WO2007117607A2 - Quinazolines for pdk1 inhibition - Google Patents
Quinazolines for pdk1 inhibition Download PDFInfo
- Publication number
- WO2007117607A2 WO2007117607A2 PCT/US2007/008592 US2007008592W WO2007117607A2 WO 2007117607 A2 WO2007117607 A2 WO 2007117607A2 US 2007008592 W US2007008592 W US 2007008592W WO 2007117607 A2 WO2007117607 A2 WO 2007117607A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- alkyl
- heteroaryl
- amino
- compound
- Prior art date
Links
- 0 C*(C**1(C)C)CC1OC Chemical compound C*(C**1(C)C)CC1OC 0.000 description 3
- UZRXHHMTKCJKTQ-UHFFFAOYSA-N CC(C)N(CC1)CCC1O Chemical compound CC(C)N(CC1)CCC1O UZRXHHMTKCJKTQ-UHFFFAOYSA-N 0.000 description 1
- XFHJQRZBIUMSES-UHFFFAOYSA-N CCCN(CCO)S(c(cc1)ccc1Nc1nc(cc(cc2)O)c2cn1)(=O)=O Chemical compound CCCN(CCO)S(c(cc1)ccc1Nc1nc(cc(cc2)O)c2cn1)(=O)=O XFHJQRZBIUMSES-UHFFFAOYSA-N 0.000 description 1
- AYOXOMXNWRXDGY-UHFFFAOYSA-N COc1cc2nc(Cl)ncc2cc1 Chemical compound COc1cc2nc(Cl)ncc2cc1 AYOXOMXNWRXDGY-UHFFFAOYSA-N 0.000 description 1
- JBORNNNGTJSTLC-UHFFFAOYSA-N Cc(ccc(OC)c1)c1[N+]([O-])=O Chemical compound Cc(ccc(OC)c1)c1[N+]([O-])=O JBORNNNGTJSTLC-UHFFFAOYSA-N 0.000 description 1
- UTAQOVYPSZIDTK-UHFFFAOYSA-N FC(c1cccnc1F)(F)F Chemical compound FC(c1cccnc1F)(F)F UTAQOVYPSZIDTK-UHFFFAOYSA-N 0.000 description 1
- UKHCBGWJQNMNEA-UHFFFAOYSA-N FC(c1cccnc1NCCN1CCCC1)(F)F Chemical compound FC(c1cccnc1NCCN1CCCC1)(F)F UKHCBGWJQNMNEA-UHFFFAOYSA-N 0.000 description 1
- WRXNJTBODVGDRY-UHFFFAOYSA-N NCCN1CCCC1 Chemical compound NCCN1CCCC1 WRXNJTBODVGDRY-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/86—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in position 4
- C07D239/94—Nitrogen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D451/00—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof
- C07D451/02—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing not further condensed 8-azabicyclo [3.2.1] octane or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane; Cyclic acetals thereof
- C07D451/04—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing not further condensed 8-azabicyclo [3.2.1] octane or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane; Cyclic acetals thereof with hetero atoms directly attached in position 3 of the 8-azabicyclo [3.2.1] octane or in position 7 of the 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring system
- C07D451/06—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D453/00—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids
- C07D453/02—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids containing not further condensed quinuclidine ring systems
Definitions
- the present invention generally relates to small molecule inhibitors of 3- phosphoinositide-dependent kinase (PDKl).
- the compounds can be used as therapeutics in the treatment of cellular proliferative diseases.
- PDKl (3-Phopsphoinositide-dependent kinase 1) is a serine/threonine kinase belonging to the AGC kinase super family. PDKl was first identified as the upstream kinase responsible for activating protein kinase B/AKT in the presence of phosphoinositide lipids (PIP3). PDKl activates AKT by phosphorylating a specific residue (threonine 308) located in the activation loop of this kinase.
- PDKl is responsible for phosphorylating the activation-loop of many AGC kinases including p90 ribosomal S6 kinase (RSK), protein kinase C family members (PKC), p70 ribosomal S6 kinase (70S6K), and the serum and glucocorticoid-induced protein kinase (SGK).
- RSK ribosomal S6 kinase
- PLC protein kinase C family members
- 70S6K p70 ribosomal S6 kinase
- SGK serum and glucocorticoid-induced protein kinase
- AKT is highly activated in a large percentage of common tumor types including melanoma, breast, lung, prostate and ovarian cancers.
- RSK levels are elevated in prostate cancers, and an RSK-specif ⁇ c inhibitor (SLOlOl) has recently been shown to inhibit the proliferation of multiple prostate cancer cell lines.
- SLOlOl RSK-specif ⁇ c inhibitor
- PKC ⁇ has been shown to play an important role in regulating apoptosis and promoting survival of glioma cells.
- the human PDKl gene encodes a 556 amino acid protein with an amino-terminal catalytic domain and a non-catalytic carboxy terminal containing a pleckstrin homology domain (PH).
- PH pleckstrin homology domain
- the PH domain of PDKl is required for the binding OfPIP 3 lipids produced by PI3kinase (PI3K). PDKl binding OfPIP 3 lipids results in membrane co-
- PDKl activates AKT by phosphorylating tihreonine308.
- PDKl can activate other AGC kinases independent of PIP3 lipids by binding directly to a conserved motif found on these targets. Because PDKl regulates two distinct classes of downstream signaling substrates (PI3K-de ⁇ endent and independent targets), inhibitors of this enzyme could have important therapeutic value in a variety of human cancers.
- PDKl inhibitors could be efficacious in tumors in which the PI3K signaling pathway is upregulated due to activating mutations, amplification of PI3K itself or its upstream receptor tyrosine kinases, or deletion of PTEN, the phosphatase the counteracts PI3K activity.
- the finding that mice expressing half the normal amount of PTEN are protected from developing a wide range of tumors by reducing PDKl expression levels supports this idea.
- PDKl inhibitors could be useful in treating cancers driven by PHVindependent PDKl signaling pathways (e.g. K-ras or H-ras driven cancers).
- PDKl is a central activator of several signaling pathways that are frequently altered in human cancers making it an attractive target for therapeutic intervention.
- U.S. Patent No. 6,982,260 discloses quinazoline compounds for inhibition of cyclin- dependent kinases, and International Application PCT/IB2004/000091 discloses 2- aminopyridine substituted heterocycles as inhibitors for cellular proliferation.
- This invention is directed to the discovery of novel compounds for PDKl inhibition and use of these compounds to treat a variety of diseases or disorders involving cellular proliferation.
- the present invention provides PDKl, Cdkl, and/or Cdk2 inhibitors that are useful as therapeutic agents, for the treatment of diseases and disorders characterized by abnormal cellular proliferation, for example cancers of the prostate, lung, colon, breast, among others. jfr ⁇ z ⁇ zi ⁇ .u ⁇ z
- the instant invention provides compounds that have the Formula I:
- one of W 1 or W 2 is R 1 and the other is -L-A 1 ;
- L is a covalent bond, carbonyl, carbonylamino, aminocarbonyl, -O-, -S-, -SO-, -SO 2 -, -NH-, C 1-3 alkyl, substituted C 1-3 alkyl, or an alkyl interrupted with -O-, -S-, -SO-, -SO 2 -, - NH-, carbonyl, carbonylamino, or aminocarbonyl;
- a 1 is aryl, substituted aryl, heteroaryl, substituted heteroaryl, heterocyclyl, or substituted heterocyclyl;
- Y is H, C 1 - S alkyl, halo, cyano, nitro, or amino;
- R 1 is selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, alkoxy, substituted alkoxy, acyl, acylamino, acyloxy, amino, substituted amino, aminocarbonyl, aminothiocarbonyl, aminocarbonylamino, aminothiocarbonylamino, aminocarbonyloxy, aminosulfonyl, aminosulfonyloxy, aminosulfonylamino, amidino, carboxyl, carboxyl ester, (carboxyl ester)amino, (carboxyl ester)oxy, cyano, halo, hydroxy, nitro, SO 3 H, sulfonyl, substituted sulfonyl, sulfonyloxy, thioacyl, thiol, alkylthio, substituted alkylthio, aryl, substituted ary
- R 2 and R 3 are independently selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, alkoxy, substituted alkoxy, acyl, acylamino, acyloxy, amino, substituted amino, aminocarbonyl, aminothiocarbonyl, aminocarbonylamino, aminothiocarbonylamino, aminocarbonyloxy, aminosulfonyl, aminosulfonyloxy, aminosulfonylamino, amidino, carboxyl, carboxyl ester, (carboxyl ester)amino, (carboxyl ester)oxy, cyano, halo, hydroxy, nitro, SO 3 H, sulfonyl, substituted sulfonyl, sulfonyloxy, thioacyl, thiol, alkylthio, substituted alkylthio, aryl,
- R 4 is aryl, substituted aryl, heteroaryl, substituted heteroaryl, cycloalkyl, substituted cycloalkyl, heterocyclyl, or substituted heterocyclyl; provided when R 4 is heteroaryl or substituted heteroaryl, W 2 is not aryl or heteroaryl; or a stereoisomer, tautomer, or pharmaceutically acceptable salt thereof.
- the compounds of the invention have the Formula I:
- one of W 1 or W 2 is R 1 and the other is -L-A 1 ;
- L is a covalent bond, carbonyl, carbonylamino, aminocarbonyl, -O-, -S-, -SO-, -SO 2 -, -NH-, Ci-3 alkyl, substituted Ci -3 alkyl, or an alkyl interrupted with -O-, -S-, -SO-, -SO 2 -, - NH-, carbonyl, carbonylamino, or aminocarbonyl;
- a 1 is hydroxyl, amino, alkyl, substituted alkyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, cyclyl, or substituted cyclyl, heterocyclyl, or substituted heterocyclyl, provided whenW 2 is hydroxyl or methoxy, A 1 is not isopropyl or cyclopentyl;
- Y is H, Ci-3 alkyl, halo, cyano, nitro, or amino;
- R 1 is selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, alkoxy, substituted alkoxy, acyl, acylamino, acyloxy, amino, substituted amino, aminocarbonyl, aminothiocarbonyl, aminocarbonylamino, aminothiocarbonylamino, aminocarbonyloxy, aminosulfonyl, aminosulfonyloxy, aminosulfonylamino, amidino, carboxyl, carboxyl ester, (carboxyl ester)amino, (carboxyl ester)oxy, cyano, halo, hydroxy, nitro, SO3H, sulfonyl, substituted sulfonyl, sulfonyloxy, thioacyl, thiol, alkylthio, substituted alkylthio, aryl, substituted ary
- R 2 and R 3 are independently selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, alkoxy, substituted alkoxy, acyl, acylamino, acyloxy, amino, substituted amino, aminocarbonyl, aminothiocarbonyl, arninocarbonylamino, aminothiocarbonylamino, aminocarbonyloxy, aminosulfonyl, aminosulfonyloxy, aminosulfonylamino, arnidino, carboxyl, carboxyl ester, (carboxyl ester)amino, (carboxyl ester)oxy, cyano, halo, hydroxy, nitro, SO 3 H, sulfonyl, substituted sulfonyl, sulfonyloxy, thioacyl, thiol, alkylthio, substituted alkylthio,
- compounds of the invention have the Formula II:
- X is O or NR 6 ;
- Y is H, Ci . 3 alkyl, halo, cyano, nitro, or amino;
- R 5a and R 5b are each independently H, halo, hydroxy, alkyl, substituted alkyl, alkoxy, substituted alkoxy, amino, or substituted amino;
- R 6 is H, acyl, substituted carbonyl, sulfonyl, alkyl, or substituted alkyl; or R 5a and R 6 are taken together to form a bridging alkylene moiety; or R 5a and R 5b are taken together to form a bridging alkylene moiety;
- m and n are independently 0, 1 or 2;
- R 1 is selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl. substituted alkynyl, alkoxy, substituted alkoxy, acyl, acylamino, acyloxy, amino, substituted amino, aminocarbonyl, aminothiocarbonyl, aminocarbonylamino, aminothiocarbonylamino, aminocarbonyloxy, aminosulfonyl, aminosulfonyloxy, aminosulfonylamino, amidino, carboxyl, carboxyl ester, (carboxyl ester)amino, (carboxyl ester)oxy, cyano, halo, hydroxy, nitro, SO 3 H, sulfonyl, substituted sulfonyl, jrruz ⁇ .4 i ⁇ .uuuz
- R 2 and R 3 are independently selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, alkoxy, substituted alkoxy, acyl, acylamino, acyloxy, amino, substituted amino, aminocarbonyl, aminothiocarbonyl, aminocarbonylamino, aminothiocarbonylamino, aminocarbonyloxy
- R 4 is aryl, substituted aryl, heteroaryl, substituted heteroaryl, cycloalkyl, substituted cycloalkyl, heterocyclyl, or substituted heterocyclyl; or a stereoisomer, tautomer, or pharmaceutically acceptable salt thereof.
- the compounds of the invention have the Formula III:
- X is O or NR 6 ;
- Y is H, Ci- 3 alkyl, halo, cyano, nitro, or amino;
- R 5a and R 5b are each independently H, halo, hydroxy, alkyl, substituted alkyl, alkoxy, substituted alkoxy, amino, or substituted amino;
- R 6 is H, acyl, substituted carbonyl, sulfonyl, alkyl, or substituted alkyl; or R 5a and R 6 are taken together to form a bridging alkylene moiety; or R 5a and R 5b are taken together to form abridging alkylene moiety;
- m and n are independently 0, 1 or 2;
- R 1 is selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, alkoxy, substituted alkoxy, acyl, acylamino, acyloxy, amino, substituted amino, aminocarbonyl, aminothiocarbonyl, aminocarbonylamino, aminothiocarbonylamino, aminocarbonyl
- R 2 and R 3 are independently selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, alkoxy, substituted alkoxy, acyl, acylamino, acyloxy, amino, substituted amino, aminocarbonyl, aminothiocarbonyl, aminocarbonylamino, aminothiocarbonylamino, aminocarbonyloxy, aminosulfonyl, aminosulfonyloxy, aminosulfonylamino, amidino, carboxyl, carboxyl ester, (carboxyl ester)amino, (carboxyl ester)oxy, cyano, halo, hydroxy, nitro, SO3H, sulfonyl, substituted sulfonyl, sulfonyloxy, thioacyl, thiol, alkylthio, substituted alkylthio, aryl,
- the invention provides compositions, preferably pharmaceutical compositions, that contain one or more compounds of the invention.
- the invention provides methods of treating diseases or disorders that are characterized by, inter alia, abnormal cellular proliferation, for example cancer and/or precancerous lesions, that employ compounds of the invention.
- the invention further provides methods of inhibiting tumor cell growth in a subject, that employ compounds of the invention.
- the invention further provides methods of manufacturing compounds and compositions described herein, and method for the use of the quinazolines of the invention in methods for manufacturing medicaments for use in the methods of the invention.
- compounds, such as those of Formula I can be used in the manufacture of a medicament for inhibiting PDKl . rr ⁇ z»zi». ⁇ uuz
- the invention provides the use of the compounds of the invention, in the manufacture of medicament for PDKl inhibition, and/ or for treatment of one or more of the aforementioned diseases or disorders.
- the present invention provides compounds that have the
- one of W 1 or W 2 is R 1 and the other is -L-A 1 ;
- L is a covalent bond, carbonyl, carbonylamino, aminocarbonyl, -O-, -S-, -SO-, -SO 2 - J -NH-, Ci-3 alkyl, substituted C 1 -3 alkyl, or an alkyl interrupted with -O-, -S-, -SO-, -SO 2 -, - NH-, carbonyl, carbonylamino, or aminocarbonyl;
- a 1 is aryl, substituted aryl, heteroaryl, substituted heteroaryl, heterocyclyl, or substituted heterocyclyl;
- Y is H, C 1-3 alkyl, halo, cyano, nitro, or amino;
- R 1 is selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, alkoxy, substituted alkoxy, acyl, acylamino, acyloxy, amino, substituted amino, aminocarbonyl, aminothiocarbonyl, aminocarbonylamino, aminothiocarbonylamino, aminocarbonyloxy, aminosulfonyl, aminosulfonyloxy, aminosulforiylamino, amidino, carboxyl, carboxyl ester, (carboxyl ester)amino, (carboxyl ester)oxy, cyano, halo, hydroxy, nitro, SO3H, sulfonyl, substituted sulfonyl, sulfonyloxy, thioacyl, thiol, alkylthio, substituted alkylthio, aryl, substituted ary
- R 2 and R 3 are independently selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, alkoxy, substituted alkoxy, acyl, acylamino, acyloxy, amino, substituted amino, aminocarbonyl, aminothiocarbonyl, aminocarbonylamino, aminothiocarbonylamino, aminocarbonyloxy, aminosulfonyl, aminosulfonyloxy, aminosulfonylamino, amidino, carboxyl, carboxyl ester, (carboxyl ester)amino, (carboxyl ester)oxy, cyano, halo, hydroxy, nitro, SO 3 H, sulfonyl, substituted sulfonyl, sulfonyloxy, thioacyl, thiol, alkylthio, substituted alkylthio, aryl,
- R 4 is aryl, substituted aryl, heteroaryl, substituted heteroaryl, cycloalkyl, substituted cycloalkyl, heterocyclyl, or substituted heterocyclyl; or a stereoisomer, tautomer, or pharmaceutically acceptable salt thereof.
- the quinazoline compounds of this invention inhibit of PDKl activity or inhibit cell proliferation.
- the quinazoline compounds have low IC 50 values with regard to inhibition of PDKl activity (i.e., the concention of a compound that is required for 50% inhibition of PDKl activity) or low EC50 with regard to inhibition of cell proliferation (i.e., the concentration of a compound which is required to induce inhibitory response against cell proliferation halfway between the baseline and maximum).
- some quinazoline compounds exhibit IC50 or ECso valaues of about 25 ⁇ M or less, about 10 ⁇ M or less, about 1 ⁇ M or less, or about 0.1 ⁇ M or even less according to the PDKl kinase alpha screen assay and cell proliferation described herein.
- -L-A 1 is a heterocyclyloxy group having the structure:
- X is O or NR 6 ;
- R 5a and R 515 are each independently H, halo, hydroxy, alkyl, substituted alkyl, alkoxy, 30 substituted alkoxy, amino, or substituted amino;
- R 6 is H, acyl, substituted carbonyl, sulfonyl, alkyl, or substituted alkyl; or R 5a and R 6 are taken together to form a bridging alkylene moiety; or R 5a and R 5b are taken together to form a bridging alkylene moiety; m and n are independently 0, 1 or 2.
- W 1 is -L-A 1 .
- W 2 is -L-A 1 .
- R 4 is substituted phenyl.
- -N(RsOi)(RsO 2 ) forms -NH2, -NH-alkyl, -NH-alkyl substituted with alkoxy, -NH-cycloalkyl, morpholino, - NH-(alkyl)-pyrrolidinyl or piperizinyl optionally substituted with alkyl.
- -N(RsOi)(RsO 2 ) forms -NH2, -NH-alkyl, -NH-alkyl substituted with alkoxy, -NH-cycloalkyl, morpholino, - NH-(alkyl)-pyrrolidinyl or piperizinyl optionally substituted with alkyl.
- -N(R 5 Oi)(R 5 O 2 ) forms -NH 2 , -NH-CH(CH 3 ) 2 , -NH-(CH 2 ) 2 -O-CH 35 -NH-cyclopropyl, morpholin-4-yl, 4-methyl-piperizine-l-yI, or -NH-(CH 2 ) 2 -pyrrolidin-l-yl.
- -N(R 5 Oi)(RsO 2 ) forms -NH 2 , -NH-alkyl, -NH-alkyl substituted with alkoxy, or - NH-cycloalkyl.
- R 2 is H or halogen.
- R 3 is H.
- W 2 is H, halogen, cyano, heteroaryl, substituted heteroaryl, phenyl, substituted phenyl, or a group of formula:
- each R 50I and each R 502 is independently selected from H, alkyl, substituted alkyl, alkoxyalkyl, cycloalkyl and heterocyclylalkyL
- W 2 is H, halogen or cyano.
- R 501 and R 502 are each independently selected from H and alkyl.
- W 2 is a 5- or 6-membered heteroaryl group having 1 or 2 heteroatoms independently selected from O, S and N, that is optionally substituted with up to three substituents selected from alkyl, alkoxy and -N(R 501 )(R 502 ); wherein R 501 and R 5 02 are independently selected from H, alkyl, substituted alkyl, alkoxyalkyl, cycloalkyl and heterocyclylalkyl; or R 50 I and R 5025 taken together, form a heterocyclyl group that is optionally substituted with up to three groups independently selected from Ci. 3 alkyl, hydroxyl, halo, alkoxy, amino, and substituted amino.
- W 2 is a heteroaryl group selected from pyridinyl, pyrimidinyl, pyrazolyl, oxazolyl and thiazolyl, the heteroaryl group being optionally substituted with up to three substituents selected from alkyl, alkoxy and — N(R 501 )(R 502 ); wherein R 501 and R 502 are independently selected from H, alkyl, substituted alkyl, alkoxyalkyl, cycloalkyl and heterocyclylalkyl; or R501 and Rso2, taken together, form a heterocyclyl group that is optionally substituted with up to three groups independently selected from C 1-J alkyl, hydroxyl, halo, alkoxy, amino, and substituted amino, hi some such embodiments, R 501 and R 502 are each independently selected from H and alkyl.
- W 2 is a group of formula:
- R 6 is H or alkyl.
- W 1 is H, heteroaryl, substituted heteroaryl, or a group of formula:
- W 1 is a group of formula:
- X is NR 6 .
- X is NR 6
- R 5b is H
- R 6 and R 5b together form an alkylene bridge, for example -(CHb) Z -.
- X is NR 6 , and R 5a and R 5b together form an alkylene bridge, for example -(CHa) 2 -.
- X is NR 6 , and R 6 is H or alkyl.
- X is NR 6 , m and n are each 1 , and R 5a and R 5b are each H. In some such embodiments, R 6 is H or alkyl.
- X is NR 6 , m is 1 and n is 0, and R 5a and R 5b are each H. In some such embodiments, R 6 is H or alkyl.
- X is NR 6
- m is 2 and n is 0, and R 5a and R 5b are each H.
- R 6 is H or alkyl.
- X is NR 6 , m and n are each 0, and R 5a and R 5b are each H. In some such embodiments, R 6 is H or alkyl.
- Y is H or Cj. 3 alkyl. In other embodiments Y is H Or-CH 3 . In more particular embodiments, Y is H.
- W 1 is heteroaryl or substituted heteroaryl.
- W 1 is heteroaryl selected from pyridinyl, pyrimidinyl, pyrazolyl, oxazolyl and thiazolyl, the heteroaryl group being optionally substituted with up to three substituents selected from alkyl, alkoxy, -N(R 50 ⁇ (R 502 ) and heterocyclyl; wherein R 501 and R 502 are independently selected from H, alkyl, substituted alkyl, alkoxyalkyl, cycloalkyl and heterocyclylalkyl; or R501 and R5 0 2, taken together, form a heterocyclyl group that is optionally substituted with up to three groups independently selected from Ci -3 alkyl, hydroxyl, halo, alkoxy, amino, and substituted amino.
- W 1 is pyridinyl optionally substituted with up to three substituents selected from alkyl, alkoxy, -N(R 501 )(R 502 ) and heterocyclyl; wherein Rso t and Rso 2 are independently selected from H, alkyl, substituted alkyl, alkoxyalkyl, cycloalkyl and heterocyclylalkyl; or Rs 01 and Rso2 » taken together, form a heterocyclyl group that is optionally substituted with up to three groups independently selected from C J-3 alkyl, hydroxyl, halo, alkoxy, amino, and substituted amino.
- W 1 is pyridinyl optionally substituted with heterocyclyl, for example a group of formula:
- R 6 is H or alkyl; and R5 01 and R 502 are independently selected from H, alkyl, substituted alkyl, alkoxyalkyl, cycloalkyl and heterocyclylalkyl; or R 50I and R 502 , taken together, form a heterocyclyl group that is optionally substituted with up to three groups independently selected from C1-3 alkyl, hydroxyl, halo, alkoxy, amino, and substituted amino.
- W 1 is pyridinyl optionally substituted with up to three substituents selected from alkyl, alkoxy, -N(R 501 J(R 502 ) and heterocyclyl; or W 1 is a group of formula:
- R 4 is substituted phenyl
- W 2 is H, halogen, cyano, heteroaryl, substituted heteroaryl, phenyl, substituted phenyl, or a group of formula:
- W 1 is H, heteroaryl, substituted heteroaryl, or a group of formula:
- W 1 is a group of formula:
- heteroaryl selected from pyridinyl, pyrimidinyl, pyrazolyl, oxazolyl and thiazolyl, the heteroaryl group being optionally substituted with up to three substituents selected from alkyl, alkoxy, -N(R 501 )(R 502 ) and heterocyclyl; wherein each R 501 and each R 502 is independently selected from H, alkyl, substituted alkyl, alkoxyalkyl, cycloalkyl and heterocyclylalkyl.
- R 2 is H or halogen
- R 3 is H.
- compounds of the invention have the Formula II:
- X is O or NR 6 ;
- Y is H, Ci - 3 alkyl, halo, cyano, nitro, or amino;
- R 5a and R 5b are each independently H, halo, hydroxy, alkyl, substituted alkyl, alkoxy, substituted alkoxy, amino, or substituted amino;
- R 6 is H, acyl, substituted carbonyl, sulfonyl, alkyl, or substituted alkyl; or R 5a and R 6 are taken together to form a bridging alkylene moiety; or R 5a and R 5b are taken together to form a bridging alkylene moiety;
- m and n are independently 0, 1 or 2;
- R 1 is selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, alkoxy, substituted alkoxy, acyl, acylamino, acyloxy, amino, substituted amino, aminocarbonyl, arninothiocarbonyl, aminocarbonylamino, aminothiocarbonylamino, aminocarbonyloxy, aminosulfonyl, aminosulfonyloxy, aminosulfonylamino, amidino, carboxyl, carboxyl ester, (carboxyl ester)amino, (carboxyl ester)oxy, cyano, halo, hydroxy, nitro, SO 3 H, sulfonyl, substituted sulfonyl, sulfonyloxy, thioacyl, thiol, alkylthio, substituted alkylthio, aryl, substitute
- R 2 and R 3 are independently selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, alkoxy, substituted alkoxy, acyl, acylamino, acyloxy, amino, substituted amino, aminocarbonyl, aminothiocarbonyl, aminocarbonylamino, aminothiocarbonylamino, aminocarbonyloxy, aminosulfonyl, aminosulfonyloxy, aminosulfonylamino, amidino, carboxyl, carboxyl ester,
- R 4 is aryl, substituted aryl, heteroaryl, substituted heteroaryl, cycloalkyl, substituted cycloalkyl, heterocyclyl, or substituted heterocyclyl; or a stereoisomer, tautomer, or pharmaceutically acceptable salt thereof.
- R 4 is substituted phenyl.
- X 1 is SO 2 -
- -N(R 50 ')(R 502 ) forms -NH 2 , -NH- alkyl, -NH-alkyl substituted with alkoxy, -NH-cycloalkyl, morpholino, - NH-( alkyl)-pyrrolidinyl or piperizinyl optionally substituted with alkyl.
- -N(R 501 )(R 502 ) forms -NH 2 , -NH-CH(CH 3 ) 2 , -NH-(CH 2 ) 2 -O-CH 3 , -NH- cyclopropyl, morpholin-4-yl, 4-rnethyl-piperizine- 1 -yl, or -NH-(CH 2 ) 2 -pyrrolidin- 1 -yl.
- - N(R 501 )(R 502 ) forms -NH 2 , -NH-alkyl, -NH-alkyl substituted with alkoxy, or -NH-cycloalkyl.
- Y is H or C 1 . 3 alkyl. In other embodiments Y is H Or-CH 3 . In more particular embodiments, Y is H. In some embodiments, R 2 is H or halogen. In some further embodiments, R 3 is H.
- R 1 is selected from H, alkyl, and substituted alkyl.
- compounds of the invention have the Formula HI:
- X is O or NR 6 ;
- Y is H, Ci . 3 alkyl, halo, cyano, nitro, or amino; rjruza_j ⁇ .uuuz
- R 5a and R 5b are each independently H, halo, hydroxy, alkyl, substituted alkyl, alkoxy, substituted alkoxy, amino, or substituted amino;
- R 6 is H, acyl, substituted carbonyl, sulfonyl, alkyl, or substituted alkyl; or R 5a and R 6 are taken together to form a bridging alkylene moiety; or R 5a and R 5b are taken together to form a bridging alkylene moiety; m and n are independently 0, 1 or 2;
- R 1 is selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, alkoxy, substituted alkoxy, acyl, acylamino, acyloxy, amino, substituted amino, aminocarbonyl, aminothiocarbonyl, aminocarbonylamino, aminothiocarbonylamino, aminocarbonyloxy, aminosulfonyl, aminosulfonyloxy, aminosulfonylamino, amidino, carboxyl, carboxyl ester, (carboxyl ester)amino, (carboxyl ester)oxy, cyano, halo, hydroxy, nitro, SO3H, sulfonyl, substituted sulfonyl, sulfonyloxy, thioacyl, thiol, alkylthio, substituted alkylthio, aryl, substituted ary
- R 2 and R 3 are independently selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, alkoxy, substituted alkoxy, acyl, acylamino, acyloxy, amino, substituted amino, aminocarbonyl, aminothiocarbonyl, aminocarbonylamino, aminothiocarbonylamino, aminocarbonyloxy, aminosulfonyl, aminosulfonyloxy, aminosulfonylamino, amidino, carboxyl, carboxyl ester, (carboxyl ester)amino, (carboxyl ester)oxy, cyano, halo, hydroxy, nitro, SO 3 H, sulfonyl, substituted sulfonyl, sulfonyloxy, thioacyl, thiol, alkylthio, substituted alkylthio, aryl,
- R 4 is aryl, substituted aryl, heteroaryl, substituted heteroaryl, cycloalkyl, substituted cycloalkyl, heterocyclyl, or substituted heterocyclyl; or a stereoisomer, tautomer, or pharmaceutically acceptable salt thereof, hi some such embodiments, R 4 is substituted phenyl.
- R 4 is phenyl substituted with a group of formula -Xi-N(R 501 )(R 502 ); wherein X 1 is SO 2 or C(O); and R 501 and R 502 are independently selected from H, alkyl, substituted alkyl, alkoxyalkyl, cycloalkyl and heterocyclylalkyl; or R 501 and R 502 , taken together, form a heterocyclyl group that is optionally substituted with up to three groups independently selected from Cu 3 alkyl.
- X 1 is SO 2 .
- - N(R 501 )(R 502 ) forms -NH 2 , -NH-alkyl, -NH-alkyl substituted with alkoxy, -NH-cycloalkyl, morpholino, -NH-(alkyl)-pyrrolidinyl or piperizinyl optionally substituted with alkyl.
- -N(R 501 )(R 502 ) forms -NH 2 , -NH-CH(CH 3 ) 2 , -NH-(CH 2 ) 2 -O-CH 3 , -NH-cyclopropyl, morpholin-4-yl, 4-methyl-piperizine-l-yl, or -NH-(CH 2 ) 2 -py ⁇ rolidin-l-yl.
- -N(R 501 XR 502 ) forms -NH 2 , -NH-alkyl, -NH-alkyl substituted with alkoxy, or -NH-cycloalkyL
- R 2 is H or halogen.
- R 3 is H.
- X is NR 6 .
- X is NR 6 , R 5b is H, and R 6 and R 5b together form an alkylene bridge, for example -(CHa) 2 -.
- X is NR 6
- R 5a and R 5b together form an alkylene bridge, for example -(CHa) 2 -.
- X is NR 6 , wherein R 6 is H or alkyl. Ih some embodiments, X is NR 6 , m and n are each 1 , and R 5a and R 5b are each H.
- R 6 is H or alkyl.
- X is NR 6
- m is 1 and n is 0, and R 5a and R 5b are each H.
- R 6 is H or alkyl.
- X is NR 6
- m is 2 and n is 0, and R 5a and R 5b are each H.
- R 6 is H or alkyl.
- X is NR 6 , m and n are each 0, and R 5a and R 5b are each H. hi some such embodiments, R 6 is H or alkyl.
- Y is H or Ci . 3 alkyl. In other embodiments Y is H or -CH 3 . hi more particular embodiments, Y is H.
- R 501 and R 502 are each independently selected from H and alkyl.
- R 1 is H, halogen or cyano.
- R 1 is a 5- or 6-membered heteroaryl group having 1 or 2 heteroatoms independently selected from O, S and N, that is optionally substituted with up to three substituents selected from alkyl, alkoxy and — N(R 501 )(R 502 ).
- R 1 is a heteroaryl group selected from pyridinyl, pyrimidinyl, pyrazolyl, oxazolyl and thiazolyl, the heteroaryl group being optionally substituted with up to three substituents selected from alkyl, alkoxy and — N(R 501 )(R 502 ).
- R 501 and R 502 are each independently selected from H and alkyl.
- R 4 is other than pyridinyl. In some of each of the foregoing embodiments, R 4 is other than pyridin-2-yl.
- the columns labeled "PDKl IC 50 " "CPEC50 A2780,” “CPEC50 PC3,” and “CPEC50 PC3MM” indicates the compound's activity in the PDKl Kinase Alpha Screen Assay and the Cell Proliferation Assay described below.
- the symbol “+” indicates IC 5O values or EC 50 values of 25 ⁇ M or greater (or compounds not evaluated)
- the symbol “++” indicates IC 50 values or EC50 values between less than 25 ⁇ M and greater than 10 ⁇ M
- the symbol “+++” indicates IC50 values or EC 50 values of 10 ⁇ M or less and greater than 5 ⁇ M
- inventions provide methods for inhibiting PDKl in a subject. More particularly, the present invention provides a method of inhibitng PDKl comprising administering to a human or animal subject, a quinazoline compound as described herein. Such methods include administering a compound of Formula I, II or III to the subject.
- compositions including: a compound of Formula I, II or III and a pharmaceutically acceptable carrier or excipient.
- compositions described herein are used for the treatment of cancer and reduction of tumor growth.
- the quinazoline compounds are useful in the treatment of human or animal (e.g., murine) cancers, including, for example, lung and bronchus; prostate; breast; pancreas; colon and rectum; thyroid; liver and intrahepatic bile duct; hepatocellular; gastric; glioma/glioblastoma; endometrial; melanoma; kidney and renal pelvis; urinary bladder; uterine corpus; uterine cervix; ovary; multiple myeloma; esophagus; acute myelogenous leukemia; chronic myelogenous leukemia; lymphocytic leukemia; myeloid leukemia; brain; oral cavity and pharynx; larynx; small intestine; non-Hodgkin lymphoma; melanoma; and villous colon a
- human or animal e.g
- the administration of the compound has an inhibiting effect upon tumor cell growth.
- the composition is effective at inhibiting the growth of one or more mammalian tumor cells.
- compositions that include the compounds described herein may include additives such as excipients.
- suitable pharmaceutically acceptable excipients include processing agents and drug delivery modifiers and enhancers, such as, for example, calcium phosphate, magnesium stearate, talc, monosaccharides, disaccharides, starch, gelatin, cellulose, methyl cellulose, sodium carboxymethyl cellulose, dextrose, hydroxypropyl- ⁇ -cyclodextrin, polyvinylpyrrolidinone, low melting waxes, ion exchange resins, and the like, as well as combinations of any two or more of these.
- processing agents and drug delivery modifiers and enhancers such as, for example, calcium phosphate, magnesium stearate, talc, monosaccharides, disaccharides, starch, gelatin, cellulose, methyl cellulose, sodium carboxymethyl cellulose, dextrose, hydroxypropyl- ⁇ -cyclodextrin, polyvinylpyrrolidinone, low melting wax
- compositions that include the compounds of the invention may be in any form suitable for the intended method of administration, including, for example, as a solution, a suspension, or an emulsion.
- Liquid carriers are typically used in preparing solutions, suspensions, and emulsions.
- Liquid carriers contemplated for use in the practice of the present invention include, for example, water, saline, pharmaceutically acceptable organic solvent(s), pharmaceutically acceptable oils or fats, and the like, as well as mixtures of two or more of these.
- the liquid carrier may include other suitable pharmaceutically acceptable additives such as solubilizers, emulsifiers, nutrients, buffers, preservatives, suspending agents, thickening agents, viscosity regulators, stabilizers, and the like.
- Suitable organic solvents include, for example, monohydric alcohols, such as ethanol, and polyhydric alcohols, such as glycols.
- Suitable oils include, but are not limited to, soybean oil, coconut oil, olive oil, safflower oil, cottonseed oil, and the like.
- the carrier may be an oily ester such as ethyl oleate, isopropyl myristate, and the like.
- Compositions of the present invention may also be in the form PP028218.0002
- microparticles are of microparticles, microcapsules, and the like, as well as combinations of any two or more of these.
- liposomes are generally derived from phospholipids or other lipid substances. Liposomes are formed by mono- or multilamellar hydrated liquid crystals that are dispersed in an aqueous medium. Any non-toxic, physiologically acceptable and metabolizable lipid capable of forming liposomes can be used.
- the present compositions in liposome form may include, in addition to a compound of the present invention, stabilizers, preservatives, excipients, and the like.
- Preferred lipids include phospholipids and phosphatidyl cholines (lecithins), both natural and synthetic. Methods of forming liposomes are known in the art. See, for example, Prescott, Ed., Methods in Cell Biology, Volume XIV, Academic Press, New York, N. W., p. 33 et seq (1976).
- Controlled release delivery systems may also be used, such as a diffusion controlled matrix system or an erod ⁇ ble system, as described for example in: Lee, "Diffusion-Controlled Matrix Systems", pp. 155-198 and Ron and Langer, “Erodible Systems", pp. 199-224, in
- the matrix may be, for example, a biodegradable material that can degrade spontaneously in situ and in vivo for, example, by hydrolysis or enzymatic cleavage, e.g., by proteases.
- the delivery system may be, for example, a naturally occurring or synthetic polymer or copolymer, for example in the form of a hydrogel.
- Exemplary polymers with cleavable linkages include polyesters, polyorthoesters, polyanhydrides, polysaccharides, poly(phosphoesters), polyamides, polyurethanes, poly(imidocarbonates) and poly(phosphazenes).
- the compounds of the invention may be administered enterally, orally, parenterally, sublingually, by inhalation spray, rectally, or topically in dosage unit formulations that include conventional nontoxic pharmaceutically acceptable carriers, adjuvants, and vehicles as desired.
- suitable modes of administration include oral, subcutaneous, transdermal, transmucosal, iontophoretic, intravenous, intramuscular, intraperitoneal, intranasal, subdermal, rectal, and the like.
- Topical administration may also include the use of transdermal administration such as transdermal patches or ionophoresis devices.
- parenteral as used PP028218.0002
- herein includes subcutaneous injections, intravenous, intramuscular, intrasternal injection, or infusion techniques.
- sterile injectable aqueous or oleaginous suspensions may be formulated according to the known art using suitable dispersing or wetting agents and suspending agents.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a nontoxic parenterally acceptable diluent or solvent, for example, as a solution in 1,3-propanediol.
- acceptable vehicles and solvents that may be employed are water, Ringer's solution, and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid find use in the preparation of injectables.
- Suppositories for rectal administration of the drug can be prepared by mixing the drug with a suitable nonirritating excipient such as cocoa butter and polyethylene glycols that are solid at ordinary temperatures but liquid at the rectal temperature and will, therefore, melt in the rectum and release the drug.
- a suitable nonirritating excipient such as cocoa butter and polyethylene glycols that are solid at ordinary temperatures but liquid at the rectal temperature and will, therefore, melt in the rectum and release the drug.
- Solid dosage forms for oral administration may include capsules, tablets, pills, powders, and granules.
- the active compound may be admixed with at least one inert diluent such as sucrose lactose or starch.
- Such dosage forms may also include, as is normal practice, additional substances other than inert diluents, e.g., lubricating agents such as magnesium stearate.
- the dosage forms may also include buffering agents. Tablets and pills can additionally be prepared with enteric coatings.
- Liquid dosage forms for oral administration may include pharmaceutically acceptable emulsions, solutions, suspensions, syrups, and elixirs containing inert diluents commonly used in the art, such as water.
- Such compositions may also comprise adjuvants, such as wetting agents, emulsifying and suspending agents, cyclodextrins, and sweetening, flavoring, and perfuming agents.
- Effective amounts of the compounds of the invention generally include any amount sufficient to detectably treat the disorders described herein. PP028218.0002
- Successful treatment of a subject in accordance with the invention may result in a reduction or alleviation of symptoms in a subject afflicted with a medical or biological disorder.
- treatment may halt the further progression of the disorder, or may prevent or retard development of the disorder.
- the amount of active ingredient that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host treated and the particular mode of administration. It will be understood, however, that the specific dose level for any particular patient will depend upon a variety of factors including the activity of the specific compound employed, the age, body weight, general health, sex, diet, time of administration, route of administration, rate of excretion, drug combination, and severity of the particular disease undergoing therapy.
- the therapeutically effective amount for a given situation can be readily determined by routine experimentation and is within the skill and judgment of the ordinary clinician.
- ICso value The concentration of an inhibitor that causes a 50 % reduction in a measured activity.
- quinazolines (as pertaining to quinazolines of the present invention), indicates compounds having the general structure of Formula I, II or III as described herein.
- the quinazolines include the compounds listed in Tables 1-5, infra.
- Modeulating refers to inducing or suppressing.
- a “disease associated with cellular proliferation” includes, but is not limited to cancers, for example cancers of the prostate, lung, colon and breast, neuro-fibromatosis, atherosclerosis, pulmonary fibrosis, arthritis, psoriasis, glomerulonephritis, restenosis, proliferative diabetic PP028218.0002
- PDR retinopathy
- an effective amount of a compound to treat a disorder may be an amount necessary to cause reduction or alleviation of symptoms in a subject afflicted with a medical or biological disorder.
- treatment may halt the further progression of the disorder, or may prevent or retard development of the disorder.
- the effective amount may vary, depending, for example, upon the condition treated, weight of the subject and severity of the disease. One of skill in the art can readily determine the effective amount empirically without undue experimentation.
- an effective amount for treatment refers to an amount sufficient to palliate, ameliorate, stabilize, reverse, slow or delay progression of a condition such as a disease state.
- a “subject” or “patient” is meant to describe a human or vertebrate animal including a dog, cat, horse, cow, pig, sheep, goat, monkey, rat, mouse, and other mammals.
- ester refers to esters, which hydrolyze in vivo and include those that break down readily in the human body to leave the parent compound or a salt thereof.
- Suitable ester groups include, for example, those derived from pharmaceutically acceptable aliphatic carboxylic acids, particularly alkanoic, alkenoic, cycloalkanoic and alkanedioic acids, in which each alkyl or alkenyl moiety advantageously has not more than 6 carbon atoms.
- Representative examples of particular esters include, but are not limited to, formates, acetates, propionates, butyrates, acrylates and ethylsuccinates.
- the compounds of the present invention can be used in the form of salts as in "pharmaceutically acceptable salts" derived from inorganic or organic acids.
- These salts include but are not limited to the following: acetate, adipate, alginate, citrate, aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, camphorate, camphorsulfonate, digluconate, cyclopentanepropionate, dodecylsulfate, ethanesulfonate, glucoheptanoate, glycerophosphate, hemisulfate, heptanoate, hexanoate, fumarate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethanesulfonate, lactate, maleate, methanesulfonate, nicotinate, 2-napthalenesulfonate, oxalate, pamoate, pe
- the basic nitrogen-containing groups can be quaternized with such agents as lower alkyl halides, such as methyl, ethyl, propyl, and butyl chloride, bromides, and iodides; dialkyl sulfates like dimethyl, diethyl, dibutyl, and diamyl sulfates, long chain halides such as decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides, aralkyl halides like benzyl and phenethyl bromides, and others. Water or oil-soluble or dispersible products are thereby obtained.
- lower alkyl halides such as methyl, ethyl, propyl, and butyl chloride, bromides, and iodides
- dialkyl sulfates like dimethyl, diethyl, dibutyl, and diamyl sulfates
- long chain halides such
- Alkyl refers to saturated aliphatic hydrocarbyl groups having from 1 to 10 carbon atoms and preferably 1 to 6 carbon atoms. This term includes, by way of example, linear and branched hydrocarbyl groups such as methyl (CH 3 -), ethyl (CH 3 CH2-), »-propyl (CHaCH 2 CH 2 -), isopropyl ((CHa) 2 CH-), n-butyl (CH 3 CH 2 CH 2 CH 2 -), isobutyl ((CH 3 ) 2 CHCH 2 -) 5 sec-butyl ((CH 3 )(CH 3 CH 2 )CH-), f-butyl ((CH 3 ) 3 C-), n-pentyl (CH 3 CH 2 CH 2 CH 2 CH 2 -), andneopentyl ((CH 3 ) 3 CCH 2 -).
- Substituted alkyl refers to an alkyl group having from 1 to 5, preferably 1 to 3, or more preferably 1 to 2 substituents selected from the group consisting of alkoxy, substituted alkoxy, acyl, acylamino, acyloxy, amino, substituted amino, aminocarbonyl, aminothiocarbonyl, aminocarbonylamino, aminothiocarbonylamino, aminocarbonyloxy, aminosulfonyl, aminosulfonyloxy, aminosulfonylamino, amidino, aryl, substituted aryl, aryloxy, substituted aryloxy, arylthio, substituted arylthio, carboxyl, carboxyl ester, (carboxyl ester)amino, (carboxyl ester)oxy, cyano, cycloalkyl, substituted cycloalkyl, cycloalkyloxy, substituted cycloalkyloxy, cycloalkylthio,
- Alkyl interrupted with -O-, -S-, -SO-, -SO2-, -NH-, carbonyl, carbonylamino, or aminocarbonyl refers to a C2-10 alkyl group in which an -O-, -S-, -SO-, -SO2-, -NH-, carbonyl, carbonylamino, or aminocarbonyl is inserted between the carbon atoms of the alkyl group.
- an ethylene radical interrupted with -O- is -C-O-C-.
- Alkoxy refers to the group -O-alkyl wherein alkyl is defined herein. Alkoxy includes, byway of example, methoxy, ethoxy, w-propoxy, isopropoxy, «-butoxy, t-butoxy, sec-butoxy, and «-pentoxy.
- Substituted alkoxy refers to the group -O-(substituted alkyl) wherein substituted alkyl is defined herein.
- Acyl refers to the groups H-C(O)-, alkyl-C(O)-, substituted alkyl-C(O)-, alkenyl-C(O)-, substituted alkenyl-C(O)-, alkynyl-C(O)-, substituted alkynyl-C(OK cycloalkyl-C(O)-, substituted cycloalkyl-C(O)-, cycloalkenyl-C(0)-, substituted cycloalkenyl-C(O)-, aryl-C(O)-, substituted aryl-C(O)-, heteroaryl-C(O)-, substituted heteroaryl-C(O)-, heterocyclic-C(O)-, and substituted heterocyclic-C(O)-, wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, al
- Acylamino refers to the groups -NRC(O)alkyl, -NRC(O)substituted alkyl, -NRC(O)cycloalkyl, -NRC(O)substituted cycloalkyl, -NRC(O)cycloalkenyl, -NRC(O)substituted cycloalkenyl, -NRC(O)alkenyl, -NRC(O)substituted alkenyl, -NRC(O)alkynyl, - NRC(O)substituted alkynyl, -NRC(O)aryl, -NRC(O)substituted aryl, -NRC(O)heteroaryl, -NRC(O)substituted heteroaryl, -NRC(0)hetero cyclic, and -NRC(O) substituted heterocyclic wherein R is hydrogen or alkyl
- Acyloxy refers to the groups alkyl-C(O)O-, substituted alkyl-C(O)O-, alkenyl-C(O)O-, substituted alkenyl-C(O)O-, alkynyl-C(O)O-, substituted alkynyl-C(O)O ⁇ , aryl-C(O)O-, substituted aryl-C(O)O-, cycloalkyl-C(O)O-, substituted cycloalkyl-C(O)O-, cycloalkenyl-C(O)O-, substituted cycloalkenyl-C(O)O-, heteroaryl-C(O)O-, substituted PP028218.0002
- Amino refers to the group -NH 2 .
- Substituted amino refers to the group -NR'R" where R' and R" are independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, heteroaryl, substituted heteroaryl, heterocyclic, substituted heterocyclic, -S ⁇ 2-alkyl, -SCh-substituted alkyl, -S ⁇ 2-alkenyl, -SCh-substituted alkenyl, -S ⁇ 2 -cycloalkyl, -SC> 2 -substituted cylcoalkyl, -S ⁇ 2 -cycloalkenyl, -SCh-substituted cylcoalkenyl,-SO 2
- R' is hydrogen and R" is alkyl
- the substituted amino group is sometimes referred to herein as alkylamino.
- R' and R" are alkyl
- the substituted amino group is sometimes referred to herein as dialkylamino.
- a monosubstituted amino it is meant that either R' or R" is hydrogen but not both.
- a disubstituted amino it is meant that neither R' nor R" are hydrogen.
- Aminocarbonyl refers to the group -C(O)N R 10 R 11 where R 10 and R 11 are independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic and where R 10 and R 11 are optionally joined together with the nitrogen bound thereto to form a heterocyclic or substituted heterocyclic group, and wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, PP028218.0002
- substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, heterocyclic and substituted heterocyclic are as defined herein.
- Aminothiocarbonyl refers to the group -C(S)NR 10 R 11 where R 10 and R 11 are independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic and where R 10 and R 1 ' are optionally joined together with the nitrogen bound thereto to form a heterocyclic or substituted heterocyclic group, and wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, substituted
- Aminocarbonylamino refers to the group -NRC(O)NR 10 R 11 where R is hydrogen or alkyl and R 10 and R 11 are independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic and where R 10 and R 11 are optionally joined together with the nitrogen bound thereto to form a heterocyclic or substituted heterocyclic group, and wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl
- Aminothiocarbonylamino refers to the group -NRC(S)NR 10 R 11 where R is hydrogen or alkyl and R 10 and R 11 are independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic and where R 10 and R 11 are optionally joined together with the nitrogen bound thereto to form a heterocyclic or substituted heterocyclic group, and wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloal
- heteroaryl substituted heteroaryl, heterocyclic and substituted heterocyclic are as defined herein.
- Aminocarbonyloxy refers to the group -0-C(O)NR 10 R 11 where R 10 and R n are independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic and where R 10 and R 11 are optionally joined together with the nitrogen bound thereto to form a heterocyclic or substituted heterocyclic group, and wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, substituted
- Aminosulfonyl refers to the group -SO 2 NR 10 R 11 where R 10 and R 11 are independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic and where R 10 and R 11 are optionally joined together with the nitrogen bound thereto to form a heterocyclic or substituted heterocyclic group, and wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, substituted substituted
- Aminosulfonyloxy refers to the group -0-SO 2 NR 10 R 11 where R 10 and R 11 are independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic and where R 10 and R 11 are optionally joined together with the nitrogen bound thereto to form a heterocyclic or substituted heterocyclic group, and wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, substituted
- Aminosulfonylamino refers to the group -NR-SO 2 NR 10 R 1 ' where R is hydrogen or alkyl and R 10 and R 11 are independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkyenyl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic and where R 10 and R 1 * are optionally joined together with the nitrogen bound thereto to form a heterocyclic or substituted heterocyclic group, and wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted
- Aryl or “Ar” refers to a monovalent aromatic carbocyclic group of from 6 to 14 carbon atoms having a single ring (e.g., phenyl) or multiple condensed rings (e.g., naphthyl or anthryl) which condensed rings may or may not be aromatic (e.g., 2-benzoxazolinone, 2H-1,4- benzoxazin-3(4H)-one-7-yl, and the like) provided that the point of attachment is at an aromatic carbon atom.
- Preferred aryl groups include phenyl and naphthyl.
- Substituted aryl refers to aryl groups which are substituted with 1 to 5, preferably 1 to
- substituents selected from the group consisting of alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, alkoxy, substituted alkoxy, acyl, acylamino, acyloxy, amino, substituted amino, aminocarbonyl, aminothiocarbonyl, aminocarbonylamino, aminothiocarbonylamino, aminocarbonyloxy, aminosulfonyl, PP028218.0002
- Aryloxy refers to the group -O-aryl, where aryl is as defined herein, that includes, by way of example, phenoxy and naphthoxy . '
- Substituted aryloxy refers to the group -O-(substituted aryl) where substituted aryl is as defined herein.
- Arylthio refers to the group — S-aryl, where aryl is as defined herein.
- Substituted arylthio refers to the group -S-(substituted aryl), where substituted aryl is as defined herein.
- Alkenyl refers to alkenyl groups having from 2 to 6 carbon atoms and preferably 2 to 4 carbon atoms and having at least 1 and preferably from 1 to 2 sites of alkenyl unsaturation. Such groups are exemplified, for example, by vinyl, allyl, and but-3-en-l-yl.
- Substituted alkenyl refers to alkenyl groups having from 1 to 3 substituents, and preferably 1 to 2 substituents, selected from the group consisting of alkoxy, substituted alkoxy, acyl, acylamino, acyloxy, amino, substituted amino, aminocarbonyl, aminothiocarbonyl, aminocarbonylamino, aminothiocarbonylamino, aminocarbonyloxy, aminosulfonyl, aminosulfonyloxy, aminosulfonylamino, amidino, aryl, substituted aryl, aryloxy, substituted aryloxy, arylthio, substituted arylthio, carboxyl, carboxyl ester, (carboxyl ester)amino, (carboxyl ester)oxy, cyano, cycloalkyl, substituted cycloalkyl, cycloalkyloxy, substituted cycloalkyloxy, cycloalkylthio,
- cycloalkenyloxy substituted cycloalkenyloxy, cycloalkenylthio, substituted cycloalkenylthio, guanidino, substituted guanidino, halo, hydroxy, heteroaryl, substituted heteroaryl, heteroaryloxy, substituted heteroaryloxy, heteroarylthio, substituted heteroarylthio, heterocyclic, substituted heterocyclic, heterocyclyloxy, substituted heterocyclyloxy, heterocyclylthio, substituted heterocyclylthio, nitro, SO 3 H, substituted sulfonyl, sulfonyloxy, thioacyl, thiol, alkylthio, and substituted alkylthio, wherein said substituents are defined herein and with the proviso that any hydroxy substitution is not attached to a vinyl (unsaturated) carbon atom.
- Alkynyl refers to alkynyl groups having from 2 to 6 carbon atoms and preferably 2 to 3 carbon atoms and having at least 1 and preferably from 1 to 2 sites of alkynyl unsaturation.
- Substituted alkynyl refers to alkynyl groups having from 1 to 3 substituents, and preferably 1 to 2 substituents, selected from the group consisting of alkoxy, substituted alkoxy, acyl, acylamino, acyloxy, amino, substituted amino, aminocarbonyl, aminothiocarbonyl, aminocarbonylamino, aminothiocarbonylamino, aminocarbonyloxy, aminosulfonyl, aminosulfonyloxy, aminosulfonylamino, amidino, aryl, substituted aryl, aryloxy, substituted aryloxy, arylthio, substituted arylthio, carboxyl, carboxyl ester, (carboxyl ester)amino, (carboxyl ester)oxy, cyano, cycloalkyl, substituted cycloalkyl, cycloalkyloxy, substituted cycloalkyloxy, cycloalkyloxy
- Carboxyl or “carboxy” refers to -COOH or salts thereof.
- Carboxyl ester or “carboxy ester” refers to the groups -C(O)O-alkyl, -C(O)O-substituted alkyl, -C(O)O-alkenyl, -C(O)O-substituted alkenyl, -C(O)O-alkynyl, PP028218.0002
- (Carboxyl ester)amino refers to the group -NR ⁇ C(O)O-alkyl, substituted -NR-C(O)O-alkyl, -NR-C(O)O-alkenyl, -NR-C(O)O-substituted alkenyl, -NR-C(O)O-alkynyl, ' -NR-C(O)O-substituted alkynyl, -NR-C(O)O-aryl, -NR-C(O)O-substituted aryl,
- R is alkyl or hydrogen, and wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic
- (Carboxyl ester)oxy refers to the group -O-C(O)O-alkyl, substituted -O-C(O)O-alkyl, -O-C(O)O-alkenyl, -O-C(O)O-substituted alkenyl, -O-C(O)O-alkynyl, -O-C(O)O-substituted alkynyl, -O-C(O)O-aryl, -O-C(O)O-substituted aryl, -0-C(0)0-cycloalkyl,
- Cyano refers to the group -CN.
- Cycloalkyl refers to cyclic alkyl groups of from 3 to 10 carbon atoms having single or multiple cyclic rings including fused, bridged, and spiro ring systems. Examples of suitable PP028218.0002
- cycloalkyl groups include, for instance, adamantyl, cyclopropyl, cyclobutyl, cyclopentyl, and cyclooctyl.
- Substituted cycloalkyl and “substituted cycloalkenyl” refers to a cycloalkyl or cycloalkenyl group having from 1 to 5 or preferably 1 to 3 substituents selected from the group consisting of oxo, thione, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, alkoxy, substituted alkoxy, acyl, acylarnino, acyloxy, amino, substituted amino, aminocarbonyl, aminothiocarbonyl, aminocarbonylamino, aminothiocarbonylamino, aminocarbonyloxy, aminosulfonyl, aminosulfonyloxy, aminosulfonylamino, amidino, aryl, substituted aryl, aryloxy, substituted aryloxy, arylthio, substituted arylthio, carboxyl, carboxyl, carb
- Cycloalkyloxy refers to -O-cycloalkyl.
- Substituted cycloalkyloxy refers to -O-(substituted cycloalkyl).
- Cycloalkylthio refers to -S-cycloalkyl.
- Substituted cycloalkylthio refers to -S-(substituted cycloalkyl).
- Cycloalkenyloxy refers to -O-cyeloalkenyl.
- Substituted cycloalkenyloxy refers to -O-(substituted cycloalkenyl).
- Cycloalkenylthio refers to — S-cycloalkenyl.
- Substituted cycloalkenylthio refers to -S-(substituted cycloalkenyl).
- Halo or halogen refers to fluoro, chloro, bromo and iodo.
- Heteroaryl refers to an aromatic group of from 1 to 10 carbon atoms and 1 to 4 heteroatoms selected from the group consisting of oxygen, nitrogen and sulfur within the ring.
- Such heteroaryl groups can have a single ring (e.g. , pyridinyl or furyl) or multiple condensed rings (e.g., indolizinyl or benzothienyl) wherein the condensed rings may or may not be aromatic and/or contain a heteroatom provided that the point of attachment is through an atom of the aromatic heteroaryl group.
- the nitrogen and/or the sulfur ring atom(s) of the heteroaryl group are optionally oxidized to provide for the N-oxide (N- »O), sulfinyl, or sulfonyl moieties.
- Preferred heteroaryls include pyridinyl, py ⁇ rolyl, indolyl, thiophenyl, and furanyl.
- Substituted heteroaryl refers to heteroaryl groups that are substituted with from 1 to 5, preferably 1 to 3, or more preferably 1 to 2 substituents selected from the group consisting of the same group of substituents defined for substituted aryl.
- Heteroaryloxy refers to -O-heteroaryl.
- Substituted heteroaryloxy refers to the group — O-(substituted heteroaryl).
- Heteroarylthio refers to the group — S-heteroaryl.
- Substituted heteroarylthio refers to the group -S-(substituted heteroaryl).
- Heterocycle or “heterocyclic” or “heterocycloalkyl” or “heterocyclyl” refers to a saturated or unsaturated group having a single ring or multiple condensed rings, including fused bridged and spiro ring systems, from 1 to 10 carbon atoms and from 1 to 4 hetero atoms selected from the group consisting of nitrogen, sulfur or oxygen within the ring wherein, in fused ring systems, one or more the rings can be cycloalkyl., aryl or heteroaryl provided that the point of attachment is through the non-aromatic ring.
- the nitrogen and/or sulfur atom(s) of the heterocyclic group are optionally oxidized to provide for the N-oxide, sulfinyl, sulfonyl moieties.
- Substituted heterocyclic or “substituted heterocycloalkyl” or “substituted heterocyclyl” refers to heterocyclyl groups that are substituted with from 1 to 5 or preferably 1 to 3 of the same substituents as defined for substituted cycloalkyl.
- Heterocyclyloxy refers to the group -O-heterocycyl.
- Substituted heterocyclyloxy refers to the group -O-(substituted heterocycyl).
- Heterocyclylth ⁇ o refers to the group — S-heterocycyl.
- Substituted heterocyclylthio refers to the group — S-(substituted heterocycyl).
- heterocycle and heteroaryls include, but are not limited to, azetidine, pyrrole, imidazole, pyrazole, pyridine, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, dihydroindole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthylpyridine, quinoxaline, quinazoline, cinnoline, pteridine, carbazole, carboline, phenanthridine, acridine, phenanthroline, isothiazole, phenazine, isoxazole, phenoxazine, phenothiazine, imidazolidine, imidazoline, piperidine, piperazine, indoline, phthalimide, 1,2,3,4- tetrahydroisoquinoline, 4,5,6,
- Niro refers to the group -NO 2 .
- Oxo refers to the atom (O) or (-O " ).
- “Spirocycloalkyl” refers to divalent cyclic groups from 3 to 10 carbon atoms having a cycloalkyl ring with a spiro union (the union formed by a single atom which is the only common member of the rings) as exemplified by the following structure:
- Sulfonyl refers to the divalent group -S(O) 2 -.
- Substituted sulfonyl refers to the group -S ⁇ 2 -alkyl, -SO 2 -substituted alkyl, -SO 2 - alkenyl, -S ⁇ 2-substituted alkenyl, -SOa-cycloalkyl, -S ⁇ 2 -substituted cylcoalkyl, -SO 2 - cycloalkenyl, -S ⁇ 2-substituted cylcoalkenyl, -S ⁇ 2-aryl, -SO 2 -substituted aryl, -S ⁇ 2 -heteroaryl, -S ⁇ 2-substituted heteroaryl, -S0 2 -heterocyclic, -S ⁇ 2 -substituted heterocyclic, wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, substituted
- “Sulfonyloxy” refers to the group -OSO 2 -alkyl, -OSO 2 -substituted alkyl, -OSO 2 -alkenyl, -OSO 2 -substituted alkenyl, -OSO 2 -cycloalkyl, -OSO 2 -substituted cylcoalkyl, -OSO 2 - cycloalkenyl, -OSO 2 -substituted cylcoalkenyl.-OSO ⁇ -aryl, -OSO ⁇ -substituted aryl, -OSO 2 - heteroaryl, -OSOa-s ⁇ bstituted heteroaryl, -OSO 2 -heterocyclic, -OSO 2 -substituted heterocyclic, wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted al
- Thioacyl refers to the groups H-C(S)-, alkyl-C(S)-, substituted alkyl-C(S)-, alkenyl-C(S)-, substituted alkenyl-C(S)-, alkynyl-C(S)-, substituted alkynyl-C(S)-, cycloalkyl-C(S)-, substituted cycloalkyl-C(S)-, cycloalkenyl-C(S)-, substituted PP028218.0002
- Thiol refers to the group -SH.
- Alkylthio refers to the group -S-alkyl wherein alkyl is as defined herein.
- Substituted alkylthio refers to the group -S-(substituted alkyl) wherein substituted alkyl is as defined herein.
- substituents of compounds of the invention are disclosed in groups or in ranges. It is specifically intended that the invention include each and every individual subcombination of the members of such groups and ranges.
- the term "Ci-e alkyl” is specifically intended to individually disclose methyl, ethyl, C 3 alkyl (propyl and isopropyl), C 4 alkyl, Cs alkyl, and Ce alkyl.
- Stereoisomer or “stereoisomers” refer to compounds that differ in the chirality of one or more stereocenters. Stereoisomers include enantiomers and diastereomers.
- protected or a “protecting group” with respect to hydroxyl groups, amine groups, and sulfhydryl groups refers to forms of these functionalities which are protected from undesirable reaction with a protecting group known to those skilled in the art such as those set forth in Protective Groups in Organic Synthesis, Greene, T. W., John Wiley & Sons, New York, NY, (1 st Edition, 1981) which can be added or removed using the procedures set forth therein.
- Examples of protected hydroxyl groups include, but are not limited to, silyl ethers such as those obtained by reaction of a hydroxyl group with a reagent such as, but not limited to, t- butyldimethyl-chlorosilane, trimethylchlorosilane, triisopropylchlorosilane, triethylchlorosilane; substituted methyl and ethyl ethers such as, but not limited to methoxymethyl ether, methythiomethyl ether, benzyloxymethyl ether, t-butoxymethyl ether, 2-methoxyethoxymethyl ether, tetrahydropyranyl ethers, 1-ethoxyethyl ether, allyl ether, benzyl ether; esters such as, but not limited to, benzoylformate, formate, acetate, trichloroacetate, and trifluoracetate.
- a reagent such as, but not limited to
- protected amine groups include, but are not limited to, benzyl or dibenzyl, amides such as, formamide, acetamide, trifluoroacetamide, and benzamide; imides, such as phthalimide, and dithiosuccinimide; and others.
- a protecting group for amines is a benzyl group.
- protected sulfhydryl groups include, but are not limited to, thioethers such as S-benzyl thioether, and S-4-picolyl thioether; substituted S-methyl derivatives such as hemithio, dithio and aminothio acetals; and others.
- Quinazoline compounds of Formula I, II or III may exhibit the phenomenon of tautomerism, and the formula drawings within this specification can represent only one of the possible tautomeric forms. It is to be understood that the invention encompasses any tautomeric form which possesses immunomodulatory activity and is not to be limited merely to any one tautomeric form utilized within the formula drawings.
- Quinazolines of Formula I, II or III also may exist in solvated as well as unsolvated forms such as, for example, hydrated forms.
- the invention encompasses both solvated and unsolvated forms which possess immunomodulatory activity.
- the invention also includes isotopically-labeled quinazoline compounds, that are structurally identical to those disclosed above, except that one or more atom is/are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- isotopes that can be incorporated into compounds of the PP028218.0002
- isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, sulfur, fluorine and chlorine such as 2 H, 3 H, 13 C, 14 C, 15 N, 18 0, 17 0, 31 P, 32 P, 35 S, 18 F and 36 Cl, respectively.
- Compounds of the present invention, tautomers thereof, prodrugs thereof, and pharmaceutically acceptable salts of the compounds and of the prodrugs that contain the aforementioned isotopes and/or other isotopes of other atoms are within the scope of this invention.
- Certain isotopically- labeled compounds of the present invention for example those into which radioactive isotopes such as 3 H and 14 C are incorporated, are useful in drug and/or substrate tissue distribution assays.
- Tritiated, i.e., 3 H, and carbon-14, i.e., 14 C, isotopes are particularly preferred for their ease of preparation and detectability. Further, substitution with heavier isotopes such as deuterium, i.e., 2 H, may afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements and, hence, may be preferred in some circumstances.
- Isotopically labeled compounds of this invention and prodrugs thereof can generally be prepared by carrying out known or referenced procedures and by substituting a readily available isotopically labeled reagent for a non-isotopically labeled reagent.
- the compounds of the invention are useful in pharmaceutical compositions for human or veterinary use where inhibition of PDKl is indicated, for example, in the treatment of cellular proliferative diseases such as tumor and/or cancerous cell growth mediated by PDKl .
- the compounds are useful in the treatment of human or animal (e.g., murine) cancers, including, for example, lung and bronchus; prostate; breast; pancreas; colon and rectum; thyroid; liver and intrahepatic bile duct; hepatocellular; gastric; glioma/glioblastoma; endometrial; melanoma; kidney and renal pelvis; urinary bladder; uterine corpus; uterine cervix; ovary; multiple myeloma; esophagus; acute myelogenous leukemia; chronic myelogenous leukemia; lymphocytic leukemia; myeloid leukemia; brain; oral cavity and pharynx; larynx;
- the invention provides methods for manufacture of PDKl inhibitor compounds. It is further contemplated that, in addition to the compounds of Formulas I-III, intermediates, and their corresponding methods of syntheses are included within the scope of the embodiments. PP028218.0Q02
- the present invention provides for compounds of Formula I, II, or III for inhibition of Cdkl and/or Cdk2. Another embodiment provides a method of treating cancer responsive to inhibition of Cdkl, comprising administering a compound of Formula I, II, or III. Another embodiment provides a method of treating cancer responsive to inhibition of Cdk2, comprising administering a compound of Formula I, II, or III.
- the invention provides methods of inhibiting phosphorylation of Akt comprising administering a compound of Formula I, II, or III to a human in need thereof. Another embodiment provides a method of treating cancer responsive to inhibition of phosphorylation of Akt, comprising administering a compound of Formula I, II, or III. Another embodiment provides a method of inhibiting phosphorylation of Akt comprising contacting a cell with a compound of Formula I, II, or III. hi further embodiments, the invention provides methods of inhibiting PDKl comprising orally administering a compound of Formula I, II, or III to a human in need thereof.
- the human is suffering from cancer, hi a more particular embodiment the cancer is responsive to treatment with a compound that inhibits phosphorylation of PDKl .
- the compound is orally bioavailable.
- the IC 50 value of the compound is less than or equal to about 1 mM with respect to PDKl .
- the IC 50 value is less than or equal to about 100 ⁇ M, is less than or equal to about 25 ⁇ M, is less than or equal to about 10 ⁇ M, is less than or equal to about 1 ⁇ M, is less than or equal to about 0.1 ⁇ M, is less than or equal to about 0.050 ⁇ M, or is less than or equal to about 0.010 ⁇ M.
- a method of reducing PDKl activity in a human or animal subject is provided. In the method, a compound of the any of the aforementioned embodiments is administered in an amount effective to reduce PDKl activity.
- the IC 50 value of the compound is between about 1 nM to about 10 nM. In other such embodiments, the IC50 value is between about 10 nM to about 50 nM, between about 50 nM to about 100 nM, between about 100 nM to about 1 ⁇ M, between about 1 ⁇ M to about 25 ⁇ M, or is between about 25 ⁇ M to about 100 ⁇ M.
- Another embodiment provides methods of treating a PDKl -mediated disorder.
- an effective amount of a PDKl inhibitor compound is administered to a patient (e.g., a human or animal subject) in need thereof to mediate (or modulate) PDKl activity.
- the PDKl -mediated disorder is cancer.
- Still another embodiment provides methods of treating diseases characterized by
- abnormal cellular proliferation includes, for example, any disease or disorder characterized by excessive or pathologically elevated cell growth such as is characteristic of various cancers and non-cancer proliferative disorders.
- Example cancers include, for example, lung cancer, bronchial cancer, prostate cancer, breast cancer, pancreatic cancer, colon cancer, rectal cancer, colorectal cancer, thyroid cancer, liver cancer, intrahepatic bile duct cancer, hepatocellular cancer, gastric cancer, glioma/glioblastoma, endometrial cancer, melanoma, kidney cancer, renal pelvic cancer, urinary bladder cancer; uterine corpus cancer; uterine cervical cancer, ovarian cancer, multiple myeloma, esophageal cancer, acute myelogenous leukemia, chronic myelogenous leukemia, lymphocytic leukemia, myeloid leukemia, brain cancer, oral cavity cancer, and pharyngeal cancer, laryngeal cancer, small intestinal cancer, non-Hodgkin lymphoma, and villous colon adenoma.
- Example non-cancer proliferative disorders include neuro-fibromatosis, atherosclerosis, pulmonary fibrosis, arthritis, psoriasis, glomerulonephritis, restenosis, proliferative diabetic retinopathy (PDR), hypertrophic scar formation, inflammatory bowel disease, transplantation rejection, angiogenesis, and endotoxic shock. . .
- the invention provides pharmaceutical compositions including at least one compound of Formula I, II, or III, together with one or more pharmaceutically acceptable carriers suitable for administration to a human or animal subject, either alone or together with other agents, for example, anticancer agents.
- the invention provides methods of treating human or animal subjects suffering from a cellular proliferative disease, such as cancer. Ih some such embodiments, the invention provides methods of treating a human or animal subject in need of such treatment, comprising administering to the subject a therapeutically effective amount of a compound of Formula I, II, or III, either alone or in combination with other anticancer agents.
- compositions will either be formulated together as a combination PP028218.0002
- Anticancer agents for use with the preferred embodiments include, but are not limited to, one or more of the following set forth below:
- kinase inhibitors for use as anticancer agents in conjunction with the compositions of the preferred embodiments include inhibitors of Epidermal Growth Factor Receptor (EGFR) kinases such as small molecule quinazolines, for example gef ⁇ tinib (US 5457105, US 5616582, and US 5770599), ZD-6474 (WO 01/32651), erlotinib (Tarceva®, US 5,747,498 and WO 96/30347), and lapatinib (US 6,727,256 and WO 02/02552); Vascular Endothelial Growth Factor Receptor (VEGFR) kinase inhibitors, including SU-11248 (WO 01/60814), SU 5416 (US 5,883,113 and WO 99/61422), SU 6668 (US 5,883,113 and WO 99/61422), CHIR-258 (US 6,605,617 and US 6,774,237), vatalanib or PTK-787 (US 6,258,
- Estrogen-targeting agents for use in anticancer therapy in conjunction with the compositions of the preferred embodiments include Selective Estrogen Receptor Modulators (SERMs) including tamoxifen, toremifene, raloxifene; aromatase inhibitors including Arimidex® or anastrozole; Estrogen Receptor Downregulators (ERDs) including Faslodex® or fulvestrant.
- SERMs Selective Estrogen Receptor Modulators
- ESDs Estrogen Receptor Downregulators
- Androgen-targeti ⁇ g agents for use in anticancer therapy in conjunction with the compositions of the preferred embodiments include flutamide, bicalutamide, finasteride, aminoglutetharnide, ketoconazole, and corticosteroids.
- flutamide bicalutamide
- finasteride aminoglutetharnide
- ketoconazole ketoconazole
- corticosteroids corticosteroids
- inhibitors for use as anticancer agents in conjunction with the compositions of the preferred embodiments include protein farnesyl transferase inhibitors including u ' pifarn ⁇ b or R- 115777 (US 2003134846 and WO 97/21701), BMS-214662, AZD-3409, and FTI-277; topoisomerase inhibitors including merbarone and difiomotecan (BN-80915); mitotic kinesin spindle protein (KSP) inhibitors including SB-743921 and MKI-833; proteasome modulators such as bortezomib or Velcade® (US 5,780,454), XL-784; and cyclooxygenase 2 (COX-2) inhibitors including non-steroidal antiinflammatory drugs I (NSAIDs).
- protein farnesyl transferase inhibitors including u ' pifarn ⁇ b or R- 115777 (US 2003134846 and WO 97/21701), BMS-214662, AZ
- cancer chemotherapeutic drugs for use as anticancer agents in conjunction with the compositions of the preferred embodiments include anastrozole (Arimidex®), bicalutamide (Casodex®), bleomycin sulfate (Blenoxane®), busulfan (Myleran®), busulfan injection (Busulfex®), capecitabine (Xeloda®), N4-pentoxycarbonyl-5-deoxy-5-fluorocytidine, carboplatin (Paraplatin®), carmustine (BiCNU®), chlorambucil (Leukera ⁇ ®), cisplatin (Platinol®), cladribine (Leustatin®), cyclophosphamide (Cytoxan® or Neosar®), cytarabine, cytosine arabinoside (Cytosar-U®), cytarabine liposome injection (DepoCy
- thioguanine, thiotepa, tirapazamine Terazone®
- topotecan hydrochloride for injection Hycamptin®
- vinblastine Velban®
- vincristine Oncovin®
- vinorelbine® vinorelbine
- Alkylating agents for use in conjunction with the compositions of the preferred embodiments for anticancer therapeutics include VNP-40101M or cloretizine, oxaliplatin (US 4,169,846, WO 03/24978 and WO 03/04505), glufosfamide, mafosfamide, etopophos (US 5,041,424), prednimustine; treosulfan; busulfan, irofluven (acylfulvene), penclomedine, pyrazoloacridine (PD-115934); O6-benzylguanine, decitabine (5-aza-2-deoxycytidine), brostallicin, mitomycin C (MitoExtra), TLK-286 (Telcyta®), temozolomide, trabectedin (US 5,478,932), AP-5280 (Platinate formulation of Cisplatin), porfuromycin, and clearazide (meclorethamine) .
- Chelating agents for use in conjunction with the compositions of the preferred embodiments for anticancer therapeutics include tetrathiomolybdate (WO 01/60814); RP-697, Chimeric T84.66 (cT84.66), gadofosveset (Vasovist®), deferoxamine, and bleomycin optionally in combination with electorporation (EPT).
- Biological response modifiers such as immune modulators, for use in conjunction with the compositions of the preferred embodiments for anticancer therapeutics include staurosprine and macrocyclic analogs thereof, including UCN-Ol, CEP-701 and midostaurin (see WO
- interleukins specifically IL-2 or aldesleukin as well as IL-I, IL-3, DL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-IO, IL-11, IL-12, and active biological variants thereof having amino acid sequences greater than 70% of the native human sequence; altretamine (Hexalen®); SU 101 or lefiunomide (WO 04/06834 and US 6,331,555); imidazoquinolines such as resiquimod and imiquimod (US 4,689,338, 5,389,640, 5,268,376,
- Anticancer vaccines for use in conjunction with the compositions of the preferred embodiments include Avicine® ⁇ Tetrahedron Lett. 26:2269-70 (1974)); oregovomab
- Theratope® (STn-KJLH); Melanoma Vaccines; GI-4000 series (GI-4014, GI-401S, and GI-4016), which are directed to five mutations in the Ras protein; GlioVax-1 ; MelaVax; Advexin® or INGN-201 (WO 95/12660); Sig/E7/LAMP-1, encoding HPV-16 E7; MAGE-3 Vaccine or M3TK (WO 94/05304); HER-2VAX; ACTIVE, which stimulates T-cells specific for tumors; GM-CSF cancer vaccine; and Listeria monocytogenes-based vaccines.
- Anticancer agents for use in conjunction with the compositions of the preferred embodiments also include antisense compositions, such as AEG-35156 (GEM-640); AP-12009 and AP-11014 (TGF-beta2-specific antisense oligonucleotides); AVI-4126; AVI-4557; AVI- 4472; oblimersen (Genasense®); JFS2; aprinocarsen (WO 97/297S0); GTI-2040 (R2 ribonucleotide reductase mRNA antisense oligo) (WO 98/05769); GTI-2501 (WO 98/05769); liposome-encapsulated c-Raf antisense oligodeoxynucleotides (LErafAON) (WO 98/43095); and Sirna-027 (RNAi-based therapeutic targeting VEGFR-I mRNA).
- AEG-35156 GEM-640
- Example compounds and their analogs are easily synthesized by one skilled in the art from procedures described herein, as well as in patents or patent applications listed herein which are all hereby incorporated by reference in their entireties and for all purposes as if fully set forth herein.
- the compounds and/or intermediates were characterized by high performance liquid chromatography (HPLC) using a Waters Millenium chromatography system with a 2695 Separation Module (Milford, MA).
- HPLC high performance liquid chromatography
- the analytical columns were reversed phase Phenomenex Luna Cl 8 -5 ⁇ , 4.6 x 50 mm, from Alltech (Deerf ⁇ eld, IL).
- a gradient elution was PP028218.0002
- TLC thin layer chromatography
- glass or plastic backed silica gel plates such as, for example, Baker-Flex Silica Gel 1B2-F flexible sheets.
- TLC results were readily detected visually under ultraviolet light, or by employing well known iodine vapor and other various staining techniques.
- Mass spectrometric analysis was performed on one of two LCMS instruments: a Waters
- GCMS analysis is performed on a Hewlett Packard instrument (HP6890 Series gas chromatograph with a Mass Selective Detector 5973; injector volume: 1 ⁇ L; initial column temperature: 5O 0 C; final column temperature: 250 0 C; ramp time: 20 minutes; gas flow rate: 1 mL/min; column: 5% phenyl methyl siloxane, Model No. HP 190915-443, dimensions: 30.0 m x 25 m x 0.25 m).
- NMR Nuclear magnetic resonance
- Preparative separations are carried out using a Flash 40 chromatography system and KP-SiI, 6OA (Biotage, Charlottesville, VA), or by flash column chromatography using silica gel (230-400 mesh) packing material, or by HPLC using a Waters 2767 Sample Manager, C- 18 reversed phase column, 30X50 mm, flow 75 rnL/min.
- Typical solvents employed for the Flash 40 Biotage system and flash column chromatography are dichloromethane, methanol, ethyl acetate, hexane, acetone, aqueous ammonia (or ammonium hydroxide), and triethyl amine.
- Typical solvents employed for the reverse phase HPLC are varying concentrations of acetonitrile and water with 0.1% trifluoroacetic acid.
- the reaction flask was washed out with water (3 x 100 mL) and EtOAc (4 x 50 mL). After stirring ca. 30 minutes, mixture was filtered to collect the light solid precipitate. The solid filter cake was washed with portions of water and EtOAc to give a white solid. The solid was dried on the frit and in vacuo to give the product in 99% purity and 95% yield (41 g, 233 mmole).
- Step 3 Preparation of 2-chloro-8-methoxyquinazoline Neat POCI3 (400 mL) was added to 8-methoxyquinazolin-2-ol (5.0 g, 28.4 mmole) with stirring and cooling over an ice bath under argon. After ca. 1 minute, the reaction was removed from the ice bath and stirred at RT for ca. 20 minutes until a fine yellow suspension was formed. The reaction, fitted with a reflux condenser, was heated in an oil bath at 140-145 0 C. After about 1 hour, the reaction turned clear and colorless. LCMS showed that the reaction was complete. The POCI 3 was evaporated under reduced pressure, and dried in vacuo.
- the product was extracted into CH 2 Cl 2 .
- the emulsion that formed was filtered through celite, organic layer was washed with brine, dried over Na 2 SO ⁇ concentrated giving 12.0 g (98% yield) of the title compound.
- Step 3 Preparation of 2-Hydroxy-7-methoxyquinazoline 2-Formyl-5-methoxyaniline (11.93 g, 79.5 mmol, 1.Oeq) and urea ( 38.Og, 0.636 mol, 8.0 eq) were mixed, ground into a fine powder and placed into a IL 2-neck flask equipped with a mechanical stirrer and an air condenser. The flask was placed into a preheated (160 0 C) oil bath. The reaction mixture melted and was stirred at 170-180 0 C for 1 hour until it solidified forming yellowish-brown solid.
- the reaction mixture was cooled to RT, crushed, mixed with 70 mL of H 2 O and stirred for ⁇ 1 hour at RT.
- the solid was filtered, washed with 150 mL of H 2 O and 20 mL of ice cold Et 2 O.
- the yellow solid thus obtained was dried over P4O10 under high vacuum for several hours and the crude material (12.57 g) was used for the next step without purification.
- Step 4 Preparation of 2-Chloro-7-methoxyquinazoline
- the crude 2-Hydroxy-7-methoxyquinazoline (12.48 g) was mixed with 170 mL of POCI 3 , and the reaction mixture was stirred under reflux conditions for 6 hours. POCI 3 was removed using a PP028218.0002
- Triphenylphosphine (0.49 g, 1.86 mmol), di-t-butylazodicarboxylate (0.42 g, 1.86 mmol) and 1- isoprpopylpiperidin-4-ol (0.54 g, 3.74 mmol) were mixed in anhydrous THF (6 mL) and stirred PP028218.0002
- Step 1 To a suspension of 2-amino-3-fluorobenzoic acid (5 g, 32.2 mmol) in chloroform (200 mL) was added dropwise bromine (1.1 equiv.) in chloroform (125 mL) solution. The mixture was stirred at room temperature for 16 hrs. The resulting white solid was collected by filtration and washed thoroughly with methylene chloride until the filtrate was colorless. The solid was air-dried to give 9.6 g of white powder as HBr salt of 2-amino-5-bromo-3-fluorobenzoic acid (95% yield). ES/MS m/z 234/236 (MH 4 ). PP028218.0002
- Step 2 To the above intermediate (30.6 mmol) in THF (100 mL) at 0 C was added boran tetrahydrofuran complex solution (1 M in THF, 129 mL, 4 equiv.). The mixture was stirred at room temperature for 40 hrs. The solvent was removed in vacuo and the excess reagent was quenched by addition of water (30 mL) slowly. The pH ( ⁇ 3) was adjusted by adding sodium bicarbonate (sat. aq.) to pH 7. Extracted with methylene chloride. The organic extracts were combined, washed with brine, dried with sodium sulfate and concentrated to give a crude material as white solide. ES/MS m/z 220/222 (MH + ).
- Step 3 To the above intermediate (30.6 mmol) in dichloromethane (450 mL) was added manganese dioxide (MnO 2 , 22 g, 258 mmol). The mixture was stirred at room temperature under argon for 18 hrs. The mixture was filtered through celite pad and washed thoroughly with dichloromethane. The filtrated was concentrated in vacuo to give crude product (2-amino-5- bromo-3-fluorophenyi)methanol (5.6 g) which was used for the next step without further purification. ES/MS m/z 218/220 (MH + ).
- Step 4 A mixture of (2-amino-5-bromo-3-fluorophenyl)methanol (5.6 g, 23.7 mmol, obtained from step 3) and urea (21 g, 15 equiv.) was heated to 175 0 C with vigorous stirring for 15 min. The reaction was cooled to room temperature and water was added. The solid was collected by filtration. Air-dried to give 2-hydroxyquinazoline as a light brown solid.
- Step 5 To the above crude material was added phosphooxychloride (POCI 3 , 20 mL) and heated to 110 0 C for 30 min. The resulting mixture was cooled to room temperature and concentrated in vacuo to nearly dryness. Ice water was added and pH was adjusted to ⁇ 6 using sodium bicarbonate. Extraction with dichloromethane followed by drying with sodium sulfate and concentrated in vacuo yielded desired product 6-bromo-2-chloro-8-fluoroquuiazoline as light brown powder (1.63 g).
- phosphooxychloride POCI 3 , 20 mL
- Step 1 To a suspension of 2-amino-3-chlorobenzoic acid (2 g, 11.6 mmol) in chloroform (120 mL) was added dropwise bromine (1.1 equiv.) in chloroform (12 mL) solution. The mixture was stirred at room temperature for 16 hrs. The resulting white solid was collected by filtration and washed thoroughly with methylene chloride until the filtrate was colorless. The solid was air- dried to give 3.35 g of white powder as HBr salt of 2-amino-5-bromo-3-chlorobenzoic acid (87% yield). ES/MS m/z 250/252 (MH + ).
- Step 2 To the above intermediate (3.35 g, 10.1 mmol) in THF (40 mL) at 0 0 C was added borane tetrahydrofuran complex solution (1 M in THF, 40 mL, 4 equiv.). The mixture was stirred at room temperature for 18 hrs. Additional borane tetrahydrofuran (20 mL) was added and continued reaction for another 24 hrs until the complete conversion of the starting material. The solvent was removed in vacuo and the excess reagent was quenched by addition of ethanol (20 mL) slowly. Water was added and the pH ( ⁇ 3) was adjusted by adding sodium bicarbonate (sat. aq.) to pH 7. Extracted with methylene chloride. The organic extracts were combined, washed with brine, dried with sodium sulfate and concentrated to give a crude material as white solide. ES/MS m/z 236/238 (MH + ).
- Step 3 To the above intermediate (10.1 mmol) in dichloromethane (80 mL) was added manganese dioxide (MnO 2 , 6 g, 70 mmol). The mixture was stirred at room temperature under argon for 40 hrs. Additional manganese dioxide (6 g) was added and the reaction was continued for another 20 hrs until the complete conversion of the starting material. The mixture was filtered through celite pad and washed thoroughly with dichloromethane. The filtrated was concentrated in vacuo to give crude product (2-amino-5-bromo-3-chlorophenyl)methanol (3.3 g, orange solid) which was used for the next step without further purification. ES/MS m/z 234/236 (MH + ). PP028218.0002
- Step 4 A mixture of (2-amino-5-bromo-3-chlorophenyl)methanol (3.3 g, obtained from step 3) and urea (10.5 g, 15 equiv.) was heated to 180 0 C with vigorous stirring for 1 hr. The reaction was cooled to room temperature and water was added. The solid was collected by filtration. Air-dried to give 2-hydroxyquinazoline as a yellow powder ( 2.18 g, crude). ES/MS m/z 259/261 (MH 4 ).
- Step 5 To the above crude material was added phosphooxychloride (POCb 3 25 mL) and heated to 130 0 C for 30 min. The resulting mixture was cooled to room temperature and concentrated in vacuo to nearly dryness. Ice water was added and pH was adjusted to ⁇ 8 using sodium bicarbonate. Extraction with dichloromethane followed by drying with sodium sulfate and concentrated in vacuo yielded desired product 6-bromo-2,8-dichloroquinazoline as brown foam (1.4 g). This material was used in other chemical medications without further purification.
- phosphooxychloride POCb 3 25 mL
- Step 2 Preparation of 4-(8-isopropoxyquinazolin-2-ylamino)-N-methylbenzamide To a 0.30 M solution of the product from Step 1 in 2-propanol was added 4-amino-N- methylbenzamide (1.0 eq). The reaction was stirred at 100 °C for 14 h. The mixture was concentrated and purified by reverse-phase HPLC and lyophilized to give the desired product as the trifluoroacetic acid salt. ES/MS m/z 337 (MH + ).
- Step 2 Preparation of 8-bromo-6-(trifluoromethyl)-l,4-dihydroquinazoline-2,4-diol 2-Amino-3-bromo-5-(trifluoromethyl)benzaldehyde (1.74 g, 6.49 mmol, 1.00 eq) and urea (5.85 g, 97.4 mmol, 15.0 eq) were stirred at 190 0 C for 3 h. The resulting solid was returned to ambient temperature, stirred in water (60 mL) for 20 min, and filtered. This was repeated for a total of three washes. The solid was dried in a desiccator to give 3.79 g of the desired product as an off-white solid. ES/MS m/z 311 ,313 (MH + ).
- Phosphorus oxychloride (20 mL) was added. The mixture was stirred at 110 0 C for 1.5 h. Volatiles were removed under reduced pressure. Ice water was added, and the pH was adjusted to 6-7 with aqueous sodium hydroxide and sodium bicarbonate. The precipitate was filtered off, rinsed with water, and dried under high vacuum. The crude material was triturated with THF.
- Step 4 Preparation of 4-(8-broino-6-(trifluoromethyl)q ⁇ inazolin-2-ylamino)benzenesulfonamide
- sulfanilamide 1.0 eq
- the reaction was stirred at 110 0 C for 14 h.
- the hydrochloride was collected by vacuum filtration and then stirred in aqueous sodium bicarbonate.
- the solid was collected by vacuum filtration and rinsed with water.
- the light yellow solid was dried in a desiccator to give 343 mg of the desired product.
- Step 1 Preparation of 2-Chloro-8-(l-isopropylpiperidin-4-yloxy)quinazoline
- triphenylphosphine 1.5 eq
- di-tert- butylazodicarboxylate 1.5 eq
- 4- Hydroxy-1-isopropylpiperidine 4.0 eq
- 2-Chloroquinazolin-8-ol 1.0 eq
- the mixture was stirred an additional 2 h.
- Step 3 Preparation of morpholino(4-(8-(piperidin-4-yloxy)quinazolm- 2-ylamino)phenyl)methanone
- the product from Step 2 was dissolved in enough 1:1 DCM-.TFA to make a 0.20M solution. The mixture was stirred for 30 min at ambient temperature and concentrated. The crude product was purified by reverse-phase HPLC and lyophilized to give the desired product as its trifluoroacetic acid salt.
- Step 2-3 Preparation of 6-ethynyl-8-(l-isopropylpiperidin-4-yloxy)-N-(3- morpholinophenyl)quinazolin-2-amine
- step 1 The product of step 1 was subjected to subjected to Sonogashira and desilylation reaction (see the scheme above) to give the title compound.
- Step 1 Preparation of 2,6-dibromo -8-(N-Boc-piperidin-4-yloxy)quinazoline
- diethylazodicarboxylate 2.0 eq
- diethylazodicarboxylate 2.0 eq
- N-Tert-butyl-4-Hydroxy-l- piperidine carboxylate 4.0 eq
- 2,6-Dibromo-8-hydroxyquinazoline 1.0 eq
- the mixture was stirred an additional 24 h.
- the crude mixture was concentrated, purified by flash chromatography (2:1 hexanes:EtOAc), and concentrated to give the desired product.
- Step 3a Sonogashira & desilylation
- the product from Step 2 was subjected to Sonogashira and desilylation reaction (see the scheme above) and carried on to Step 4 without purification.
- Step 4 Preparation of 6-ethynyl-N-(4-morpholinophenyl)-8-(piperidin-4-yloxy)quinazolin-2- amine
- the product from Step 3 was dissolved in enough 1 : 1 DCMrTFA to make a 0.20M solution. The mixture was stirred for 30 min at ambient temperature and concentrated. The crude product was purified by reverse-phase HPLC and lyophilized to give the desired product as its trifluoroacetic acid salt.
- Step 1 To 2-Amino-5-fluorobenzoic acid (5 g, 32.2 mmol) in chloroform (90 mL) was added bromine (1.82 mL, 35.4 mmol) in chloroform (10 mL) solution dropwise via an additional funnel. The mixture was stirred at room temperature for 16 hrs. and LC/MS showed about 50% conversion of the starting material. Additional bromine (1.8 mL) was added to the reaction and continued stirring for another 24 hrs. The resulting white precipitate was collected by filtration, washed thoroughly with dichloromethane and air-dried to give 2-amino-3-bromo-5- fluorobenzoic acid as its HBr salt. ES/MS m/z 234/236 (MH + ).
- Pd(dppf)2C12CH2C12 (0.05eq) was added to a 0.1 M mixture of 4-(8-bromo-6-fluoroquinazolin- 2-ylamino)benzensulfonamide (leq), 6-fluoropyridin-3-ylboronic acid (3eq), potassium carbonate/water (2.0M, 2eq) in DME.
- the mixture was microwaved at 120 0 C for 20min. Reaction mixture was diluted with ethyl acetate and washed with water, brine, dried and PP028218.0002
- Manganese (IV) oxide (4eq) was added to a 0.18M solution of (4-bromo-2-nitrophenyl)methanol in dichloromethane. The suspension was stirred at ambient temperature under Argon for 12 h. The reaction mixture was filtered through celite and filter cake was washed with dichloromethane. The combined filtrate was concentrated to give brown color solid in 78% yield.
- Step 6 4-(7-Bromoquinazolm-2-ylarnino)-N-(3-(pyrrolidin-l-yl)propyl) benzenesulfonamide
- a 0. IM suspension of 7-bromo-2-chloroquinazoline in isopropanol was added 4-amino-N-(3- (pyrrolidin-l-yl)propyl)benzenesulfonamide (l.leq), followed by the addition of 4.0M HCl in dioxane (1. leq) .
- the reaction mixture was heated to 120 0 C in an oil bath for 1 h. LCMS showed that reaction was complete under the condition.
- Step 7 Preparation of 4-(7-(l-isobutyl-lH-pyrazol-4 r yl)qumazolin-2-ylamino)-N-(3-Q)yi ⁇ olidin- 1 -yl)propyl)benzenesulfonamide
- Pd(dppf)2C12CH2C12 (0.05eq) was added to a 0.05M mixture of 4-(7-bromoquinazolin-2- ylamino)-N-(3-(pyrrolidin-l-yl)propyl) benzenesulfonamide (leq), l-isobutyl-4-(4,4,5,5- PP028218.0002
- Step 1 4-(6-Bromo-7-methoxyquinazolin-2-ylamino)-N-isopropylbenzamide
- A- amino-N-isopropylbenzamide (l.Oeq).
- the reaction mixture was heated to 100 0 C in an oil bath for 14 h.
- Reaction mixture was diluted with ethyl acetate and filtered to collect desired product.
- Step 2 4-(6-Bromo-7-hydroxyquinazolin-2-ylamino)-N-isopropylbenzamide
- a 0.15M suspension of 4-(6-bromo-7-methoxyquinazolin-2-ylamino)-N-isopropylbenzarnide (1.Oeq) and sodium thiomethoxide (4.0eq) in DMF was heated to 80 0 C in an oil bath for 12 h. The mixture was partitioned between ethyl acetate and water. The pH of aqueous phase was adjusted to 5 by adding saturated ammonium hydrochloride.
- Step 1 6-Bromo-7-methoxy-N-(4-morpholinophenyl)quinazolm-2-amine
- 4-morpholinoaniline (1.Oeq)
- the reaction mixture was heated to 120 0 C in an oil bath for 5 h.
- LCMS showed that reaction was complete under the condition.
- the reaction mixture was allowed to cool to room temperature and was filtered.
- the filter cake was rinsed with ethyl acetate and allowed air dry. Desired product was a brown solid as HCl salt.
- Boc-4-hydroxypiperidine (3.Oeq) was added, and the mixture was stirred at ambient temperature PP028218.0002
- 6-Bromo-2-(4-moipholmophenylamino)quinazolin-7-ol (l.Oeq, a 0.12M solution in THF) was added to reaction flask. The mixture was stirred an additional 15 h at ambient temperature. Solvent was removed under reduced pressure and the residue was purified by Biotage using 2% methanol in dichloromethane to give tert-butyl 4-(6-bromo-2-(4- morpholinophenylamino)quinazolin-7-yloxy)piperidine-l-carboxy. ES/MS m/z 584/586 (MH + ).
- Step 1 Preparation of tert-butyl 4-(2-chJoroquinazolin-8-yloxy)piperidine-l-carboxylate
- triphenylphosphine (2 eq) in THF was added di-ethyl-azodicarboxylate (2 eq).
- tert-butyl4-hydroxypiperidine-l- carboxylate (6 eq) was added.
- the mixture was stirred 15 minutes at ambient temperature.
- 4-(8- hydroxyquinazolin-2-ylamino)benzenesulfonamide (1.0 eq) was added. The mixture was stirred overnight at ambient temperature. The reaction goes to completion.
- Step 3 Preparation of 4-(8-(l-isobutylpiperidine-4-yioxy)quinazolin- 2ylamino)b enzenesulfonamide
- isobutyraldehyde (2eq) isobutyraldehyde (2eq) and a few drops of acetic acid.
- the mixture was stirred for lOmins and to it was added sodiumtriacetoxyborohydride (1.5eq) and the mixture was stirred for Ih.
- the reductive amination went to completion, by LC/MS.
- Step 1 Preparation of 2-(4-sulfamoylphenylamino) quinazolin-7-yltrifluoromethane sulfonate
- phenyltrifluoromethanesulfonate 1.2eq
- DIEA 2.5eq
- Step 2 Preparation of 4-(7-(6-fluoropyridin3-yl)quinazolin-2-ylamino)benzenesulfonaniide.
- 2-(4-sulfanioylphenylainino)quinazolin-7-yltrifluoroniethane sulfonate (leq) in DME was added 2M sodium carbonate solution and 6-fluoropyridin-3ylboronic acid (3eq) and Pd(dppf) 2 Cl 2 .CH 2 Cl 2 (0.05eq) and the mixture was micro waved for 10 min at 120 0 C.
- the reaction mixture was then partitioned between ethyl acetate and water.
- the organic layer was concentrated to yield 4-(7-(6-fluoropyridin3-yl)quinazolin-2-ylamino)ben2enesulfonamide.
- ES/MS m/z 396(MH + ).
- Step 3 Preparation of 4-(7-(6-morpholinopyridin-3yl)quinazolin-2ylarnino-benzene sulfonamide
- 4-(7-(6-fluoropyridin3-yl)qumazolin-2-ylamino)benzenesulfonamide was added morpholine.
- the solution was heated at 80 0 C for 3h.
- SNAR goes to completion and the product was purified on prep HPLC to yield 4-(7-(6-morpholinopyridin-3yl)quinazolin-2ylamino-benzene sulfonamide in 50% yield.
- ES/MS m/z 463(MH + ). PP028218.0002
- Step 4 Preparation of methyl 2-(4-sulfamoylphenylamino)quinazolin-7-carboxylate To methyl 2-chloroquinazoin-7-carboxylate (leq) was added sulfanilde (leq) and isopropanol and the mixture was heated to 90 0 C for 2h. The reaction went to completion. The reaction PP028218.0002
- Step 5 Preparation of methyl 2-(4-sulfamoylphenylamino)quinazolin-7-carboxylic acid
- 2-(4-sulfamoylphenyl amino)quinazolin-7-carboxylate was added 2N sodium hydroxide (4eq) and methanol and the mixture was heated to 80 0 C for lOmin. The saponification went to completion.
- the reaction mixture was concentrated and IN HCl was added to precipitate methyl 2-(4-sulfamoylphenylamino)quinazolin-7-carboxylic acid as the HCl salt in quantitative yield.
- ES/MS m/z 344(MH + ).
- Step 2 Preparation of 5-chloro-2-(4-mo ⁇ holinophenylamino)quinazolin-8-ol
- 2-dichloroquinazolin-8-ol leq
- 4- morpholinoaniline leq
- the SNAR went to completion by LC/MS and on concentration yielded 5-chloro-2-(4-morpholinophenylamino) quinazolin-8-ol in quantitative yield.
- Step 3 The tert-butyl4-(methylsulfonyloxy)piperidine-l -carboxylate used in this step was made as follows:
- Step 1 Preparation of l-(4-methoxy-2-aminophenyl)ethanone To a 1:1 mixture of ethanol and acetic acid was added l-(4-methoxy-2-nitrophenyl)ethanone (leq) and Fe dust (3eq)was added in portions. The reduction was complete in Ih. The reaction mixture was filtered and then concentrated and partitioned between ethyl acetate and water. The organic layer was washed with saturated sodium bicarbonate and dried and concentrated to give methyl l-(4-methoxy-2-aminophenyl)ethanone in 85 % yield. ES/MS m/z 196(MH + ).
- Step 4 Preparation of 4-(7-methoxy-4-methylquinazolin-2-ylamino)benzenesulfonamide
- 2-chloro-7-methoxy-4-methylquinazoline(l eq ) in isopropanol was added sulfanilide (1 eq) and the mixture was heated to 90 0 C for 16h. The solid that precipitated was filtered and collected to give 4-(7-methoxy-4-methylquinazolin-2-ylamino)benzene sulfonamide.
- ES/MS m/z 345(MH + ).
- Step 1 Preparation of 7-methoxy-N- (4-(morpholinosulfonyl) phenyl) quinazolin-2-amine
- 2-chloro-7-methoxyquinazoline (leq) and 4-(M ⁇ holinosulfonyl) Aniline (leq) in 2-propanol was heated at 8O 0 C overnight.
- Product was precipitated in the reaction mixture.
- the precipitate was filtered, washed and dried under vacuum to provide 7-methoxy-N- (4- PP028218.0002
- Step 2 Preparation of 2-(4-(mo ⁇ holinosulfonyl)phenylamino)quinazolin-7-ol
- a mixture of 7-methoxy-N- (4-(morpholinosulfonyl) phenyl) quinazolin-2-amine (leq) and sodium thiomethoxide (4eq) in NMP was heated at 80 0 C for 4h.
- Reaction mixture was diluted with water and acidified with IN HCl.
- Compound was extracted with ethyl acetate.
- the organic layer was washed with satd. NaHCOa, brine and dried over sodium sulfate.
- Step 1 Preparation of 2-(4-(morpholinosulfonyl)phenylamino) quinazolin-7-yltrifluoromethane sulfonate For preparation see example 39 step 1.
- Step 1 Preparation of N- (4-fluoro-3-methylphenyl)-7-methoxy quinazolin-2-amine See example 37, step 1 for synthesis.
- Step 3 Preparation of tert-butyl 4-(2-(ethylsulfonyl) quinazolin-7-yloxy) piperidine-1 - carboxylate
- a solution of tert -butyl 4-(2-(ethylthio) quinazolin-7-yloxy) piperidine-1- carboxylate (leq) in THF was added a solution of oxone in water (5ml) at 0 0 C.
- the reaction mixture was stirred for 30min at 0 0 C then warmed to room temperature and stirred for 4h.
- the reaction was quenched with satd. sodium thiosulfate solution and basified with IN NaOH.
- Step 4 Preparation of tert-butyl 4-(2-(2-oxo-2, 3-dihydro-lH-benzo[d]imidazol-5-ylamino) quinazolin-7-yloxy) piperidine-1- carboxylate
- a solution of tert-butyl 4-(2-(ethylsulfonyl) quinazolin-7-yloxy) piperidine-1- carboxylate (leq) and 5-amino-l, 3-dihydro-benzimidazole-2-one (5eq) in acetonitrile was heated in sealed tube at 110 0 C for 48h. The product was filtered, washed and dried. A brown solid, yield 50%. ES/MS m/z All.5 (MH + ).
- Step 1 Preparation of tert-butyl 2-(4-sulfamoylphenylamino) quinazolin-7-yl) carbamate
- 2-(4-sulfamoylphenylamino) quinazoline-7-carboxylic acid (leq) in toluene was added diphenylphosphorylazide (1.2eq), tert-butanol (lOeq) and triethylamine (2eq).
- the reaction mixture was heated at 70 0 C for 30min then 100 0 C for overnight.
- the reaction mixture was concentrated and purified by semi-prep HPLC to provide pure product as a yellow solid in 35% yield.
- Step 1 Preparation of 5-brorno-2-chloroquinazolin-8-ol Solid NBS (leq) was added to a stirred solution of 2-chloroquinazolin-8-ol (leq) (See example 1 for synthesis) in NMP at 0 0 C under argon. After stirring at 0 0 C for 30min, LCMS showed that the reaction was complete. The formation of both 5-bromo (Major) and 7-bromo (Minor) isomers PP028218.0002
Abstract
Description



















































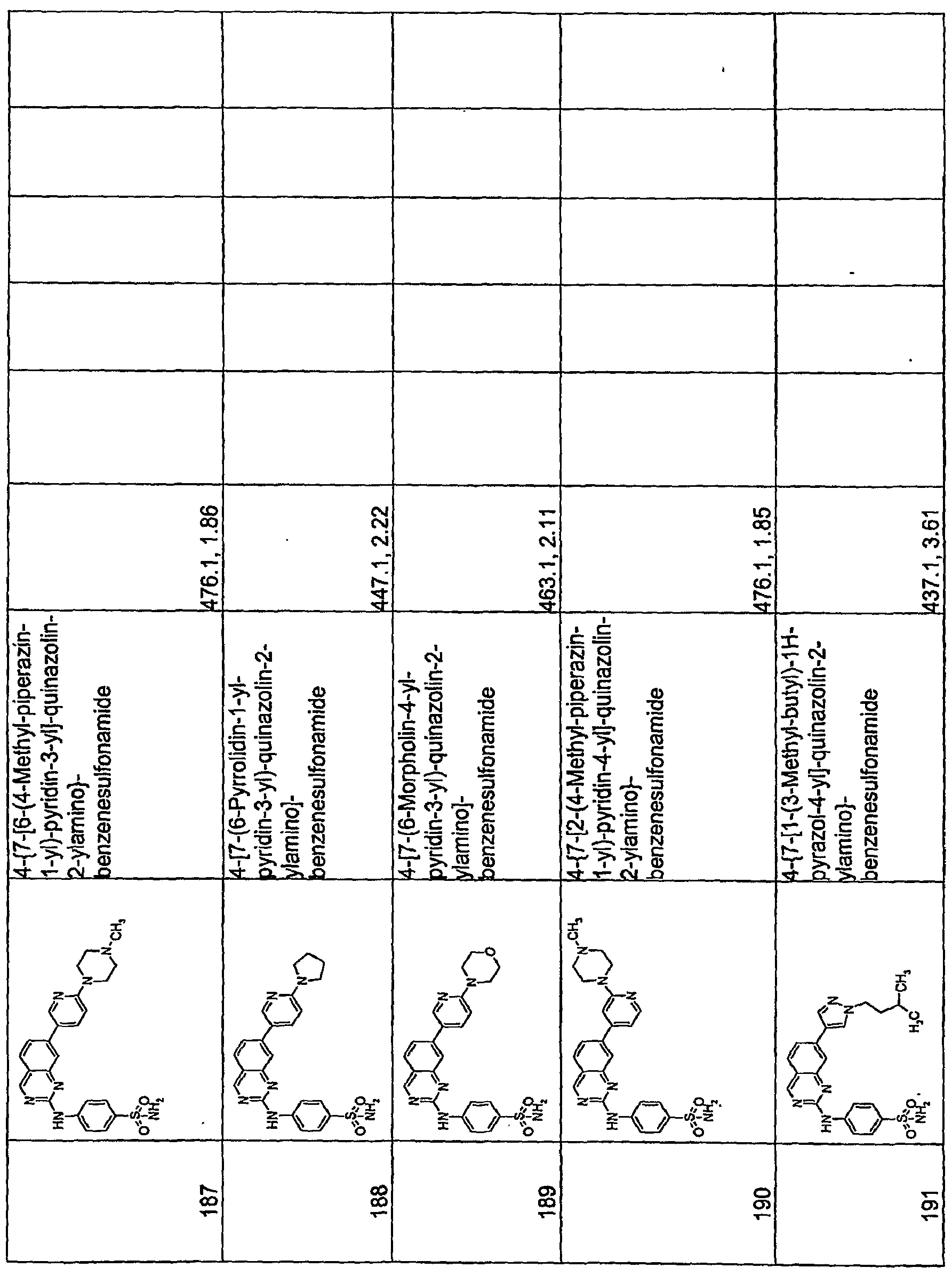



































































































































































































































































































Claims




Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002648529A CA2648529A1 (en) | 2006-04-06 | 2007-04-05 | Quinazolines for pdk1 inhibition |
| EP07755008A EP2004615A2 (en) | 2006-04-06 | 2007-04-05 | Quinazolines for pdk1 inhibition |
| MX2008012852A MX2008012852A (en) | 2006-04-06 | 2007-04-05 | Quinazolines for pdk1 inhibition. |
| US12/295,967 US7932262B2 (en) | 2006-04-06 | 2007-04-05 | Quinazolines for PDK1 inhibition |
| EA200870415A EA200870415A1 (en) | 2006-04-06 | 2007-04-05 | QUINAZOLINS TO INHIBIT PDK 1 |
| JP2009504315A JP2009532486A (en) | 2006-04-06 | 2007-04-05 | Quinazolines for PDK1 inhibition |
| BRPI0710521-5A BRPI0710521A2 (en) | 2006-04-06 | 2007-04-05 | quinazines for pdk1 inhibition |
| AU2007235339A AU2007235339A1 (en) | 2006-04-06 | 2007-04-05 | Quinazolines for PDK1 inhibition |
| SM200800059T SMP200800059B (en) | 2006-04-06 | 2007-04-05 | Chinazolines for the inhibition of pdk1 |
| TNP2008000375A TNSN08375A1 (en) | 2006-04-06 | 2008-09-25 | Quinazolines for pdk1 inhibition |
| NO20084665A NO20084665L (en) | 2006-04-06 | 2008-11-05 | Quinazolines for PDK1 inhibition |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US79030406P | 2006-04-06 | 2006-04-06 | |
| US60/790,304 | 2006-04-06 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| WO2007117607A2 true WO2007117607A2 (en) | 2007-10-18 |
| WO2007117607A3 WO2007117607A3 (en) | 2007-12-21 |
| WO2007117607A9 WO2007117607A9 (en) | 2008-03-06 |
Family
ID=38476139
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/008592 WO2007117607A2 (en) | 2006-04-06 | 2007-04-05 | Quinazolines for pdk1 inhibition |
Country Status (22)
| Country | Link |
|---|---|
| US (1) | US7932262B2 (en) |
| EP (1) | EP2004615A2 (en) |
| JP (1) | JP2009532486A (en) |
| KR (1) | KR20080111114A (en) |
| CN (1) | CN101454294A (en) |
| AR (1) | AR060358A1 (en) |
| AU (1) | AU2007235339A1 (en) |
| BR (1) | BRPI0710521A2 (en) |
| CA (1) | CA2648529A1 (en) |
| CR (1) | CR10427A (en) |
| EA (1) | EA200870415A1 (en) |
| EC (1) | ECSP088823A (en) |
| GT (1) | GT200800202A (en) |
| MA (1) | MA30358B1 (en) |
| MX (1) | MX2008012852A (en) |
| NO (1) | NO20084665L (en) |
| PE (1) | PE20080059A1 (en) |
| SM (1) | SMP200800059B (en) |
| TN (1) | TNSN08375A1 (en) |
| TW (1) | TW200808739A (en) |
| WO (1) | WO2007117607A2 (en) |
| ZA (1) | ZA200808307B (en) |
Cited By (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008079988A2 (en) * | 2006-12-22 | 2008-07-03 | Novartis Ag | Quinazolines for pdk1 inhibition |
| WO2009084695A1 (en) | 2007-12-28 | 2009-07-09 | Carna Biosciences Inc. | 2-aminoquinazoline derivative |
| WO2009007984A3 (en) * | 2007-07-11 | 2009-10-15 | Hetero Drugs Limited | An improved process for erlotinib hydrochloride |
| WO2009131173A1 (en) | 2008-04-23 | 2009-10-29 | 協和発酵キリン株式会社 | 2-aminoquinazoline derivative |
| WO2009153313A1 (en) * | 2008-06-20 | 2009-12-23 | Novartis Ag | 2 -arylaminoquinazolines for treating proliferative diseases |
| WO2009155121A3 (en) * | 2008-05-30 | 2010-02-18 | Amgen Inc. | Inhibitors of pi3 kinase |
| EP2167092A2 (en) * | 2007-06-14 | 2010-03-31 | GlaxoSmithKline LLC | Quinazoline derivatives as pi3 kinase inhibitors |
| US7923450B2 (en) * | 2008-01-11 | 2011-04-12 | Hoffmann-La Roche Inc. | Modulators for amyloid beta |
| EP2381943A1 (en) * | 2008-12-29 | 2011-11-02 | Fovea Pharmaceuticals | Substituted quinazoline compounds |
| US8273882B2 (en) | 2008-05-23 | 2012-09-25 | Novartis Ag | Quinoxaline carboxamide derivatives as protein tyrosine kinase inhibitors |
| WO2013050527A1 (en) | 2011-10-05 | 2013-04-11 | H. Lundbeck A/S | Quinazoline derivatives as pde10a enzyme inhibitors |
| US20160304471A1 (en) * | 2015-02-17 | 2016-10-20 | Neupharma, Inc. | Certain chemical entities, compositions, and methods |
| KR20170023158A (en) | 2014-09-01 | 2017-03-02 | 닛뽕소다 가부시키가이샤 | Method for producing 2-amino-substituted benzaldehyde compound |
| EP3078660A4 (en) * | 2013-12-06 | 2017-05-17 | Carna Biosciences Inc. | Novel quinazoline derivative |
| US9834552B2 (en) | 2012-09-07 | 2017-12-05 | Cancer Research Technology Limited | Inhibitor compounds |
| US9849139B2 (en) | 2013-08-23 | 2017-12-26 | Neupharma, Inc. | Substituted quinazolines for inhibiting kinase activity |
| US9902721B2 (en) | 2014-02-28 | 2018-02-27 | Cancer Research Technology Limited | N2-phenyl-pyrido[3,4-d]pyrimidine-2, 8-diamine derivatives and their use as Mps1 inhibitors |
| CN109320511A (en) * | 2018-10-26 | 2019-02-12 | 广安凯特制药有限公司 | A kind of high-purity Pa Boxini intermediate product and preparation method thereof |
| US10344033B2 (en) | 2015-10-02 | 2019-07-09 | Sentinel Oncology Limited | 2-aminoquinazoline derivatives as P70S6 kinase inhibitors |
| US10544106B2 (en) | 2016-08-15 | 2020-01-28 | Neupharma, Inc. | Certain chemical entities, compositions, and methods |
| WO2021180032A1 (en) * | 2020-03-13 | 2021-09-16 | Guangzhou Maxinovel Pharmaceuticals Co., Ltd. | Novel Therapeutic Methods |
| US11207321B2 (en) | 2017-06-20 | 2021-12-28 | The Institute Of Cancer Research: Royal Cancer Hospital | Methods and medical uses |
| WO2022222951A1 (en) * | 2021-04-22 | 2022-10-27 | 浙江海正药业股份有限公司 | Aromatic condensed ring derivative, preparation method therefor and use thereof |
| US11684620B2 (en) | 2015-02-10 | 2023-06-27 | Astex Therapeutics Ltd | Pharmaceutical compositions comprising N-(3,5-dimethoxyphenyl)-N′-(1-methylethyl)-N-[3-(1-methyl-1H-pyrazol-4-yl)quinoxalin-6-yl]ethane-1,2-diamine |
| WO2023218241A1 (en) * | 2022-05-13 | 2023-11-16 | Voronoi Inc. | Heteroaryl derivative compounds, and pharmaceutical composition comprising thereof |
| EP4025585A4 (en) * | 2019-06-05 | 2024-01-03 | Advanced Tech For Novel Therapeutics Llc | Modified peptides and associated methods of use |
| WO2024020380A1 (en) * | 2022-07-18 | 2024-01-25 | Iambic Therapeutics, Inc. | Quinazoline compounds and methods of use |
| US11918576B2 (en) | 2014-03-26 | 2024-03-05 | Astex Therapeutics Ltd | Combination of an FGFR inhibitor and a CMET inhibitor |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20130141500A (en) * | 2010-09-29 | 2013-12-26 | 크리스탈지노믹스(주) | Novel aminoquinazoline compound having a protein-kinase inhibiting action |
| KR101360948B1 (en) * | 2012-01-17 | 2014-02-12 | 한국과학기술연구원 | Novel 2-aminoquinazolin derivatives and the pharmaceutical composition for treating tumor comprising the same as an active ingredient |
| ES2750236T3 (en) * | 2013-12-09 | 2020-03-25 | UCB Biopharma SRL | Fused bicyclic heteroaromatic derivatives as modulators of TNF activity |
| TWI699355B (en) | 2014-12-24 | 2020-07-21 | 美商基利科學股份有限公司 | Quinazoline compounds |
| CN107207498B (en) | 2014-12-24 | 2020-03-24 | 吉利德科学公司 | Fused pyrimidine compounds for the treatment of HIV |
| JP6356919B2 (en) | 2014-12-24 | 2018-07-11 | ギリアード サイエンシーズ, インコーポレイテッド | Isoquinoline compounds for the treatment of HIV |
| CN113999180B (en) * | 2015-07-03 | 2024-03-26 | 上海医药集团股份有限公司 | Quinazoline compound, intermediate, preparation method, pharmaceutical composition and application |
| CA2993096C (en) * | 2015-07-21 | 2022-04-12 | Guangzhou Maxinovel Pharmaceuticals Co., Ltd. | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof |
| CN105753793B (en) * | 2016-01-19 | 2018-09-04 | 河北医科大学 | Fragrant formyl urea coupling quinazoline compounds and preparation method thereof, pharmaceutical composition and medicinal usage |
| GB201705263D0 (en) * | 2017-03-31 | 2017-05-17 | Probiodrug Ag | Novel inhibitors |
| RU2761824C2 (en) | 2018-08-03 | 2021-12-13 | Закрытое Акционерное Общество "Биокад" | Cdk8/19 inhibitors |
| CN110872243B (en) * | 2018-08-31 | 2023-03-31 | 四川科伦博泰生物医药股份有限公司 | Sulfonamide compound, preparation method and application thereof |
| EP4240361A1 (en) * | 2020-11-09 | 2023-09-13 | Merck Sharp & Dohme LLC | 7-azole substituted 2-aminoquinazoline inhibitors of hpk1 |
| CN114533733A (en) * | 2021-12-31 | 2022-05-27 | 北京鑫开元医药科技有限公司 | Isoquinoline-1, 3-diamine analogue pharmaceutical preparation and preparation method thereof |
| CN113999206B (en) * | 2021-12-31 | 2022-04-12 | 北京鑫开元医药科技有限公司 | Isoquinoline-1, 3-diamine analogue, preparation method, pharmaceutical composition and application thereof |
| WO2023138412A1 (en) * | 2022-01-20 | 2023-07-27 | Insilico Medicine Ip Limited | Fused pyrimidin-2-amine compounds as cdk20 inhibitors |
| CN116514728A (en) * | 2022-01-28 | 2023-08-01 | 赛诺哈勃药业(成都)有限公司 | Quinazoline derivative and application thereof |
| CN116672450A (en) * | 2022-02-23 | 2023-09-01 | 智宠制药(北京)有限公司 | Use of HPK1 inhibitors for the treatment of interferon-related disorders |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003064397A1 (en) | 2002-01-25 | 2003-08-07 | Vertex Pharmaceuticals Incorporated | Indazole compounds useful as protein kinase inhibitors |
| WO2005030776A1 (en) | 2003-09-23 | 2005-04-07 | Vertex Pharmaceuticals Incorporated | Pyrazolopyrrole derivatives as protein kinase inhibitors |
Family Cites Families (78)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US73044A (en) | 1868-01-07 | John c | ||
| US134846A (en) | 1873-01-14 | Half his eight to william h | ||
| US171303A (en) | 1875-12-21 | Improvement in corn-stalk knives | ||
| JPS6041077B2 (en) * | 1976-09-06 | 1985-09-13 | 喜徳 喜谷 | Cis platinum(2) complex of 1,2-diaminocyclohexane isomer |
| US4323581A (en) * | 1978-07-31 | 1982-04-06 | Johnson & Johnson | Method of treating carcinogenesis |
| IL73534A (en) * | 1983-11-18 | 1990-12-23 | Riker Laboratories Inc | 1h-imidazo(4,5-c)quinoline-4-amines,their preparation and pharmaceutical compositions containing certain such compounds |
| WO1988007045A1 (en) | 1987-03-09 | 1988-09-22 | Kyowa Hakko Kogyo Co., Ltd. | Derivatives of physiologically active substance k-252 |
| US4904768A (en) * | 1987-08-04 | 1990-02-27 | Bristol-Myers Company | Epipodophyllotoxin glucoside 4'-phosphate derivatives |
| WO1989007105A1 (en) | 1988-02-04 | 1989-08-10 | Kyowa Hakko Kogyo Co., Ltd. | Staurosporin derivatives |
| US5238944A (en) * | 1988-12-15 | 1993-08-24 | Riker Laboratories, Inc. | Topical formulations and transdermal delivery systems containing 1-isobutyl-1H-imidazo[4,5-c]quinolin-4-amine |
| US4929624A (en) * | 1989-03-23 | 1990-05-29 | Minnesota Mining And Manufacturing Company | Olefinic 1H-imidazo(4,5-c)quinolin-4-amines |
| US5389640A (en) * | 1991-03-01 | 1995-02-14 | Minnesota Mining And Manufacturing Company | 1-substituted, 2-substituted 1H-imidazo[4,5-c]quinolin-4-amines |
| US6410010B1 (en) | 1992-10-13 | 2002-06-25 | Board Of Regents, The University Of Texas System | Recombinant P53 adenovirus compositions |
| US5268376A (en) * | 1991-09-04 | 1993-12-07 | Minnesota Mining And Manufacturing Company | 1-substituted 1H-imidazo[4,5-c]quinolin-4-amines |
| EP0540185A1 (en) | 1991-10-10 | 1993-05-05 | Schering Corporation | 4'-(N-substituted-N-oxide)staurosporine derivatives |
| US5266575A (en) * | 1991-11-06 | 1993-11-30 | Minnesota Mining And Manufacturing Company | 2-ethyl 1H-imidazo[4,5-ciquinolin-4-amines |
| EP0610433A1 (en) | 1991-11-08 | 1994-08-17 | The University Of Southern California | Compositions containing k-252 compounds for potentiation of neurotrophin activity |
| GB9300059D0 (en) * | 1992-01-20 | 1993-03-03 | Zeneca Ltd | Quinazoline derivatives |
| US5948898A (en) | 1992-03-16 | 1999-09-07 | Isis Pharmaceuticals, Inc. | Methoxyethoxy oligonucleotides for modulation of protein kinase C expression |
| IL105325A (en) * | 1992-04-16 | 1996-11-14 | Minnesota Mining & Mfg | Immunogen/vaccine adjuvant composition |
| US5621100A (en) * | 1992-07-24 | 1997-04-15 | Cephalon, Inc. | K-252a derivatives for treatment of neurological disorders |
| US5756494A (en) | 1992-07-24 | 1998-05-26 | Cephalon, Inc. | Protein kinase inhibitors for treatment of neurological disorders |
| EP0658113B1 (en) | 1992-08-31 | 2004-10-20 | Ludwig Institute For Cancer Research | Isolated nonapeptide derived from mage-3 gene and presented by hla-a1, and uses thereof |
| DE69331228D1 (en) | 1992-09-21 | 2002-01-10 | Kyowa Hakko Kogyo Kk | Heilmittel für thrombozytopenia |
| HU225646B1 (en) | 1992-10-28 | 2007-05-29 | Genentech Inc | Hvegf receptors as vascular endothelial cell growth factor antagonists |
| US5395937A (en) * | 1993-01-29 | 1995-03-07 | Minnesota Mining And Manufacturing Company | Process for preparing quinoline amines |
| CZ288182B6 (en) * | 1993-07-15 | 2001-05-16 | Minnesota Mining & Mfg | Imidazo[4,5-c]pyridine-4-amines and pharmaceutical preparations based thereon |
| US5352784A (en) * | 1993-07-15 | 1994-10-04 | Minnesota Mining And Manufacturing Company | Fused cycloalkylimidazopyridines |
| US5478932A (en) * | 1993-12-02 | 1995-12-26 | The Board Of Trustees Of The University Of Illinois | Ecteinascidins |
| DE69434294T2 (en) | 1993-12-23 | 2005-12-29 | Eli Lilly And Co., Indianapolis | Protein kinase C inhibitors |
| US5525613A (en) * | 1994-06-16 | 1996-06-11 | Entropin, Inc. | Covalently coupled benzoylecgonine ecgonine and ecgonidine |
| US5587459A (en) | 1994-08-19 | 1996-12-24 | Regents Of The University Of Minnesota | Immunoconjugates comprising tyrosine kinase inhibitors |
| US6083903A (en) * | 1994-10-28 | 2000-07-04 | Leukosite, Inc. | Boronic ester and acid compounds, synthesis and uses |
| US5482936A (en) * | 1995-01-12 | 1996-01-09 | Minnesota Mining And Manufacturing Company | Imidazo[4,5-C]quinoline amines |
| DK0817775T3 (en) | 1995-03-30 | 2001-11-19 | Pfizer | quinazoline |
| GB9508538D0 (en) * | 1995-04-27 | 1995-06-14 | Zeneca Ltd | Quinazoline derivatives |
| US6331555B1 (en) * | 1995-06-01 | 2001-12-18 | University Of California | Treatment of platelet derived growth factor related disorders such as cancers |
| US5747498A (en) * | 1996-05-28 | 1998-05-05 | Pfizer Inc. | Alkynyl and azido-substituted 4-anilinoquinazolines |
| US5880141A (en) * | 1995-06-07 | 1999-03-09 | Sugen, Inc. | Benzylidene-Z-indoline compounds for the treatment of disease |
| WO1997007081A2 (en) | 1995-08-11 | 1997-02-27 | Yale University | Glycosylated indolocarbazole synthesis |
| FR2741881B1 (en) | 1995-12-01 | 1999-07-30 | Centre Nat Rech Scient | NOVEL PURINE DERIVATIVES HAVING IN PARTICULAR ANTI-PROLIFERATIVE PRORIETES AND THEIR BIOLOGICAL APPLICATIONS |
| NZ320244A (en) | 1995-12-08 | 1999-06-29 | Janssen Pharmaceutica Nv | Farnesyl protein transferase inhibiting (imidazol-5-yl)methyl-2-quinolinone derivatives |
| KR100447918B1 (en) | 1996-07-25 | 2005-09-28 | 동아제약주식회사 | Flavones and flavanone compounds with protective gastrointestinal tract including large intestine |
| EP0917569B1 (en) | 1996-08-02 | 2005-11-09 | GeneSense Technologies Inc. | Antitumor antisense sequences directed against r1 and r2 components of ribonucleotide reductase |
| CO4950519A1 (en) * | 1997-02-13 | 2000-09-01 | Novartis Ag | PHTHALAZINES, PHARMACEUTICAL PREPARATIONS THAT UNDERSTAND THEM AND THE PROCESS FOR THEIR PREPARATION |
| US6126965A (en) | 1997-03-21 | 2000-10-03 | Georgetown University School Of Medicine | Liposomes containing oligonucleotides |
| JP2001509483A (en) | 1997-07-12 | 2001-07-24 | カンサー リサーチ キャンペーン テクノロジー リミテッド | Cyclin-dependent kinase inhibitor purine derivatives |
| GB9800569D0 (en) * | 1998-01-12 | 1998-03-11 | Glaxo Group Ltd | Heterocyclic compounds |
| EA005032B1 (en) | 1998-05-29 | 2004-10-28 | Сьюджен, Инк. | Pyrrole substituted 2-indolinone (variants), pharmaceutical composition (variants), method for modulation of catalytic activity method for treating and preventing a protein kinase related disorders in an organism |
| US20030083242A1 (en) | 1998-11-06 | 2003-05-01 | Alphonse Galdes | Methods and compositions for treating or preventing peripheral neuropathies |
| DK1189641T5 (en) | 1999-06-25 | 2011-04-11 | Genentech Inc | Humanized ANTI-ErbB2 antibodies and treatment with ANTI-ErbB2 antibodies |
| WO2001004125A1 (en) | 1999-07-13 | 2001-01-18 | Kyowa Hakko Kogyo Co., Ltd. | Staurosporin derivatives |
| NZ518028A (en) | 1999-11-05 | 2004-03-26 | Astrazeneca Ab | Quinazoline derivatives as VEGF inhibitors |
| US6982260B1 (en) * | 1999-11-22 | 2006-01-03 | Warner-Lambert Company | Quinazolines and their use for inhibiting cyclin-dependent kinase enzymes |
| EP1235815A1 (en) * | 1999-11-22 | 2002-09-04 | Warner-Lambert Company | Quinazolines and their use for inhibiting cyclin-dependent kinase enzymes |
| ES2290117T3 (en) | 2000-02-15 | 2008-02-16 | Sugen, Inc. | PROTEIN QUINASE 2-INDOLIN INHIBITORS REPLACED WITH PIRROL. |
| DE60123939T2 (en) | 2000-04-12 | 2007-05-31 | Genaera Corp. | A PROCESS FOR THE PREPARATION OF 7.ALPHA.-HYDROXY 3-AMINO-SUBSTITUTED STEROL COMPOUNDS, WITHOUT PROTECTION OF THE 7.ALPHA.-HYDROXY GROUP |
| TWI307339B (en) | 2000-06-30 | 2009-03-11 | Glaxo Group Ltd | Quinazoline ditosylate salt comprounds |
| IL154618A0 (en) * | 2000-09-11 | 2003-09-17 | Chiron Corp | Quinolinone derivatives |
| US6677450B2 (en) | 2000-10-06 | 2004-01-13 | Bristol-Myers Squibb Company | Topoisomerase inhibitors |
| AU2002245272B2 (en) | 2001-01-16 | 2006-06-29 | Regeneron Pharmaceuticals, Inc. | Isolating cells expressing secreted proteins |
| WO2002062826A1 (en) | 2001-02-07 | 2002-08-15 | Vadim Viktorovich Novikov | Method for producing peptides |
| EP1404689A1 (en) | 2001-07-02 | 2004-04-07 | Debiopharm S.A. | Oxaliplatin active substance with a very low content of oxalic acid |
| KR100484504B1 (en) | 2001-09-18 | 2005-04-20 | 학교법인 포항공과대학교 | Inclusion compound comprising curcurbituril derivatives as host molecule and pharmaceutical composition comprising the same |
| US20030134846A1 (en) * | 2001-10-09 | 2003-07-17 | Schering Corporation | Treatment of trypanosoma brucei with farnesyl protein transferase inhibitors |
| US6927036B2 (en) * | 2002-02-19 | 2005-08-09 | Xero Port, Inc. | Methods for synthesis of prodrugs from 1-acyl-alkyl derivatives and compositions thereof |
| PL402389A1 (en) | 2002-03-29 | 2013-04-02 | Novartis Ag | Method for inhibiting Raf kinase activity in humans or inanimals |
| US6900342B2 (en) * | 2002-05-10 | 2005-05-31 | Dabur India Limited | Anticancer taxanes such as paclitaxel, docetaxel and their structural analogs, and a method for the preparation thereof |
| US6727272B1 (en) | 2002-07-15 | 2004-04-27 | Unitech Pharmaceuticals, Inc. | Leflunomide analogs for treating rheumatoid arthritis |
| US7148342B2 (en) | 2002-07-24 | 2006-12-12 | The Trustees Of The University Of Pennyslvania | Compositions and methods for sirna inhibition of angiogenesis |
| EP1587473A4 (en) | 2002-12-27 | 2008-08-13 | Novartis Vaccines & Diagnostic | Thiosemicarbazones as anti-virals and immunopotentiators |
| DE602004021558D1 (en) | 2003-01-17 | 2009-07-30 | Warner Lambert Co | 2-AMINOPYRIDINE SUBSTITUTED HETEROCYCLES AS INHIBITORS OF CELLULAR PROLIFERATION |
| ES2391770T3 (en) | 2003-01-21 | 2012-11-29 | Novartis Vaccines And Diagnostics, Inc. | Use of triptantrin compounds for immune potentiation |
| ES2423800T3 (en) | 2003-03-28 | 2013-09-24 | Novartis Vaccines And Diagnostics, Inc. | Use of organic compounds for immunopotentiation |
| RU2415857C2 (en) | 2004-09-14 | 2011-04-10 | Новартис Вэксинс Энд Диагностикс Инк. | Imidazoquinoline compounds |
| WO2006118256A1 (en) | 2005-04-28 | 2006-11-09 | Kyowa Hakko Kogyo Co., Ltd. | 2-aminoquinazoline derivatives |
| WO2006137421A1 (en) | 2005-06-21 | 2006-12-28 | Kyowa Hakko Kogyo Co., Ltd. | Agent for topical administration |
| JO2660B1 (en) | 2006-01-20 | 2012-06-17 | نوفارتيس ايه جي | PI-3 Kinase inhibitors and methods of their use |
-
2007
- 2007-04-04 TW TW096112158A patent/TW200808739A/en unknown
- 2007-04-04 AR ARP070101448A patent/AR060358A1/en unknown
- 2007-04-05 CN CNA2007800195027A patent/CN101454294A/en active Pending
- 2007-04-05 SM SM200800059T patent/SMP200800059B/en unknown
- 2007-04-05 WO PCT/US2007/008592 patent/WO2007117607A2/en active Application Filing
- 2007-04-05 AU AU2007235339A patent/AU2007235339A1/en not_active Abandoned
- 2007-04-05 JP JP2009504315A patent/JP2009532486A/en not_active Withdrawn
- 2007-04-05 US US12/295,967 patent/US7932262B2/en not_active Expired - Fee Related
- 2007-04-05 KR KR1020087026508A patent/KR20080111114A/en not_active Application Discontinuation
- 2007-04-05 EA EA200870415A patent/EA200870415A1/en unknown
- 2007-04-05 EP EP07755008A patent/EP2004615A2/en not_active Withdrawn
- 2007-04-05 CA CA002648529A patent/CA2648529A1/en not_active Abandoned
- 2007-04-05 BR BRPI0710521-5A patent/BRPI0710521A2/en not_active IP Right Cessation
- 2007-04-05 MX MX2008012852A patent/MX2008012852A/en not_active Application Discontinuation
- 2007-04-09 PE PE2007000418A patent/PE20080059A1/en not_active Application Discontinuation
-
2008
- 2008-09-25 TN TNP2008000375A patent/TNSN08375A1/en unknown
- 2008-09-29 ZA ZA200808307A patent/ZA200808307B/en unknown
- 2008-10-01 GT GT200800202A patent/GT200800202A/en unknown
- 2008-10-15 EC EC2008008823A patent/ECSP088823A/en unknown
- 2008-10-27 MA MA31336A patent/MA30358B1/en unknown
- 2008-11-05 NO NO20084665A patent/NO20084665L/en not_active Application Discontinuation
- 2008-11-05 CR CR10427A patent/CR10427A/en not_active Application Discontinuation
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003064397A1 (en) | 2002-01-25 | 2003-08-07 | Vertex Pharmaceuticals Incorporated | Indazole compounds useful as protein kinase inhibitors |
| WO2005030776A1 (en) | 2003-09-23 | 2005-04-07 | Vertex Pharmaceuticals Incorporated | Pyrazolopyrrole derivatives as protein kinase inhibitors |
Cited By (57)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008079988A2 (en) * | 2006-12-22 | 2008-07-03 | Novartis Ag | Quinazolines for pdk1 inhibition |
| WO2008079988A3 (en) * | 2006-12-22 | 2008-10-16 | Novartis Vaccines & Diagnostic | Quinazolines for pdk1 inhibition |
| EP2167092A4 (en) * | 2007-06-14 | 2012-07-25 | Glaxosmithkline Llc | Quinazoline derivatives as pi3 kinase inhibitors |
| EP2167092A2 (en) * | 2007-06-14 | 2010-03-31 | GlaxoSmithKline LLC | Quinazoline derivatives as pi3 kinase inhibitors |
| JP2010532320A (en) * | 2007-06-14 | 2010-10-07 | グラクソスミスクライン・リミテッド・ライアビリティ・カンパニー | Quinazoline derivatives as PI3 kinase inhibitors |
| WO2009007984A3 (en) * | 2007-07-11 | 2009-10-15 | Hetero Drugs Limited | An improved process for erlotinib hydrochloride |
| US8389531B2 (en) | 2007-07-11 | 2013-03-05 | Hetero Drugs Limited | Process for erlotinib hydrochloride |
| WO2009084695A1 (en) | 2007-12-28 | 2009-07-09 | Carna Biosciences Inc. | 2-aminoquinazoline derivative |
| US7923450B2 (en) * | 2008-01-11 | 2011-04-12 | Hoffmann-La Roche Inc. | Modulators for amyloid beta |
| WO2009131173A1 (en) | 2008-04-23 | 2009-10-29 | 協和発酵キリン株式会社 | 2-aminoquinazoline derivative |
| US8815901B2 (en) | 2008-05-23 | 2014-08-26 | Novartis Ag | Quinoline carboxamide derivatives as protein tyrosine kinase inhibitors |
| US8273882B2 (en) | 2008-05-23 | 2012-09-25 | Novartis Ag | Quinoxaline carboxamide derivatives as protein tyrosine kinase inhibitors |
| US8536175B2 (en) | 2008-05-23 | 2013-09-17 | Novartis Ag | Quinoxaline carboxamide derivatives as protein tyrosine kinase inhibitors |
| US8674099B2 (en) | 2008-05-23 | 2014-03-18 | Novartis Ag | Quinoxaline carboxamide derivatives as protein tyrosine kinase inhibitors |
| AU2009260447B2 (en) * | 2008-05-30 | 2012-03-29 | Amgen Inc. | Inhibitors of PI3 kinase |
| WO2009155121A3 (en) * | 2008-05-30 | 2010-02-18 | Amgen Inc. | Inhibitors of pi3 kinase |
| US8415376B2 (en) | 2008-05-30 | 2013-04-09 | Amgen Inc. | Inhibitors of PI3 kinase |
| WO2009153313A1 (en) * | 2008-06-20 | 2009-12-23 | Novartis Ag | 2 -arylaminoquinazolines for treating proliferative diseases |
| CN102333533A (en) * | 2008-12-29 | 2012-01-25 | 佛维雅制药公司 | Substituted quinazoline compounds |
| JP2012514020A (en) * | 2008-12-29 | 2012-06-21 | フォーヴィア・ファーマシューティカルズ | Substituted quinazoline compounds |
| EP2381943A1 (en) * | 2008-12-29 | 2011-11-02 | Fovea Pharmaceuticals | Substituted quinazoline compounds |
| WO2013050527A1 (en) | 2011-10-05 | 2013-04-11 | H. Lundbeck A/S | Quinazoline derivatives as pde10a enzyme inhibitors |
| US11897877B2 (en) | 2012-09-07 | 2024-02-13 | Cancer Research Technology Limited | Inhibitor compounds |
| US11046688B2 (en) | 2012-09-07 | 2021-06-29 | Cancer Research Technology Limited | Inhibitor compounds |
| US10479788B2 (en) | 2012-09-07 | 2019-11-19 | Cancer Research Technology Limited | Compounds that inhibit MPS1 kinase |
| RU2673079C2 (en) * | 2012-09-07 | 2018-11-22 | Кэнсэр Ресерч Текнолоджи Лимитед | Inhibitor compounds |
| US9834552B2 (en) | 2012-09-07 | 2017-12-05 | Cancer Research Technology Limited | Inhibitor compounds |
| US9890157B2 (en) | 2012-09-07 | 2018-02-13 | Cancer Research Technology Limited | Inhibitor compounds |
| US9849139B2 (en) | 2013-08-23 | 2017-12-26 | Neupharma, Inc. | Substituted quinazolines for inhibiting kinase activity |
| AU2019201461B2 (en) * | 2013-08-23 | 2020-11-12 | Neupharma, Inc. | Certain chemical entities, compositions, and methods |
| AU2014308616B2 (en) * | 2013-08-23 | 2018-12-06 | Neupharma, Inc. | Certain chemical entities, compositions, and methods |
| US10172868B2 (en) | 2013-08-23 | 2019-01-08 | Neupharma, Inc. | Substituted quinazolines for inhibiting kinase activity |
| US11865120B2 (en) | 2013-08-23 | 2024-01-09 | Neupharma, Inc. | Substituted quinazolines for inhibiting kinase activity |
| US11304957B2 (en) | 2013-08-23 | 2022-04-19 | Neupharma, Inc. | Substituted quinazolines for inhibiting kinase activity |
| US10653701B2 (en) | 2013-08-23 | 2020-05-19 | Neupharma, Inc. | Substituted quinazolines for inhibiting kinase activity |
| US9682961B2 (en) | 2013-12-06 | 2017-06-20 | Carna Biosciences, Inc. | Quinazoline derivative |
| EP3078660A4 (en) * | 2013-12-06 | 2017-05-17 | Carna Biosciences Inc. | Novel quinazoline derivative |
| US10399974B2 (en) | 2014-02-28 | 2019-09-03 | Cancer Research Technology Limited | N2-phenyl-pyrido[3,4-D]pyrimidine-2, 8-diamine derivatives and their use as Mps1 inhibitors |
| US9902721B2 (en) | 2014-02-28 | 2018-02-27 | Cancer Research Technology Limited | N2-phenyl-pyrido[3,4-d]pyrimidine-2, 8-diamine derivatives and their use as Mps1 inhibitors |
| US11918576B2 (en) | 2014-03-26 | 2024-03-05 | Astex Therapeutics Ltd | Combination of an FGFR inhibitor and a CMET inhibitor |
| KR20170023158A (en) | 2014-09-01 | 2017-03-02 | 닛뽕소다 가부시키가이샤 | Method for producing 2-amino-substituted benzaldehyde compound |
| US10047037B2 (en) | 2014-09-01 | 2018-08-14 | Nippon Soda Co., Ltd. | Method for producing 2-amino-substituted benzaldehyde compound |
| US11684620B2 (en) | 2015-02-10 | 2023-06-27 | Astex Therapeutics Ltd | Pharmaceutical compositions comprising N-(3,5-dimethoxyphenyl)-N′-(1-methylethyl)-N-[3-(1-methyl-1H-pyrazol-4-yl)quinoxalin-6-yl]ethane-1,2-diamine |
| US10947201B2 (en) * | 2015-02-17 | 2021-03-16 | Neupharma, Inc. | Certain chemical entities, compositions, and methods |
| US20160304471A1 (en) * | 2015-02-17 | 2016-10-20 | Neupharma, Inc. | Certain chemical entities, compositions, and methods |
| EP3868751A1 (en) | 2015-10-02 | 2021-08-25 | Sentinel Oncology Limited | 2-aminoquinazoline derivatives as p70s6 kinase inhibitors |
| US10730882B2 (en) | 2015-10-02 | 2020-08-04 | Sentinel Oncology Limited | 2-aminoquinazoline derivatives as P70S6 kinase inhibitors |
| US10344033B2 (en) | 2015-10-02 | 2019-07-09 | Sentinel Oncology Limited | 2-aminoquinazoline derivatives as P70S6 kinase inhibitors |
| US10544106B2 (en) | 2016-08-15 | 2020-01-28 | Neupharma, Inc. | Certain chemical entities, compositions, and methods |
| US11208388B2 (en) | 2016-08-15 | 2021-12-28 | Neupharma, Inc | Certain chemical entities, compositions, and methods |
| US11207321B2 (en) | 2017-06-20 | 2021-12-28 | The Institute Of Cancer Research: Royal Cancer Hospital | Methods and medical uses |
| CN109320511A (en) * | 2018-10-26 | 2019-02-12 | 广安凯特制药有限公司 | A kind of high-purity Pa Boxini intermediate product and preparation method thereof |
| EP4025585A4 (en) * | 2019-06-05 | 2024-01-03 | Advanced Tech For Novel Therapeutics Llc | Modified peptides and associated methods of use |
| WO2021180032A1 (en) * | 2020-03-13 | 2021-09-16 | Guangzhou Maxinovel Pharmaceuticals Co., Ltd. | Novel Therapeutic Methods |
| WO2022222951A1 (en) * | 2021-04-22 | 2022-10-27 | 浙江海正药业股份有限公司 | Aromatic condensed ring derivative, preparation method therefor and use thereof |
| WO2023218241A1 (en) * | 2022-05-13 | 2023-11-16 | Voronoi Inc. | Heteroaryl derivative compounds, and pharmaceutical composition comprising thereof |
| WO2024020380A1 (en) * | 2022-07-18 | 2024-01-25 | Iambic Therapeutics, Inc. | Quinazoline compounds and methods of use |
Also Published As
| Publication number | Publication date |
|---|---|
| CN101454294A (en) | 2009-06-10 |
| WO2007117607A9 (en) | 2008-03-06 |
| SMP200800059B (en) | 2009-09-07 |
| PE20080059A1 (en) | 2008-03-05 |
| MX2008012852A (en) | 2008-11-28 |
| CA2648529A1 (en) | 2007-10-18 |
| US20100048561A1 (en) | 2010-02-25 |
| TNSN08375A1 (en) | 2009-12-29 |
| AR060358A1 (en) | 2008-06-11 |
| SMAP200800059A (en) | 2008-11-12 |
| NO20084665L (en) | 2009-01-06 |
| BRPI0710521A2 (en) | 2012-03-13 |
| AU2007235339A8 (en) | 2008-11-20 |
| WO2007117607A3 (en) | 2007-12-21 |
| ZA200808307B (en) | 2010-01-27 |
| KR20080111114A (en) | 2008-12-22 |
| CR10427A (en) | 2009-01-27 |
| ECSP088823A (en) | 2008-12-30 |
| JP2009532486A (en) | 2009-09-10 |
| EP2004615A2 (en) | 2008-12-24 |
| TW200808739A (en) | 2008-02-16 |
| EA200870415A1 (en) | 2009-02-27 |
| GT200800202A (en) | 2009-06-25 |
| AU2007235339A1 (en) | 2007-10-18 |
| MA30358B1 (en) | 2009-04-01 |
| US7932262B2 (en) | 2011-04-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007117607A2 (en) | Quinazolines for pdk1 inhibition | |
| WO2008079988A9 (en) | Quinazolines for pdk1 inhibition | |
| AU2008213808B2 (en) | PI 3-kinase inhibitors and methods of their use | |
| JP5160637B2 (en) | Compounds and compositions as c-kit and PDGFR kinase inhibitors | |
| EP2456440B1 (en) | Quinolinone pde2 inhibitors | |
| CA2727251A1 (en) | 2 -arylaminoquinazolines for treating proliferative diseases | |
| BRPI0707816A2 (en) | pi - 3 kinase inhibitors and methods of use | |
| BRPI0718677A2 (en) | COMPOUNDS AND COMPOSITIONS AS PROTEIN KINASE INHIBITORS | |
| CN102083828A (en) | Heterocyclic compounds and compositions as C-KIT and PDGFR kinase inhibitors | |
| TW200911249A (en) | TRPV1 antagonists and uses thereof | |
| EP2148874A1 (en) | Pyrimidine derivatives and compositions as c-kit and pdgfr kinase inhibitors | |
| AU2017289021B2 (en) | Novel pyrazole derivative as ALK5 inhibitor and uses thereof | |
| KR20200013718A (en) | Heteroaromatic Compounds as Banin Inhibitors | |
| CN110156754B (en) | Inhibition of protein kinase by trisubstituted pyrimidine derivative | |
| CN105408332B (en) | Substituted triazolopyridines | |
| CN104968654B (en) | Substituted 2-aminopyridine albuminoid kinase inhibitor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780019502.7 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07755008 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 194283 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12008502155 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 571670 Country of ref document: NZ Ref document number: 2007235339 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009504315 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008101638 Country of ref document: EG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2008/012852 Country of ref document: MX Ref document number: 2648529 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4156/KOLNP/2008 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007755008 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2007235339 Country of ref document: AU Date of ref document: 20070405 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020087026508 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10967 Country of ref document: GE Ref document number: CR2008-010427 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 08118444 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200870415 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12295967 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: PI0710521 Country of ref document: BR Kind code of ref document: A2 Effective date: 20081006 |