disulfide-bonded dimers
- Figure 2 shows schematic structures of some embodiments of the composition of the 0 invention that shows a single unit of the pharmacologically active toxin peptide.
- Figure 2A shows a single chain molecule and can also represent the DNA construct for the molecule.
- Figure 2B shows a dimer in which the linker-peptide portion is present on only one chain of the dimer.
- Figure 2C shows a dimer having the peptide portion on both chains.
- the dimer of Figure 2C will form spontaneously in certain host cells upon expression of a DNA construct encoding the single chain 5 shown in Figure 2A. In other host cells, the cells could be placed in conditions favoring formation of dimers or the dimers can be formed in vitro.
- Figure 3 shows exemplary nucleic acid and amino acid sequences (SEQ ID NOS: 1 and 2, respectively) of human IgGI Fc that is optimized for mammalian expression and can be used in this invention.
- O Figure 4 shows exemplary nucleic acid and amino acid sequences (SEQ ID NOS: 3 and
- Figure 5A shows the amino acid sequence of the mature ShK peptide (SEQ ID NO: 5), which can be encoded for by a nucleic acid sequence containing codons optimized for expression 5 in mammalian cell, bacteria or yeast.
- Figure 5B shows the three disulfide bonds (--S-S-) formed by the six cysteines within the ShK peptide (SEQ ID NO: 10),
- Figure 6 shows an alignment of the voltage-gated potassium channel inhibitor Stichodactyla helianthus (ShK) with other closely related members of the sea anemone toxin O family.
- the sequence of the 35 amino acid mature ShK toxin (Accession #P29187) isolated from the venom of Stichodactyla helianthus is shown aligned to other closely related members of the sea anemone family.
- the consensus sequence and predicted disulfide linkages are shown, with highly conserved residues being shaded.
- HmK peptide toxin sequence shown (Swiss-Protein Accession #097436) is of the immature precursor from the Magnificent sea anemone (Radianthus maqnifica; Heteractis maqnifica) and the putative signal peptide is underlined.
- the mature HmK peptide toxin would be predicted to be 35 amino acids in length and span residues 40 through 74.
- AeK is the mature peptide toxin, isolated from the venom of the sea anemone Actinia equine (Accession #P81897).
- FIG. 6A shows the amino acid alignment (SEQ ID NO: 10) of ShK to other members of the sea anemone family of toxins, HmK (SEQ ID NO: 6 (Mature Peptide), (SEQ ID NO: 542, Signal and Mature Peptide portions)), AeK (SEQ ID NO: 7), AsKs (SEQ ID NO: 8), and BgK (SEQ ID NO: 9).
- Figure 6B shows a disulfide linkage map for this family having 3 disulfide linkages (C1-C6, C2-C4, C3-C5).
- Figure 7 shows an amino acid alignment of the alpha-scorpion toxin family of potassium channel inhibitors.
- BmKKI SEQ ID NO: 11; BmKK4, SEQ ID NO: 12; PBTxI, SEQ ID NO: 14; Tc32, SEQ ID NO: 13; BmKK6, SEQ ID NO: 15; P01, SEQ ID NO: 16; Pi2, SEQ ID NO: 17; Pi3, SEQ ID NO: 18; Pi4, SEQ ID NO: 19; MTX, SEQ ID NO: 20; Pi1, SEQ ID NO: 21; HsTxI, SEQ ID NO: 61; AgTx2, SEQ ID NO: 23; KTX1, SEQ ID NO: 24; OSK1, SEQ ID NO: 25; BmKTX, SEQ ID NO: 22; HgTXI, SEQ ID NO: 27; MgTx, SEQ ID NO: 28; C11Tx1, SEQ ID NO: 29; NTX, SEQ ID NO: 30; Tc30, SEQ ID NO
- Figure 8 shows an alignment of toxin peptides converted to peptibodies in this invention (Apamin, SEQ ID NO: 68; HaTxI 1 SEQ ID NO: 494; ProTxi, SEQ ID NO: 56; PaTx2, SEQ ID NO: 57; ShK[2-35], SEQ ID NO: 92; ShK[1-35], SEQ ID NO: 5; HmK, SEQ ID NO: 6; ChTx (K32E), SEQ ID NO: 59; ChTx 1 SEQ ID NO: 36; IbTx, SEQ ID NO: 38; 0SK1 (E16K, K20D), SEQ ID NO: 296; 0SK1 , SEQ ID NO: 25; AgTx2, SEQ ID NO: 23; KTX1 , SEQ ID NO: 24; MgTx, SEQ ID NO: 28; NTX, SEQ ID NO: 30; MTX, SEQ ID NO: 20; Pi2, SEQ ID NO: 17; H
- Figure 9 shows disulfide arrangements within the toxin family. The number of disulfides and the disulfide bonding order for each subfamily is indicated. A partial list of toxins that fall within each disulfide linkage category is presented. Figure 10 illustrates that solution structures of toxins reveal a compact structure.
- Solution structures of native toxins from sea anemone (ShK), scorpion (MgTx, MTX, HsTxI), marine cone snail (wGVIA) and tarantula (HaTxI) indicate the 28 to 39 amino acid peptides all form a compact structure.
- the toxins shown have 3 or 4 disulfide linkages and fall within 4 of the 6 subfamilies shown in Figure 9.
- Figure 11A-C shows the nucleic acid (SEQ ID NO: 69 and SEQ ID NO: 1358) and encoded amino acid (SEQ ID NO:70, SEQ ID NO:1359 and SEQ ID NO: 1360) sequences of residues 5131-6660 of pAMG21ampR-Fc-pep.
- the sequences of the Fc domain (SEQ ID NOS: 71 and 72) exclude the five C-terminal glycine residues.
- This vector enables production of peptibodies in which the peptide-linker portion is at the C-terminus of the Fc domain.
- Figure 11 D shows a circle diagram of a peptibody bacterial expression vector pAMG21ampR-Fc-pep having a chloramphenicol acetyltransferase gene (cat; "CmR" site) that is replaced with the peptide-linker sequence.
- Figure 12A-C shows the nucleic acid (SEQ ID NO: 73 and SEQ ID NO: 1361) and encoded amino acid (SEQ ID NO:74, SEQ ID NO: 1362 and SEQ ID NO: 1363) sequences of residues 5131-6319 of pAMG21ampR-Pep-Fc.
- the sequences of the Fc domain (SEQ ID NOS: 75 and 76) exclude the five N-terminal glycine residues.
- This vector enables production of 5 peptibodies in which the peptide-linker portion is at the N-terminus of the Fc domain.
- Figure 12D shows a circle diagram of a peptibody bacterial expression vector having a zeocin resistance (ble; "ZeoR”) site that is replaced with the peptide-linker sequence.
- ZeoR zeocin resistance
- Figure 12E-F shows the nucleic acid (SEQ ID NO:1339) and encoded amino acid sequences of pAMG21ampR-Pep-Fc (SEQ ID NO:1340, SEQ ID NO:1341, and SEQ ID NO:1342). 0
- the sequences of the Fc domain (SEQ ID NOS: 75 and 76) exclude the five N-terminal glycine residues. This vector enables production of peptibodies in which the peptide-linker portion is at the N-terminus of the Fc domain.
- Figure 13A is a circle diagram of mammalian expression vector pCDNA3.1(+) CMVi.
- Figure 13B is a circle diagram of mammalian expression vector pCDNA3.1(+)CMVi-Fc- 5 2xG4S-Activin RIIb that contains a Fc region from human IgGI, a 10 amino acid linker and the activin RIIb gene.
- Figure 13C is a circle diagram of the CHO expression vector pDSRa22 containing the Fc- L10-ShK[2-35] coding sequence.
- Figure 14 shows the nucleotide and encoded amino acid sequences (SEQ. ID. NOS: 77 O and 78, respectively) of the molecule identified as "Fc-LI 0-ShK[1 -35]" in Example 1 hereinafter.
- the L10 linker amino acid sequence (SEQ ID NO: 79) is underlined.
- Figure 15 shows the nucleotide and encoded amino acid sequences (SEQ. ID. NOS: 80 and 81, respectively) of the molecule identified as "Fc-LI 0-ShK[2-35]" in Example 2 hereinafter.
- the same L10 linker amino acid sequence (SEQ ID NO: 79) as used in Fc-LI 0-ShK[1 -35] ( Figure 5 14) is underlined.
- Figure 16 shows the nucleotide and encoded amino acid sequences (SEQ. ID. NOS: 82 and 83, respectively) of the molecule identified as "Fc-L25-ShK[2-35]" in Example 2 hereinafter.
- the L25 linker amino acid sequence (SEQ ID NO: 84) is underlined.
- Figure 17 shows a scheme for N-terminal PEGylation of ShK peptide (SEQ ID NO: 5 and O SEQ ID N0:10) by reductive amination, which is also described in Example 32 hereinafter.
- Figure 18 shows a scheme for N-terminal PEGylation of ShK peptide (SEQ ID NO: 5 and SEQ ID NO:10) via amide formation, which is also described in Example 34 hereinafter.
- Figure 19 shows a scheme for N-terminal PEGylation of ShK peptide (SEQ ID NO: 5 and SEQ ID NO:10) by chemoselective oxime formation, which is also described in Example 33 hereinafter.
- Figure 2OA shows a reversed-phase HPLC analysis at 214 nm and Figure 2OB shows electrospray mass analysis of folded ShK[2-35], also described as folded-"Des-Arg1-ShK" (Peptide
- Figure 21 shows reversed phase HPLC analysis at 214 nm of N-terminally PEGylated ShK[2-35], also referred to as N-Terminally PEGylated-"Des-Arg1-ShK".
- Figure 22A shows a reversed-phase HPLC analysis at 214 nm of folded ShK[1-35], also referred to as "ShK".
- Figure 22B shows electrospray mass analysis of folded ShK[1-35], also referred to as "ShK”.
- Figure 23 illustrates a scheme for N-terminal PEGylation of ShK[2-35] (SEQ ID NO: 92 and SEQ ID NO: 58, also referred to as "Des-Arg1-ShK” or "ShK d1") by reductive amination, which is also described in Example 31 hereinafter.
- Figure 24A shows a western blot of conditioned medium from HEK 293 cells transiently transfected with Fc-LI 0-ShK[1 -35].
- Lane 1 molecular weight markers
- Lane 2 15 ⁇ l Fc-LIO-ShK
- Lane 3 10 ⁇ l Fc-LIO-ShK
- Lane 4 5 ⁇ l Fc-LIO-ShK
- Lane 5 molecular weight markers
- Lane 6 blank
- Lane 7 15 ⁇ l No DNA control
- Lane 8 10 ⁇ l No DNA control
- Lane 9 5 ⁇ l No DNA control
- Lane 10 molecular weight markers.
- Figure 24B shows a western blot of with 15 ⁇ l ofconditioned medium from clones of Chinese Hamster Ovary (CHO) cells stably transfected with Fc-L-ShK[I -35], Lanes 1 - 15 were loaded as follows: blank, BB6, molecular weight markers, BB5, BB4, BB3, BB2, BB1, blank, BD6, BD5, molecular weight markers, BD4, BD3, BD2.
- Figure 25A shows a western blot of a non-reducing SDS-PAGE gel containing conditioned medium from 293T cells transiently transfected with Fc-L-SmIIIA.
- Figure 25B shows a western blot of a reducing SDS-PAGE gel containing conditioned medium from 293T cells transiently transfected with Fc-L-SmIIIA.
- Figure 26A shows a Spectral scan of 10 ⁇ l purified Fc-LI 0-ShK[1 -35] product from stably transfected CHO cells diluted in 700 ⁇ l PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1-cm path length quartz cuvette.
- Figure 26B shows Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final Fc-LI 0-ShK[1 -35] product.
- Lanes 1 - 12 were loaded as follows: Novex Mark12 wide range protein standards, 0.5 ⁇ g product non-reduced, blank, 2.0 ⁇ g product non-reduced, blank, 10 ⁇ g product non-reduced, Novex Mark12 wide range protein standards, 0.5 ⁇ g product reduced, blank, 2.0 ⁇ g product reduced, blank, and 10 ⁇ g product reduced.
- Figure 26C shows size exclusion chromatography on 20 ⁇ g of the final Fc-LI 0-ShK[1 -35] product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH 2 PO 4 , 250 mM NaCl, and pH 6,9 at 1 ml/min observing the absorbance at 280 nm.
- Figure 26D shows a MALDI mass spectral analysis of the final sample of Fc-L10-ShK[1- 35] analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ⁇ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses.
- Figure 27A shows a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final purified Fc-LI 0-ShK[2-35] product from stably transfected CHO cells.
- Lane 1 - 12 were loaded as follows: Novex Mark12 wide range protein standards, 0.5 ⁇ g product non-reduced, blank, 2.0 ⁇ g product non-reduced, blank, 10 ⁇ g product non-reduced, Novex Mark12 wide range protein standards, 0.5 ⁇ g product reduced, blank, 2.0 ⁇ g product reduced, blank, and 10 ⁇ g product reduced.
- Figure 27B shows size exclusion chromatography on 50 ⁇ g of the purified Fc-LI 0-ShK[2- 35] injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH 2 PO 4 , 250 mM NaCI, and pH 6.9 at 1 ml/min observing the absorbance at 280 nm,
- Figure 27C shows a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of Fc- L5-ShK[2-35] purified from stably transfected CHO cells.
- Lane 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 ⁇ g product non-reduced, blank, 2.0 ⁇ g product non- reduced, blank, 10 ⁇ g product non-reduced, Novex Mark12 wide range protein standards, 0.5 ⁇ g product reduced, blank, 2,0 ⁇ g product reduced, blank, and 10 ⁇ g product reduced.
- Figure 27D shows a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of Fc- L25-ShK[2-35] purified from stably transfected CHO cells.
- Lane 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 ⁇ g product non-reduced, blank, 2.0 ⁇ g product non-reduced, blank, 10 ⁇ g product non-reduced, Novex Mark12 wide range protein standards, 0.5 ⁇ g product reduced, blank, 2.0 ⁇ g product reduced, blank, and 10 ⁇ g product reduced.
- Figure 27E shows a spectral scan of 10 ⁇ l of the Fc-LI 0-ShK[2-35] product diluted in 700 ⁇ l PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
- Figure 27F shows a MALDI mass spectral analysis of the final sample of Fc-LI 0-ShK[2- 35] analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from about 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses.
- Figure 27G shows a spectral scan of 10 ⁇ l of the Fc-L5-ShK[2-35] product diluted in 700 ⁇ l PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
- Figure 27H shows the size exclusion chromatography on 50 mg of the final Fc-L5-ShK[2- 35] product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
- Figure 271 shows a MALDI mass spectral analysis of the final sample of Fc-L5-ShK[2-35] analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV.
- Figure 27J shows a Spectral scan of 20 ⁇ l of the product diluted in 700 ⁇ l PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
- Figure 27K shows the size exclusion chromatography on 50 ⁇ g of the final Fc-L25-ShK[2- 35] product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH 2 PO 4 , 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
- Figure 27L shows a MALDI mass spectral analysis of the final sample of Fc-L25-ShK[2-
- Lane 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 ⁇ g product non-reduced, blank, 2.0 ⁇ g product non- reduced, blank, 10 ⁇ g product non-reduced, Novex Mark12 wide range protein standards, 0.5 ⁇ g product reduced, blank, 2.0 ⁇ g product reduced, blank, and 10 ⁇ g product reduced.
- Figure 28B shows size exclusion chromatography on 45 ⁇ g of purified Fc-LI 0-KTX1 injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH 2 PO 4 , 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
- Figure 28C shows a Spectral scan of 20 ⁇ l of the Fc-LI 0-KTX1 product diluted in 700 ⁇ l PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
- Figure 28D shows a MALDI mass spectral analysis of the final sample of Fc-L10-KTX1 analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ⁇ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses.
- Figure 29A shows a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of Fc- L-AgTx2 purified and refolded from bacterial cells.
- Lane 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 ⁇ g product non-reduced, blank, 2.0 ⁇ g product non- reduced, blank, 10 ⁇ g product non-reduced, Novex Mark12 wide range protein standards, 0.5 ⁇ g product reduced, blank, 2.0 ⁇ g product reduced, blank, and 10 ⁇ g product reduced.
- Figure 29B shows a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of Fc- L10-HaTx1 purified and refolded from bacterial cells.
- Lane 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 ⁇ g product non-reduced, blank, 2.0 ⁇ g product non- reduced, blank, 10 ⁇ g product non-reduced, Novex Mark12 wide range protein standards, 0.5 ⁇ g product reduced, blank, 2.0 ⁇ g product reduced, blank, and 10 ⁇ g product reduced, spectral scan of the purified material.
- Figure 29C shows a Spectral scan of 20 ⁇ l of the Fc-LI 0-AgTx2 product diluted in 700 ⁇ l
- Figure 29D shows the Size exclusion chromatography on 20 ⁇ g of the final Fc-LI 0-AgTx2 product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH 2 PCM, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
- Figure 29E shows a MALDI mass spectral analysis of the final sample of Fc-LI 0-AgTx2 analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from about 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses.
- Figure 29F shows a Spectral scan of 20 ⁇ l of the Fc-LIO-HaTxI product diluted in 700 ⁇ l PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
- Figure 29G shows the size exclusion chromatography on 20 ⁇ g of the final Fc-UO-HaTxI product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
- Figure 29H shows a MALDl mass spectral analysis of the final sample of Fc-LI 0-HaTxI analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from - 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses.
- Figure 3OA shows Fc-L10-ShK[1-35] purified from CHO cells produces a concentration dependent block of the outward potassium current recorded from HEK293 cell stably expressing the human Kv1.3 channel.
- Figure 3OB shows the time course of potassium current block by Fc-LI 0-ShK[1-35] at various concentrations.
- Figure 3OC shows synthetic ShK[I -35] (also referred to as "ShK” alone) produces a concentration dependent block of the outward potassium current recorded from HEK293 cell stably expressing human Kv1.3 channel.
- Figure 3OD shows the time course of ShK[1-35] block at various concentrations.
- Figure 32A shows Fc-L-KTXI peptibody purified from bacterial cells producing a concentration dependent block of the outward potassium current as recorded from HEK293 cell stably expressing human Kv1.3 channel.
- Figure 32B shows the time course of potassium current block by Fc-LI 0-KTX1 at various 5 concentrations.
- Figure 33 shows by immunohistochemistry that CHO-derived Fc-LI 0-ShK[1 -35] peptibody stains HEK 293 cells stably transfected with human Kv1.3 ( Figure 33A), whereas untransfected HEK 293 cells are not stained with the peptibody ( Figure 33B).
- Figure 34 shows results of an enzyme-immunoassay using fixed HEK 293 cells stably 0 transfected with human Kv1.3.
- Figure 34A shows the CHO-derived Fc-LI 0-ShK[1 -35] (referred to here simply as "Fc-LIO-ShK”) peptibody shows a dose-dependent increase in response, whereas the CHO-Fc control ("Fc control”) does not
- Figure 34B shows the Fc-LI 0-ShK[1 -35] peptibody (referred to here as "Fc-ShK”) does not elicit a response from untransfected HEK 293 cells using similar conditions and also shows other negative controls.
- Figure 35 shows the CHO-derived Fc-LI 0-ShK[1 -35] peptibody shows a dose-dependent inhibition of IL-2 (Figure 35A) and IFN ⁇ (Figure 35B) production from PMA and ⁇ CD3 antibody stimulated human PBMCs.
- the peptibody shows a novel pharmacology exhibiting a complete inhibition of the response, whereas the synthetic ShK[I -35] peptide alone shows only a partial inhibition.
- O Figure 36 shows the mammalian-derived Fc-LI 0-ShK[1 -35] peptibody inhibits T cell proliferation ( 3 H-thymidine incorporation) in human PBMCs from two normal donors stimulated with antibodies to CD3 and CD28.
- Figure 36A shows the response of donor 1 and Figure 36B the response of donor 2.
- Pre-incubation with the anti-CD32 (FcgRII) blocking antibody did not alter the sensitivity toward the peptibody.
- Figure 37 shows the purified CHO-derived Fc-LI 0-ShK[1 -35] peptibody causes a dose- dependent inhibition of IL-2 ( Figure 37A) and IFN ⁇ ( Figure 37B) production from ⁇ CD3 and ⁇ CD28 antibody stimulated human PBMCs.
- Figure 38A shows the PEGylated ShK[2-35] synthetic peptide produces a concentration dependent block of the outward potassium current recorded from HEK293 cell stably expressing O human Kv1 ,3 channel and the time course of potassium current block at various concentrations is shown in Figure 38B.
- Figure 39A shows a spectral scan of 50 ⁇ l of the Fc-LI 0-ShK(1-35) product diluted in 700 ⁇ l PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
- Figure 39B shows a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final Fc-LI 0-ShK(1-35) product.
- Lane 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 ⁇ g product non-reduced, blank, 2.0 ⁇ g product non-reduced, blank, 10 ⁇ g product non-reduced, Novex Mark12 wide range protein standards, 0.5 ⁇ g product reduced, blank, 2.0 ⁇ g product reduced, blank, and 10 ⁇ g product reduced.
- Figure 39C shows the Size exclusion chromatography on 50 ⁇ g of the final Fc-LIO- ShK(1-35) product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH 2 PO 4 , 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
- Figure 40A shows a Spectral scan of 20 ⁇ l of the Fc-LI 0-ShK(2-35) product diluted in 700 ⁇ l PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
- Figure 40B shows a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final Fc-L10-ShK(2-35) product.
- Lanes 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 ⁇ g product non-reduced, blank, 2.0 ⁇ g product non-reduced, blank, 10 ⁇ g product non-reduced, Novex Mark12 wide range protein standards, 0.5 ⁇ g product reduced, blank, 2.0 ⁇ g product reduced, blank, and 10 ⁇ g product reduced.
- Figure 40C shows the size exclusion chromatography on 50 ⁇ g of the final Fc-LI 0-ShK(2-
- Figure 4OD shows a MALDI mass spectral analysis of the final sample of Fc-LI 0-ShK(2- 35) analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of
- Figure 41 A shows spectral scan of 50 ⁇ l of the Fc-LI 0-OSK1 product diluted in 700 ⁇ l Formulation Buffer using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
- Figure 41 B shows Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final Fc-L10 ⁇ OSK1 product.
- Lanes 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 ⁇ g product non-reduced, blank, 2.0 ⁇ g product non-reduced, blank, 10 ⁇ g product non-reduced, Novex Mark12 wide range protein standards, 0.5 ⁇ g product reduced, blank, 2.0 ⁇ g product reduced, blank, and 10 ⁇ g product reduced.
- Figure 41 C shows size exclusion chromatography on 123 ⁇ g of the final Fc-LI 0-OSK1 product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaHaPQt, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
- Figure 41 D shows liquid chromatography - mass spectral analysis of approximately 4 ⁇ g of the final Fc-LI 0-OSK1 sample using a Vydac C4 column with part of the effluent directed into a LCQ ion trap mass spectrometer. The mass spectrum was deconvoluted using the Bioworks software provided by the mass spectrometer manufacturer.
- Figure 42A-B shows nucleotide and amino acid sequences (SEQ ID NO: 1040 and SEQ
- Figure 43A-B shows nucleotide and amino acid sequences (SEQ ID NO: 1042 and SEQ ID NO: 1043, respectively) of Fc-LI 0-OSK1[K7S].
- Figure 44A-B shows nucleotide and amino acid sequences (SEQ ID NO: 1044 and SEQ ID NO: 1045, respectively) of Fc-LI 0-OSK1 [E16K.K20D].
- Figure 45A-B shows nucleotide and amino acid sequences (SEQ ID NO: 1046 and SEQ ID NO: 1047, respectively) of Fc-L10-OSK1[K7S,E16K,K20D].
- Figure 46 shows a Western blot (from tris-glycine 4-20% SDS-PAGE) with anti-human Fc antibodies.
- Lanes 1 - 6 were loaded as follows: 15 ⁇ l of Fc-LI 0-OSK1 [K7S.E16K,K20D];15 ⁇ l of FC-L10-OSK1 [E16K.K20D]; 15 ⁇ l of Fc-LI 0-OSK1 [K7S];15 ⁇ l of Fc-LI 0-OSK1 ;15 ⁇ l of "No DNA" control; molecular weight markers
- Figure 47 shows a Western blot (from tris-glycine 4-20% SDS-PAGE) with anti-human Fc antibodies. Lanes 1-5 were loaded as follows: 2 ⁇ l of Fc-LI 0-OSK1; 5 ⁇ l of Fc-LI 0-OSK1;10 ⁇ l of Fc-LI 0-OSK1; 20ng Human IgG standard; molecular weight markers.
- Figure 48 shows a Western blot (from tris-glycine 4-20% SDS-PAGE) with anti-human Fc antibodies.
- Lanes 1-13 were loaded as follows: 20 ng Human IgG standard; D1; C3; C2; B6; B5; B2; B1 ; A6; A5; A4; A3; A2 (5 ⁇ l of clone-conditioned medium loaded in lanes 2-13).
- Figure 49A shows a spectral scan of 50 ⁇ l of the Fc-LI 0-OsK1 product diluted in 700 ⁇ l PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
- Figure 49B shows Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final Fc-LI 0-OsK1 product.
- Lane 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 ⁇ g product non-reduced, blank, 2.0 ⁇ g product non-reduced, blank, 10 ⁇ g product non-reduced, Novex Mark12 wide range protein standards, 0.5 ⁇ g product reduced, blank, 2.0 ⁇ g product reduced, blank, and 10 ⁇ g product reduced.
- Figure 49C shows Size exclusion chromatography on 149 ⁇ g of the final Fc-LI 0-OsK1 product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH 2 PO 4 , 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
- Figure 49D shows MALDI mass spectral analysis of the final sample of Fc-LI 0-OsK1 analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse), The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ⁇ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses.
- Figure 5OA shows a spectral scan of 50 ⁇ l of the Fc-LI 0-OsK1 (K7S) product diluted in 700 ⁇ l PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
- Figure 5OB shows Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final Fc-LI 0-OsK1(K7S) product.
- Lane 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 ⁇ g product non-reduced, blank, 2.0 ⁇ g product non-reduced, blank, 10 ⁇ g product non-reduced, Novex Mark12 wide range protein standards, 0.5 ⁇ g product reduced, blank, 2.0 ⁇ g product reduced, blank, and 10 ⁇ g product reduced.
- Figure 5OC shows size exclusion chromatography on 50 ⁇ g of the final Fc-LIO- OsK1 (K7S) product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
- Figure 5OD shows MALDI mass spectral analysis of a sample of the final product Fc-LI 0- OsK1 (K7S) analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ⁇ 200 laser shots.
- Figure 51 A shows a spectral scan of 50 ⁇ l of the Fc-LI 0-OsK1(E16K, K20D) product diluted in 700 ⁇ l PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
- Figure 51 B shows Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final Fc-LI 0-OsK1(E16K, K20D) product.
- Lane 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 ⁇ g product non-reduced, blank, 2.0 ⁇ g product non-reduced, blank, 10 ⁇ g product non-reduced, Novex Mark12 wide range protein standards, 0.5 ⁇ g product reduced, blank, 2.0 ⁇ g product reduced, blank, and 10 ⁇ g product reduced.
- Figure 51 C shows size exclusion chromatography on 50 ⁇ g of the final Fc-LIO- OsK1(E16K, K20D) product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH 2 PQt, 250 mM NaCl, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
- Figure 51 D shows MALDl mass spectral analysis of a sample of the final product Fc-LI 0- OsK1(E16K, K20D) analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ⁇ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses.
- Figure 52A shows a spectral scan of 50 ⁇ l of the Fc-LI 0-OsK1(K7S, E16K, K20D) product diluted in 700 ⁇ l PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
- Figure 52B shows Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final Fc-LI 0-OsK1(K7S, E16K, K20D) product.
- Lanes 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 ⁇ g product non-reduced, blank, 2.0 ⁇ g product non-reduced, blank, 10 ⁇ g product non-reduced, Novex Mark12 wide range protein standards, 0.5 ⁇ g product reduced, blank, 2.0 ⁇ g product reduced, blank, and 10 ⁇ g product reduced.
- Figure 52C shows size exclusion chromatography on 50 ⁇ g of the final Fc-LI 0-
- OsK1(K7S, E16K, K20D) product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH 2 PO 4 , 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
- Figure 52D shows MALDI mass spectral analysis of a sample of the final product Fc-LIO- OsK1 (K7S, E16K, K20D) analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ⁇ 200 laser shots.
- Figure 53 shows inhibition of the outward potassium current recorded from HEK293 cell stably expressing human Kv1.3 channel by synthetic Osk1, a 38-residue toxin peptide of the Asian scorpion Orthochirus scrobiculosus venom.
- Figure 53A shows a concentration dependent block of the outward potassium current recorded from HEK293 cell stably expressing human Kv1.3 channel by the synthetic 0sk1 toxin peptide.
- Figure 53B shows the time course of the synthetic Osk1 toxin peptide block at various concentrations.
- Figure 54 shows that modification of the synthetic 0SK1 toxin peptide by fusion to the Fc- fragment of an antibody (0SK1 -peptibody) retained the inhibitory activity against the human Kv1.3 channel
- Figure 54A shows a concentration dependent block of the outward potassium current recorded from HEK293 cells stably expressing human Kv1.3 channel by 0SK1 linked to a human IgGI Fc-fragment with a linker chain length of 10 amino acid residues (Fc-L10-OSK1).
- the fusion construct was stably expressed in Chinese Hamster Ovarian (CHO) cells.
- Figure 54B shows the time course of the Fc-L10-OSK1 block at various concentrations.
- Figure 55 shows that a single amino-acid residue substitution of the OSK1 -peptibody retained the inhibitory activity against the human Kv1.3 channel.
- Figure 55A shows a concentration dependent block of the outward potassium current recorded from HEK293 cell stably expressing human Kv1.3 channel by 0SK1 -peptibody with a single amino acid substitution (lysine to serine at the 7 th position from N-terminal, [K7S]) and linked to a human IgGI Fc-fragment with a linker chain length of 10 amino acid residues (Fc-LI 0-OSK1[K7S]).
- the fusion construct was stably expressed in Chinese Hamster Ovarian (CHO) cells.
- Figure 55B shows the time course of potassium current block by Fc-LI 0-OSK1 [K7S] at various concentrations.
- Figure 56 shows that a two amino-acid residue substitution of the 0SK1 -peptibody retained the inhibitory activity against the human Kv1.3 channel.
- Figure 56A shows a concentration dependent block of the outward potassium current recorded from HEK293 cell stably expressing human Kv1.3 channel by 0SK1 -peptibody with two amino acid substitutions (glutamic acid to lysine and lysine to aspartic acid at the 16 th and 20 th position from N-terminal respectively, [E16KK20D]) and linked to a human IgGI Fc-fragment with a linker chain length of 10 amino acid residues (Fc-LI 0-OSK1[E16KK20D]).
- the fusion construct was stably expressed in Chinese Hamster Ovarian (CHO) cells.
- Figure 56B shows the time course of potassium current block by
- Figure 57 shows that a triple amino-acid residue substitution of the OSK1-peptibody retained the inhibitory activity against the human Kv1.3 channel, but the potency of inhibition was significantly reduced when compared to the synthetic 0SK1 toxin peptide.
- Figure 57A shows a concentration dependent block of the outward potassium current recorded from HEK293 cell stably expressing human Kv1.3 channel by 0SK1 -peptibody with triple amino acid substitutions (lysine to serine, glutamic acid to lysine and lysine to aspartic acid at the 7 th , 16 th and 20 th position from N- terminal respectively, [K7SE16KK20D]) and linked to a human IgGI Fc-fragment with a linker chain length of 10 amino acid residues (Fc-LI 0-OSK1[K7SE16KK20D]).
- the fusion construct was stably expressed in Chinese Hamster Ovarian (CHO) cells.
- Figure 57B shows the time course of potassium current block by Fc-LI 0-OSK1 [K7SE16KK20D] at various concentrations.
- Figure 58 shows Standard curves for ShK ( Figure 58A) and 2OK PEG-ShK[I -35] ( Figure 58B) containing linear regression equations for each Standard at a given percentage of serum.
- Figure 59 shows the pharmacokinetic profile in rats of 2OK PEG ShK[I -35] molecule after
- Figure 60 shows Kv1.3 inhibitory activity in serum samples (5%) of rats receiving a single equal molar IV injection of Kv1.3 inhibitors ShK versus 2OK PEG-ShK[I -35].
- Figure 62 shows that treatment with PEG-ShK ameliorated disease in rats in the adoptive transfer EAE model.
- Figure 63 shows that treatment with PEG-ShK prevented loss of body weight in the adoptive transfer EAE model. Rats were weighed on days -1, 4, 6, and 8 (for surviving rats). Mean ⁇ sem values are shown.
- Figure 64 shows that thapsigargin-induced IL-2 production in human whole blood was suppressed by the Kv1.3 channel inhibitors ShK[1-35] and Fc-LI 0-ShK[2-35]. The calcineurin inhibitor cyclosporine A also blocked the response. The BKCa channel inhibitor iberiotoxin (IbTx) showed no significant activity. The response of whole blood from two separate donors is shown in
- Figure 65 shows that thapsigargin-induced IFN-g production in human whole blood was suppressed by the Kv1.3 channel inhibitors ShK[1-35] and Fc-LI 0-ShK[2-35].
- the calcineurin inhibitor cyclosporine A also blocked the response.
- the BKCa channel inhibitor iberiotoxin (IbTx) showed no significant activity.
- the response of whole blood from two separate donors is shown in
- Figure 66 shows that thapsigargin-induced upregulation of CD40L on T cells in human whole blood was suppressed by the Kv1.3 channel inhibitors ShK[I -35] and Fc-LI 0-ShK[1 -35] (Fc- ShK).
- the calcineurin inhibitor cyclosporine A (CsA) also blocked the response.
- Figure 66A shows results of an experiment looking at the response of total CD4+ T cells.
- Figure 66B shows results of an experiment that looked at total CD4+ T cells, as well as CD4+CD45+ and
- CD4+CD45- T cells CD4+CD45- T cells.
- the BKCa channel inhibitor iberiotoxin (IbTx) and the KvH channel inhibitor dendrotoxin-K (DTX-K) showed no significant activity.
- Figure 67 shows that thapsigargin-induced upregulation of the IL-2R on T cells in human whole blood was suppressed by the Kv1.3 channel inhibitors ShK[1-35] and Fc-LI 0-ShK[1-35] (Fc-
- FIG. 67A shows results of an experiment looking at the response of total CD4+ T cells.
- Figure 67B shows results of an experiment that looked at total CD4+ T cells, as well as CD4+CD45+ and CD4+CD45- T cells.
- the BKCa channel inhibitor iberiotoxin (IbTx) and the Kv1.1 channel inhibitor dendrotoxin-K (DTX-K) showed no significant activity.
- Figure 68 shows cation exchange chromatograms of PEG-peptide purification on SP
- Figure 69 shows RP-HPLC chromatograms on final PEG-peptide pools to demonstrate purity of PEG-Shk purity >99% ( Figure 69A) and PEG-0sk1 purity >97% ( Figure 69B).
- Figure 70 shows the amino acid sequence (SEQ ID NO: 976) of an exemplary FcLoop-L2- OsK1-L2 having three linked domains: Fc N-terminal domain (amino acid residues 1-139); OsK1 (underlined amino acid residues 142-179); and Fc C-terminal domain (amino acid residues 182-270).
- Figure 71 shows the amino acid sequence (SEQ ID NO: 977) of an exemplary FcLoop-L2- ShK-L2 having three linked domains: Fc N-terminal domain (amino acid residues 1-139); ShK (underlined amino acid residues 142-176); and Fc C-terminal domain (amino acid residues 179- 267).
- Figure 72 shows the amino acid sequence (SEQ ID NO: 978) of an exemplary Fcl_oop-L2- ShK-L4 having three linked domains: Fc N-terminal domain (amino acid residues 1-139); ShK 5 (underlined amino acid residues 142-176); and Fc C-terminal domain (amino acid residues 181- 269).
- Figure 73 shows the amino acid sequence (SEQ ID NO: 979) of an exemplary FcLoop-L4- OsK1-L2 having three linked domains: Fc N-terminal domain (amino acid residues 1-139); OsK1 (underlined amino acid residues 144-181); and Fc C-terminal domain (amino acid residues 0 184-272).
- Figure 74 shows that the 2OK PEGylated ShK[I -35] provided potent blockade of human Kv1.3 as determined by whole cell patch clamp electrophysiology on HEK293/KV1.3 cells.
- the data represents blockade of peak current.
- Figure 75 shows schematic structures of some other exemplary embodiments of the 5 composition of matter of the invention.
- "X 2 " and "X 3 " represent toxin peptides or linker-toxin peptide combinations (i.e., -(L)rP-(L)g-) as defined herein.
- an additional X 1 domain and one or more additional PEG moieties are also encompassed in other embodiments.
- the specific embodiments shown here are as follows:
- Figure 75C 1 Figure 75D, Figure 75G and Figure 75H show a single chain molecule and O can also represent the DNA construct for the molecule.
- Figure 75A, Figure 75B 1 Figure 75E and Figure 75F show doubly disulfide-bonded Fc dimers (in position F 2 );
- Figure 75A and Figure 75B show a dimer having the toxin peptide portion on both chains in position X 3 ;
- Figure 75E and Figure 75F show a dimer having the toxin peptide portion on both chains In position X 2 .
- Figure 76A shows a spectral scan of 50 ⁇ l of the ShK[2-35]-Fc product diluted in 700 ⁇ l
- Figure 76B shows Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final ShK[2-35]-Fc product.
- Lanes 1 - 12 were loaded as follows: Novex Mark12 wide range 0 protein standards, 0.5 ⁇ g product non-reduced, blank, 2.0 ⁇ g product non-reduced, blank, 10 ⁇ g product non-reduced, Novex Mark12 wide range protein standards, 0.5 ⁇ g product reduced, blank, 2.0 ⁇ g product reduced, blank, and 10 ⁇ g product reduced.
- Figure 76C shows size exclusion chromatography on 70 ⁇ g of the final ShK[2-35]-Fc product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PU4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
- Figure 76D shows LC-MS analysis of the final ShK[2-35]-Fc sample using an Agilent 1100 HPCL running reverse phase chromatography, with the column effluent directly coupled to an electrospray source of a Thermo Finnigan LCQ ion trap mass spectrometer. Relevant spectra were summed and deconvoluted to mass data with the Bioworks software package.
- Figure 77A shows a spectral scan of 20 ⁇ l of the met-ShK[1-35]-Fc product diluted in 700 ⁇ l PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
- Figure 77B shows Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final met-ShK[1-35]-Fc product.
- Lanes 1 - 12 were loaded as follows: Novex Mark12 wide range protein standards, 0.5 ⁇ g product non-reduced, blank, 2.0 ⁇ g product non-reduced, blank, 10 ⁇ g product non-reduced, Novex Mark12 wide range protein standards, 0.5 ⁇ g product reduced, blank, 2.0 ⁇ g product reduced, blank, and 10 ⁇ g product reduced.
- Figure 77C shows size exclusion chromatography on 93 ⁇ g of the final met-ShK[1-35]-Fc product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH 2 PU 4 , 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
- Figure 77D shows MALDI mass spectral analysis of the final met-ShK[1-35]-Fc sample analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ⁇ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses.
- half-life extending moiety refers to a pharmaceutically acceptable moiety, domain, or "vehicle” covalently linked or conjugated to the toxin peptide, that prevents or mitigates in vivo proteolytic degradation or other activity-diminishing chemical modification of the toxin peptide, increases half-life or other pharmacokinetic properties such as but not limited to increasing the rate of absorption, reduces toxicity, improves solubility, increases biological activity and/or target selectivity of the toxin peptide with respect to a target ion channel of interest, increases manufacturability, and/or reduces immunogenicity of the toxin peptide, compared to an unconjugated form of the toxin peptide.
- PEGylated peptide is meant a peptide or protein having a polyethylene glycol (PEG) moiety covalently bound to an amino acid residue of the peptide itself or to a peptidyl or non- peptidyl linker (including but not limited to aromatic linkers) that is covalently bound to a residue of the peptide.
- PEG polyethylene glycol
- polyethylene glycol or “PEG” is meant a polyalkylene glycol compound or a derivative thereof, with or without coupling agents or derivatization with coupling or activating moieties (e.g., with aldehyde, hydroxysuccinimidyl, hydrazide, thiol, triflate, tresylate, azirdine, oxirane, orthopyridyl disulphide, vinylsulfone, iodoacetamide or a maleimide moiety).
- useful PEG includes substantially linear, straight chain PEG, branched PEG, or dendritic PEG. (See, e.g., Merrill, US Patent No. 5,171,264; Harris et al., Multiarmed, monofunctional, polymer for coupling to molecules and surfaces, US Patent No. 5,932,462; Shen, N-maleimidyl polymer derivatives, US Patent No. 6,602,498).
- Peptibody refers to molecules of Formula I in which F 1 and/or F 2 is an immunoglobulin Fc domain or a portion thereof, such as a CH2 domain of an Fc 1 or in which the toxin peptide is inserted into a human IgGI Fc domain loop, such that F 1 and F 2 are each a portion of an Fc domain with a toxin peptide inserted between them (See, e.g., Figures 70-73 and Example 49 herein).
- Peptibodies of the present invention can also be PEGylated as described further herein, at either an Fc domain or portion thereof, or at the toxin peptide(s) portion of the inventive composition, or both.
- native Fc refers to molecule or sequence comprising the sequence of a non- antigen-binding fragment resulting from digestion of whole antibody, whether in monomeric or multimeric form.
- the original immunoglobulin source of the native Fc is preferably of human origin and can be any of the immunoglobulins, although IgGI or lgG2 are preferred.
- Native Fc's are made up of monomeric polypeptides that can be linked into dimeric or multimeric forms by covalent (i.e., disulfide bonds) and non-covalent association.
- the number of intermolecular disulfide bonds between monomeric subunits of native Fc molecules ranges from 1 to 4 depending on class (e.g., IgG, IgA, IgE) or subclass (e.g., IgGI, lgG2, IgGS, IgAI, lgGA2).
- class e.g., IgG, IgA, IgE
- subclass e.g., IgGI, lgG2, IgGS, IgAI, lgGA2
- native Fc is a disulfide-bonded dimer resulting from papain digestion of an IgG (see Ellison et al. (1982), Nucleic Acids Res. 10: 4071-9).
- native Fc as used herein is generic to the monomeric, dimeric, and multimeric forms.
- Fc variant refers to a molecule or sequence that is modified from a native Fc but still comprises a binding site for the salvage receptor, FcRn.
- FcRn a binding site for the salvage receptor
- WO 97/34631 published 25 September 1997; WO 96/32478, corresponding to US Pat. No. 6,096,891, issued August 1, 2000, hereby incorporated by reference in its entirety; and WO 04/110472.
- Fc variant includes a molecule or sequence that is humanized from a non-human native Fc.
- a native Fc comprises sites that can be removed because they provide structural features or biological activity that are not required for the fusion molecules of the present invention.
- Fc variant includes a molecule or sequence that lacks one or more native Fc sites or residues that affect or are involved in (1) disulfide bond formation, (2) incompatibility with a selected host cell (3) N-terminal heterogeneity upon expression in a selected host cell, (4) glycosylate , (5) interaction with complement, (6) binding to an Fc receptor other than a salvage receptor, or (7) antibody-dependent cellular cytotoxicity (ADCC).
- ADCC antibody-dependent cellular cytotoxicity
- Fc domain encompasses native Fc and Fc variant molecules and sequences as defined above. As with Fc variants and native Fc's, the term “Fc domain” includes molecules in monomeric or multimeric form, whether digested from whole antibody or produced by other means.
- multimer as applied to Fc domains or molecules comprising Fc domains refers to molecules having two or more polypeptide chains associated covalently, noncovalently, or by both covalent and non-covalent interactions.
- IgG molecules typically form dimers; IgM 1 pentamers; IgD, dimers; and IgA, monomers, dimers, trimers, or tetramers.
- One skilled in the art can form multimers by exploiting the sequence and resulting activity of the native Ig source of the Fc or by derivatizing (as defined below) such a native Fc,
- dimer as applied to Fc domains or molecules comprising Fc domains refers to molecules having two polypeptide chains associated covalently or non-covalently.
- exemplary 5 dimers within the scope of this invention are as shown in Figure 2.
- the terms "derivatizing” and “derivative” or “derivatized” comprise processes and resulting compounds respectively in which (1) the compound has a cyclic portion; for example, cross-linking between cysteinyl residues within the compound; (2) the compound is cross-linked or has a cross- linking site; for example, the compound has a cysteinyl residue and thus forms cross-linked dimers 0 in culture or in vivo; (3) one or more peptidyl linkage is replaced by a non-peptidyl linkage; (4) the N-terminus is replaced by -NRR i 1 NRC(O)R i , -NRC(O)OR i , -NRS(O) 2 R 1 , -NHC(O)NHR, a succinimide group, or substituted or unsubstituted benzyloxycarbonyl-NH-, wherein R and R 1 and the ring substituents are as defined hereinafter; (5) the C-terminus is replaced by -C(
- peptide refers to molecules of 2 to about 80 amino acid residues, with molecules of about 10 to about 60 amino acid residues preferred and those of about 30 to about 50 amino acid residuess most preferred.
- Exemplary peptides can be randomly generated by any O known method, carried in a peptide library (e.g., a phage display library), or derived by digestion of proteins.
- a peptide library e.g., a phage display library
- additional amino acids can be included on either or both of the N- or C- termini of the given sequence. Of course, these additional amino acid residues should not significantly interfere with the functional activity of the composition.
- Toxin peptides include 5 peptides having the same amino acid sequence of a naturally occurring pharmacologically active peptide that can be isolated from a venom, and also include modified peptide analogs of such naturally occurring molecules. Examples of toxin peptides useful in practicing the present invention are listed in Tables 1-32.
- the toxin peptide (“P", or equivalent ⁇ shown as “P'” in Figure 2) comprises at least two intrapeptide disulfide bonds, as shown, for example, in Figure 9.
- this invention concerns molecules comprising: a) C 1 -C 3 and C 2 -C 4 disulfide bonding in which C 1 , C 2 , C 3 , and C 4 represent the order in which cysteine residues appear in the primary sequence of the toxin peptide stated conventionally with the N-terminus of the peptide on the left, with the first and third cysteines in the amino acid sequence forming a disulfide bond, and the second and fourth cysteines forming a disulfide bond.
- toxin peptides with such a C 1 - C 3 , C 2 -C 4 disulfide bonding pattern include, but are not limited to, apamin peptides, ⁇ - conopeptides, PnIA peptides, PnIB peptides, and Mil peptides, and analogs of any of 5 the foregoing.
- C 1 -C 6 , C 2 -C 4 and C 3 -C 5 disulfide bonding in which, as described above, C 1 , C 2 , C 3 , C 4 , C 5 and C 6 represent the order of cysteine residues appearing in the primary sequence of the toxin peptide stated conventionally with the N-terminus of the peptide(s) on the left, with the first and sixth cysteines in the amino acid sequence 0 forming a disulfide bond, the second and fourth cysteines forming a disulfide bond, and the third and fifth cysteines forming a disulfide bond.
- toxin peptides with such a C 1 -C 6 , C 2 -C 4 , C 3 -C 5 disulfide bonding pattern include, but are not limited to, ShK, BgK, HmK, AeKS, AsK, and DTX1, and analogs of any of the foregoing.
- C 1 -C 4 , C 2 -C 5 and C 3 -C 6 disulfide bonding in which, as described above, C 1 , C 2 , C 3 , C 4 , 5 C 5 and C 6 represent the order of cysteine residues appearing in the primary sequence of the toxin peptide stated conventionally with the N-terminus of the peptide(s) on the left, with the first and fourth cysteines in the amino acid sequence forming a disulfide bond, the second and fifth cysteines forming a disulfide bond, and the third and sixth cysteines forming a disulfide bond,
- Examples of toxin peptides with such a C 1 -C 4 , C 2 - O C 5 , C 3 -C s disulfide bonding pattern include, but are not limited to, ChTx, MgTx, 0SK1 ,
- C 1 , C 2 , C 3 , C 4 , C 5 , C 6 , C 7 5 and C 8 represent the order of cysteine residues appearing in the primary sequence of the toxin peptide stated conventionally with the N-terminus of the peptide(s) on the left, with the first and fifth cysteines in the amino acid sequence forming a disulfide bond, the second and sixth cysteines forming a disulfide bond, the third and seventh cysteines forming a disulfide bond, and the fourth and eighth cysteines forming a O disulfide bond.
- Examples of toxin peptides with such a C 1 -C 5 , C 2 -C 6 , C 3 -C 7 , C 4 -C 8 disulfide bonding pattern include, but are not limited to, Anuoroctoxin (AnTx), PM, HsTxI, MTX (P12A, P20A), and Pi4 peptides, and analogs of any of the foregoing.
- Anuoroctoxin AnTx
- PM HsTxI
- MTX P12A, P20A
- Pi4 peptides and analogs of any of the foregoing.
- C 1 C, C 7 and C 8 represent the order of cysteine residues appearing in the primary sequence of the toxin peptide stated conventionally with the N-terminus of the peptide(s) on the left, with the first and fourth cysteines in the amino acid sequence forming a disulfide 5 bond, the second and sixth cysteines forming a disulfide bond, the third and seventh cysteines forming a disulfide bond, and the fifth and eighth cysteines forming a disulfide bond.
- toxin peptides with such a C 1 -C 4 , C 2 -C 6 , C 3 -C 7 , C 5 -C 8 disulfide bonding pattern include, but are not limited to, Chlorotoxin, Bm-12b, and, and analogs of either.
- C 1 -C 5 , C 2 -C 6 , C 3 -C 4 , and C 7 -C 8 disulfide bonding in which C 1 , C 2 , C 3 , C 4 , C 5 , C 6 , C 7 and C 8 represent the order of cysteine residues appearing in the primary sequence of the toxin peptide stated conventionally with the N-terminus of the peptide(s) on the left, with the first and fifth cysteines in the amino acid sequence forming a disulfide bond, the second and sixth cysteines forming a disulfide bond, the third and fourth 5 cysteines forming a disulfide bond, and the seventh and eighth cysteines forming a disulfide bond.
- toxin peptides with such a C 1 -C 5 , C 2 -C 6 , C 3 -C 4 , C 7 -C 8 disulfide bonding pattern include, but are not limited to, Maurotoxin peptides and analogs thereof.
- randomized refers to fully random O sequences (e.g., selected by phage display methods) and sequences in which one or more residues of a naturally occurring molecule is replaced by an amino acid residue not appearing in that position in the naturally occurring molecule.
- Exemplary methods for identifying peptide sequences include phage display, E. coli display, ribosome display, yeast-based screening, RNA- peptide screening, chemical screening, rational design, protein structural analysis, and the like.
- pharmaceutically active means that a substance so described is determined to have activity that affects a medical parameter (e.g., blood pressure, blood cell count, cholesterol level) or disease state (e.g., cancer, autoimmune disorders).
- pharmacologically active peptides comprise agonistic or mimetic and antagonistic peptides as defined below.
- -mimetic peptide and “-agonist peptide” refer to a peptide having biological O activity comparable to a naturally occurring toxin peptide molecule, e.g., naturally occurring ShK toxin peptide. These terms further include peptides that indirectly mimic the activity of a naturally occurring toxin peptide molecule, such as by potentiating the effects of the naturally occurring molecule.
- -antagonist peptide or “inhibitor peptide” refers to a peptide that blocks or in some way interferes with the biological activity of a receptor of interest, or has biological activity comparable to a known antagonist or inhibitor of a receptor of interest (such as, but not limited to, an ion channel).
- acidic residue refers to amino acid residues in D- or L-form having sidechains comprising acidic groups. Exemplary acidic residues include D and E.
- amide residue refers to amino acids in D- or L-form having sidechains comprising amide derivatives of acidic groups.
- Exemplary residues include N and Q.
- aromatic residue refers to amino acid residues in D- or L-form having sidechains comprising aromatic groups.
- exemplary aromatic residues include F, Y, and W.
- basic residue refers to amino acid residues in D- or L-form having sidechains comprising basic groups.
- Exemplary basic residues include H, K, R 1 N-methyl-arginine, ⁇ - aminoarginine, ⁇ -methyl-arginine, 1-methyl-histidine, 3-methyl-histidine, and homoarginine (hR) residues.
- hydrophilic residue refers to amino acid residues in D- or L-form having sidechains comprising polar groups.
- hydrophilic residues include C, S, T, N, Q, D, E, K, and citrulline (Cit) residues.
- nonfunctional residue refers to amino acid residues in D- or L-form having sidechains that lack acidic, basic, or aromatic groups
- Exemplary nonfunctional amino acid residues include M, G, A, V, 1, L and norleucine (NIe).
- neutral polar residue refers to amino acid residues in D- or L-form having sidechains that lack basic, acidic, or polar groups.
- exemplary neutral polar amino acid residues include A, V, L, I, P 1 W, M, and F.
- polar hydrophobic residue refers to amino acid residues in D- or L-form having sidechains comprising polar groups.
- exemplary polar hydrophobic amino acid residues include T,
- hydrophobic residue refers to amino acid residues in D- or L-form having sidechains that lack basic or acidic groups.
- exemplary hydrophobic amino acid residues include A, V, L, I, P, W, M, F, T, G, S, Y, C, Q, and N.
- the amino acid sequence of the toxin peptide is modified in one or more ways relative to a native toxin peptide sequence of interest, such as, but not limited to, a native ShK or 0SK1 sequence, their peptide analogs, or any other toxin peptides having amino acid sequences as set for in any of Tables 1-32.
- the one or more useful modifications can include amino acid additions or insertions, amino acid deletions, peptide truncations, amino acid substitutions, and/or chemical derivatization of amino acid residues, accomplished by known chemical techniques. Such modifications can be, for example, for the purpose of enhanced potency, selectivity, and/or proteolytic stability, or the like.
- ShK analogs can be designed to remove protease cleavage sites (e.g., trypsin cleavage sites at K or R residues and/or chymotrypsin cleavage sites at F, Y, or W residues) in a ShK peptide- or ShK analog- containing composition of the invention, based partially on alignment mediated mutagenesis using HmK (see, e.g., Figure 6) and molecular modeling.
- protease cleavage sites e.g., trypsin cleavage sites at K or R residues and/or chymotrypsin cleavage sites at F, Y, or W residues
- protease is synonymous with “peptidase”.
- Proteases comprise two groups of enzymes: the endopeptidases which cleave peptide bonds at points within the protein, and the exopeptidases, which remove one or more amino acids from either N- or C-terminus respectively.
- proteinase is also used as a synonym for endopeptidase.
- the four mechanistic classes of proteinases are: serine proteinases, cysteine proteinases, aspartic proteinases, and metallo- proteinases. In addition to these four mechanistic classes, there is a section of the enzyme nomenclature which is allocated for proteases of unidentified catalytic mechanism. This indicates that the catalytic mechanism has not been identified.
- Cleavage subsite nomenclature is commonly adopted from a scheme created by Schechter and Berger (Schechter I. & Berger A., On the size of the active site in proteases. I. Papain, Biochemical and Biophysical Research Communication, 27:157 (1967); Schechter I. & Berger A., On the active site of proteases. 3. Mapping the active site of papain; specific inhibitor peptides of papain, Biochemical and Biophysical Research Communication, 32:898 (1968)). According to this model, amino acid residues in a substrate undergoing cleavage are designated P1.P2, P3, P4 etc. in the N-terminal direction from the cleaved bond.
- the residues in the C-terminal direction are designated PV, P2', P3', P4'. etc.
- the skilled artisan is aware of a variety of tools for identifying protease binding or protease cleavage sites of interest.
- the PeptideCutter software tool is available through the ExPASy (Expert Protein Analysis System) proteomics server of the Swiss Institute of Bioinformatics (SIB; www.expasy.org/tools/peptidecutter). PeptideCutter searches a protein sequence from the SWISS-PROT and/or TrEMBL databases or a user-entered protein sequence for protease cleavage sites. Single proteases and chemicals, a selection or the whole list of proteases and chemicals can be used.
- a third option for output is a map of cleavage sites.
- the sequence and the cleavage sites mapped onto it are grouped in blocks, the size of which can be chosen by the user.
- Other tools are also known for determining protease cleavage sites. (E.g., Turk, B. et al, Determination of protease cleavage site motifs using mixture-based oriented peptide libraries, Nature Biotechnology, 19:661-667 (2001); Barrett A. et al., Handbook of proteolytic enzymes, Academic Press (1998)).
- the serine proteinases include the chymotrypsin family, which includes mammalian protease enzymes such as chymotrypsin, trypsin or elastase or kallikrein.
- the serine proteinases exhibit different substrate specificities, which are related to amino acid substitutions in the various enzyme subsites interacting with the substrate residues. Some enzymes have an extended interaction site with the substrate whereas others have a specificity restricted to the P1 substrate residue.
- a proline residue in position PV normally exerts a strong negative influence on trypsin cleavage.
- Chymotrypsin preferentially cleaves at a W, Y or F in position P1 (high specificity) and to a lesser extent at L, M or H residue in position P1. (Keil, 1992). Exceptions to these rules are the following: When W is found in position P1, the cleavage is blocked when M or P are found in position PV at the same time. Furthermore, a proline residue in position PV nearly fully blocks the cleavage independent of the amino acids found in position P1. When an M residue is found in position P1 , the cleavage is blocked by the presence of a Y residue in position PV. Finally, when H is located in position P1 , the presence of a D, M or W residue also blocks the cleavage.
- Membrane metallo-endopeptidase (NEP; neutral endopeptidase, kidney-brush-border neutral proteinase, enkephalinase, EC 3.4.24.11) cleaves peptides at the amino side of hydrophobic amino acid residues.
- NEP neutral endopeptidase
- Thrombin preferentially cleaves at an R residue in position P1.
- the natural substrate of thrombin is fibrinogen.
- Optimum cleavage sites are when an R residue is in position P1 and GIy is in position P2 and position PV.
- hydrophobic amino acid residues are found in position P4 and position P3, a praline residue in position P2, an R residue in position P1, and non-acidic amino acid residues in position P1' and position P2'.
- a very important residue for its natural substrate fibrinogen is a D residue in P10.
- Caspases are a family of cysteine proteases bearing an active site with a conserved amino acid sequence and which cleave peptides specifically following D residues. (Earnshaw WC et al., Mammalian caspases: Structure, activation, substrates, and functions during apoptosis, Annual Review of Biochemistry, 68:383-424 (1999)).
- the Arg-C proteinase preferentially cleaves at an R residue in position P1.
- the cleavage behavior seems to be only moderately affected by residues in position PV. (Keil, 1992).
- the Asp- N endopeptidase cleaves specifically bonds with a D residue in position P1 1 . (Keil, 1992).
- a toxin peptide amino acid sequence modified from a naturally occurring toxin peptide amino acid sequence includes at least one amino acid residue inserted or substituted therein, relative to the amino acid sequence of the native toxin peptide sequence of interest, in which the inserted or substituted amino acid residue has a side chain comprising a nucleophilic orelectrophilic reactive functional group by which the peptide is conjugated to a linker or half-life extending moiety.
- nucleophilic orelectrophilic reactive functional group examples include, but are not limited to, a thiol, a primary amine, a seleno, a hydrazide, an aldehyde, a carboxylic acid, a ketone, an aminooxy, a masked (protected) aldehyde, or a masked (protected) keto functional group
- amino acid residues having a side chain comprising a nucleophilic reactive functional group include, but are not limited to, a lysine residue, an ⁇ , ⁇ -diaminopropionic acid residue, an ⁇ , ⁇ -diaminobutyric acid residue, an ornithine residue, a cysteine, a homocysteine, a glutamic acid residue, an aspartic acid residue, or a selenocysteine residue.
- amino acid substitution in an amino acid sequence is typically designated herein with a one-letter abbreviation for the amino acid residue in a particular position, followed by the numerical amino acid position relative to the native toxin peptide sequence of interest, which is then followed by the one-letter symbol for the amino acid residue substituted in.
- T30D symbolizes a substitution of a threonine residue by an aspartate residue at amino acid position 30, relative to a hypothetical native toxin peptide sequence.
- R18hR or “R18Cit” indicates a substitution of an arginine residue by a homoarginine or a citrulline residue, respectively, at amino acid position 18, relative to the hypothetical native toxin peptide
- An amino acid position within the amino acid sequence of any particular toxin peptide (or peptide analog) described herein may differ from its position relative to the native sequence, i.e., as determined in an alignment of the N-terminal or C-terminal end of the peptide's amino acid sequence with the N- terminal or C-terminal end, as appropriate, of the native toxin peptide sequence.
- amino acid position 1 of the sequence SCIDTIPKSRCTAFQCKHSMKYRLSFCRKTCGTC (ShK(2- 35); SEQ ID NO:92)
- amino acid position 2 of native ShK(1-35) corresponds to amino acid position 2 relative to the native sequence
- amino acid position 34 of SEQ ID NO:92 corresponds to amino acid position 35 relative to the native sequence (SEQ ID N0:5).
- amino acid substitutions encompass, non-canonical amino acid residues, which can include naturally rare (in peptides or proteins) amino acid residues or unnatural amino acid residues.
- Non-canonical amino acid residues can be incorporated into the peptide by chemical peptide synthesis rather than by synthesis in biological systems, such as recombinantly expressing cells, or alternatively the skilled artisan can employ 0 known techniques of protein engineering that use recombinantly expressing cells. (See, e.g., Link et al., Non-canonical amino acids in protein engineering, Current Opinion in Biotechnology, 14(6):603-609 (2003)).
- non-canonical amino acid residue refers to amino acid residues in D- or L-form that are not among the 20 canonical amino acids generally incorporated into naturally occurring proteins, for example, ⁇ -amino acids, homoamino acids, cyclic amino acids and 5 amino acids with derivatized side chains.
- Examples include (in the L-form or D-form): citrulline (Cit), homocitrulline (hCit), /V-methylcitrulIine (NMeCit), /V-methylhomocitrulline (NMeHoCit), ornithine (Om or O), ⁇ /-Methylornithine (NMeOrn), sarcosine (Sar), homolysine (hK or Hlys), homoarginine (hR or hArg), homoglutamine (hQ), ⁇ /-methylarginine (NMeR), ⁇ /-methylleucine (NMeL), ⁇ f-methylhomolysine (NMeHoK), N-methylglutamine (NMeQ), norleucine (NIe), norvaline O (Nva), 1 ,2,3,4-tetrahydroisoquinoline (Tic), nitrophenylalanine (nitrophe), aminophenylalanine
- aminobutyric acid (aminophe), benzylphenyalanine (benzylphe), ⁇ -carboxyglutamic acid ( ⁇ -carboxyglu), hydroxyproline (hydroxypro), p-carboxyl-phenylalanine (Cpa), ⁇ -aminoadipic acid (Aad), acetylarginine (acetylarg), ⁇ , ⁇ -diaminopropionoic acid (Dpr), ⁇ , ⁇ -diaminobutyric acid (Dab), diaminopropionic acid (Dap), ⁇ -(1 -Naphthyl)-alanine (1NaI), ⁇ -(2-Naphthyl)-alanine (2Na1), 5 cyclohexylalanine (Cha), 4-methyl-phenylalanine (MePhe), ⁇ , ⁇ -diphenyl-alanine (BiPhA), aminobutyric
- peptide portions of the inventive compositions can also be chemically derivatized at one or more amino acid residues.
- Peptides that contain derivatized amino acid residues can be synthesized by known organic chemistry techniques.
- “Chemical derivative” or 5 "chemically derivatized” in the context of a peptide refers to a subject peptide having one or more residues chemically derivatized by reaction of a functional side group.
- Such derivatized molecules include, for example, those molecules in which free amino groups have been derivatized to form amine hydrochlorides, p-toluene sulfonyl groups, carbobenzoxy groups, t-butyloxycarbonyl groups, chloroacetyl groups orformyl groups.
- Free carboxyl groups may be derivatized to form salts, 0 methyl and ethyl esters or other types of esters or hydrazides.
- Free hydroxyl groups may be derivatized to form O-acyl or O-alkyl derivatives.
- the imidazole nitrogen of histidine may be derivatized to form N-im-benzylhistidine.
- chemical derivatives are those peptides which contain one or more naturally occurring amino acid derivatives of the twenty canonical amino acids, whether in L- or D- form. For example, 4-hydroxyproline may be substituted for proline; 5- 5 hydroxylysine maybe substituted for lysine; 3-methylhistidine may be substituted for histidine; homoserine may be substituted for serine; and ornithine may be substituted for lysine.
- basic residues e.g., lysine
- residues nonfunctional residues preferred.
- Such molecules will be less basic than the molecules from which they are derived and otherwise retain O the activity of the molecules from which they are derived, which can result in advantages in stability and immunogenicity; the present invention should not, however, be limited by this theory.
- physiologically acceptable salts of the inventive compositions are also encompassed, including when the inventive compositons are referred to herein as "molecules" or “compounds.”
- physiologically acceptable salts is meant any salts that are known or later 5 discovered to be pharmaceutically acceptable. Some examples are: acetate; trifluoroacetate; hydrohalides, such as hydrochloride and hydrobromide; sulfate; citrate; maleate; tartrate; glycolate; gluconate; succinate; mesylate; besylate; and oxalate salts.
- moieties include the "Fc" domain of an antibody, as is used in Enbrel ® (etanercept) , as well as biologically suitable polymers (e.g., polyethylene glycol, or "PEG”), as is used in Neulasta ® (pegfilgrastim).
- PEG polyethylene glycol
- molecules of this invention peptides of about 80 amino acids or less with at least two intrapeptide disulfide bonds— possess therapeutic advantages when covalently attached to half-life extending moieties.
- Molecules of the present invention can further comprise an additional pharmacologically active, covalently bound peptide, which can be bound to the half-life extending moiety (F 1 and/or F 2 ) or to the peptide portion (P).
- Embodiments of the inventive compositions containing more than one half-life extending moiety (F 1 and F 2 ) include those in which F 1 and F 2 are the same or different half-life extending moieties. Examples (with or without a linker between each domain) include structures as illustrated in Figure 75 as well as the following embodiments (and others described herein and in the working Examples):
- 20KPEG - toxin peptide - HSA consistent with the formula [(F 1 ) r (X 2 )i-(F 2 )i]; 20KPEG - Fc domain- toxin peptide, consistent with the formula [(F 1 )i-(F 2 )i-(X 3 )i]; 20KPEG - Fc CH2 domain- toxin peptide, consistent with the formula [(F 1 )i-(F 2 )i-(X 3 )i]; and 20KPEG - HSA - toxin peptide, consistent with the formula [(F 1 )r(F 2 )i-(X 3 )i],
- Toxin peptides Any number of toxin peptides (i.e., "P”, or equivalents shown as “P 1 " in Figure 2) can be used in conjunction with the present invention. Of particular interest are the toxin peptides ShK, HmK, MgTx, AgTx2, OsK1 (also referred to as “OSK1”), Agatoxins, and HsTxI, as well as modified analogs of these, and other peptides that mimic the activity of such toxin peptides. As stated herein above, if more than one toxin peptide "P” is present in the inventive composition, "P” can be independently the same or different from any other toxin peptide(s) also present in the inventive composition.
- both of the toxin peptides, "P” can be the same peptide analog of ShK, different peptide analogs of ShK, or one can be a peptide analog of ShK and the other a peptide analog of OSK1.
- other peptides of interest are especially useful in molecules having additional features over the molecules of structural Formula I.
- the molecule of Formula I further comprises an additional pharmacologically active, covalently bound peptide, which is an agonistic peptide, an antagonistic peptide, or a targeting peptide; this peptide can be conjugated to F 1 or F 2 or P,
- agonistic peptides have activity agonistic to the toxin peptide but are not required to exert such activity by the same mechanism as the toxin peptide.
- Peptide antagonists are also useful in embodiments of the invention, with a preference for those with activity that can be complementary to the activity of the toxin peptide, Targeting peptides are also of interest, such as peptides that direct the molecule to particular cell types, organs, and the like.
- Phage display is useful in generating toxin peptides for use in the present invention, Affinity selection from libraries of random peptides can be used to identify peptide ligands for any site of any gene product.
- peptides as described in Table 2 can be prepared as described in U.S. Pat. No. 6,077,680 issued June 20, 2000 to Kem et al., which is hereby incorporated by reference in its entirety.
- Other peptides of Table 2 can be prepared by techniques known in the art.
- ShK(L5) SEQ ID NO: 950
- X s15 , X s21 , X s22 , X s23 and X s27 each independently refer to nonfunctional amino acid residues.
- X h6 , X h22 , X h26 are each independently nonfunctional residues.
- Peptides as described in Table 5 can be prepared as described in U. S, Pat. No. 6,689,749, issued February 10, 2004 to Lebrun et al., which is hereby incorporated by reference in its entirety.
- any of the sequences set forth in Table 7, can also be derivatized at either its N-terminal or C-terminal with a fatty acid having from 4 to 10 carbon atoms and from 0 to 2 carbon-carbon double bonds, or a derivative thereof such as an ⁇ -amino-fatty acid.
- a fatty acid having from 4 to 10 carbon atoms and from 0 to 2 carbon-carbon double bonds, or a derivative thereof such as an ⁇ -amino-fatty acid.
- fatty acids include valeric acid or (for the C-terminal) ⁇ -amino-valeric acid
- X m19 and X m34 are each independently nonfunctional residues.
- toxin peptide (P) portions of the molecule comprises a Kv1.3 antagonist peptide.
- Kv1.3 antagonist peptide sequences include those having any amino acid sequence set forth in Table 1 , Table 2, Table 3, Table 4, Table 5, Table 6, Table 7, Table 8, Table 9, Table 10, and/or Table 11 herein above;
- inventive compositions include at least one toxin peptide (P) that is an IKCaI antagonist peptide.
- Useful IKCaI antagonist peptides include Maurotoxin (MTx), ChTx.peptides and peptide analogs of either of these, examples of which include those having any amino acid sequence set forth in Table 12, Table 13, and/or Table 14;
- Other embodiments of the inventive composition include at least one toxin peptide (P) that is a
- SKCa inhibitor peptide useful SKCa inhibitor peptides include, Apamin, ScyTx, BmP05, P01, P05, Tamapin, TsK, and peptide analogs of any of these, examples of which include those having any amino acid sequence set forth in Table 15;
- inventive composition include at least one toxin peptide (P) that is an apamin peptide, and peptide analogs of apamin, examples of which include those having any amino acid sequence set forth in Table 16;
- inventive composition include at least one toxin peptide (P) that is a Scyllotoxin family peptide, and peptide analogs of any of these, examples of which include those having any amino acid sequence set forth in Table 17;
- inventive composition include at least one toxin peptide (P) that is a
- BKCa inhibitor peptide examples of which include those having any amino acid sequence set forth in Table 18;
- inventive compositions include at least one toxin peptide (P) that is a Slotoxin family peptide, and peptide analogs of any of these, examples of which include those having any amino acid sequence set forth in Table 19;
- inventive compositions include at least one toxin peptide (P) that is a Martentoxin peptide, and peptide analogs thereof, examples of which include those having any amino acid sequence set forth in Table 20;
- inventive composition include at least one toxin peptide (P) that is a N-type Ca 2 * channel inhibitor peptide, examples of which include those having any amino acid sequence set forth in Table 21 ;
- inventive composition include at least one toxin peptide (P) that is a ⁇ MVIIA peptide, and peptide analogs thereof, examples of which include those having any amino acid sequence set forth in Table 22;
- inventive composition include at least one toxin peptide (P) that is a coGVIA peptide, and peptide analogs thereof, examples of which include those having any amino acid sequence set forth in Table 23;
- inventive composition include at least one toxin peptide (P) that is a Ptu 1 peptide, and peptide analogs thereof, examples of which include those having any amino acid sequence set forth in Table 24;
- inventive composition include at least one toxin peptide (P) that is a ProTxi peptide, and peptide analogs thereof, examples of which include those having any amino acid sequence set forth in Table 25;
- inventive composition include at least one toxin peptide (P) that is a BeKMI peptide, and peptide analogs thereof, examples of which include those having any amino acid sequence set forth in Table 26;
- inventive composition include at least one toxin peptide (P) that is a Na + channel inhibitor peptide, examples of which include those having any amino acid sequence set forth in Table 27;
- inventive composition include at least one toxin peptide (P) that is a
- Cl- channel inhibitor peptide examples of which include those having any amino acid sequence set forth in Table 28;
- inventive composition include at least one toxin peptide (P) that is a Kv2.1 inhibitor peptide, examples of which include those having any amino acid sequence set forth in Table 29;
- inventive compositions include at least one toxin peptide (P) that is a Kv4.2/Kv4.3 inhibitor peptide, examples of which include those having any amino acid sequence setforth in Table 30;
- inventive compositions include at least one toxin peptide (P) that is a nACHR inhibitor peptide, examples of which include those having any amino acid sequence set forth in Table 31; and
- inventions of the inventive composition include at least one toxin peptide (P) that is an Agatoxin peptide, a peptide analog thereof or other calcium channel inhibitor peptide, examples of which include those having any amino acid sequence set forth in Table 32.
- Half-life extending moieties This invention involves the presence of at least one half-life extending moiety (F 1 and/or F 2 in Formula I) attached to a peptide through the N-terminus, C-terminus or a sidechain of one of the intracalary amino acid residues. Multiple half-life extending moieties can also be used; e.g., Fc's at each terminus or an Fc at a terminus and a PEG group at the other terminus or at a sidechain.
- the Fc domain can be PEGylated (e.g., in accordance with the formulae F 1 -F 2 -(L) f -P; P-(L) g -F 1 -F 2 ; or P-(L) g -F i -F 2 -(L) f -P).
- the half-life extending moiety can be selected such that the inventive composition achieves a sufficient hydrodynamic size to prevent clearance by renal filtration in vivo.
- a half-life extending moiety can be selected that is a polymeric macromolecule, which is substantially straight chain, branched-chain, or dendritic in form.
- a half-life extending moiety can be selected such that, in vivo, the inventive composition of matter will bind to a serum protein to form a complex, such that the complex thus formed avoids substantial renal clearance.
- the half-life extending moiety can be, for example, a lipid; a cholesterol group (such as a steroid); a carbohydrate or oligosaccharide; or any natural or synthetic protein, polypeptide or peptide that binds to a salvage receptor.
- Exemplary half-life extending moieties that can be used, in accordance with the present invention, include an immunoglobulin Fc domain, or a portion thereof, or a biologically suitable polymer or copolymer, for example, a polyalkylene glycol compound, such as a polyethylene glycol or a polypropylene glycol,
- a polyalkylene glycol compound such as a polyethylene glycol or a polypropylene glycol
- Other appropriate polyalkylene glycol compounds include, but are not limited to, charged or neutral polymers of the following types: dextran, polylysine, colominic acids or other carbohydrate based polymers, polymers of amino acids, and biotin derivatives.
- half-life extending moiety examples include a copolymer of ethylene glycol, a copolymer of propylene glycol, a carboxymethylcellulose, a polyvinyl pyrrolidone, a poly-1 ,3-dioxolane, a poly-1 ,3,6-trioxane, an ethylene/maleic anhydride copolymer, a polyaminoacid (e.g., polylysine), a dextran n-vinyl pyrrolidone, a poly n-vinyl pyrrolidone, a propylene glycol homopolymer, a propylene oxide polymer, an ethylene oxide polymer, a polyoxyethylated polyol, a polyvinyl alcohol, a linear or branched glycosylated chain, a polyacetal, a long chain fatty acid, a long chain hydrophobic aliphatic group, an immunoglobin, ethylene glycol, a cop
- exemplary embodiments of the inventive compositions also include HSA fusion constructs such as but not limited to: HSA fusions with ShK, OSK1 , or modified analogs of those toxin peptides. Examples include HSA-L10-ShK(2-35); HSA-L10-OsK1(1-38); HSA-L10-ShK(2- 35); and HSA-L10-O
- peptide ligands or small (organic) molecule ligands that have binding affinity for a long half- life serum protein under physiological conditions of temperature, pH, and ionic strength.
- examples include an albumin-binding peptide or small molecule ligand, a transthyretin-binding peptide or small molecule ligand, a thyroxine-binding globulin-binding peptide or small molecule ligand, an antibody-binding peptide or small molecule ligand, or another peptide or small molecule that has an affinity for a long half-life serum protein.
- a “long half-life serum protein” is one of the hundreds of different proteins dissolved in mammalian blood plasma, including so-called “carrier proteins” (such as albumin, transferrin and haptoglobin), fibrinogen and other blood coagulation factors, complement components, immunoglobulins, enzyme inhibitors, precursors of substances such as angiotensin and bradykinin and many other types of proteins.
- carrier proteins such as albumin, transferrin and haptoglobin
- fibrinogen and other blood coagulation factors such as albumin, transferrin and haptoglobin
- complement components such as immunoglobulins, enzyme inhibitors, precursors of substances such as angiotensin and bradykinin and many other types of proteins.
- the invention encompasses the use of any single species of pharmaceutically acceptable half-life extending moiety, such as, but not limited to, those described herein, or the use of a combination of two or more different half-life extending moieties, such as PEG and immunoglobulin Fc domain or a CH2 domain of Fc, albumin (e.g., HSA), an albumin-binding protein, transthyretin or TBG.
- any single species of pharmaceutically acceptable half-life extending moiety such as, but not limited to, those described herein, or the use of a combination of two or more different half-life extending moieties, such as PEG and immunoglobulin Fc domain or a CH2 domain of Fc, albumin (e.g., HSA), an albumin-binding protein, transthyretin or TBG.
- an Fc domain or portion thereof, such as a CH2 domain of Fc is used as a half-life extending moiety.
- the Fc domain can be fused to the N- terminal (e.g., in accordance with the formula F 1 -(L)rP) or C-terminal (e.g., in accordance with the formula P-(L) 9 -F 1 ) of the toxin peptides or at both the N and C termini (e.g., in accordance with the formulae F 1 -(L)f-P-(L) g -F 2 or P-(L) g -F 1 -(L)f-P).
- a peptide linker sequence can be optionally included between the Fc domain and the toxin peptide, as described herein.
- Examples of the ⁇ 5 formula P-(LJrP include: Fc-LI 0-ShK(K22A)[2-35]; Fc-L10-ShK(R1K/K22A)[1-35]; Fc-LIO-
- Examples of the formula P-(L) 9 -F 1 include: ShK(1-35)-L10-Fc; OsK1(1-38)-L10-Fc; Met-ShK(1-35)-L10-Fc; ShK(2- 35)-L10-Fc; Gly-ShK(1-35)-L10-Fc; Osk1(1-38)-L10-Fc; and any other working examples described 0 herein.
- Fc variants are suitable half-life extending moieties within the scope of this invention.
- a native Fc can be extensively modified to form an Fc variant in accordance with this invention, provided binding to the salvage receptor is maintained; see, for example WO 97/34631, WO 96/32478, and WO 04/110472.
- One can remove these sites by, for example, substituting or deleting residues, inserting residues into the site, or truncating portions containing the site.
- the inserted or substituted residues can also be altered amino acids, such as peptidomimetics or D-amino acids.
- Fc variants can be desirable for a number of reasons, several of which are described below.
- Exemplary Fc O variants include molecules and sequences in which:
- cysteine-containing segment at the N-terminus can be truncated or cysteine residues can be deleted or substituted with other amino acids (e.g., 5 alanyl, seryl).
- cysteine residues can be deleted or substituted with other amino acids (e.g., 5 alanyl, seryl).
- one can truncate the N-terminal 20-amino acid segment of SEQ ID
- a native Fc is modified to make it more compatible with a selected host cell. For example, one 0 can remove the PA sequence near the N-terminus of a typical native Fc 1 which can be recognized by a digestive enzyme in E. coli such as proline iminopeptidase. One can also add an N-terminal methionine residue, especially when the molecule is expressed recombinantly in a bacterial cell such as E. coli.
- the Fc domain of SEQ ID NO: 2 ( Figure 4) is one such Fc variant.
- a portion of the N-terminus of a native Fc is removed to prevent N-terminal heterogeneity when expressed in a selected host cell. For this purpose, one can delete any of the first 20
- Residues that are typically glycosylated can confer cytolytic response. Such residues can be deleted or substituted with unglycosylated residues (e.g., alanine).
- a native Fc can have sites for interaction with certain white blood cells that are not required for the 5 fusion molecules of the present invention and so can be removed.
- ADCC site is removed.
- ADCC sites are known in the art; see, for example, Molec. Immunol. 29 (5): 633-9 (1992) with regard to ADCC sites in IgGl These sites, as well, are not required for the fusion molecules of the present invention and so can be removed.
- the native Fc When the native Fc is derived from a non-human antibody, the native Fc can be humanized. O Typically, to humanize a native Fc, one will substitute selected residues in the non-human native Fc with residues that are normally found in human native Fc. Techniques for antibody humanization are well known in the art.
- Preferred Fc variants include the following.
- the leucine at position 15 can be substituted with glutamate; the glutamate at position 99, with alanine; and the lysines at 5 positions 101 and 103, with alanines.
- phenyalanine residues can replace one or more tyrosine residues.
- An alternative half-life extending moiety would be a protein, polypeptide, peptide, antibody, antibody fragment, or small molecule (e.g., a peptidomimetic compound) capable of binding to a salvage receptor.
- a half-life extending moiety a 0 polypeptide as described in U.S. Pat. No. 5,739,277, issued April 14, 1998 to Presta etal.
- Peptides could also be selected by phage display for binding to the FcRn salvage receptor.
- salvage receptor-binding compounds are also included within the meaning of "half-life extending moiety" and are within the scope of this invention.
- Such half-life extending moieties should be selected for increased half-life (e.g., by avoiding sequences recognized by proteases) and decreased immunogenicity (e.g., by favoring non-immunogenic sequences, as discovered in antibody humanization).
- polymer half-life extending moieties can also be used for F 1 and F 2 .
- Various means for attaching chemical moieties useful as half-life extending moieties are currently available, see, e.g., Patent Cooperation Treaty ("PCT") International Publication No. WO 96/11953, entitled “N-Terminally Chemically Modified Protein Compositions and Methods," herein incorporated by reference in its entirety.
- PCT Patent Cooperation Treaty
- the polymer half-life extending moiety is polyethylene glycol (PEG), as F 1 and/or F 2 , but it should be understood that the inventive composition of matter, beyond positions F 1 and/or F 2 , can also include one or more PEGs conjugated at other sites in the molecule, such as at one or more sites on the toxin peptide. Accordingly, some embodiments of the inventive composition of matter further include one or more 5 PEG moieties conjugated to a non-PEG half-life extending moiety, which is F 1 and/or F 2 , or to the toxin peptide(s) ( P), or to any combination of any of these.
- PEG polyethylene glycol
- an Fc domain or portion thereof (as F1 and/or F2) in the inventive composition can be made mono-PEGylated, di- PEGylated, or otherwise multi-PEGylated, by the process of reductive alkylation.
- PEG polyethylene glycol
- PEGylation of proteins and peptides include increased solubility, resistance to proteolytic degradation, and reduced immunogenicity of the therapeutic polypeptide.
- the merits of protein PEGylation are evidenced by the commercialization of several PEGylated proteins including PEG-Adenosine deaminase (AdagenTM/Enzon Corp,), PEG-L-asparaginase (OncasparTM/Enzon Corp.), PEG-lnterferon ⁇ -2b O (PEG-lntronTM/Schering/Enzon), PEG-lnterferon ⁇ -2a (PEGASYSTM/Roche) and PEG-G-CSF
- the PEG groups are generally attached to the peptide portion of the composition of the invention via acylation or reductive alkylation through a reactive group on the PEG moiety (e.g., an aldehyde, amino, thiol, or ester group) to a reactive group on the inventive compound (e.g., an aldehyde, amino, or ester group).
- a reactive group on the PEG moiety e.g., an aldehyde, amino, thiol, or ester group
- a reactive group on the inventive compound e.g., an aldehyde, amino, or ester group
- a useful strategy for the PEGylation of synthetic peptides consists of combining, through forming a conjugate linkage in solution, a peptide and a PEG moiety, each bearing a special functionality that is mutually reactive toward the other.
- the peptides can be easily prepared with conventional solid phase synthesis (see, for example, Figures 5 and 6 and the accompanying text herein).
- the peptides are "preactivated” with an appropriate functional group at a specific site.
- the precursors are purified and fully characterized prior to reacting with the PEG moiety.
- Ligation of the peptide with PEG usually takes place in aqueous phase and can be easily monitored by reverse phase analytical HPLC.
- the PEGylated peptides can be easily purified by preparative HPLC and characterized by analytical HPLC, amino acid analysis and laser desorption mass spectrometry.
- PEG is a well-known, water soluble polymer that is commercially available or can be prepared by ring-opening polymerization of ethylene glycol according to methods well known in the art (Sandler and Karo, Polymer Synthesis, Academic Press, New York, Vol. 3, pages 138-161).
- PEG is used broadly to encompass any polyethylene glycol molecule, in mono-, bi-, or poly- functional form, without regard to size or to modification at an end of the PEG, and can be represented by the formula;
- n 20 to 2300 and X is H or a terminal modification, e.g., a C 1 - 4 alkyl.
- a PEG used in the invention terminates on one end with hydroxy or methoxy, i.e., X is H or CH3 ("methoxy PEG"). It is noted that the other end of the PEG, which is shown in formula (II) terminating in OH, covalently attaches to an activating moiety via an ether oxygen bond, an amine linkage, or amide linkage.
- PEG When used in a chemical structure, the term "PEG” includes the formula (II) above without the hydrogen of the hydroxyl group shown, leaving the oxygen available to react with a free carbon atom of a linker to form an ether bond. More specifically, in order to conjugate PEG to a peptide, the peptide must be reacted with PEG in an "activated” form.
- Activated PEG can be represented by the formula:
- PEG PEG-(A) (Xl)
- PEG PEG (defined supra) covalently attaches to a carbon atom of the activation moiety (A) to form an ether bond, an amine linkage, or amide linkage
- A contains a reactive group which can react with an amino, imino, or thiol group on an amino acid residue of a peptide or a linker moiety covalently attached to the peptide.
- Activated PEG such as PEG-aldehydes or PEG-aldehyde hydrates
- PEG-aldehydes or PEG-aldehyde hydrates can be chemically synthesized by known means or obtained from commercial sources, e.g., Shearwater Polymers, (Huntsville, Al) orEnzon, Inc. (Piscataway, N.J.).
- PEG- aldehyde compound e.g., a methoxy PEG-aldehyde
- PEG-propionaldehyde which is commercially available from Shearwater Polymers (Huntsville, Al).
- PEG-propionaldehyde is represented by the formula PEG-CH 2 CH 2 CHO.
- Other examples of useful activated PEG are PEG acetaldehyde hydrate and PEG bis aldehyde hydrate, which latter yields a bifunctionally activated structure.
- PEG-maleimide compound such as, but not limited to, a methoxy PEG-maleimide, such as maleimido monomethoxy PEG, are particularly useful for generating the PEG-conjugated peptides of the invention.
- a PEG-maleimide compound such as, but not limited to, a methoxy PEG-maleimide, such as maleimido monomethoxy PEG, are particularly useful for generating the PEG-conjugated peptides of the invention.
- a polyethylene glycol) vinyl sulfone is another useful activated PEG for generating the PEG-conjugated peptides of the present invention by conjugation at thiolated amino acid residues, e.g., at C residues.
- PEG poly(ethylene glycol) vinyl sulfone
- Bioconj. Chem., 7:363-368 (1996); see also Harris, Functionalization of polyethylene glycol for formation of active sulfone-terminated PEG derivatives for binding to proteins and biologically compatible materials, U.S. Patent Nos. 5,446,090; 5,739,208; 5,900,461; 6,610,281 and 6,894,025; and Harris, Water soluble active sulfones of poly(ethylene glycol), WO 95/13312 A1).
- PEG-N-hydroxysuccinimide ester compound for example, methoxy PEG-N-hydroxysuccinimidyl (NHS) ester.
- NHS methoxy PEG-N-hydroxysuccinimidyl
- Heterobifunctionally activated forms of PEG are also useful. (See, e.g., Thompson et al.,
- a toxin peptide or, a fusion protein comprising the toxin peptide is reacted by known chemical techniques with an activated PEG compound, such as but not limited to, a thiol- activated PEG compound, a diol-activated PEG compound, a PEG-hydrazide compound, a PEG- oxyamine compound, or a PEG-bromoacetyl compound.
- an activated PEG compound such as but not limited to, a thiol- activated PEG compound, a diol-activated PEG compound, a PEG-hydrazide compound, a PEG- oxyamine compound, or a PEG-bromoacetyl compound.
- N-terminal PEGylation Methods for N-terminal PEGylation are exemplified herein in Examples 31-34, 45 and 47- 48, but these are in no way limiting of the PEGylation methods that can be employed by one skilled in the art.
- any molecular mass for a PEG can be used as practically desired, e.g., from about 1 ,000 or 2,000 Daltons (Da) to about 100,000 Da (n is 20 to 2300),
- the combined or total molecular mass of PEG used in a PEG-conjugated peptide of the present invention is from about 3,000 Da or 5,000 Da, to about 50,000 Da or 60,000 Da (total n is from 70 to 1 ,400), more preferably from about 10,000 Da to about 40,000 Da (total n is about 230 to about 910).
- the most preferred combined mass for PEG is from about 20,000 Da to about 30,000 Da (total n is about 450 to about 680).
- the number of repeating units "n" in the PEG is approximated for the molecular mass described in Daltons. It is preferred that the combined molecular mass of PEG on an activated linker is suitable for pharmaceutical use. Thus, the combined molecular mass of the PEG molecule should not exceed about 100,000 Da.
- Polysaccharide polymers are another type of water-soluble polymer that can be used for protein modification.
- Dextrans are polysaccharide polymers comprised of individual subunits of glucose predominantly linked by ⁇ 1-6 linkages. The dextran itself is available in many molecular weight ranges, and is readily available in molecular weights from about 1 kD to about 70 kD.
- Dextran is a suitable water-soluble polymer for use in the present invention as a half-life extending moiety by itself or in combination with another half-life extending moiety (e.g., Fc). See, for example, WO 96/11953 and WO 96/05309. The use of dextran conjugated to therapeutic or diagnostic immunoglobulins has been reported; see, for example, European Patent Publication No. 0 315456, which is hereby incorporated by reference in its entirety. Dextran of about 1 kD to about
- dextran is used as a half-life extending moiety in accordance with the present invention.
- Linkers Any "linker” group or moiety (i.e., "-(L)r” or “-(L) 9 -” in Formulae I-IX) is optional. When present, its chemical structure is not critical, since it serves primarily as a spacer. As stated herein above, the linker moiety (-(L) f - and/or -(L) 9 -), if present, can be independently the same or different from any other linker, or linkers, that may be present in the inventive composition. For example, an "(L)f' can represent the same moiety as, or a different moiety from, any other "(L)f” or any "(L) 9 " in accordance with the invention.
- the linker is preferably made up of amino acids linked together by peptide bonds.
- the linker is made up of from 1 to about 30 amino acids linked by peptide bonds, wherein the amino acids are selected from the 20 naturally occurring amino acids. Some of these amino acids can be glycosylated, as is well understood by those in the art.
- a useful linker sequence constituting a sialylation site is X1X2NX4X5G (SEQ ID NO: 637), wherein X 1 , X 21 X 4 and X 5 are each independently any amino acid residue.
- the 1 to 20 amino acids are selected from glycine, alanine, proline, asparagine, glutami ⁇ e, and lysine.
- a linker is made up of a majority of amino acids that are sterically unhindered, such as glycine and alanine.
- preferred linkers include polyglycines (particularly (GIy) 4 , (Gly)s), poJy(Gly-Ala), and polyalanines.
- linkers are those identified herein as "L5" (GGGGS; SEQ ID NO: 638), “L10” (GGGGSGGGGS; SEQ ID NO:79), “L25” GGGGSGGGGSGGGGSGGGGSGGGGS; SEQ ID NO:84) and any linkers used in the working examples hereinafter.
- the linkers described herein, however, are exemplary; linkers within the scope of this invention can be much longer and can include other residues.
- acidic residues for example, glutamate or aspartate residues, are placed in the amino acid sequence of the linker moiety (L). Examples include the following peptide linker sequences:
- GGEGGG (SEQ ID NO: 639); GGEEEGGG (SEQ ID NO: 640);
- GEEEG (SEQ ID NO: 641); GEEE (SEQ ID NO: 642); GGDGGG (SEQ ID NO: 643); GGDDDGG (SEQ ID NO: 644); GDDDG (SEQ ID NO: 645);
- GGGGSDDSDEGSDGEDGGGGS SEQ ID NO: 647
- WEWEW SEQ ID NO: 648
- FEFEF SEQ ID NO: 649
- EEEWWW SEQ ID NO: 650
- the linker constitutes a phosphorylation site, e.g., X 1 X 2 YX 3 )C 1 G (SEQ ID NO: 654), wherein Xi, X 2 .X3 and X4 are each independently any amino acid residue; X1X2SX3X 4 G (SEQ ID NO: 655), wherein Xi, X 2 ,X3 and X 4 are each independently any amino acid residue; or X 1 X 2 TX3X 4 G (SEQ ID NO: 656), wherein Xi, X 2 ,X3 and X4 are each independently any amino acid residue.
- Non-peptide linkers are also possible.
- These alkyl linkers can further be substituted by any non- sterically hindering group such as lower alkyl (e.g., Ci-C ⁇ ) lower acyl, halogen (e.g., CI, Br), CN, NH2, phenyl, etc.
- An exemplary non-peptide linker is a PEG linker, (XII)
- n is such that the linker has a molecular weight of 100 to 5000 kD, preferably 100 to 500 kD.
- the peptide linkers can be altered to form derivatives in the same manner as described above. Derivatives. The inventors also contemplate derivatizing the peptide and/or half-life extending moiety portion of the compounds. Such derivatives can improve the solubility, absorption, biological half-life, and the like of the compounds. The moieties can alternatively eliminate or attenuate any undesirable side-effect of the compounds and the like. Exemplary derivatives include compounds in which: 1. The compound or some portion thereof is cyclic.
- the peptide portion can be modified to contain two or more Cys residues (e.g., in the linker), which could cyclize by disulfide bond formation. 2.
- the compound is cross-linked or is rendered capable of cross-linking between molecules.
- the peptide portion can be modified to contain one Cys residue and thereby be able to form an intermolecular disulfide bond with a like molecule.
- the compound can also be cross-linked through its C-terminus, as in the molecule shown below.
- Non-peptidyl linkages replace one or more peptidyl [-C(O)NR-] linkages.
- Exemplary non-peptidyl linkages are -CH2-carbamate [-CHh-OC(O)NR-], phosphonate , -CH 2 -sulfonamide [-CH 2 -S(O) 2 NR-], urea [-NHC(O)NH-], -CH 2 -secondary amine, and alkylated peptide [- C(O)NR 6 - wherein R s is lower alkyl].
- the N-terminus is chemically derivatized. Typically, the N-terminus can be acylated or modified to a substituted amine.
- Exemplary N-terminal derivative groups include -NRR 1 (other than -NH 2 ), -NRC(O)R 1 ,
- R and R 1 are each independently hydrogen or lower alkyl and wherein the phenyl ring can be substituted with 1 to 3 substituents selected from the group consisting of C1-C4 alkyl, C1-C4 alkoxy, chloro, and bromo.
- the free C-terminus is derivatized. Typically, the C-terminus is esterified or amidated.
- Exemplary C-terminal derivative groups include, for example, -C(O)R 2 wherein R 2 is lower alkoxy Or-NR 3 R 4 wherein R 3 and R 4 are independently hydrogen or CI-C B alkyl (preferably d- C 4 alkyl).
- a disulfide bond is replaced with another, preferably more stable, cross-linking moiety (e.g., an alkylene). See, e.g., Bhatnagar etjL (1996), J. Med. Chem. 39: 3814-9; Alberts et_al. (1993) Thirteenth Am. Pep. Syrnp., 357-9.
- another, preferably more stable, cross-linking moiety e.g., an alkylene
- One or more individual amino acid residues are modified.
- Various derivatizing agents are known to react specifically with selected sidechains or terminal residues, as described in detail below.
- Lysinyl residues and amino terminal residues can be reacted with succinic or other carboxylic acid anhydrides, which reverse the charge of the lysinyl residues.
- suitable reagents for derivatizing alpha-amino-containing residues include imidoesters such as methyl picolinimidate; pyridoxal phosphate; pyridoxal; chloroborohydride; trinitrobenzenesulfonic acid; O-methylisourea; 2,4 pentanedione; and transaminase-catalyzed reaction with glyoxylate.
- Arginyi residues can be modified by reaction with any one or combination of several conventional reagents, including phenylglyoxal, 2,3-butanedione, 1,2-cyclohexanedione, and ninhydrin. 5 Derivatization of arginyi residues requires that the reaction be performed in alkaline conditions because of the high pKa of the guanidine functional group. Furthermore, these reagents can react with the groups of lysine as well as the arginine epsilon-amino group.
- aspartyl and glutamyl residues can be converted 5 to asparaginyl and glutaminyl residues by reaction with ammonium ions.
- Glutaminyl and asparaginyl residues can be deamidated to the corresponding glutamyl and aspartyl residues. Alternatively, these residues are deamidated under mildly acidic conditions. Either form of these residues falls within the scope of this invention.
- Cysteinyl residues can be replaced by amino acid residues or other moieties either to eliminate O disulfide bonding or, conversely, to stabilize cross-linking. See, e.g., Bhatnagaretaj. (1996), J. Med. Chem. 39: 3814-9.
- Derivatization with bifunctional agents is useful for cross-linking the peptides or their functional derivatives to a water-insoluble support matrix or to other macromolecular half-life extending moieties.
- Commonly used cross-linking agents include, e.g., 1,1-bis(diazoacetyl)-2-phenylethane, glutaraldehyde, 5 N-hydroxysuccinimide esters, for example, esters with 4-azidosalicylic acid, homobifunctional imidoesters, including disuccinimidyl esters such as 3,3'-dithiobis(succinimidylpropionate), and bifunctional maleimides such as bis-N-maleimido-1,8-octane, Derivatizing agents such as methyl-3-[(p- azidophenyl)dithio]propioimidate yield photoactivatable intermediates that are capable of forming crosslinks in the presence of light.
- Carbohydrate (oligosaccharide) groups can conveniently be attached to sites that are known to be glycosylation sites in proteins.
- O-linked oligosaccharides are attached to serine (Ser) or threonine (Thr) residues while N-linked oligosaccharides are attached to asparagine (Asn) residues when they are part of the sequence Asn-X-Ser/Thr, where X can be any amino acid except proline.
- X is preferably one of the 19 naturally occurring amino acids other than proline.
- the structures of N-linked and O-Iinked oligosaccharides and the sugar residues found in each 5 type are different.
- sialic acid is usually the terminal residue of both N-linked and O-linked oligosaccharides and, by virtue of its negative charge, can confer acidic properties to the glycosylated compound.
- site(s) can be incorporated in the linker of the compounds of this invention and are preferably glycosylated by a cell during recombinant production of the 0 polypeptide compounds (e.g., in mammalian cells such as CHO, BHK 1 COS). However, such sites can further be glycosylated by synthetic or semi-synthetic procedures known in the art.
- Compounds of the present invention can be changed at the DNA level, as well.
- the DNA sequence of any portion of the compound can be changed to codons more compatible with the chosen host cell.
- optimized codons are known in the art. Codons can be substituted to eliminate restriction sites or to include silent restriction sites, O which can aid in processing of the DNA in the selected host cell.
- the half-life extending moiety, linker and peptide DNA sequences can be modified to include any of the foregoing sequence changes.
- a process for preparing conjugation derivatives is also contemplated.
- Tumor cells for example, exhibit epitopes not found on their normal counterparts. Such epitopes include, for 5 example, different post-translational modifications resulting from their rapid proliferation.
- one aspect of this invention is a process comprising: a) selecting at least one randomized peptide that specifically binds to a target epitope; and b) preparing a pharmacologic agent comprising (i) at least one half-life extending O moiety (Fc domain preferred), (ii) at least one amino acid sequence of the selected peptide or peptides, and (iii) an effector molecule.
- the target epitope is preferably a tumor-specific epitope or an epitope specific to a pathogenic organism.
- the effector molecule can be any of the above-noted conjugation partners and is preferably a radioisotope.
- the present invention also relates to nucleic acids, expression vectors and host cells useful in producing the polypeptides of the present invention.
- Host cells can be eukaryotic cells, with mammalian cells preferred and CHO cells most preferred.
- Host cells can also be prokaryotic cells, with E. coli cells most preferred.
- the compounds of this invention largely can be made in transformed host cells using 0 recombinant DNA techniques.
- a recombinant DNA molecule coding for the peptide is prepared. Methods of preparing such DNA molecules are well known in the art. For instance, sequences coding for the peptides could be excised from DNA using suitable restriction enzymes. Alternatively, the DNA molecule could be synthesized using chemical synthesis techniques, such as the phosphoramidate method. Also, a combination of these techniques could be used 5
- the invention also includes a vector capable of expressing the peptides in an appropriate host.
- the vector comprises the DNA molecule that codes for the peptides operatively linked to appropriate expression control sequences.
- Expression control sequences include promoters, activators, enhancers, operators, ribosomal binding sites, start O signals, stop signals, cap signals, polyadenylation signals, and other signals involved with the control of transcription or translation.
- the resulting vector having the DNA molecule thereon is used to transform an appropriate host. This transformation can be performed using methods well known in the art,
- Any of a large number of available and well-known host cells can be used in the practice 5 of this invention.
- the selection of a particular host is dependent upon a number of factors recognized by the art. These include, for example, compatibility with the chosen expression vector, toxicity of the peptides encoded by the DNA molecule, rate of transformation, ease of recovery of the peptides, expression characteristics, bio-safety and costs. A balance of these factors must be struck with the understanding that not all hosts can be equally effective for the O expression of a particular DNA sequence.
- useful microbial hosts include bacteria (such as E.
- coli sp. coli sp.
- yeast such as Saccharomvces sp.
- other fungi insects, plants, mammalian (including human) cells in culture, or other hosts known in the art.
- the transformed host is cultured and purified.
- Host cells can be cultured under conventional fermentation conditions so that the desired compounds are expressed. Such fermentation conditions are well known in the art.
- the peptides are purified from culture by methods well known in the art.
- the compounds can also be made by synthetic methods. Solid phase synthesis is the preferred technique of making individual peptides since it is the most cost-effective method of making small peptides.
- solid phase synthesis techniques include the use of protecting groups, linkers, and solid phase supports, as well as specific protection and deprotection reaction conditions, linker cleavage conditions, use of scavengers, and other aspects of solid phase peptide synthesis.
- Suitable techniques are well known in the art.
- compositions of the present invention are prepared by synthetic or recombinant techniques, suitable protein purification techniques can also be involved, when applicable.
- the toxin peptide portion and/or the half-life extending portion, or any other portion can be prepared to include a suitable isotopic label (e.g., 125 1, 14 C, 13 C 1 35 S, 3 H, 2 H, 13 N, 15 N, 18 0, 17 O, efc.), for ease of quantification or detection.
- the compounds of this invention have pharmacologic activity resulting from their ability to bind to proteins of interest as agonists, mimetics or antagonists of the native ligands of such proteins of interest.
- Heritable diseases that have a known linkage to ion channels cover various fields of medicine, some of which include neurology, nephrology, myology and cardiology.
- a list of inherited disorders attributed to ion channels includes:
- cystic fibrosis Cho channel; CFTR
- KCNQ4 hereditary hearing loss
- SCNN1 hereditary hypertension
- IKCaI sickle cell anemia
- glaucoma BKCa
- Block of BKCa potassium channels can reduce intraocular fluid secretion and increase smooth muscle contraction, possibly leading to lower intraocular pressure and neuroprotection in the eye.
- multiple sclerosis Kv, KCa
- Kv diabetes, type I
- - type I diabetes is an autoimmune disease that is characterized by abnormal glucose, protein and lipid metabolism and is associated with insulin deficiency or resistance.
- Kv1.3-expressing T-lymphocytes attack and destroy pancreatic islets leading to loss of beta-cells.
- Block of Kv1.3 decreases inflammatory cytokines.
- block of Kv1.3 facilitates the translocation of GLUT4 to the plasma membrane, thereby increasing insulin sensitivity.
- Kv obesity
- restenosis KCa, Ca 2+
- KCa vascular smooth muscle cells
- neointimal thickening and vascular restenosis Excessive neointimal vascular smooth muscle cell proliferation is associated with elevated expression of IKCaI . Therefore, block of IKCaI could represent a therapeutic strategy to prevent
- renal incontinence is associated with overactive bladder smooth muscle cells.
- Calcium-activated potassium channels are expressed in bladder smooth muscle cells, 5 where they control the membrane potential and indirectly control the force and frequency of cell contraction. Openers of calcium-activated potassium channels therefore provide a mechanism to dampen electrical and contractile activity in bladder, leading to reduced urge to urinate.
- N-type voltage-gated calcium channels are key regulators of nociceptive neurotransmission in the spinal cord.
- Ziconotide a peptide blocker of N-type calcium channels reduces nociceptive neurotransmission and is approved worldwide for the symptomatic alleviation of severe chronic pain in humans. Novel blockers of nociceptor-specific N-type calcium channels would be improved analgesics with reduced side-effect profiles.
- potassium channel genes are amplified and protein subunits are upregulated in many cancerous condition. Consistent with a pathophysiological role for potassium channel upregulation, potassium channel blockers have been shown to suppress proliferation of uterine cancer cells and hepatocarcinoma cells, presumably through inhibition of calcium influx and effects on calcium-dependent gene expression. • a variety of neurological, cardiovascular, metabolic and autoimmune diseases.
- Both agonists and antagonists of ion channels can achieve therapeutic benefit.
- Therapeutic benefits can result, for example, from antagonizing Kv1.3, IKCaI, SKCa, BKCa, N- type or T-type Ca 2+ channels and the like. Small molecule and peptide antagonists of these channels have been shown to possess utility in vitro and in vivo. Limitations in production efficiency and pharmacokinetics, however, have largely prevented clinical investigation of inhibitor peptides of ion channels.
- compositions of this invention incorporating peptide antagonists of the voltage-gated potassium channel Kv1.3 are useful as immunosuppressive agents with therapeutic value for autoimmune diseases.
- such molecules are useful in treating multiple sclerosis, type 1 diabetes, psoriasis, inflammatory bowel disease, and rheumatoid arthritis.
- Kv1.3 have been examined in a variety of preclinical animal models of inflammation. Small molecule and peptide inhibitors of Kv1.3 have been shown to block delayed type hypersensitivity responses to ovalbumin [C. Beeton et al. (2005) MoI. Pharmacol. 67, 1369] and tetanus toxoid [G.C. Koo et al. (1999) Clin. Immunol. 197, 99]. In addition to suppressing inflammation in the skin, inhibitors also reduced antibody production [G.C. Koo et al. (1997) J. Immunol. 158, 5120].
- Kv1.3 antagonists have shown efficacy in a rat adoptive-transfer experimental autoimmune encephalomyelitis (AT-EAE) model of multiple sclerosis (MS).
- AT-EAE experimental autoimmune encephalomyelitis
- MS multiple sclerosis
- the Kv1.3 channel is overexpressed on myelin-specific T cells from MS patients, lending further support to the utility Kv1.3 inhibitors may provide in treating MS.
- Inflammatory bone resorption was also suppressed by Kv1.3 inhibitors in a preclinical adoptive-transfer model of periodontal disease [P. Valverde et al. (2004) J. Bone Mineral Res. 19, 155].
- inhibitors additionally blocked antibody production to a bacterial outer membrane protein, - one component of the bacteria used to induce gingival inflammation.
- Kv1.3 channel is expressed on all subsets of T cells and B cells, but effector memory T cells and class-switched memory B cells are particularly dependent on Kv1.3 [H. Wulff et al. (2004) J. Immunol. 173, 776].
- Kv1.3 is also being investigated for the treatment of obesity and diabetes.
- Breast cancer specimens [M. Abdul et al. (2003)Anticancer Res. 23, 3347] and prostate cancer cell lines [S.P. Fraser et al. (2003) Pflugers Arch.446, 559] have also been shown to express Kv1.3, and Kv1.3 blockade may be of utility for treatment of cancer.
- disorders that can be treated in accordance with the inventive method of treating an autoimmune disorder, involving Kv1.3 inhibitor toxin peptide(s), include multiple sclerosis, type 1 diabetes, psoriasis, inflammatory bowel disease, contact-mediated dermatitis, rheumatoid arthritis, psoriatic arthritis, asthma, allergy, restinosis, systemic sclerosis, fibrosis, scleroderma, glomerulonephritis, Sjogren syndrome, inflammatory bone resorption, transplant rejection, graft-versus-host disease, and systemic lupus erythematosus (SLE) and other forms of lupus.
- SLE systemic lupus erythematosus
- T cells that express the calcium-activated potassium of intermediate conductance IKCaI include T cells, B cells, mast cells and red blood cells (RBCs).
- T cells and RBCs from mice deficient in IKCaI show defects in volume regulation [T. Begenisich et al. (2004) J. Biol. Chem.279, 47681], Preclinical and clinical studies have demonstrated IKCaI inhibitors utility in treating sickle cell anemia [J. W. Stacker et al. (2003) Blood 101, 2412; www.icagen.com].
- Blockers of the IKCaI channel have also been shown to block EAE, indicating they may possess utility in treatment of MS [E. P. Reich et al. (2005) Eur. J. Immunol. 35, 1027].
- IgE-mediated histamine production from mast cells is also blocked by IKCaI inhibitors [S. Mark Duffy et al. (2004) J. Allergy Clin. Immunol. 114, 66], therefore they may also be of benefit in treating asthma.
- the iKCai channel is overexpressed on activated T and B lymphocytes [H. Wulff et al. (2004) J. Immunol. 173, 776] and thus may show utility in treatment of a wide variety of immune disorders.
- IKCaI inhibitors have also shown efficacy in a rat model of vascular restinosis and thus might represent a new therapeutic strategy to prevent restenosis after angioplasty [R. Kohler et al.
- IKCaI antagonists are of utility in treatment of tumor angiogenesis since inhibitors suppressed endothelial cell proliferation and angionenesis in vivo [1. Grgic et al. (2005) Arterioscler. Thromb. Vase. Biol. 25, 704].
- the IKCaI channel is upregulated in pancreatic tumors and inhibitors blocked proliferation of pancreatic tumor cell lines [H. Jager et al. (2004) MoI Pharmacol. 65, 630].
- IKCaI antagonists may also represent an approach to attenuate acute brain damage caused by traumatic brain injury [F. Mauler (2004) Eur. J. Neurosci.
- IKCaI inhibitors include multiple sclerosis, asthma, psoriasis, contact-mediated dermatitis, rheumatoid & psoriatic arthritis, inflammatory bowel disease, transplant rejection, graft-versus-host disease, Lupus, restinosis, pancreatic cancer, tumor angiogenesis and traumatic brain injury.
- molecules of this invention incorporating peptide antagonists of the calcium- activated potassium channel of intermediate conductance, IKCa can be used to treat immune dysfunction, multiple sclerosis, type 1 diabetes, psoriasis, inflammatory bowel disease, contact- mediated dermatitis, rheumatoid arthritis, psoriatic arthritis, asthma, allergy, restinosis, systemic sclerosis, fibrosis, scleroderma, glomerulonephritis, Sjogren syndrome, inflammatory bone resorption, transplant rejection, graft-versus-host disease, and lupus.
- the present invention includes a method of treating an autoimmune disorder, which involves administering to a patient who has been diagnosed with an autoimmune disorder, such as multiple sclerosis, type 1 diabetes, psoriasis, inflammatory bowel disease, contact- mediated dermatitis, rheumatoid arthritis, psoriatic arthritis, asthma, allergy, restinosis, systemic sclerosis, fibrosis, scleroderma, glomerulonephritis, Sjogren syndrome, inflammatory bone resorption, transplant rejection, graft-versus-host disease, or lupus, a therapeutically effective amount of the inventive composition of matter, whereby at least one symptom of the disorder is alleviated in the patient.
- an autoimmune disorder such as multiple sclerosis, type 1 diabetes, psoriasis, inflammatory bowel disease, contact- mediated dermatitis, rheumatoid arthritis, psoriatic arthritis, asthma, allergy, restinosis, systemic sclerosis, fibro
- Alleviated means to be lessened, lightened, diminished, softened, mitigated (i.e., made more mild or gentle), quieted, assuaged, abated, relieved, nullified, or allayed, regardless of whether the symptom of interest is entirely erased, eradicated, eliminated, or prevented in a particular patient.
- the present invention is further directed to a method of preventing or mitigating a relapse of a symptom of multiple sclerosis, which method involves administering to a patient, who has previously experienced at least one symptom of multiple sclerosis, a prophylactically effective amount of the inventive composition of matter, such that the at least one symptom of multiple sclerosis is prevented from recurring or is mitigated.
- compositions of matter preferred for use in practicing the inventive method of treating an autoimmune disorder and the method of preventing or mitigating a relapse of a symptom of multiple sclerosis include as P (conjugated as in Formula I), a Kv1.3 or IKCaI antagonist peptide, such as a ShK peptide, an 0SK1 peptide, a ChTx peptide and/or a Maurotoxin (MTx) peptide, or peptide analogs of any of these.
- P conjuggated as in Formula I
- a Kv1.3 or IKCaI antagonist peptide such as a ShK peptide, an 0SK1 peptide, a ChTx peptide and/or a Maurotoxin (MTx) peptide, or peptide analogs of any of these.
- the conjugated ShK peptide peptide or ShK peptide analog can comprise an amino acid sequence selected from the following:
- the conjugated OSK1 peptide peptide or OSK1 peptide analog can comprise an amino acid sequence selected from the following: SEQ ID NOS: 25, 294 through 298, 562 through 636, 980 through 1274,
- GV1INVSCKISRQCLEPCKKAGMRFGKCMNGKCHCTPK (OSK1-S7)(SEQ ID NO: 1303), or GVIINVSCKISRQCLKPCKDAGMRFGKCMNGKCHCTPK (OSK1-S7,K16,D20)(SEQ ID NO: 1308) as set forth in Table 7.
- a the conjugated MTX peptide, MTX peptide analog, ChTx peptide or ChTx peptide analog can comprise an amino acid sequence selected from:
- Kv1.3 or IKCaI inhibitor toxin peptide analog that comprises an amino acid sequence selected from:
- a patient who has been diagnosed with an autoimmune disorder such as, but not limited to multiple sclerosis, type 1 diabetes, psoriasis, inflammatory bowel disease, contact-mediated dermatitis, rheumatoid arthritis, psoriatic arthritis, asthma, allergy, restinosis, systemic sclerosis, fibrosis, scleroderma, glomerulonephritis, Sjogren syndrome, inflammatory bone resorption, transplant rejection, graft-versus-host disease, or lupus, or a patient who has previously experienced at least one symptom of multiple sclerosis, are well- recognizable and/or diagnosed by the skilled practitioner, such as a physician, familiar with autoimmune disorders and their symptoms.
- an autoimmune disorder such as, but not limited to multiple sclerosis, type 1 diabetes, psoriasis, inflammatory bowel disease, contact-mediated dermatitis, rheumatoid arthritis, psoriatic arthritis, asthma, allergy, restinosis, systemic
- symptoms of multiple sclerosis can include the following: visual symptoms, such as, optic neuritis (blurred vision, eye pain, loss of color vision, blindness); diplopia (double vision); nystagmus (jerky eye movements); ocular dysmetria (constant under- or overshooting eye movements); internuclear ophthalmoplegia (lack of coordination between the two eyes, nystagmus, diplopia); movement and sound phosphenes (flashing lights when moving eyes or in response to a sudden noise); afferent pupillary defect (abnormal pupil responses); motor symptoms, such as, paresis, monoparesis, paraparesis, hemiparesis, quadraparesis (muscle weakness - partial or mild paralysis); plegia, paraplegia, hemiplegia, tetraplegia, quadraplegia (paralysis - total or near total loss of muscle strength); spasticity (loss of muscle tone causing stiffness
- bowel, bladder and sexual symptoms such as, frequent micturation, bladder spasticity 0 (urinary urgency and incontinence); flaccid bladder, detrusor-sphincter dyssynergia (urinary hesitancy and retention); erectile dysfunction (male and female impotence); anorgasmy (inability to achieve orgasm); retrograde ejaculation (ejaculating into the bladder); frigidity (inability to become sexually aroused); constipation (infrequent or irregular bowel movements); fecal urgency (bowel urgency); fecal incontinence (bowel incontinence); 5 cognitive symptoms, such as, depression; cognitive dysfunction (short-term and
- multiple sclerosis enumerated above, are merely illustrative and are not intended to be an exhaustive description of all possible symptoms experienced by a single patient or by several sufferers in composite, and to which the present invention is directed.
- Those skilled 5 in the art are aware of various clinical symptoms and constellations of symptoms of autoimmune disorders suffered by individual patients, and to those symptoms are also directed the present inventive methods of treating an autoimmune disorder or of preventing or mitigating a relapse of a symptom of multiple sclerosis.
- the therapeutically effective amount, prophylactically effective amount, and dosage O regimen involved in the inventive methods of treating an autoimmune disorder or of preventing or mitigating a relapse of a symptom of multiple sclerosis will be determined by the attending physician, considering various factors which modify the action of therapeutic agents, such as the age, condition, body weight, sex and diet of the patient, the severity of the condition being treated, time of administration, and other clinical factors.
- the daily amount or regimen should be in the range of about 1 to about 10,000 micrograms ( ⁇ g) of the vehicle-conjugated peptide per kilogram (kg) of body mass, preferably about 1 to about 5000 ⁇ g per kilogram of body mass, and most preferably about 1 to about 1000 ⁇ g per kilogram of body mass.
- Molecules of this invention incorporating peptide antagonists of the voltage-gated potassium channel Kv2.1 can be used to treat type Il diabetes.
- Molecules of this invention incorporating peptide antagonists of the M current can be used to treat Alzheimer's disease and enhance cognition.
- Molecules of this invention incorporating peptide antagonists of the voltage-gated potassium channel Kv4.3 can be used to treat Alzheimer's disease.
- Molecules of this invention incorporating peptide antagonists of the calcium-activated potassium channel of small conductance, SKCa can be used to treat epilepsy, memory, learning, neuropsychiatric, neurological, neuromuscular, and immunological disorders, schizophrenia, bipolar disorder, sleep apnea, neurodegeneration, and smooth muscle disorders.
- Molecules of this invention incorporating N-type calcium channel antagonist peptides are useful in alleviating pain.
- Peptides with such activity e.g., ZiconotideTM, ⁇ -conotoxin-MVIIA have been clinically validated.
- T-type calcium channel antagonist peptides are useful in alleviating pain.
- T-type calcium channels are found at extremely high levels in the cell bodies of a subset of neurons in the DRG; these are likely mechanoreceptors adapted to detect slowly-moving stimuli (Shin et al., Nature Neuroscience 6:724-730, 2003), and T-type channel activity is likely responsible for burst spiking (Nelson et al., J Neurosci 25:8766-8775, 2005).
- Inhibition of T-type channels by either mibefradil or ethosuximide reverses mechanical allodynia in animals induced by nerve injury (Dogrul et al., Pain 105:159-168,
- Molecules of this invention incorporating peptide antagonists of the Nav1 (TTXs-type) 5 channel can be used to alleviate pain. Local anesthetics and tricyclic antidepressants with such activity have been clinically validated. Such molecules of this invention can in particular be useful as muscle relaxants.
- Molecules of this invention incorporating peptide antagonists of the Navi(TTX R -type) channel can be used to alleviate pain arising from nerve and or tissue injury, 0 Molecules of this invention incorporating peptide antagonists of glial & epithelial cell Ca 2+ - activated chloride channel can be used to treat cancer and diabetes.
- Molecules of this invention incorporating peptide antagonists of NMDA receptors can be used to treat pain, epilepsy, brain and spinal cord injury.
- Molecules of this invention incorporating peptide antagonists of nicotinic receptors can be 5 used as muscle relaxants. Such molecules can be used to treat pain, gastric motility disorders, urinary incontinence, nicotine addiction, and mood disorders.
- Molecules of this invention incorporating peptide antagonists of 5HT3 receptor can be used to treat Nausea, pain, and anxiety.
- Molecules of this invention incorporating peptide antagonists of the norepinephrine O transporter can be used to treat pain, anti-depressant, learning, memory, and urinary incontinence.
- Molecules of this invention incorporating peptide antagonists of the Neurotensin receptor can be used to treat pain.
- the compounds of the present invention can be useful in diagnosing diseases characterized by dysfunction of their associated protein of interest.
- a method of detecting in a biological sample a protein of interest e.g., a receptor
- the biological samples include tissue specimens, intact cells, or extracts thereof.
- the compounds of this invention can be used as part of a diagnostic kit to detect the presence of their associated O proteins of interest in a biological sample. Such kits employ the compounds of the invention having an attached label to allow for detection. The compounds are useful for identifying normal or abnormal proteins of interest.
- compositions can also be employed, alone or in combination with other molecules in the treatment of disease.
- Pharmaceutical Compositions can also be employed, alone or in combination with other molecules in the treatment of disease.
- the present invention also provides pharmaceutical compositions comprising the inventive composition of matter and a pharmaceutically acceptable carrier.
- Such pharmaceutical compositions can be configured for administration to a patient by a wide variety of delivery routes, e.g., an intravascular delivery route such as by injection or infusion, subcutaneous, intramuscular, intraperitoneal, epidural, or intrathecal delivery routes, or for oral, enteral, pulmonary (e.g., inhalant), intranasal, transmucosal (e.g., sublingual administration), transdermal or other delivery routes and/or forms of administration known in the art.
- the inventive pharmaceutical compositions may be prepared in liquid form, or may be in dried powder form, such as lyophilized form.
- the pharmaceutical compositions can be configured, for example, as tablets, troches, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsions, hard or soft capsules, syrups, elixirs or enteral formulas.
- the "pharmaceutically acceptable carrier” is any physiologically tolerated substance known to those of ordinary skill in the art useful in formulating pharmaceutical compositions, including, any pharmaceutically acceptable diluents, excipients, dispersants, binders, fillers, glidants, anti-frictional agents, compression aids, tablet-disintegrating agents (disintegrants), suspending agents, lubricants, flavorants, odorants, sweeteners, permeation or penetration enhancers, preservatives, surfactants, solubilizers, emulsifiers, thickeners, adjuvants, dyes, coatings, encapsulating material(s), and/or other additives singly or in combination.
- Such pharmaceutical compositions can include diluents of various buffer content (e.g., Tris-HCl, acetate, phosphate), pH and ionic strength; additives such as detergents and solubilizing agents ⁇ e.g., Tween ® 80, Polysorbate 80), anti-oxidants (e.g., ascorbic acid, sodium metabisulfite), preservatives (e.g., Thimersol ® , benzyl alcohol) and bulking substances ⁇ e.g., lactose, mannitol); incorporation of the material into particulate preparations of polymeric compounds such as polylactic acid, polyglycolic acid, etc. or into liposomes.
- buffer content e.g., Tris-HCl, acetate, phosphate
- additives such as detergents and solubilizing agents ⁇ e.g., Tween ® 80, Polysorbate 80
- anti-oxidants e.g., ascorbic acid, sodium metabisul
- Hyaluronic acid can also be used, and this can have the effect of promoting sustained duration in the circulation.
- Such compositions can influence the physical state, stability, rate of in vivo release, and rate of in vivo clearance of the present proteins and derivatives. See, e.g., Remington's Pharmaceutical Sciences, 18th Ed. (1990, Mack Publishing Co., Easton, PA 18042) pages 1435-1712, which are herein incorporated by reference in their entirety.
- the compositions can be prepared in liquid form, or can be in dried powder, such as lyophilized form. Implantable sustained release formulations are also useful, as are transdermal or transmucosal formulations.
- diluents can include carbohydrates, especially, mannitol, ⁇ -lactose, anhydrous lactose, cellulose, sucrose, modified dextrans and starch.
- Certain inorganic salts may also be used as fillers, including calcium triphosphate, magnesium carbonate and sodium chloride.
- Some commercially available diluents are Fast-Flo, Emdex, STA-Rx 1500, Emcompress and Avicell.
- a variety of conventional thickeners are useful in creams, ointments, suppository and gel configurations of the pharmaceutical composition, such as, but not limited to, alginate, xanthan gum, or petrolatum, may also be employed in such configurations of the pharmaceutical composition of the present invention.
- a permeation or penetration enhancer such as polyethylene glycol monolaurate, dimethyl sulfoxide, N-vinyl-2-pyrrolidone, N-(2-hydroxyethyl)-pyrrolidone, or 3-hydroxy-N-methyl-2- pyrrolidone can also be employed.
- Useful techniques for producing hydrogel matrices are known.
- biodegradable hydrogel matrices for the controlled release of pharmacologically active agents, U.S. Patent No.4,925,677; Shah et al., Biodegradable pH/thermosensitive hydrogels for sustained delivery of biologically active agents, WO 00/38651 A1).
- biodegradable gel matrices can be formed, for example, by crosslinking a proteinaceous component and a polysaccharide or mucopolysaccharide component, then loading with the inventive composition of matter to be delivered.
- Liquid pharmaceutical compositions of the present invention that are sterile solutions or suspensions can be administered to a patient by injection, for example, intramuscularly, intrathecally, epidurally, intravascularly (e.g., intravenously or intraarterially), intraperitoneally or subcutaneously.
- injection for example, intramuscularly, intrathecally, epidurally, intravascularly (e.g., intravenously or intraarterially), intraperitoneally or subcutaneously.
- intravascularly e.g., intravenously or intraarterially
- Sterile solutions can also be administered by intravenous infusion.
- the inventive composition can be included in a sterile solid pharmaceutical composition, such as a lyophilized powder, which can be dissolved or suspended at a convenient time before administration to a patient using sterile water, saline, buffered saline or other appropriate sterile injectable medium.
- a sterile solid pharmaceutical composition such as a lyophilized powder
- Implantable sustained release formulations are also useful embodiments of the inventive pharmaceutical compositions.
- the pharmaceutically acceptable carrier being a biodegradable matrix implanted within the body or under the skin of a human or non-human vertebrate, can be a hydrogel similar to those described above. Alternatively, it may be formed from a poly-alpha- amino acid component.
- Sidman, Biodegradable, implantable drug delivery device, and process for preparing and using same, U.S. Patent No. 4,351 ,337) Other techniques for making implants for delivery of drugs are also known and useful in accordance with the present invention.
- the pharmaceutically acceptable carrier is a finely divided solid, which is in admixture with finely divided active ingredient(s), including the inventive composition.
- a powder form is useful when the pharmaceutical composition is configured as an inhalant.
- Zeng et al. Method of preparing dry powder inhalation compositions, WO 2004/017918; Trunk et al., Salts of the CGRP antagonist BIBN4096 and inhalable powdered medicaments containing them, U.S. Patent No. 6,900,317.
- diluents could include carbohydrates, especially mannitol, ⁇ -lactose, anhydrous lactose, cellulose, sucrose, modified dextrans and starch.
- Certain inorganic salts can also be used as fillers including calcium triphosphate, magnesium carbonate and sodium chloride.
- Some commercially available diluents are Fast-FloTM, EmdexTM, STA-RxTM 1500, EmcompressTM and AvicellTM.
- Disintegrants can be included in the formulation of the pharmaceutical composition into a solid dosage form. Materials used as disintegrants include but are not limited to starch including the commercial disintegrant based on starch, ExplotabTM.
- Sodium starch glycolate, AmberliteTM, sodium carboxymethylcelluiose, ultramylopectin, sodium alginate, gelatin, orange peel, acid carboxymethyl cellulose, natural sponge and bentonite can all be used.
- Insoluble cationic exchange resin is another form of disintegrant.
- Powdered gums can be used as disintegrants and as binders and these can include powdered gums such as agar, Karaya ortragacanth. Alginic acid and its sodium salt are also useful as disintegrants.
- Binders can be used to hold the therapeutic agent together to form a hard tablet and include materials from natural products such as acacia, tragacanth, starch and gelatin. Others include methyl cellulose (MC), ethyl cellulose (EC) and carboxymethyl cellulose (CMC). Polyvinyl pyrrolidone (PVP) and hydroxypropy I methyl cellulose (HPMC) could both be used in alcoholic solutions to granulate the therapeutic.
- MC methyl cellulose
- EC ethyl cellulose
- CMC carboxymethyl cellulose
- PVP polyvinyl pyrrolidone
- HPMC hydroxypropy I methyl cellulose
- Lubricants can be used as a layer between the therapeutic and the die wall, and these can include but are not limited to; stearic acid including its magnesium and calcium salts, polytetrafluoroethylene (PTFE), liquid paraffin, vegetable oils and waxes. Soluble lubricants can also be used such as sodium lauryl sulfate, magnesium lauryl sulfate, polyethylene glycol of various molecular weights, Carbowax 4000 and 6000. Glida ⁇ ts that might improve the flow properties of the drug during formulation and to aid rearrangement during compression might be added.
- the glidants can include starch, talc, pyrogenic silica and hydrated silicoaluminate.
- surfactant might be added as a wetting agent.
- Surfactants can include anionic detergents such as sodium lauryl sulfate, dioctyl sodium sulfosuccinate and dioctyl sodium sulfonate.
- anionic detergents such as sodium lauryl sulfate, dioctyl sodium sulfosuccinate and dioctyl sodium sulfonate.
- Cationic detergents might be used and could include benzalkonium chloride or benzethonium chloride.
- nonionic detergents that could be included in the formulation as surfactants are lauromacrogol 400, polyoxyl 40 stearate, polyoxyethylene hydrogenated castor oil 10, 50 and 60, glycerol monostearate, polysorbate 40, 60, 65 and 80, sucrose fatty acid ester, methyl cellulose and carboxymethyl cellulose. These surfactants could be present in the formulation of the protein or derivative either alone or as a mixture in different ratios,
- Oral dosage forms are also useful.
- oral dosage forms of the inventive compositionss are also useful.
- the composition can be chemically modified so that oral delivery is efficacious.
- the chemical modification contemplated is the attachment of at least one moiety to the molecule itself, where said moiety permits (a) inhibition of proteolysis; and (b) uptake into the blood stream from the stomach or intestine.
- the increase in overall stability of the compound and increase in circulation time in the body Moieties useful as covalently attached half- life extending moieties in this invention can also be used for this purpose.
- moieties include: PEG, copolymers of ethylene glycol and propylene glycol, carboxymethyl cellulose, dextran, polyvinyl alcohol, polyvinyl pyrrolidone and polyproline. See, for example, Abuchowski and Davis (1981), Soluble Polymer-Enzyme Adducts, Enzymes as Drugs (Hocenberg and Roberts, eds.), Wiley-lnterscience, New York, NY, pp 367-83; Newmark, elaj. (1982), J. Appl. Biochem, 4:185-9.
- Other polymers that could be used are poly-1 ,3-dioxolane and poly-1,3,6- tioxocane.
- Preferred for pharmaceutical usage, as indicated above, are PEG moieties.
- a salt of a modified aliphatic amino acid such as sodium N-(8-[2-hydroxybenzoyl] amino) caprylate (SNAC)
- SNAC sodium N-(8-[2-hydroxybenzoyl] amino) caprylate
- the pharmaceutically acceptable carrier can be a liquid and the pharmaceutical composition is prepared in the form of a solution, suspension, emulsion, syrup, elixir or pressurized composition.
- the active ingredient(s) e.g., the inventive composition of matter
- a pharmaceutically acceptable liquid carrier such as water, an organic solvent, a mixture of both, or pharmaceutically acceptable oils or fats.
- the liquid carrier can contain other suitable pharmaceutical additives such as detergents and/or solubilizers (e.g., Tween 80, Polysorbate 80), emulsifiers, buffers at appropriate pH (e.g., Tris-HCI, acetate, phosphate), adjuvants, anti-oxidants (e.g., ascorbic acid, sodium metabisulfite), preservatives (e.g., Thimersol, benzyl alcohol), sweeteners, flavoring agents, suspending agents, thickening agents, bulking substances (e.g., lactose, mannitol), colors, viscosity regulators, stabilizers, electrolytes, osmolutes or osmo-regulators.
- additives e.g., Tween 80, Polysorbate 80
- emulsifiers e.g., buffers at appropriate pH (e.g., Tris-HCI, acetate, phosphate), adjuvants, anti-oxid
- Solid dosage forms include tablets, capsules, pills, troches or lozenges, cachets or pellets.
- liposomal or proteinoid encapsulation can be used to formulate the present compositions (as, for example, proteinoid microspheres reported in U.S. Patent No.4,925,673).
- Liposomal encapsulation can be used and the liposomes can be derivatized with various polymers (e.g., U.S. Patent No. 5,013,556).
- the formulation will include the inventive compound, and inert ingredients that allow for protection against the stomach environment, and release of the biologically active material in the intestine.
- the composition of this invention can be included in the formulation as fine multiparticulates in the form of granules or pellets of particle size about 1 mm.
- the formulation of the material for capsule administration could also be as a powder, lightly compressed plugs or even as tablets.
- the therapeutic could be prepared by compression.
- Colorants and flavoring agents can all be included.
- the protein (or derivative) can be formulated (such as by liposome or microsphere encapsulation) and then further contained within an edible product, such as a refrigerated beverage containing colorants and flavoring agents.
- the active ingredient(s) are mixed with a pharmaceutically acceptable carrier having the necessary compression properties in suitable proportions and compacted in the shape and size desired.
- a pharmaceutically acceptable carrier having the necessary compression properties in suitable proportions and compacted in the shape and size desired.
- the powders and tablets preferably contain up to 99% of the active ingredient(s).
- Suitable solid carriers include, for example, calcium phosphate, magnesium stearate, talc, sugars, lactose, dextrin, starch, gelatin, cellulose, polyvinylpyrrolidine, low melting waxes and ion exchange resins.
- Controlled release formulation can be desirable.
- the composition of this invention could be incorporated into an inert matrix that permits release by either diffusion or leaching mechanisms e.g., gums.
- Slowly degenerating matrices can also be incorporated into the formulation, e.g., alginates, polysaccharides.
- Another form of a controlled release of the compositions of this invention is by a method based on the OrosTM therapeutic system (Alza Corp.), i.e., the drug is enclosed in a semipermeable membrane which allows water to enter and push drug out through a single small opening due to osmotic effects. Some enteric coatings also have a delayed release effect.
- coatings can be used for the formulation. These include a variety of sugars that could be applied in a coating pan.
- the therapeutic agent could also be given in a film-coated tablet and the materials used in this instance are divided into 2 groups.
- the first are the nonenteric materials and include methylcellulose, ethyl cellulose, hydroxyethyl cellulose, methylhydroxy-ethyl cellulose, hydroxypropyl cellulose, hydroxypropyl-methyl cellulose, sodium carboxymethyl cellulose, providone and the polyethylene glycols.
- the second group consists of the enteric materials that are commonly esters of phthalic acid.
- Film coating can be carried out in a pan coater or in a fluidized bed or by compression coating.
- Pulmonary delivery forms Pulmonary delivery of the inventive compositions is also useful.
- the protein (or derivative) is delivered to the lungs of a mammal while inhaling and traverses across the lung epithelial lining to the blood stream.
- Adjei et al. Pharma. Res. (1990) 7: 565-9
- Adjei et aL (1990), Internatl. J. Pharmaceutics 63: 135-44 (leuprolide acetate); Braquet etjl. (1989), J. Cardiovasc. Pharmacol. 13 (suppl.5): s.143-146 (endothelin-1); Hubbard et a
- nebulizers used for pulmonary delivery of therapeutic products, including but not limited to nebulizers, metered dose inhalers, and powder inhalers, all of which are familiar to those skilled in the art.
- Some specific examples of commercially available devices suitable for the practice of this invention are the Ultravent nebulizer, manufactured by Mallinckrodt, Inc., St. Louis, Missouri; the Acorn Il nebulizer, manufactured by Marquest Medical Products, Englewood, Colorado; the Ventolin metered dose inhaler, manufactured by Glaxo Inc., Research Triangle Park, North Carolina; and the Spinhaler powder inhaler, manufactured by Fisons Corp., Bedford, Massachusetts.
- each formulation is specific to the type of device employed and can involve the use of an appropriate propellant material, in addition to diluents, adjuvants and/or carriers useful in therapy,
- the inventive compound should most advantageously be prepared in particulate form with an average particle size of less than 10 ⁇ m (or microns), most preferably 0.5 to 5 ⁇ m, for most effective delivery to the distal lung.
- Pharmaceutically acceptable carriers include carbohydrates such as trehalose, mannitol, xylitol, sucrose, lactose, and sorbitol.
- Other ingredients for use in formulations can include DPPC, DOPE, DSPC and DOPC.
- Natural or synthetic surfactants can be used. PEG can be used (even apart from its use in derivatizing the protein or analog).
- Dextrans such as cyclodextran, can be used.
- Bile salts and other related enhancers can be used. Cellulose and cellulose derivatives can be used. Amino acids can be used, such as use in a buffer formulation.
- liposomes are contemplated.
- microcapsules or microspheres inclusion complexes, or other types of carriers.
- Formulations suitable for use with a nebulizer will typically comprise the inventive compound dissolved in water at a concentration of about 0.1 to 25 mg of biologically active protein per mL of solution.
- the formulation can also include a buffer and a simple sugar (e.g., for protein stabilization and regulation of osmotic pressure).
- the nebulizer formulation can also contain a surfactant, to reduce or prevent surface induced aggregation of the protein caused by atomization of the solution in forming the aerosol.
- Formulations for use with a metered-dose inhaler device will generally comprise a finely divided powder containing the inventive compound suspended in a propellant with the aid of a surfactant.
- the propellant can be any conventional material employed for this purpose, such as a chlorofluorocarbon, a hydrochlorofluorocarbon, a hydrofluorocarbon, or a hydrocarbon, including trichlorofluoromethane, dichlorod if I uoromethane, dichlorotetrafluoroethanol, and 1,1,1,2- tetrafluoroethane, or combinations thereof.
- Suitable surfactants include sorbitan trioleate and soya lecithin. Oleic acid can also be useful as a surfactant.
- Formulations for dispensing from a powder inhaler device will comprise a finely divided dry powder containing the inventive compound and can also include a bulking agent, such as lactose, sorbitol, sucrose, mannitol, trehalose, orxylitol in amounts which facilitate dispersal of the powder from the device, e.g., 50 to 90% by weight of the formulation.
- a bulking agent such as lactose, sorbitol, sucrose, mannitol, trehalose, orxylitol in amounts which facilitate dispersal of the powder from the device, e.g., 50 to 90% by weight of the formulation.
- intranasal delivery of the inventive composition of matter and/or pharmaceutical compositions is also useful, which allows passage thereof to the blood stream directly after administration to the inside of the nose, without the necessity for deposition of the product in the lung.
- Formulations suitable for intransal administration include those with dextran orcyclodextran, and intranasal delivery devices are known. (See, e.g. Freezer, Inhaler, U.S. Patent No.4,083,368).
- the inventive composition is configured as a part of a pharmaceutically acceptable transdermal or transmucosal patch or a troche.
- Transdermal patch drug delivery systems for example, matrix type transdermal patches, are known and useful for practicing some embodiments of the present pharmaceutical compositions.
- Chien et al. Transdermal estrogen/progestin dosage unit, system and process, U.S. Patent Nos.4,906,169 and 5,023,084; Cleary et al., Diffusion matrix for transdermal drug administration and transdermal drug delivery devices including same, U.S.
- a variety of pharmaceutically acceptable systems for transmucosal delivery of therapeutic agents are also known in the art and are compatible with the practice of the present invention.
- Buccal delivery of the inventive compositions is also useful.
- Buccal delivery formulations are known in the art for use with peptides.
- known tablet or patch systems configured for drug delivery through the oral mucosa (e.g., sublingual mucosa), include some embodiments that comprise an inner layer containing the drug, a permeation enhancer, such as a bile salt or fusidate, and a hydrophilic polymer, such as hydroxypropyl cellulose, hydroxypropyl methylcellulose, hydroxyethyl cellulose, dextran, pectin, polyvinyl pyrrolidone, starch, gelatin, or any number of other polymers known to be useful for this purpose.
- a permeation enhancer such as a bile salt or fusidate
- a hydrophilic polymer such as hydroxypropyl cellulose, hydroxypropyl methylcellulose, hydroxyethyl cellulose, dextran, pectin, polyvinyl pyrrolidone, starch, gelatin, or any number of other polymers known to be useful for this purpose.
- This inner layer can have one surface adapted to contact and adhere to the moist mucosal tissue of the oral cavity and can have an opposing surface adhering to an overlying non-adhesive inert layer.
- a transmucosal delivery system can be in the form of a bilayer tablet, in which the inner layer also contains additional binding agents, flavoring agents, or fillers.
- Some useful systems employ a non-ionic detergent along with a permeation enhancer.
- Transmucosal delivery devices may be in free form, such as a cream, gel, or ointment, or may comprise a determinate form such as a tablet, patch or troche.
- delivery of the inventive composition can be via a transmucosal delivery system comprising a laminated composite of, for example, an adhesive layer, a backing layer, a permeable membrane defining a reservoir containing the inventive composition, a peel seal disc underlying the membrane, one or more heat seals, and a removable release liner.
- a transmucosal delivery system comprising a laminated composite of, for example, an adhesive layer, a backing layer, a permeable membrane defining a reservoir containing the inventive composition, a peel seal disc underlying the membrane, one or more heat seals, and a removable release liner.
- the dosage regimen involved in a method for treating the above-described conditions will be determined by the attending physician, considering various factors which modify the action of drugs, e.g. the age, condition, body weight, sex and diet of the patient, the severity of any infection, time of administration and other clinical factors.
- the daily regimen should be in the range of 0.1- 1000 micrograms of the inventive compound per kilogram of body weight, preferably 0.1-150 micrograms per kilogram.
- compositions described above can be prepared as described below. These examples are not to be construed in any way as limiting the scope of the present invention.
- Fc-LI Q-Sh KM -351 mammalian expression Fc-LIO-ShK[I -35], also referred to as Tc-2xL-ShK[1-35]", an inhibitor of Kv1.3.
- a DNA sequence coding for the Fc region of human IgGI fused in-frame to a linker sequence and a monomer of the Kv1.3 inhibitor peptide ShK[I -35] was constructed as described below. Methods for expressing and purifying the peptibody from mammalian cells (HEK 293 and Chinese Hamster Ovary cells) are disclosed herein.
- the expression vector pcDNA3.1(+)CMVi ( Figure 13A) was constructed by replacing the CMV promoter between MIuI and Hindi 11 in pcDNA3.1 (+) with the CMV promoter plus intron (Invitrogen).
- the expression vector pcDNA3.1(+)CMVi-hFc-ActivinRIIB ( Figure 13B) was generated by cloning a Hindlll-Notl digested PCR product containing a 5' Kozak sequence, a signal peptide and the human Fc-linker— ActivinRIIB fusion protein with the large fragment of Hindlll-Notl digested pcDNA3.1(+)CMVi.
- the nucleotide and amino acid sequence of the human IgGI Fc region in pcDNA3.1(+)CMVi-hFc-ActivinRIIB is shown in Figure 3.
- This vector also has a GGGGSGGGGS ("L10"; SEQ ID NO:79) linker split by a BamHl site thus enabling with the oligo below formation of the 10 amino acid linker between Fc and the ShK[1-35] peptide (see Figure 14) for the final Fc-LI 0-ShK[1 -35] nucleotide and amino acid sequence ( Figure 14 and SEQ ID NO: 77 and SEQ ID NO:78).
- the Fc-L10-ShK[1-35] expression vector was constructing using PCR stategies to generate the full length ShK gene linked to a four glycine and one serine amino acid linker (lower case letters here indicate linker sequence of L-form amino acid residues) with two stop codons and flanked by BamHl and Notl restriction sites as shown below.
- HerculaseTM polymerase (Stratagene) at 94°C-30sec, 50°C-30sec, and 72°C-1 min for 30 cycles.
- cat gcg gcc get cat tag cag gtg ccg cag gtc ttg egg cag aag etc agg egg tac ttc atg ctg tgc ttg cac tgg aag g //SEQ ID NO: 660
- the resulting PCR products were resolved as the 150bp bands on a one percent agarose gel.
- the 150bp PCR product was digested with BamHl and Notl (Roche) restriction enzymes and agarose gel purified by Gel Purification Kit (Qiagen).
- the pcDNA3.1(+)CMVi- hFc-ActivinRIIB vector ( Figure 13B ) was digested with BamHl and Notl restriction enzymes and the large fragment was purified by Gel Purification Kit.
- the gel purified PCR fragment was ligated to the purified large fragment and transformed into XL-1blue bacteria (Stratagene).
- DNAs from transformed bacterial colonies were isolated and digested with BamHI and Notl restriction enzyme digestion and resolved on a one percent agarose gel. DNAs resulting in an expected pattern were submitted for sequencing. Although, analysis of several sequences of clones yielded a 100% percent match with the above sequence, only one clone was selected for large scaled plasmid purification.
- the DNA from Fc-2xL-ShK in pcDNA3.1(+)CMVi clone was resequenced to confirm the Fc and linker regions and the sequence was 100% identical to the predicted coding sequence, which is shown in Figure 14.
- HEK-293 cells used in transient transfection expression of Fc-2xL-ShK[1-35] in pcDNA3.1(+)CMVi protein were cultured in growth medium containing DMEM High Glucose (Gibco), 10% fetal bovine serum (FBS from Gibco) and 1X non-essential amino acid (NEAA from Gibco).
- DMEM High Glucose Gibco
- FBS 10% fetal bovine serum
- NEAA non-essential amino acid
- the conditioned medium was concentrated 5OX by running 30ml through Centriprep YM-10 filter (Amicon) and further concentrated by a Centricon YM-10 (Amicon) filter.
- Various amounts of concentrated medium were mixed with an in-house 4x Loading Buffer (without B-mercaptoethanol) and electrophoresed on a Novex 4-20% tris-glycine gel using a Novex Xcell Il apparatus at 101 V746mA for 2 hours in a 5x Tank buffer solution (0.123 Tris Base, 0.96M Glycine) along with 1OuI of BenchMark Pre-Stained Protein ladder (Invitrogen).
- Electroblot buffer 35mM Tris base, 20%methanol, 192mM glycine
- a PVDF membrane from Novex (Gat. No. LC2002, 0.2um pores size) was soaked in methanol for 30 seconds to activate the PVDF, rinsed with deionized water, and soaked in Electroblot buffer.
- the pre-soaked gel was blotted to the PVDF membrane using the XCeII Il Blot module according to the manufacturer instructions (Novex) at 4OmA for 2 hours.
- the blot was first soaked in a 5% milk (Carnation) in Tris buffered saline solution pH7.5 (TBS) for 1 hour at room temperature and incubated with 1:500 dilution in TBS with 0.1%Tween-20 (TBST Sigma) and 1% milk buffer of the HRP- conjugated murine anti-human Fc antibody (Zymed Laboratores Cat, no. 05-3320) for two hours shaking at room temperature. The blot was then washed three times in TBST for 15 minutes per wash at room temperature. The primary antibody was detected using Amersham Pharmacia Biotech's ECL western blotting detection reagents according to manufacturer's instructions. Upon ECL detection, the western blot analysis displayed the expected size of 66kDa under non-reducing gel conditions ( Figure 24A).
- AM1 CHOd- (Amgen Proprietary) cells used in the stable expression of Fc-LI 0-ShK[1 -35] protein were cultured in AM1 CHOd- growth medium containing DMEM High Glucose, 10% fetal bovine serum, 1x hypoxantine/thymidine (HT from Gibco) and 1X NEAA. 6.5ug of pcDNA3.1(+)CMVi-Fc-ShK plasmid was also transfected into AM1 CHOd- cells using Fugene 6.
- the transfected cells were plated into twenty 15cm dishes and selected using DMEM high glucose, 10%FBS, 1xHT, IxNEAA and Geneticin (800ug/mi G418 from Gibco) for thirteen days. Forty-eight surviving colonies were picked into two 24-welI plates. The plates were allowed to grow up for a week and then replicated for freezing. One set of each plate was transferred to AM1 CHOd- growth medium without 10% FBS for 48 hours and the conditioned media were harvested. Western Blot analysis similar to the transient Western blot analysis with detection by the same anti-human Fc antibody was used to screen 15ul of conditioned medium for expressing stable CHO clones.
- the BB6 clone was scaled up into ten roller bottles (Corning) using AM1 CHOd- growth medium and grown to confluency as judged under the microscope. Then, the medium was exchanged with a serum-free medium containing to 50% DMEM high glucose and 50% Ham's F12 (Gibco) with 1xHT and IxNEAA and let incubate for one week. The conditioned medium was harvested at the one-week incubation time, filtered through 0.45 ⁇ m filter (Corning) and frozen. Fresh serum-free medium was added and incubated for an additional week. The conditioned serum-free medium was harvested like the first time and frozen.
- Fractions were then analyzed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE, and the fractions containing the desired product were pooled based on these data.
- the pooled material was then concentrated to about 3.4 ml using a Pall Life Sciences Macrosep 1OK Omega centrifugal ultra-filtration device and then filtered though a Costar 0.22 ⁇ m cellulose acetate syringe filter.
- a spectral scan was then conducted on 10 ⁇ l of the filtered material diluted in 700 ⁇ l PBS using a Hewlett Packard 8453 spectrophotometer (Figure 26A).
- the concentration of the filtered material was determined to be 5.4 mg/ml using a calculated molecular mass of 32,420 g/mol and extinction coefficient of 47,900 M 1 cm 1 .
- the macromolecular state of the product was then determined using size exclusion chromatography on 20 ⁇ g of the product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH 2 PO 4 , 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm ( Figure 26C).
- the product was then subject to mass spectral analysis by diluting 1 ⁇ l of the sample into 10 ⁇ l of sinapinic acid (10 mg per ml in 0.05% trifluroacetic acid, 50% acetonitrile) .
- the resultant solution (1 ⁇ l) was spotted onto a MALDI sample plate.
- the sample was allowed to dry before being analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ⁇ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses ( Figure 26D) and confirmed (within experimental error) the integrity of the purified peptibody. The product was then stored at -80 0 C.
- the sequences of the primers used to generate Fc-L5-ShK[2-35], also referred to as "Fc- 1 ⁇ L-ShK[2-35]", are shown below: cat gga tec age tgc ate gac ace atc//sEQ ID NO: 661; cat gcg gcc get cat tag c// SEQ ID NO:662;
- the sequences of the primers used to generate Fc-LI 0-ShK[2-35] also referred to as "Fc-
- Fc-L25-ShK[2-35] The sequences of the primers used to generate Fc-L25-ShK[2-35], also referred to as "Fc- 5xL-ShK[2-35]", are shown below: cat gga tec ggg ggt ggg ggt tct ggg ggt ggg ggt tct gga gga gga age gga gga gga gga age age tgc a//sEQ ID NO:665; cat gcg gcc get cat tag cag gtg C//SEQ ID NO: 566;
- PCR products were digested with BamHI and Notl (Roche) restriction enzymes and agarose gel purified by Gel Purification Kit. At the same time, the pcDNA3.1(+)CMVi-hFc-
- ActivinRIIB vector was digested with BamHI and Notl restriction enzymes and the large fragment was purified by Gel Purification Kit. Each purified PCR product was ligated to the large fragment and transformed into XL-1 blue bacteria. DNAs from transformed bacterial colonies were isolated and subjected to BamHI and Notl restriction enzyme digestions and resolved on a one percent agarose gel. DNAs resulting in an expected pattern were submitted for sequencing. Although, analysis of several sequences of clones yielded a 100% percent match with the above sequence, only one clone was selected for large scaled plasmid purification. The DNA from this clone was resequenced to confirm the Fc and linker regions and the sequence was 100% identical to the expected sequence.
- Plasmids containing the Fc-1xL-Shk[2-35], Fc-2xL-Shk[2-35] and Fc-5xL-Shk[2-35] inserts in pcDNA3.1(+)CMVi vector were digested with Xba1 and Xho1 (Roche) restriction enzymes and gel purified. The inserts were individually ligated into Not1 and Sail (Roche) digested pDSR ⁇ -22 (Amgen Proprietary) expression vector. Integrity of the resulting constructs were confirmed by DNA sequencing.
- the final plasmid DNA expression vector constructs were pDSR ⁇ -22-Fc-1xL-Shk[2- 35], pDSR ⁇ -22- Fc-2xL-Shk[2-35] ( Figure 13C and Figure 15) and pDSR ⁇ -22- Fc-5xL-Shk[2-35] ( Figure 16) and contained 5, 10 and 25 amino acid linkers, respectively.
- AM-1/D CHOd- (Amgen Proprietary) cells were plated into a T-175 cm sterile tissue culture flask, to allow 70-80% confluency on the day of transfection.
- the cells had been maintained in the AM-1/D CHOd- culture medium containing DMEM High Glucose, 5% FBS, 1X Glutamine Pen/Strep (Gibco), 1X HT, 1X NEAA's and 1X sodium pyruvate (Gibco).
- OptiMEM in a 50ml conical tube and incubate for five minutes.
- LF2000 210 ⁇ l
- the diluted DNA and LF2000 were mixed together and incubated for 20 minutes at room temperature.
- the cells were washed one time with PBS and then 30ml OptiMEM without antibiotics were added to the cells.
- the OptiMEM was aspirated off, and the cells were incubated with 12ml of DNA/LF2000 mixture for 6 hours or overnight in the 37 0 C incubator with shaking. Twenty-four hours post transfection, the cells were split 1:5 into AM-1/D CHOd- culture medium and at differing dilutions for colony selection.
- DHFR selection medium containing 10% Dialyzed FBS (Gibco) in DMEM High Glucose, plus 1X Glutamine Pen/Strep, 1X NEAA's and 1X Na Pyr to allow expression and secretion of protein into the cell medium.
- the selection medium was changed two times a week until the colonies are big enough to pick.
- the pDSRa22 expression vector contains a DHFR expression cassette, which allows transfected cells to grow in the absence of hypoxanthine and thymidine.
- the five T-175 pools of the resulting colonies were scaled up into roller bottles and cultured under serum free conditions. The conditioned media were harvested and replaced at one-week intervals.
- the resulting 3 liters of conditioned medium was filtered through a 0.45 urn cellulose acetate filter (Corning, Acton, MA) and transferred to Protein Chemistry for purification.
- twelve colonies were selected from the 10 cm plates after 10-14 days on DHFR selection medium and expression levels evaluated by western blot using HRP conjugated anti human IgGFc as a probe.
- the three best clones expressing the highest level of each of the different linker length Fc-L-ShK[2-35] fusion proteins were expanded and frozen for future use.
- Fc-LI 0-ShK(2-35) Approximately 1 L of conditioned medium was thawed in a water bath at room temperature. The medium was loaded on to a 5 ml Amersham HiTrap Protein A column at 5 ml/min 7 0 C, and the column was washed with several column volumes of Dulbecco's phosphate buffered saline without divalent cations (PBS) and sample was eluted with a step to 100 mM glycine pH 3.0. The protein A elution pool (approximately 8.5 ml) combined with 71 ⁇ l 3 M sodium acetate and then diluted to 50 ml with water.
- PBS Dulbecco's phosphate buffered saline without divalent cations
- the diluted material was then loaded on to a 5 ml Amersham HiTrap SP-HP column in S-Buffer A (20 mM NaH 2 PO 4 , pH 7.0) at 5 ml/min 7 0 C.
- the column was then washed with several column volumes S-Buffer A, and then developed using a linear gradient from 0% to 75% S-Buffer B (20 mM NaH 2 PO 4 , 1 M NaCI, pH 7.0) at 5 ml/min followed by a step to 100% S-Buffer B at 7 0 C.
- Fractions were then analyzed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE, and the fractions containing the desired product were pooled based on these data.
- the pooled material was then filtere through a 0.22 ⁇ m cellulose acetate filter and concentrated to about 3.9 ml using a Pall Life Sciences Macrosep 10K Omega centrifugal ultra-filtration device.
- the concentrated material was then filtered though a Pall Life Sciences Acrodisc with a 0.22 ⁇ m, 25 mm Mustang E membrane at 2 ml/min room temperature.
- a spectral scan was then conducted on 10 ⁇ l of the filtered material diluted in 700 ⁇ l PBS using a Hewlett Packard 8453 spectrophotometer (Figure 27E).
- the concentration of the filtered material was determined to be 2.76 mg/ml using a calculated molecular mass of 30,008 g/mol and extinction coefficient of 36,900 M" 1 cm- 1 . Since material was found in the permeate, repeated concentration step on the permeate using a new Macrosep cartridge. The new batch of concentrated material was then filtered though a Pall Life Sciences Acrodisc with a 0.22 ⁇ m, 25 mm Mustang E membrane at 2 ml/min room temperature. Both lots of concentrated material were combined into one pool.
- a spectral scan was then conducted on 10 ⁇ l of the combined pool diluted in 700 ⁇ l PBS using a Hewlett Packard 8453 spectrophotometer.
- the concentration of the filtered material was determined to be 3.33 mg/ml using a calculated molecular mass of 30,008 g/mol and extinction coefficient of 36,900 IVr 1 cm- 1 .
- the purity of the filtered material was then assessed using a
- the macromolecular state of the product was then determined using size exclusion chromatography on 50 ⁇ g of the product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH 2 PO 4 , 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm ( Figure 27B).
- the product was then subject to mass spectral analysis by diluting 1 ⁇ l of the sample into 10 ⁇ l of sinapinic acid (10 mg per ml in 0,05% trifluroacetic acid, 50% acetonitrile) .
- the resultant solution (1 ⁇ l) was spotted onto a MALDI sample plate.
- the sample was allowed to dry before being analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse).
- the positive ion/linear mode was used, with an accelerating voltage of 25 kV.
- Each spectrum was produced by accumulating data from ⁇ 200 laser shots.
- External mass calibration was accomplished using purified proteins of known molecular masses (Figure 27F) and the experiment confirmed the itegrity of the peptibody, within experimental error.
- the product was then stored at -80 0 C.
- Figure 31 B shows that purified Fc-LI 0-ShK[2-35] potently blocks human Kv1.3 current (electrophysiology was done as described in Example 36).
- the purified Fc-LI 0-ShK[2-35] molecule also blocked IL-2 (Figure 64A and Figure 64B) and IFN-g ( Figure 65A and Figure 65B) production in human whole blood, as well as, upregulation of CD40L ( Figure 66A and Figure 66B) and IL-2R ( Figure 67A and Figure 67B) on T cells.
- Fc-L5-ShK(2-35) Approximately 1 L of conditioned medium was loaded on to a 5 ml Amersham HiTrap Protein A column at 5 ml/min 7 0 C, and the column was washed with several column volumes of Dulbecco's phosphate buffered saline without divalent cations (PBS) and sample was eluted with a step to 100 mM glycine pH 3.0.
- the protein A elution pool (approximately 9 ml) combined with 450 ⁇ l 1 M tris HCI pH 8.5 followed by 230 ⁇ l 2 M acetic acid and then diluted to 50 ml with water.
- the pH adjusted material was then filtered through a 0.22 ⁇ m cellulose acetate filter and loaded on to a 5 ml Amersham HiTrap SP-HP column in S-Buffer A (20 mM NaH 2 PO 4 , pH 7.0) at 5 ml/min 7 0 C.
- S-Buffer A (20 mM NaH 2 PO 4 , pH 7.0) at 5 ml/min 7 0 C.
- the column was then washed with several column volumes S-Buffer A, and then developed using a linear gradient from 0% to 75% S-Buffer B (20 mM NaH 2 PO 4 , 1 M NaCI, pH 7.0) at 5 ml/min followed by a step to 100% S-Buffer B at 7 0 C.
- the endotoxin level was then determined using a Charles River Laboratories Endosafe-PTS system (0.05 -5 EU/ml sensitivity) using a 92-fold dilution of the sample in Charles Rivers Endotoxin Specific Buffer BG120 yielding a result of ⁇ 1 EU/mg protein.
- the macromolecular state of the product was then determined using size exclusion chromatography on 50 ⁇ g of the product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH 2 PCM, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm (Figure 27H).
- the product was then subject to mass spectral analysis by diluting 1 ⁇ l of the sample into 10 ⁇ l of sinapinic acid (10 mg per ml in 0.05% trifluroacetic acid, 50% acetonitrile) .
- the resultant solution (1 ⁇ l) was spotted onto a MALDI sample plate.
- the sample was allowed to dry before being analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse).
- the positive ion/linear mode was used, with an accelerating voltage of 25 kV.
- Each spectrum was produced by accumulating data from ⁇ 200 laser shots.
- Fc-L25-ShK(2-35) Approximately 1 L of conditioned medium was loaded on to a 5 ml Amersham HiTrap Protein A column at 5 ml/min 7 °C, and the column was washed with several column volumes of Dulbecco's phosphate buffered saline without divalent cations (PBS) and sample was eluted with a step to 100 mM glycine pH 3.0. The protein A elution pool
- Fractions containing the main peak from the chromatogram were pooled and filtered through a 0.22 ⁇ m cellulose acetate filter. A spectral scan was then conducted on 20 ⁇ l of the combined pool diluted in 700 ⁇ l PBS using a Hewlett Packard 8453 spectrophotometer Figure 27J. The concentration of the filtered material was determined to be 1.40 mg/ml using a calculated molecular mass of 31 ,011 g/mol and extinction coefficient of 36,900 M- 1 cm- 1 . The purity of the filtered material was then assessed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE (Figure 27D).
- the endotoxin level was then determined using a Charles River Laboratories Endosafe-PTS system (0.05 - 5 EU/ml sensitivity) using a 28-fold dilution of the sample in Charles Rivers Endotoxin Specific Buffer BG120 yielding a result of ⁇ 1 EU/mg protein.
- the macromolecular state of the product was then determined using size exclusion chromatography on 50 ⁇ g of the product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH ⁇ PC ⁇ , 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm (Figure 27K).
- the product was then subject to mass spectral analysis by diluting 1 ⁇ l of the sample into 10 ⁇ l of sinapinic acid (10 mg per ml in 0.05% trifluroacetic acid, 50% acetonitrile). The resultant solution (1 ⁇ l) was spotted onto a MALDI sample plate. The sample was allowed to dry before being analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ⁇ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses (Figure 27L) and this confirmed the itegrity of the peptibody, within experimental error. The product was then stored at - 8O 0 C.
- bacterial peptibody expression vectors Description of bacterial peptibody expression vectors and procedures for cloning and expression of peptibodies.
- the cloning vector used for bacterial expression (Examples 3-30) is based on pAMG21 (originally described in U.S. Patent 2004/0044188).
- kanamycin resistance component has been replaced with ampicillin resistance by excising the DNA between the unique BstBI and Nsil sites of the vector and replacing with an appropriately digested PCR fragment bearing the beta-lactamase gene using PCR primers CCA ACA CAC TTC GAA AGA CGT TGA TCG GCA C ( SEQ I D NO : 667 ) and CAC CCA ACA ATG CAT CCT TAA AAA AAT TAC GCC C (SEQ I D NO : 668 ) with pUC19 DNA as the template source of the beta-Iactamase gene conferring resistance to ampicillin.
- the new version is called pAMG21ampR.
- Figure 11 A-C and Figure 11 D show the ds-DNA that has been added to the basic vector pAMG21ampR to permit the cloning of peptide fusions to the C-terminus of the Fc gene.
- the DNA has been introduced between the unique Ndel and BamHI sites in the pAMG21ampR vector. This entire region of DNA is shown in Figurei 1A-C.
- the coding region for Fc extends from nt 5134 to 5817 and the protein sequence appears below the DNA sequence. This is followed in frame by a glyX5 linker (nt's 5818-5832).
- a BsmBI site spans nt's 5834-5839. DNA cleavage occurs between nt's 5828 and 5829 on the upper DNA strand and between nt's 5832 and 5833 on the lower DNA strand. Digestion creates 4 bp cohesive termini as shown here. The BsmBI site is underlined.
- a second BsmBI site occurs at nt's 6643 through 6648; viz., CGTCTC.
- DNA cleavage occurs between nt's 6650 and 6651 on the upper strand and between 6654 and 6655 on the lower strand.
- Fig. 11 A-C extends from nt's 5954 to 6610.
- the peptide encoding duplexes in each example bear cohesive ends complementary to those presented by the vector.
- Figure 12A-C shows the ds-DNA sequence that has been added to the basic vector pAMG21ampR to permit the cloning of peptide fusions to the N- terminus of the Fc gene.
- the DNA has been introduced between the unique Ndel and BamHI sites in the pAMG21ampR vector
- the coding region for Fc extends from nt 5640 to 6309 and the protein sequence appears below the DNA sequence. This is preceded in frame by a glyX5 linker (nt's 5614-5628).
- a BsmBI site spans nt's 5138 to 5143; viz., GAGACG. The cutting occurs between nt's 5132 and 5133 on the upper DNA strand and between 5136 and 5137 on the lower DNA strand.
- a second BsmBI site occurs at nt's 5607 through 5612; viz., CGTCTC. Cutting occurs between nt's 5613 and 5614 on the upper strand and between 5617 and 5618 on the lower strand.
- the ble protein sequence is a dispensable zeocin resistance cassette constitutively expressing the Shigella ble protein.
- Figure 12E-F shows the ds-DNA sequence that has been added to the basic vector pAMG21ampR to permit the cloning of peptide fusions to the N-terminus of the Fc gene in which the first two codons of the peptide are to be met-gly.
- the DNA has been introduced between the unique Ndel and BamHI sites in the pAMG21 ampR vector.
- the coding region for Fc extends from nt 5632 to
- a BsmBI site spans nt's 5141 to 5146; viz., GAGACG. The cutting occurs between nt's 5135 and 5136 on the upper DNA strand and between 5139 and 5140 on the lower DNA strand. Digestion creates 4 bp cohesive termini as shown. The BsmBI site is underlined.
- a second BsmBI site occurs at nt's 5607 through 5612; viz., CGTCTC. Cutting occurs between nt's 5613 and 5614 on the upper strand and between 5617 and 5618 on the lower strand.
- cloned peptide sequences are all derived from the annealing of oligonucleotides to create a DNA duplex that is directly ligated into the appropriate vector.
- the duplex is to be inserted at the N-terminus of Fc (see, Examples 15, 16, 52, and 53 herein) the design is as follows with the ordinal numbers matching the listing of oligos in each example:
- No kinasing step is required for the phosphorylation of any of the oligos.
- a successful insertion of a duplex results in the replacement of the dispensable antibiotic resistance cassette (Zeocin resistance for pAMG21ampR-Pep-Fc and chloramphenicol resistance for pAMG21ampR- Fc-Pep).
- the resulting change in phenotype is useful for discriminating recombinant from nonrecombinant clones.
- the mix was heated to 80 0 C and allowed to cool at 0.1 degree/sec to room temperature. To this was added 10 ⁇ l of 1X ligase buffer plus 400 units of T4 DNA ligase. The sample was incubated at 14C for 20 min. Ligase was inactivated by heating at 65°C for 10 minutes. Next, 10 units of restriction endonucleases BsmBI were added follwed by incubation at 55C for one hour to cleave any reformed parental vector molecules. Fifty ul of chemically competent £ coli cells were added and held at 2C for 20 minutes followed by heat shock at 42C for 5 second.
- TGGTTCCGGTGGTGGTGG 1 T //SEQ ID NO: 676; ACCGTCTGTCCTTCTGCCGTAAAACCTGCGGTACCTGC //SEQ ID NO: 677;
- oligo duplex formed is shown below: 0 TGGTTCCGGTGGTGGTGGTTCCTGCATCGACACCATCCCGAAATCCCGTTGCACCGCTTT
- TTCCTGC //SEQ ID NO: 695 121 AAGGACGATTC //SEQ ID NO: 697
- Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
- Fc-L-KTXI Bacterial expression of Fc-L-KTXI .
- the methods to clone and express the peptibody in bacteria are described in Example 3.
- the vector used was pAMG21ampR-Fc-Pep andtheoligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-KTXl
- Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
- Fc-L-KTXI expressed in bacteria Frozen, £ coil paste (28 g) was combined with 210 ml of room temperature 5OmM tris HCI, 5 mM EDTA, pH 8.0 and was brought to about 0.1 mg/ml hen egg white lysozyme.
- the suspended paste was passed through a chilled microfluidizer twice at 12,000 PSI.
- the cell lysate was then centrifuged at 22,000 g for 20 min at 4 0 C.
- the pellet was then resuspended in 200 ml 1 % deoxycholic acid using a tissue grinder and then centrifuged at 22,000 g for 20 min at 4 0 C.
- the pellet was then resuspended in 200 ml water using a tissue grinder and then centrifuged at 22,000 g for 20 min at 4 0 C.
- the pellet (4.8 g) was then dissolved in 48 ml 8 M guanidine HCI, 50 mM tris HCI, pH 8.0.
- the dissolved pellet was then reduced by adding 30 ⁇ l 1 M dithiothreitol to 3 ml of the solution and incubating at 37 0 C for 30 minutes.
- the reduced pellet solution was then centrifuged at 14,000 g for 5 min at room temperature, and then 2.5 ml of the supernatant was transferred to 250 ml of the refolding buffer (2 M urea, 50 mM tris, 160 mM arginine HCI, 5 mM EDTA, 1 mM cystamine HCI, 4 mM cysteine, pH 8.5) at 4 0 C with vigorous stirring. The stirring rate was then slowed and the incubation was continued for 2 days at 4 0 C. The refolding solution was then filtered through a 0.22 ⁇ m cellulose acetate filter and stored at 4 0 C for 3 days.
- the stored refold was then diluted with 1 L of water and the pH was adjusted to 7.5 using 1 M H3PO 4 .
- the pH adjusted material was then loaded on to a 10 ml Amersham SP-HP HiTrap column at 10 ml/min in S-Buffer A (20 mM NaH 2 PO 4 , pH 7.3) at 7 0 C.
- the column was then washed with several column volumes of S-Buffer A, followed by elution with a linear gradient from 0% to 60% S-Buffer B (20 mM NaH 2 PO 4 , 1 M NaCl, pH 7.3) followed by a step to 100% S-Buffer B at 5 ml/min 7 0 C.
- Fractions were then analyzed using a Coomassie brilliant blue stained tris- glycine 4-20% SDS-PAGE, and the fractions containing the desired product were pooled based on these data (45 ml). The pool was then loaded on to a 1 ml Amersham rProtein A HiTrap column in PBS at 2 ml/min 7 0 C. Then column was then washed with several column volumes of PBS and eluted with 100 mM glycine pH 3.0.
- a spectral scan was then conducted on 20 ⁇ l of the combined pool diluted in 700 ⁇ l PBS using a Hewlett Packard 8453 spectrophotometer (Figure 28C).
- the concentration of the filtered material was determined to be 2.49 mg/ml using a calculated molecular mass of 30,504 g/mol and extinction coefficient of 35,410 M" 1 cm- 1 .
- the purity of the filtered material was then assessed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE (Figure 28A).
- the endotoxin level was then determined using a Charles River Laboratories Endosafe-PTS system (0.05 - 5 EU/ml sensitivity) using a 50-fold dilution of the sample in Charles Rivers Endotoxin Specific Buffer BG120 yielding a result of ⁇ 1 EU/mg protein.
- the macromolecular state of the product was then determined using size exclusion chromatography on 45 ⁇ g of the product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM Na ⁇ PO 4 , 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm ( Figure 28B).
- the product was then subject to mass spectral analysis by diluting 1 ⁇ l of the sample into 10 ⁇ l of sinapinic acid (10 mg per ml in 0.05% trifluroacetic acid, 50% acetonitrile) .
- the resultant solution (1 ⁇ l) was spotted onto a MALDI sample plate.
- the sample was allowed to dry before being analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse).
- the positive ion/linear mode was used, with an accelerating voltage of 25 kV.
- Each spectrum was produced by accumulating data from ⁇ 200 laser shots.
- Fc-L-HsTxI Bacterial expression of Fc-L-HsTI .
- the methods to clone and express the peptibody in bacteria are described in Example 3.
- the vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-HsTxI.
- Oligos used to form duplex are shown below:
- oligos above were used to form the duplex shown below: TGGTTCCGGTGGTGGTGGTTCCACCATCATCAACGTTAAATGCACCTCCCCGAAACAGTG
- Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
- Oligos used to form duplex are shown below: TGGTTCCGGTGGTGGTGGTTCCGGTGTTCCGATCAACGTTTCCTGCACCGGT //SEQ ID NO:719;
- the reduced pellet solution was then centrifuged at 14,000 g for 5 min at 5 room temperature, and then 2.5 ml of the supernatant was transferred to 250 ml of the refolding buffer (2 M urea, 50 mM tris, 160 mM arginine HCl, 5 mM EDTA, 1 mM cystamine HCl, 4 mM cysteine, pH 9.5) at 4 0 C with vigorous stirring. The stirring rate was then slowed and the incubation was continued for 2 days at 4 0 C. The refolding solution was then filtered through a 0.22 ⁇ m cellulose acetate filter and stored at -70 0 C.
- the column was then washed with several column volumes of S-Buffer A, followed by elution with a linear gradient from 0% to 60% S-Buffer B (20 mM NaH 2 PO 4 , 1 M NaCI, pH 7.3) followed by a step to 100% S-Buffer B at 5 ml/min 7 0 C.
- Fractions were then analyzed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS- PAGE, and the fractions containing the desired product were pooled based on these data (15 ml).
- the pool was then loaded on to a 1 ml Amersham rProtein A HiTrap column in PBS at 2 ml/min 7 °C.
- a spectral scan was then conducted on 20 ⁇ l of the combined pool diluted in 700 ⁇ l PBS using a Hewlett Packard 8453 spectrophotometer (Figure 29C).
- the concentration of the filtered material was determined to be 1.65 mg/ml using a calculated molecular mass of 30,446 g/mol and extinction coefficient of 35,410 M- 1 cm- 1 .
- the purity of the filtered material was then assessed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE (Figure 29A).
- the endotoxin level was then determined using a Charles River Laboratories Endosafe-PTS system (0.05 -5 EU/ml sensitivity) using a 33-fold dilution of the sample in Charles Rivers Endotoxin Specific Buffer BG120 yielding a result of ⁇ 4 EU/mg protein.
- the macromolecular state of the product was then determined using size exclusion chromatography on 20 ⁇ g of the product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH 2 PO 4 , 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm (Figure 29D).
- the product was then subject to mass spectral analysis by diluting 1 ⁇ l of the sample into 10 ⁇ l of sinapinic acid (10 mg per ml in 0.05% trifluroacetic acid, 50% acetonitrile) .
- the resultant solution (1 ⁇ l) was spotted onto a MALDI sample plate. The sample was allowed to dry before being analyzed using a
- Fc-L-OSKI Bacterial expression of Fc-L-OSKI.
- the methods used to clone and express the peptibody in bacteria were as described in Example 3.
- the vector used was pAMG21ampR-Fc- 5 Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-OSKI .
- Oligos used to form duplex are shown below: CGGTGGTGGTGGTTCCGGTGT": 0 TGCAAAAAAG //SEQ ID NO: 726;
- Fc-L-OSKI E16K, K20D
- the methods to clone and express the peptibody in bacteria are described in Example 3.
- the vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-OSK1(E16K,K20D).
- O Oligos used to form duplex are shown below: TGGTTCCGGTGGTGGTGGTTCCGGTGTTATCATCAACGTTAAATGCAAAATCTCCCGTCAGTGCCTGAAACCG TGCAAAGACG //SEQ ID NO: 733;
- Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen for later use.
- Fc-L-Anuroctoxin Bacterial expression of Fc-L-Anuroctoxin. The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-Anuroctoxin.
- Oligos used to form duplex are shown below:
- Bacterial expression of the peptibody was as described in Example 3 and paste was 5 stored frozen.
- Fc-L-Noxiustoxin or Fc-L-NTX Bacterial expression of Fc-L-Noxiustoxin or Fc-L-NTX, The methods to clone and express O the peptibody in bacteria are described in Example 3.
- the vector used was pAMG21 ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-NTX.
- Oligos used to form duplex are shown below: 5 TGGTTCCGGTGGTGGTGGTTCCACCATCATCAACGTTAAATGCACCTCCCCGAAACAGTGCTCCAAACCGTGC AAAGAACTGT //SEQ ID NO:747;
- Fc-L-Pi2 Bacterial expression of Fc-L-Pi2.
- the methods to clone and express the peptibody in bacteria are described in Example 3.
- the vector used was pAMG21 ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-Pi2.
- Oligos used to form duplex are shown below:
- GCTTCGGTCGT //SEQ ID NO : 755; CTTAACGACCGAAGCATTTGCATTTACGGTTCATGCATTTAGCG //SEQ ID NO: 756;
- Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
- Oligos used to form duplex are shown below: TATGCGTTCTTGTATTGATACTATTCCAAAATCTCGTTGTACTGCTTTTCAATGTAAACATTCTATGAAATAT CGTCTTTCTT //SEQ ID NO: 761;
- Oligos used to form duplex are shown below:
- oligos above were used to form the duplex shown below: TATGTCTTGTATTGATACTATTCCAAAATCTCGTTGTACTGCTTTTCAATGTAAACATTC
- Fc-L-ChTx Bacterial expression of Fc-L-ChTx.
- the methods to clone and express the peptibody in bacteria are described in Example 3.
- the vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-ChTx.
- Oligos used to form duplex are shown below:
- oligos shown above were used to form the duplex below: TGGTTCCGGTGGTGGTGGTTCCCAGTTCACCAACGTTTCCTGCACCACCTCCAAAGAATG
- Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
- Fc-L-MTX Bacterial expression of Fc-L-MTX.
- the methods to clone and express the peptibody in bacteria are described in Example 3.
- the vector used was pAMG21 ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-MTX.
Abstract
Description

























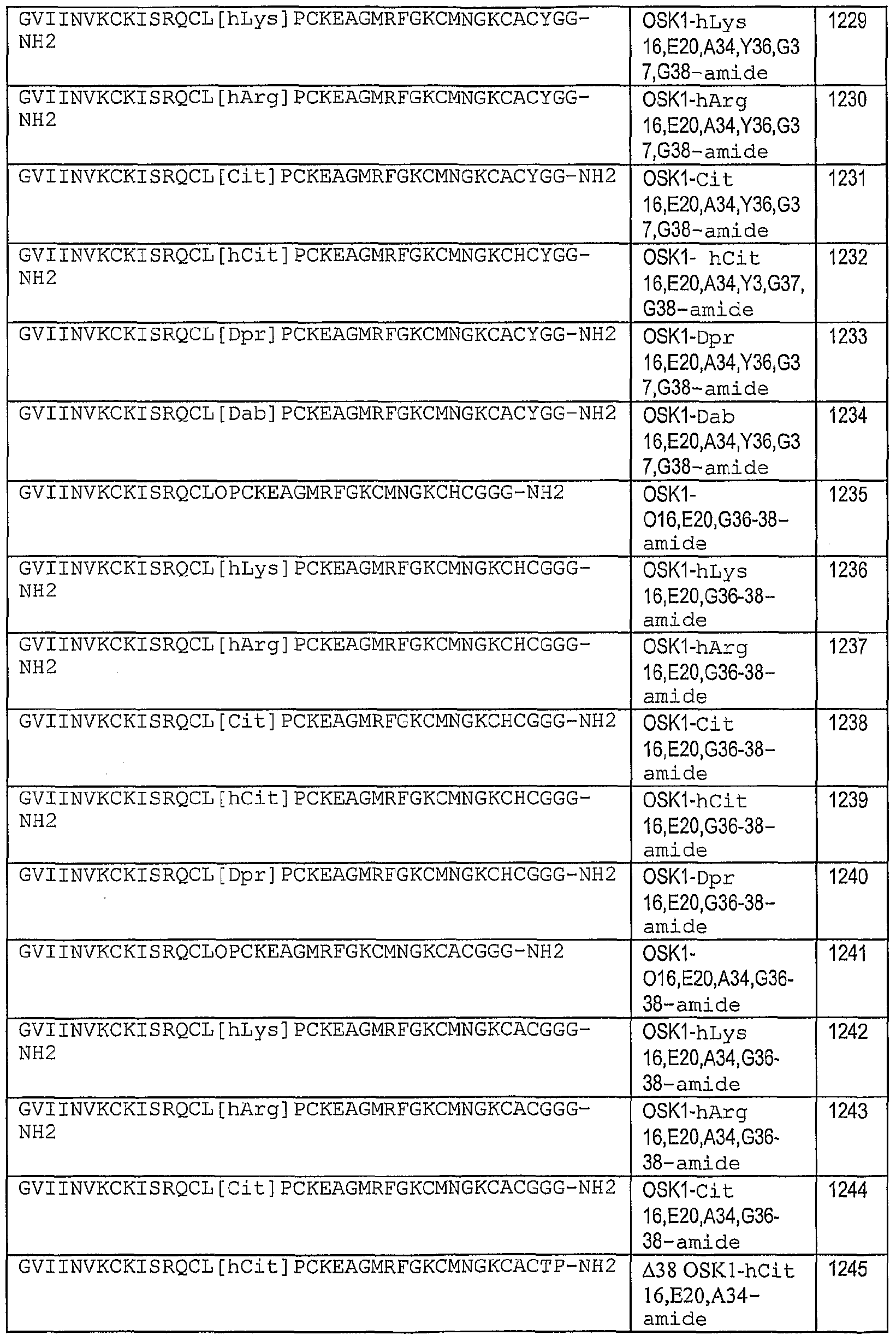





































Claims
Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MX2007013031A MX2007013031A (en) | 2005-04-22 | 2006-04-18 | Toxin peptide therapeutic agents. |
| JP2008507949A JP5087536B2 (en) | 2005-04-22 | 2006-04-18 | Toxin peptides with extended blood half-life |
| AU2006239928A AU2006239928B8 (en) | 2005-04-22 | 2006-04-18 | Toxin peptides with extended blood halflife |
| CA002604999A CA2604999A1 (en) | 2005-04-22 | 2006-04-18 | Toxin peptides with extended blood halflife |
| EA200702313A EA013470B1 (en) | 2005-04-22 | 2006-04-18 | Therapeutic agents based on toxin peptides |
| NZ562201A NZ562201A (en) | 2005-04-22 | 2006-04-18 | Toxin peptides with extended blood halflife |
| BRPI0607750-1A BRPI0607750A2 (en) | 2005-04-22 | 2006-04-18 | matter, and pharmaceutical compositions, dna, expression vector, host cell, and, use of a matter composition |
| EP06751051A EP1896080A2 (en) | 2005-04-22 | 2006-04-18 | Toxin peptides with extended blood halflife |
| IL186527A IL186527A0 (en) | 2005-04-22 | 2007-10-09 | Toxin peptide therapeutic agents |
| NO20076013A NO20076013L (en) | 2005-04-22 | 2007-11-21 | Toxin peptide therapeutics |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US67434205P | 2005-04-22 | 2005-04-22 | |
| US60/674,342 | 2005-04-22 | ||
| US11/406,454 US7833979B2 (en) | 2005-04-22 | 2006-04-17 | Toxin peptide therapeutic agents |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2006116156A2 true WO2006116156A2 (en) | 2006-11-02 |
| WO2006116156A3 WO2006116156A3 (en) | 2007-10-04 |
Family
ID=37894298
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2006/015199 WO2006116156A2 (en) | 2005-04-22 | 2006-04-18 | Toxin peptides with extended blood halflife |
Country Status (16)
| Country | Link |
|---|---|
| US (2) | US7833979B2 (en) |
| EP (1) | EP1896080A2 (en) |
| JP (2) | JP5087536B2 (en) |
| KR (2) | KR20080021606A (en) |
| AR (1) | AR053234A1 (en) |
| AU (1) | AU2006239928B8 (en) |
| BR (1) | BRPI0607750A2 (en) |
| CA (1) | CA2604999A1 (en) |
| CR (1) | CR9452A (en) |
| EA (1) | EA013470B1 (en) |
| IL (1) | IL186527A0 (en) |
| MX (1) | MX2007013031A (en) |
| NO (1) | NO20076013L (en) |
| NZ (1) | NZ562201A (en) |
| TW (1) | TW200716748A (en) |
| WO (1) | WO2006116156A2 (en) |
Cited By (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008088422A2 (en) * | 2006-10-25 | 2008-07-24 | Amgen Inc. | Toxin peptide therapeutic agents |
| WO2008139243A1 (en) * | 2007-05-14 | 2008-11-20 | Universidad Nacional Autónoma de México | Vm23 and vm24, two scorpion peptides that block human t-lymphocyte potassium channels (sub-type kv1.3) with high selectivity and decrease the in vivo dth-responses in rats |
| WO2008153745A3 (en) * | 2007-05-22 | 2009-07-16 | Amgen Inc | Compositions and methods for producing bioactive fusion proteins |
| EP2229473A2 (en) * | 2007-12-07 | 2010-09-22 | Steven A. Goldstein | Identification of toxin ligands |
| WO2010108154A2 (en) * | 2009-03-20 | 2010-09-23 | Amgen Inc. | Selective and potent peptide inhibitors of kv1.3 |
| CN102112493A (en) * | 2008-07-23 | 2011-06-29 | 韩美控股株式会社 | A polypeptide complex comprising non-peptidyl polymer having three functional ends |
| WO2012040518A2 (en) | 2010-09-22 | 2012-03-29 | Amgen Inc. | Carrier immunoglobulins and uses thereof |
| WO2012125973A2 (en) | 2011-03-16 | 2012-09-20 | Amgen Inc. | Potent and selective inhibitors of nav1.3 and nav1.7 |
| WO2013061106A1 (en) * | 2011-10-28 | 2013-05-02 | University Of Debrecen | Modified peptide toxins |
| WO2014116937A1 (en) | 2013-01-25 | 2014-07-31 | Janssen Biotech, Inc. | Kv1.3 antagonists and methods of use |
| WO2014165277A2 (en) | 2013-03-12 | 2014-10-09 | Amgen Inc. | POTENT AND SELECTIVE INHIBITORS OF Nav1.7 |
| CN104177489A (en) * | 2014-07-29 | 2014-12-03 | 暨南大学 | Gene-recombined TNF-alpha derivative RMP16 and preparation method and application thereof |
| US9102751B2 (en) | 2012-05-18 | 2015-08-11 | Janssen Biotech, Inc. | Huwentoxin-IV variants and methods of use |
| US9234015B2 (en) | 2010-02-04 | 2016-01-12 | Morphotek, Inc. | Chlorotoxin polypeptides and conjugates and uses thereof |
| US9603952B2 (en) | 2008-05-15 | 2017-03-28 | Morphotek, Inc. | Treatment of metastatic tumors |
| US9616109B2 (en) | 2014-10-22 | 2017-04-11 | Extend Biosciences, Inc. | Insulin vitamin D conjugates |
| US9624280B2 (en) | 2013-10-03 | 2017-04-18 | Janssen Biotech, Inc. | Protoxin-II variants and methods of use |
| US9789197B2 (en) | 2014-10-22 | 2017-10-17 | Extend Biosciences, Inc. | RNAi vitamin D conjugates |
| US9884124B2 (en) | 2012-05-17 | 2018-02-06 | Extend Biosciences, Inc. | Carriers for improved drug delivery |
| US9944683B2 (en) | 2010-05-11 | 2018-04-17 | Fred Hutchinson Cancer Research Center | Chlorotoxin variants, conjugates, and methods for their use |
| US10156559B2 (en) | 2012-12-10 | 2018-12-18 | Fred Hutchinson Cancer Research Center | Lipocalin fusion partners |
| EP3424955A1 (en) * | 2013-12-25 | 2019-01-09 | Daiichi Sankyo Company, Limited | Anti-trop2 antibody-drug conjugate |
| US10406202B2 (en) | 2014-10-22 | 2019-09-10 | Extend Biosciences, Inc. | Therapeutic vitamin D conjugates |
| US10414808B2 (en) | 2012-05-18 | 2019-09-17 | Janssen Biotech, Inc. | Huwentoxin-IV variants and methods of use |
| US10463714B2 (en) | 2015-04-02 | 2019-11-05 | Janssen Biotech, Inc. | Protoxin-II variants and methods of use |
| EP3496742A4 (en) * | 2016-12-01 | 2020-04-08 | University Of South Florida | Peptibodies, compositions thereof, and methods of treating atrial fibrillation |
| WO2021053194A1 (en) | 2019-09-20 | 2021-03-25 | Zealand Pharma A/S | Kv1.3 blockers |
| US10995125B2 (en) | 2015-03-03 | 2021-05-04 | Janssen Biotech, Inc. | Protoxin-II variants and methods of use |
| WO2022200374A1 (en) | 2021-03-23 | 2022-09-29 | Zealand Pharma A/S | Kv1.3 blockers |
| US11559580B1 (en) | 2013-09-17 | 2023-01-24 | Blaze Bioscience, Inc. | Tissue-homing peptide conjugates and methods of use thereof |
| US20230331827A1 (en) * | 2017-07-12 | 2023-10-19 | Maxion Therapeutics Limited | Potassium channel inhibitors |
| US11932673B2 (en) | 2017-07-12 | 2024-03-19 | Maxion Therapeutics Limited | Sodium channel inhibitors |
Families Citing this family (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9617671D0 (en) * | 1996-08-23 | 1996-10-02 | Microbiological Res Authority | Recombinant toxin fragments |
| US7192596B2 (en) * | 1996-08-23 | 2007-03-20 | The Health Protection Agency Ipsen Limited | Recombinant toxin fragments |
| GB0414272D0 (en) * | 2004-06-25 | 2004-07-28 | Cellpep Sa | OsK1 derivatives |
| EP2049138A4 (en) * | 2006-07-14 | 2011-11-09 | Georgia Tech Res Inst | Clc channel ligand |
| WO2008051383A2 (en) * | 2006-10-19 | 2008-05-02 | Amgen Inc. | Use of alcohol co-solvents to improve pegylation reaction yields |
| EP2144930A1 (en) * | 2007-04-18 | 2010-01-20 | ZymoGenetics, Inc. | Single chain fc, methods of making and methods of treatment |
| US20090252729A1 (en) * | 2007-05-14 | 2009-10-08 | Farrington Graham K | Single-chain Fc (scFc) regions, binding polypeptides comprising same, and methods related thereto |
| EP2215247B1 (en) * | 2007-11-13 | 2014-09-24 | The Scripps Research Institute | Production of cytotoxic antibody-toxin fusion in eukaryotic algae |
| WO2010033221A1 (en) | 2008-09-19 | 2010-03-25 | Nektar Therapeutics | Polymer conjugates of protegrin peptides |
| US20110171163A1 (en) * | 2008-09-19 | 2011-07-14 | Nektar Therapeutics | Polymer conjugates of ziconotide peptides |
| US20110166063A1 (en) * | 2008-09-19 | 2011-07-07 | Nektar Therapeutics | Polymer conjugates of therapeutic peptides |
| US8252228B1 (en) * | 2008-10-13 | 2012-08-28 | Abbott Cardiovascular Systems Inc. | Methods for sterilizing carriers for treatment of a kidney |
| US20120134984A1 (en) * | 2009-06-01 | 2012-05-31 | Olga Lubman | Molecules with extended half-lives and uses thereof |
| EP2501407A1 (en) | 2009-11-20 | 2012-09-26 | Amgen Inc. | Anti-orai1 antigen binding proteins and uses thereof |
| CA2807673A1 (en) | 2010-08-10 | 2012-02-16 | Xinyi Cynthia Chen | Dual function in vitro target binding assay for the detection of neutralizing antibodies against target antibodies |
| WO2012170392A2 (en) | 2011-06-06 | 2012-12-13 | Kineta One, Llc | Shk-based pharmaceutical compositions and methods of manufacturing and using the same |
| BR112014008065A2 (en) * | 2011-10-03 | 2019-09-24 | Kineta One Llc | treatment of obesity and obesity-related disorders by pharmacological targeting of potassium channels kv1.3 |
| WO2013065343A1 (en) * | 2011-10-31 | 2013-05-10 | 株式会社 島津製作所 | Peptide-hinge-free flexible antibody-like molecule |
| US20150011431A1 (en) | 2012-01-09 | 2015-01-08 | The Scripps Research Institute | Humanized antibodies |
| EP3342785B1 (en) | 2012-10-11 | 2019-12-25 | Daiichi Sankyo Company, Limited | Linkers for antibody-drug conjugates |
| US9872924B2 (en) | 2012-10-19 | 2018-01-23 | Daiichi Sankyo Company, Limited | Antibody-drug conjugate produced by binding through linker having hydrophilic structure |
| ES2875957T3 (en) | 2012-12-20 | 2021-11-11 | Amgen Inc | APJ receptor agonists and their uses |
| EA030929B1 (en) * | 2013-02-14 | 2018-10-31 | Дзе Борд Оф Трастиз Оф Дзе Юниверсити Оф Арканзас | Compositions and methods of enhancing immune responses to eimeria or limiting eimeria infection |
| US9789209B2 (en) | 2013-03-14 | 2017-10-17 | The Regents Of The University Of California, Berke | Activatable membrane-interacting peptides and methods of use |
| CN111518199A (en) | 2013-07-18 | 2020-08-11 | 图鲁斯生物科学有限责任公司 | Humanized antibodies with ultralong complementarity determining regions |
| JP2016527250A (en) * | 2013-07-22 | 2016-09-08 | キネタ ワン リミテッド ライアビリティ カンパニーKineta One,Llc | Ophthalmic use of toxin-based therapeutic peptides and pharmaceutical compositions thereof |
| WO2015061826A1 (en) * | 2013-10-28 | 2015-05-07 | Monash University | Novel scorpion toxin analogue and method for treating autoimmune diseases |
| EP4212552A1 (en) | 2014-01-31 | 2023-07-19 | Daiichi Sankyo Company, Limited | Anti-her2 antibody-drug conjugate |
| ES2754348T3 (en) | 2014-04-10 | 2020-04-17 | Daiichi Sankyo Co Ltd | Conjugate of (anti-HER2 antibody) -pharmaceutical |
| PT3129063T (en) | 2014-04-10 | 2021-04-01 | Daiichi Sankyo Europe Gmbh | Anti-her3 antibody-drug conjugate |
| GB201408135D0 (en) * | 2014-05-08 | 2014-06-25 | Conogenetix Biosciences Gmbh | Kv1.3 potassium channel antagonists |
| CA2951391C (en) | 2014-06-10 | 2021-11-02 | Amgen Inc. | Apelin polypeptides |
| JP6543342B2 (en) * | 2014-08-14 | 2019-07-10 | アルハマドシャー,マモウン,エム. | Conjugation of a pharmaceutically active agent with a transthyretin ligand via a regulatable linker to extend serum half life |
| EP3191131A4 (en) | 2014-08-21 | 2018-09-05 | The General Hospital Corporation | Tumor necrosis factor superfamily and tnf-like ligand muteins and methods of preparing and using the same |
| US9616114B1 (en) | 2014-09-18 | 2017-04-11 | David Gordon Bermudes | Modified bacteria having improved pharmacokinetics and tumor colonization enhancing antitumor activity |
| AU2015342767B2 (en) | 2014-11-07 | 2021-05-13 | Kineta Chronic Pain, Llc | Modifications and uses of conotoxin peptides |
| US20180264080A1 (en) * | 2015-01-09 | 2018-09-20 | Kineta One, Llc | TOPICAL APPLICATIONS OF Kv1.3 CHANNEL BLOCKING PEPTIDES TO TREAT SKIN INFLAMMATION |
| CN107849148B (en) | 2015-05-21 | 2023-09-19 | 哈普恩治疗公司 | Trispecific binding proteins and methods of use |
| WO2017002776A1 (en) | 2015-06-29 | 2017-01-05 | 第一三共株式会社 | Method for selectively manufacturing antibody-drug conjugate |
| US11623958B2 (en) | 2016-05-20 | 2023-04-11 | Harpoon Therapeutics, Inc. | Single chain variable fragment CD3 binding proteins |
| EA201892693A1 (en) | 2016-05-20 | 2019-04-30 | Харпун Терапьютикс, Инк. | PROTEINS CONTAINING A SINGLE-BANDY VARIABLE FRAGMENT, CONNECTING CD3 |
| US10100106B2 (en) | 2016-05-20 | 2018-10-16 | Harpoon Therapeutics, Inc. | Single domain serum albumin binding protein |
| JP7215997B2 (en) | 2016-11-23 | 2023-01-31 | ハープーン セラピューティクス,インク. | Trispecific proteins targeting prostate specific membrane antigen (PSMA) and methods of use |
| AU2017363300A1 (en) | 2016-11-23 | 2019-06-20 | Harpoon Therapeutics, Inc. | Prostate specific membrane antigen binding protein |
| US11180535B1 (en) | 2016-12-07 | 2021-11-23 | David Gordon Bermudes | Saccharide binding, tumor penetration, and cytotoxic antitumor chimeric peptides from therapeutic bacteria |
| US11129906B1 (en) | 2016-12-07 | 2021-09-28 | David Gordon Bermudes | Chimeric protein toxins for expression by therapeutic bacteria |
| JPWO2018110515A1 (en) | 2016-12-12 | 2019-10-24 | 第一三共株式会社 | Combination of antibody-drug conjugate and immune checkpoint inhibitor |
| TWI780104B (en) | 2017-01-17 | 2022-10-11 | 日商第一三共股份有限公司 | Anti gpr20 antibodies and anti gpr20 antibody-drug conjugates, as well as manufacturing method and use thereof |
| EP3589662A4 (en) | 2017-02-28 | 2020-12-30 | Harpoon Therapeutics, Inc. | Inducible monovalent antigen binding protein |
| US10668113B2 (en) | 2017-03-31 | 2020-06-02 | Vivekananda Ramana | Formulation for targeting cancer in humans and canines using venom |
| US10668130B2 (en) | 2017-03-31 | 2020-06-02 | Vivekananda Ramana | Formulation for targeting cancer in humans and canines using chlorotoxin |
| US10668112B2 (en) | 2017-03-31 | 2020-06-02 | Vivekananda Ramana | Formulation for targeting cancer in humans and canines |
| CA3063359A1 (en) | 2017-05-12 | 2018-11-15 | Harpoon Therapeutics, Inc. | Mesothelin binding proteins |
| US10730954B2 (en) | 2017-05-12 | 2020-08-04 | Harpoon Therapeutics, Inc. | MSLN targeting trispecific proteins and methods of use |
| TWI794230B (en) | 2017-05-15 | 2023-03-01 | 日商第一三共股份有限公司 | Anti cdh6 antibodies and anti cdh6 antibody drug conjugates, as well as manufacturing method thereof |
| BR112020003646A2 (en) | 2017-08-31 | 2020-09-01 | Daiichi Sankyo Company, Limited | crystals, methods for producing crystals and an antibody-drug conjugate, and, salt. |
| CN117838880A (en) | 2017-08-31 | 2024-04-09 | 第一三共株式会社 | New method for preparing antibody-drug conjugate |
| US20200262877A1 (en) * | 2017-10-09 | 2020-08-20 | Blaze Bioscience, Inc. | Ion channel-binding peptides and methods of use thereof |
| CN111465612A (en) | 2017-10-13 | 2020-07-28 | 哈普恩治疗公司 | B cell maturation antigen binding proteins |
| AU2018347582A1 (en) | 2017-10-13 | 2020-05-07 | Harpoon Therapeutics, Inc. | Trispecific proteins and methods of use |
| US20210221910A1 (en) | 2018-05-18 | 2021-07-22 | Glycotope Gmbh | Anti-muc1 antibody |
| WO2019225296A1 (en) * | 2018-05-24 | 2019-11-28 | 国立大学法人熊本大学 | Carrier for drug delivery use |
| AU2019346466A1 (en) | 2018-09-25 | 2021-05-20 | Harpoon Therapeutics, Inc. | DLL3 binding proteins and methods of use |
| EP4106806A1 (en) | 2020-02-21 | 2022-12-28 | Harpoon Therapeutics, Inc. | Flt3 binding proteins and methods of use |
| WO2022066767A2 (en) | 2020-09-23 | 2022-03-31 | Aldevron, Llc | Potent and selective inhibitors of nav1.7 |
| CN112353934B (en) * | 2020-11-23 | 2023-05-19 | 中国人民解放军军事科学院军事医学研究院 | Conotoxin pharmaceutical composition and freeze-dried preparation thereof |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006042151A2 (en) | 2004-10-07 | 2006-04-20 | The Regents Of The University Of California | Analogs of shk toxin and their uses in selective inhibition of kv1.3 potassium channels |
| WO2008088422A2 (en) | 2006-10-25 | 2008-07-24 | Amgen Inc. | Toxin peptide therapeutic agents |
| WO2010108154A2 (en) | 2009-03-20 | 2010-09-23 | Amgen Inc. | Selective and potent peptide inhibitors of kv1.3 |
Family Cites Families (84)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5206344A (en) | 1985-06-26 | 1993-04-27 | Cetus Oncology Corporation | Interleukin-2 muteins and polymer conjugation thereof |
| JPH0669959B2 (en) | 1985-09-26 | 1994-09-07 | 東燃株式会社 | Immunosuppressant containing cholera toxin as an active ingredient |
| AU607172B2 (en) | 1986-12-22 | 1991-02-28 | Cygnus, Inc. | Diffusion matrix for transdermal drug administration |
| US5023084A (en) | 1986-12-29 | 1991-06-11 | Rutgers, The State University Of New Jersey | Transdermal estrogen/progestin dosage unit, system and process |
| WO1988006451A1 (en) | 1987-02-24 | 1988-09-07 | Xoma Corporation | Immunosuppression in immunotoxin based human therapy |
| US4847325A (en) | 1988-01-20 | 1989-07-11 | Cetus Corporation | Conjugation of polymer to colony stimulating factor-1 |
| US4925677A (en) | 1988-08-31 | 1990-05-15 | Theratech, Inc. | Biodegradable hydrogel matrices for the controlled release of pharmacologically active agents |
| US4994439A (en) | 1989-01-19 | 1991-02-19 | California Biotechnology Inc. | Transmembrane formulations for drug administration |
| US5397702A (en) | 1989-03-06 | 1995-03-14 | The Regents Of The University Of California | Assay for and treatment of autoimmune diseases |
| US4994436A (en) * | 1989-03-10 | 1991-02-19 | American Air Liquide | Process for safely destroying organic binders in ceramic components during the firing process |
| US4906159A (en) | 1989-03-22 | 1990-03-06 | Caterpillar Industrial Inc. | Freely positionable load carrying attachment for an automatic guided vehicle |
| US5166322A (en) | 1989-04-21 | 1992-11-24 | Genetics Institute | Cysteine added variants of interleukin-3 and chemical modifications thereof |
| US6267964B1 (en) | 1989-08-01 | 2001-07-31 | Affibody Technology Sweden Ab | Stabilized protein or peptide conjugates able to bond albumin having extended biological half-lives |
| US5171264A (en) | 1990-02-28 | 1992-12-15 | Massachusetts Institute Of Technology | Immobilized polyethylene oxide star molecules for bioapplications |
| US6552170B1 (en) | 1990-04-06 | 2003-04-22 | Amgen Inc. | PEGylation reagents and compounds formed therewith |
| IL98932A0 (en) | 1990-07-27 | 1992-07-15 | Univ California | Assay,kits and methods based on nk+channel expression |
| JP3051145B2 (en) | 1990-08-28 | 2000-06-12 | 住友製薬株式会社 | Novel polyethylene glycol derivative modified peptide |
| US5252714A (en) | 1990-11-28 | 1993-10-12 | The University Of Alabama In Huntsville | Preparation and use of polyethylene glycol propionaldehyde |
| NZ241954A (en) | 1991-03-15 | 1994-01-26 | Amgen Inc | Compositions of g-csf for pulmonary administration. |
| AU1674292A (en) | 1991-03-15 | 1992-10-21 | Synergen, Inc. | Pegylation of polypeptides |
| FR2686899B1 (en) | 1992-01-31 | 1995-09-01 | Rhone Poulenc Rorer Sa | NOVEL BIOLOGICALLY ACTIVE POLYPEPTIDES, THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM. |
| US5792451A (en) | 1994-03-02 | 1998-08-11 | Emisphere Technologies, Inc. | Oral drug delivery compositions and methods |
| JPH0640945A (en) | 1992-07-23 | 1994-02-15 | Kureha Chem Ind Co Ltd | Fc fragment binding antitumor agent |
| US5346701A (en) | 1993-02-22 | 1994-09-13 | Theratech, Inc. | Transmucosal delivery of macromolecular drugs |
| US5494895A (en) | 1993-07-22 | 1996-02-27 | Merck & Co., Inc. | Scorpion peptide margatoxin with immunosuppressant activity |
| US5460820B1 (en) | 1993-08-03 | 1999-08-03 | Theratech Inc | Method for providing testosterone and optionally estrogen replacement therapy to women |
| US6342225B1 (en) | 1993-08-13 | 2002-01-29 | Deutshces Wollforschungsinstitut | Pharmaceutical active conjugates |
| US5919455A (en) | 1993-10-27 | 1999-07-06 | Enzon, Inc. | Non-antigenic branched polymer conjugates |
| US5643575A (en) | 1993-10-27 | 1997-07-01 | Enzon, Inc. | Non-antigenic branched polymer conjugates |
| US5446090A (en) | 1993-11-12 | 1995-08-29 | Shearwater Polymers, Inc. | Isolatable, water soluble, and hydrolytically stable active sulfones of poly(ethylene glycol) and related polymers for modification of surfaces and molecules |
| JPH09509428A (en) | 1994-02-23 | 1997-09-22 | カイロン コーポレイション | Methods and compositions for increasing the serum half-life of pharmacologically active agents |
| FR2717688B1 (en) | 1994-03-28 | 1996-07-05 | Lhd Lab Hygiene Dietetique | Transdermal matrix system for administration of an estrogen and / or an EVA-based progestin. |
| US6309853B1 (en) | 1994-08-17 | 2001-10-30 | The Rockfeller University | Modulators of body weight, corresponding nucleic acids and proteins, and diagnostic and therapeutic uses thereof |
| US5824784A (en) | 1994-10-12 | 1998-10-20 | Amgen Inc. | N-terminally chemically modified protein compositions and methods |
| MX9704550A (en) | 1994-12-22 | 1997-10-31 | Astra Ab | Aerosol drug formulations. |
| US5932462A (en) | 1995-01-10 | 1999-08-03 | Shearwater Polymers, Inc. | Multiarmed, monofunctional, polymer for coupling to molecules and surfaces |
| US5739277A (en) | 1995-04-14 | 1998-04-14 | Genentech Inc. | Altered polypeptides with increased half-life |
| US6096871A (en) | 1995-04-14 | 2000-08-01 | Genentech, Inc. | Polypeptides altered to contain an epitope from the Fc region of an IgG molecule for increased half-life |
| ATE279947T1 (en) | 1996-03-18 | 2004-11-15 | Univ Texas | IMMUNOGLOBULIN-LIKE DOMAIN WITH INCREASED HALF-LIFE TIMES |
| US5783208A (en) | 1996-07-19 | 1998-07-21 | Theratech, Inc. | Transdermal drug delivery matrix for coadministering estradiol and another steroid |
| US5763478A (en) | 1996-10-16 | 1998-06-09 | Merck & Co., Inc. | Triterpene derivatives with immunosuppressant activity |
| US6077680A (en) | 1996-11-27 | 2000-06-20 | The University Of Florida | ShK toxin compositions and methods of use |
| DE69734451T2 (en) | 1996-12-26 | 2006-07-13 | Suntory Limited | SCORPION-SPECIFIC NEUROPEPTIDES |
| US6548644B1 (en) | 1997-03-10 | 2003-04-15 | Immunex Corporation | Site protected protein modification |
| US5990237A (en) | 1997-05-21 | 1999-11-23 | Shearwater Polymers, Inc. | Poly(ethylene glycol) aldehyde hydrates and related polymers and applications in modifying amines |
| SG129211A1 (en) | 1997-11-06 | 2007-02-26 | Univ Singapore | Therapeutic molecules |
| US6703485B2 (en) | 1997-12-23 | 2004-03-09 | The Trustees Of Columbia University In The City Of New York | Brain and heart cyclic nucleotide gated ion channel compounds and uses thereof |
| US6551821B1 (en) | 1997-12-23 | 2003-04-22 | The Trustees Of Columbia University In The City Of New York | Brain cyclic nucleotide gated ion channel and uses thereof |
| JP3515072B2 (en) | 1998-01-23 | 2004-04-05 | アレクシオン ファーマシューティカルズ, インコーポレイテッド | Methods and compositions used to identify growth factor mimetics, growth factors and inhibitors |
| JP3530004B2 (en) | 1998-02-06 | 2004-05-24 | 株式会社日立ユニシアオートモティブ | Inhalation type dispenser |
| US6451986B1 (en) | 1998-06-22 | 2002-09-17 | Immunex Corporation | Site specific protein modification |
| US6660843B1 (en) * | 1998-10-23 | 2003-12-09 | Amgen Inc. | Modified peptides as therapeutic agents |
| US6818418B1 (en) | 1998-12-10 | 2004-11-16 | Compound Therapeutics, Inc. | Protein scaffolds for antibody mimics and other binding proteins |
| US6245740B1 (en) | 1998-12-23 | 2001-06-12 | Amgen Inc. | Polyol:oil suspensions for the sustained release of proteins |
| US6166322A (en) * | 1999-04-16 | 2000-12-26 | Industrial Technology Research Institute | Encapulation process for mono-and polycrystalline silicon solar cell modules |
| ES2257298T3 (en) | 1999-05-17 | 2006-08-01 | Conjuchem, Inc. | PROTECTION OF ENDOGENOUS THERAPEUTIC PEPTIDES AGAINST THE ACTIVITY OF PEPTIDASE THROUGH CONJUGATION WITH BLOOD COMPONENTS. |
| US6887470B1 (en) | 1999-09-10 | 2005-05-03 | Conjuchem, Inc. | Protection of endogenous therapeutic peptides from peptidase activity through conjugation to blood components |
| US6849714B1 (en) | 1999-05-17 | 2005-02-01 | Conjuchem, Inc. | Protection of endogenous therapeutic peptides from peptidase activity through conjugation to blood components |
| US6768002B1 (en) | 1999-06-22 | 2004-07-27 | E. I. Du Pont De Nemours And Company | Scorpion toxins |
| AU781793B2 (en) | 1999-06-22 | 2005-06-16 | E.I. Du Pont De Nemours And Company | Scorpion toxins from buthotus judaicus |
| US6096891A (en) | 1999-12-09 | 2000-08-01 | Air Products And Chemicals, Inc. | Process for the production of cyclic N,N'-dialkylureas |
| US6602498B2 (en) | 2000-02-22 | 2003-08-05 | Shearwater Corporation | N-maleimidyl polymer derivatives |
| SE0000935D0 (en) | 2000-03-21 | 2000-03-21 | Astrazeneca Ab | An inhalation device |
| US6946134B1 (en) | 2000-04-12 | 2005-09-20 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| AU2001264563A1 (en) | 2000-04-12 | 2001-10-30 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| AU2001259432B2 (en) | 2000-05-03 | 2005-04-21 | Amgen Inc. | Modified peptides, comprising an FC domain, as therapeutic agents |
| EP1177806A1 (en) | 2000-08-04 | 2002-02-06 | The Technology Partnership Public Limited Company | Dry powder inhaler |
| PL362324A1 (en) * | 2001-02-19 | 2004-10-18 | Merck Patent Gmbh | Artificial fusion proteins with reduced immunogenicity |
| AU2002335930B2 (en) | 2001-03-09 | 2005-07-28 | Morphosys Ag | Serum albumin binding moieties |
| US6858580B2 (en) * | 2001-06-04 | 2005-02-22 | Nobex Corporation | Mixtures of drug-oligomer conjugates comprising polyalkylene glycol, uses thereof, and methods of making same |
| US6861405B2 (en) | 2001-06-12 | 2005-03-01 | Yale University | Compositions and methods relating to glucose metabolism, weight control, and food intake |
| US6770625B2 (en) | 2001-09-07 | 2004-08-03 | Nobex Corporation | Pharmaceutical compositions of calcitonin drug-oligomer conjugates and methods of treating diseases therewith |
| CA2484556A1 (en) | 2001-12-21 | 2003-07-24 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| US6900317B2 (en) | 2002-02-19 | 2005-05-31 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Salts of the CGRP antagonist BIBN4096 and inhalable powdered medicaments containing them |
| TW200403058A (en) | 2002-04-19 | 2004-03-01 | Bristol Myers Squibb Co | Heterocyclo inhibitors of potassium channel function |
| GB0219512D0 (en) | 2002-08-21 | 2002-10-02 | Norton Healthcare Ltd | Inhalation compositions with high drug ratios |
| US6919426B2 (en) | 2002-09-19 | 2005-07-19 | Amgen Inc. | Peptides and related molecules that modulate nerve growth factor activity |
| WO2004043396A2 (en) | 2002-11-09 | 2004-05-27 | Nobex Corporation | Modified carbamate-containing prodrugs and methods of synthesizing same |
| US7348004B2 (en) | 2003-05-06 | 2008-03-25 | Syntonix Pharmaceuticals, Inc. | Immunoglobulin chimeric monomer-dimer hybrids |
| TWI353991B (en) | 2003-05-06 | 2011-12-11 | Syntonix Pharmaceuticals Inc | Immunoglobulin chimeric monomer-dimer hybrids |
| AU2004247042A1 (en) | 2003-06-12 | 2004-12-23 | Eli Lilly And Company | Fusion proteins |
| PL2256134T3 (en) | 2003-11-13 | 2014-06-30 | Hanmi Science Co Ltd | IgG Fc fragment for a drug carrier and method for the preparation thereof |
| GB0414272D0 (en) | 2004-06-25 | 2004-07-28 | Cellpep Sa | OsK1 derivatives |
| EP1797127B1 (en) | 2004-09-24 | 2017-06-14 | Amgen Inc. | Modified fc molecules |
-
2006
- 2006-04-17 US US11/406,454 patent/US7833979B2/en active Active
- 2006-04-18 NZ NZ562201A patent/NZ562201A/en not_active IP Right Cessation
- 2006-04-18 MX MX2007013031A patent/MX2007013031A/en active IP Right Grant
- 2006-04-18 CA CA002604999A patent/CA2604999A1/en not_active Abandoned
- 2006-04-18 BR BRPI0607750-1A patent/BRPI0607750A2/en not_active IP Right Cessation
- 2006-04-18 JP JP2008507949A patent/JP5087536B2/en not_active Expired - Fee Related
- 2006-04-18 KR KR1020077027146A patent/KR20080021606A/en active Search and Examination
- 2006-04-18 EA EA200702313A patent/EA013470B1/en not_active IP Right Cessation
- 2006-04-18 KR KR1020097027223A patent/KR20100017942A/en not_active Application Discontinuation
- 2006-04-18 AU AU2006239928A patent/AU2006239928B8/en not_active Ceased
- 2006-04-18 WO PCT/US2006/015199 patent/WO2006116156A2/en active Search and Examination
- 2006-04-18 EP EP06751051A patent/EP1896080A2/en not_active Withdrawn
- 2006-04-21 AR ARP060101572A patent/AR053234A1/en not_active Application Discontinuation
- 2006-04-24 TW TW095114588A patent/TW200716748A/en unknown
-
2007
- 2007-10-09 IL IL186527A patent/IL186527A0/en unknown
- 2007-10-19 CR CR9452A patent/CR9452A/en not_active Application Discontinuation
- 2007-11-21 NO NO20076013A patent/NO20076013L/en not_active Application Discontinuation
-
2010
- 2010-11-02 US US12/938,294 patent/US8907071B2/en active Active
-
2011
- 2011-12-19 JP JP2011276723A patent/JP5220915B2/en not_active Expired - Fee Related
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006042151A2 (en) | 2004-10-07 | 2006-04-20 | The Regents Of The University Of California | Analogs of shk toxin and their uses in selective inhibition of kv1.3 potassium channels |
| WO2008088422A2 (en) | 2006-10-25 | 2008-07-24 | Amgen Inc. | Toxin peptide therapeutic agents |
| WO2010108154A2 (en) | 2009-03-20 | 2010-09-23 | Amgen Inc. | Selective and potent peptide inhibitors of kv1.3 |
Non-Patent Citations (6)
| Title |
|---|
| BEETON ET AL., DL: MOL. PHARMACOL., vol. 67, 2005, pages 1369 - 1381 |
| BEETON ET AL.: "Targeting effector memory T cells with a selective peptide inhibitor of Kv 1.3 channnels for therapy of autoimmune diseases", MOLEC. PHARMACOL., vol. 67, no. 4, 2005, pages 1369 - 81 |
| DL: BEETON ET AL.: "Targeting effector memory T cells with a selective peptide inhibitor ofKvl.3 channnels for therapy of autoimmune diseases", MOLEC. PHARMACOL., vol. 67, no. 4, 2005, pages 1369 - 81 |
| E. DONNADIEU ET AL., J. BIOL. CHEM., vol. 267, 1991, pages 25864 - 25872 |
| G.C. KOO ET AL., CELL. IMMUNOL., vol. 197, 1999, pages 99 - 107 |
| See also references of EP1896080A2 |
Cited By (69)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7820623B2 (en) | 2006-10-25 | 2010-10-26 | Amgen Inc. | Conjugated toxin peptide therapeutic agents |
| WO2008088422A2 (en) * | 2006-10-25 | 2008-07-24 | Amgen Inc. | Toxin peptide therapeutic agents |
| WO2008088422A3 (en) * | 2006-10-25 | 2009-02-19 | Amgen Inc | Toxin peptide therapeutic agents |
| US8043829B2 (en) | 2006-10-25 | 2011-10-25 | Amgen Inc. | DNA encoding chimeric toxin peptide fusion proteins and vectors and mammalian cells for recombinant expression |
| US7910102B2 (en) | 2006-10-25 | 2011-03-22 | Amgen Inc. | Methods of using conjugated toxin peptide therapeutic agents |
| US7834164B2 (en) | 2006-10-25 | 2010-11-16 | Amgen Inc. | DNA encoding OSK1 toxin peptide analogs and vectors and cells for combinant expression |
| US7825093B2 (en) | 2006-10-25 | 2010-11-02 | Amgen Inc. | Methods of using OSK1 peptide analogs |
| US7803769B2 (en) | 2006-10-25 | 2010-09-28 | Amgen Inc. | OSK1 peptide analogs and pharmaceutical compositions |
| CN101796069B (en) * | 2007-05-14 | 2014-02-19 | 墨西哥国立自治大学 | Vm23 and Vm24, two scorpion peptides that block human T-lymphocyte potassium channels (sub-type Kv1.3) with high selectivity and decrease the in vivo DTH-responses in rats |
| KR101524517B1 (en) * | 2007-05-14 | 2015-06-10 | 유니버시다드 나시오날 오토노마 드 멕시코 | Vm23 and vm24, two scorpion peptides that block human t-lymphocyte potassium channels (sub-type kv1.3) with high selectivity and decrease the in vivo dth-responses in rats |
| WO2008139243A1 (en) * | 2007-05-14 | 2008-11-20 | Universidad Nacional Autónoma de México | Vm23 and vm24, two scorpion peptides that block human t-lymphocyte potassium channels (sub-type kv1.3) with high selectivity and decrease the in vivo dth-responses in rats |
| US8394770B2 (en) | 2007-05-14 | 2013-03-12 | Universidad Nacional Autonoma De Mexico | Vm23 and Vm24, two scorpion peptides that block human T-lymphocyte potassium channels (sub-type Kv1.3) with high selectivity and decrease the in vivo DTH-responses in rats |
| JP2010527607A (en) * | 2007-05-22 | 2010-08-19 | アムジエン・インコーポレーテツド | Compositions and methods for making biologically active fusion proteins |
| EP2738257A1 (en) | 2007-05-22 | 2014-06-04 | Amgen Inc. | Compositions and methods for producing bioactive fusion proteins |
| WO2008153745A3 (en) * | 2007-05-22 | 2009-07-16 | Amgen Inc | Compositions and methods for producing bioactive fusion proteins |
| US8420779B2 (en) | 2007-05-22 | 2013-04-16 | Amgen Inc. | Compositions and methods for producing bioactive fusion proteins |
| US8716437B2 (en) | 2007-12-07 | 2014-05-06 | Steven A. Goldstein | Identification of toxin ligands |
| EP2229473A2 (en) * | 2007-12-07 | 2010-09-22 | Steven A. Goldstein | Identification of toxin ligands |
| EP2229473A4 (en) * | 2007-12-07 | 2011-02-02 | Steven A Goldstein | Identification of toxin ligands |
| US9603952B2 (en) | 2008-05-15 | 2017-03-28 | Morphotek, Inc. | Treatment of metastatic tumors |
| CN102112493A (en) * | 2008-07-23 | 2011-06-29 | 韩美控股株式会社 | A polypeptide complex comprising non-peptidyl polymer having three functional ends |
| US11040110B2 (en) | 2008-07-23 | 2021-06-22 | Hanmi Science Co., Ltd. | Polypeptide complex comprising non-peptidyl polymer having three functional ends |
| US10071171B2 (en) | 2008-07-23 | 2018-09-11 | Hanmi Science Co., Ltd. | Polypeptide complex comprising non-peptidyl polymer having three functional ends |
| US9636420B2 (en) | 2008-07-23 | 2017-05-02 | Hanmi Science Co., Ltd. | Polypeptide complex comprising non-peptidyl polymer having three functional ends |
| CN102112493B (en) * | 2008-07-23 | 2015-04-01 | 韩美科学株式会社 | A polypeptide complex comprising non-peptidyl polymer having three functional ends |
| WO2010108154A2 (en) * | 2009-03-20 | 2010-09-23 | Amgen Inc. | Selective and potent peptide inhibitors of kv1.3 |
| WO2010108154A3 (en) * | 2009-03-20 | 2011-03-03 | Amgen Inc. | Selective and potent peptide inhibitors of kv1.3 |
| EP3385279A1 (en) | 2009-03-20 | 2018-10-10 | Amgen Inc. | Carrier immunoglobulins and uses thereof |
| US9234015B2 (en) | 2010-02-04 | 2016-01-12 | Morphotek, Inc. | Chlorotoxin polypeptides and conjugates and uses thereof |
| US10183975B2 (en) | 2010-02-04 | 2019-01-22 | Morphotek, Inc. | Chlorotoxin polypeptides and conjugates and uses thereof |
| US9637526B2 (en) | 2010-02-04 | 2017-05-02 | Morphotek, Inc. | Chlorotoxin polypeptides and conjugates and uses thereof |
| US10822381B2 (en) | 2010-05-11 | 2020-11-03 | Fred Hutchinson Cancer Research Center | Chlorotoxin variants, conjugates, and methods for their use |
| US9944683B2 (en) | 2010-05-11 | 2018-04-17 | Fred Hutchinson Cancer Research Center | Chlorotoxin variants, conjugates, and methods for their use |
| WO2012040518A2 (en) | 2010-09-22 | 2012-03-29 | Amgen Inc. | Carrier immunoglobulins and uses thereof |
| WO2012125973A2 (en) | 2011-03-16 | 2012-09-20 | Amgen Inc. | Potent and selective inhibitors of nav1.3 and nav1.7 |
| WO2013061106A1 (en) * | 2011-10-28 | 2013-05-02 | University Of Debrecen | Modified peptide toxins |
| US9884124B2 (en) | 2012-05-17 | 2018-02-06 | Extend Biosciences, Inc. | Carriers for improved drug delivery |
| US9102751B2 (en) | 2012-05-18 | 2015-08-11 | Janssen Biotech, Inc. | Huwentoxin-IV variants and methods of use |
| US10414808B2 (en) | 2012-05-18 | 2019-09-17 | Janssen Biotech, Inc. | Huwentoxin-IV variants and methods of use |
| US10156559B2 (en) | 2012-12-10 | 2018-12-18 | Fred Hutchinson Cancer Research Center | Lipocalin fusion partners |
| WO2014116937A1 (en) | 2013-01-25 | 2014-07-31 | Janssen Biotech, Inc. | Kv1.3 antagonists and methods of use |
| KR102268830B1 (en) * | 2013-01-25 | 2021-06-24 | 얀센 바이오테크 인코포레이티드 | Kv1.3 ANTAGONISTS AND METHODS OF USE |
| EP2948559A4 (en) * | 2013-01-25 | 2016-10-19 | Janssen Biotech Inc | Kv1.3 antagonists and methods of use |
| KR20150108912A (en) * | 2013-01-25 | 2015-09-30 | 얀센 바이오테크 인코포레이티드 | Kv1.3 ANTAGONISTS AND METHODS OF USE |
| US10179808B2 (en) | 2013-01-25 | 2019-01-15 | Janssen Biotech, Inc. | Kv1.3 antagonists and methods of use |
| WO2014165277A2 (en) | 2013-03-12 | 2014-10-09 | Amgen Inc. | POTENT AND SELECTIVE INHIBITORS OF Nav1.7 |
| US11559580B1 (en) | 2013-09-17 | 2023-01-24 | Blaze Bioscience, Inc. | Tissue-homing peptide conjugates and methods of use thereof |
| US9624280B2 (en) | 2013-10-03 | 2017-04-18 | Janssen Biotech, Inc. | Protoxin-II variants and methods of use |
| US10975129B2 (en) | 2013-10-03 | 2021-04-13 | Janssen Biotech, Inc. | Protoxin-II variants and methods of use |
| EP3424955A1 (en) * | 2013-12-25 | 2019-01-09 | Daiichi Sankyo Company, Limited | Anti-trop2 antibody-drug conjugate |
| CN104177489B (en) * | 2014-07-29 | 2017-03-22 | 暨南大学 | Gene-recombined TNF-alpha derivative RMP16 and preparation method and application thereof |
| CN104177489A (en) * | 2014-07-29 | 2014-12-03 | 暨南大学 | Gene-recombined TNF-alpha derivative RMP16 and preparation method and application thereof |
| US9616109B2 (en) | 2014-10-22 | 2017-04-11 | Extend Biosciences, Inc. | Insulin vitamin D conjugates |
| US10702574B2 (en) | 2014-10-22 | 2020-07-07 | Extend Biosciences, Inc. | Therapeutic vitamin D conjugates |
| US10420819B2 (en) | 2014-10-22 | 2019-09-24 | Extend Biosciences, Inc. | Insulin vitamin D conjugates |
| US10406202B2 (en) | 2014-10-22 | 2019-09-10 | Extend Biosciences, Inc. | Therapeutic vitamin D conjugates |
| US9789197B2 (en) | 2014-10-22 | 2017-10-17 | Extend Biosciences, Inc. | RNAi vitamin D conjugates |
| US11116816B2 (en) | 2014-10-22 | 2021-09-14 | Extend Biosciences, Inc. | Therapeutic vitamin d conjugates |
| US10995125B2 (en) | 2015-03-03 | 2021-05-04 | Janssen Biotech, Inc. | Protoxin-II variants and methods of use |
| US10463714B2 (en) | 2015-04-02 | 2019-11-05 | Janssen Biotech, Inc. | Protoxin-II variants and methods of use |
| US11083776B2 (en) | 2015-04-02 | 2021-08-10 | Janssen Biotech, Inc. | Protoxin-II variants and methods of use |
| US11014971B2 (en) | 2016-12-01 | 2021-05-25 | University Of South Florida | Peptibodies, compositions thereof, and methods of treating atrial fibrillation |
| EP3496742A4 (en) * | 2016-12-01 | 2020-04-08 | University Of South Florida | Peptibodies, compositions thereof, and methods of treating atrial fibrillation |
| US11932673B2 (en) | 2017-07-12 | 2024-03-19 | Maxion Therapeutics Limited | Sodium channel inhibitors |
| US20230331827A1 (en) * | 2017-07-12 | 2023-10-19 | Maxion Therapeutics Limited | Potassium channel inhibitors |
| WO2021053194A1 (en) | 2019-09-20 | 2021-03-25 | Zealand Pharma A/S | Kv1.3 blockers |
| US11780893B2 (en) | 2019-09-20 | 2023-10-10 | Zealand Pharma A/S | Kv1.3 blockers |
| US11292820B2 (en) | 2019-09-20 | 2022-04-05 | Zealand Pharma A/S | KV1.3 blockers |
| WO2022200374A1 (en) | 2021-03-23 | 2022-09-29 | Zealand Pharma A/S | Kv1.3 blockers |
Also Published As
| Publication number | Publication date |
|---|---|
| MX2007013031A (en) | 2007-12-12 |
| AU2006239928B2 (en) | 2010-12-02 |
| NO20076013L (en) | 2008-01-22 |
| EA200702313A1 (en) | 2008-06-30 |
| US20110045587A1 (en) | 2011-02-24 |
| CA2604999A1 (en) | 2006-11-02 |
| TW200716748A (en) | 2007-05-01 |
| AU2006239928B8 (en) | 2011-03-31 |
| US8907071B2 (en) | 2014-12-09 |
| EP1896080A2 (en) | 2008-03-12 |
| JP2012100671A (en) | 2012-05-31 |
| JP5087536B2 (en) | 2012-12-05 |
| EA013470B1 (en) | 2010-04-30 |
| KR20080021606A (en) | 2008-03-07 |
| WO2006116156A3 (en) | 2007-10-04 |
| NZ562201A (en) | 2011-03-31 |
| BRPI0607750A2 (en) | 2009-11-24 |
| AU2006239928A1 (en) | 2006-11-02 |
| KR20100017942A (en) | 2010-02-16 |
| JP5220915B2 (en) | 2013-06-26 |
| AR053234A1 (en) | 2007-04-25 |
| JP2008538506A (en) | 2008-10-30 |
| US20070071764A1 (en) | 2007-03-29 |
| CR9452A (en) | 2008-05-21 |
| IL186527A0 (en) | 2008-01-20 |
| US7833979B2 (en) | 2010-11-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8907071B2 (en) | Toxin peptide therapeutic agents | |
| US7825093B2 (en) | Methods of using OSK1 peptide analogs | |
| CA2687141C (en) | Compositions and methods for producing bioactive fusion proteins | |
| EP2970408B1 (en) | Potent and selective inhibitors of nav1.7 | |
| US9636418B2 (en) | Potent and selective inhibitors of NAV1.7 | |
| ZA200709701B (en) | Toxin peptide therapeutic agents | |
| AU2011213759B2 (en) | Compositions and methods for producing bioactive fusion proteins | |
| AU2011213760A1 (en) | Compositions and methods for producing bioactive fusion proteins |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680022395.9 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 562201 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 186527 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 2604999 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006239928 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2007/013031 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2008507949 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: CR2007-009452 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12007502348 Country of ref document: PH |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007/09701 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020077027146 Country of ref document: KR Ref document number: 2006751051 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 9007/DELNP/2007 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200702313 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: PI0607750 Country of ref document: BR Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020097027223 Country of ref document: KR |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) |