WO2011049191A1 - 経口投与用医薬組成物 - Google Patents
経口投与用医薬組成物 Download PDFInfo
- Publication number
- WO2011049191A1 WO2011049191A1 PCT/JP2010/068660 JP2010068660W WO2011049191A1 WO 2011049191 A1 WO2011049191 A1 WO 2011049191A1 JP 2010068660 W JP2010068660 W JP 2010068660W WO 2011049191 A1 WO2011049191 A1 WO 2011049191A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pharmaceutical composition
- weight
- solid pharmaceutical
- proline
- crystal
- Prior art date
Links
Images




Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2072—Pills, tablets, discs, rods characterised by shape, structure or size; Tablets with holes, special break lines or identification marks; Partially coated tablets; Disintegrating flat shaped forms
- A61K9/2077—Tablets comprising drug-containing microparticles in a substantial amount of supporting matrix; Multiparticulate tablets
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/401—Proline; Derivatives thereof, e.g. captopril
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1652—Polysaccharides, e.g. alginate, cellulose derivatives; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2009—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
- A61K9/2018—Sugars, or sugar alcohols, e.g. lactose, mannitol; Derivatives thereof, e.g. polysorbates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/2027—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2059—Starch, including chemically or physically modified derivatives; Amylose; Amylopectin; Dextrin
Definitions
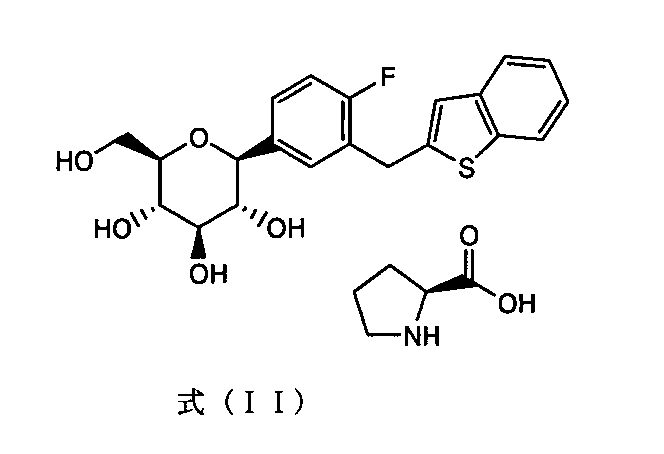
- the present invention comprises a co-crystal of (1S) -1,5-anhydro-1- [3- (1-benzothien-2-ylmethyl) -4-fluorophenyl] -D-glucitol and L-proline, (1S) -1,5-anhydro-1- [3- (1-benzothien-2-ylmethyl) -4-fluorophenyl] -D-glucitol has a good dissolution property and a solid having stable dissolution property
- the present invention relates to a pharmaceutical composition.
- the present invention also includes a co-crystal of (1S) -1,5-anhydro-1- [3- (1-benzothien-2-ylmethyl) -4-fluorophenyl] -D-glucitol and L-proline.
- (1S) -1,5-anhydro-1- [3- (1-benzothien-2-ylmethyl) -4-fluorophenyl] -D-glucitol maintains good elution and elution stability
- the present invention relates to a method for producing a solid pharmaceutical composition.
- the present invention contains a co-crystal of (1S) -1,5-anhydro-1- [3- (1-benzothien-2-ylmethyl) -4-fluorophenyl] -D-glucitol and L-proline.
- (1S) -1,5-anhydro-1- [3- (1-benzothien-2-ylmethyl) -4-fluorophenyl] -D-glucitol maintains good elution and elution stability It relates to the use of crystalline cellulose for the production of solid pharmaceutical compositions.
- C-glycoside derivative A Is a Na + -glucose cotransporter inhibitor created by Astellas Pharma Inc., for example, insulin-dependent diabetes (type 1 diabetes), non-insulin-dependent diabetes (type 2 diabetes), and insulin resistance It has been reported as a compound useful for treatment of diseases and obesity, and prevention thereof (see Patent Document 1: Example 138).
- Patent Document 2 a co-crystal of known compound A and L-proline, as a drug substance crystal used in the manufacture of a medicament, a co-crystal of L-proline having a certain quality and excellent storage stability, and this An invention relating to a pharmaceutical composition containing as an active ingredient and particularly useful as a therapeutic agent for diabetes has been disclosed (Patent Document 2).
- the crystal of the known compound A disclosed in Patent Document 1 forms an clathrate hydrate and exhibits a property of reversibly changing from an anhydride to a non-stoichiometric hydrate depending on the temperature and humidity environment. It has been difficult to maintain a certain quality as a drug substance used for pharmaceuticals. Therefore, the known compound A is provided as a co-crystal with L-proline as a drug substance crystal for use in pharmaceuticals having a certain quality and excellent storage stability.
- an object of the present invention is to provide a pharmaceutical composition having a good dissolution property, comprising an L-proline cocrystal of known compound A.
- Another object of the present invention is to provide a method for producing a pharmaceutical composition having a good dissolution property, comprising a known compound A L-proline co-crystal.
- a further object of the present invention is to produce a solid pharmaceutical composition containing a co-crystal of known compound A with L-proline, and maintaining good dissolution and stability of known compound A. It is to provide the use of crystalline cellulose to do so.
- the present inventors prepared a granulated product containing a co-crystal of a known compound A and an L-proline (1: 1) by a wet granulation method using a known agitation granulator, Thereafter, when the tablet was produced from the granulated product, the tablet had good drug dissolution immediately after the production, but the disintegration characteristics were changed, and the dissolution was decreased with time. I knew there was a problem.
- the known compound A is free due to the release of L-proline from the co-crystal structure by water used during the preparation production.
- the drug-dissolving properties are temporarily improved based on the characteristics of the free form of the known compound A, and the known compound A-free form remains in the composition over time. It was found that aggregates are produced by the action of proline and hydrogen bonds again.
- the present inventors diligently investigated a solid pharmaceutical composition having a good dissolution property while maintaining a co-crystal structure of the known compound A and L-proline.
- excipients such as D-mannitol, anhydrous calcium hydrogen phosphate, lactose, and crystalline cellulose
- the solid pharmaceutical composition containing crystalline cellulose maintained a co-crystal structure of known compound A and L-proline. And it discovered that it had favorable elution, and came to complete this invention.
- the present invention [1] (1S) -1,5-anhydro-1- [3- (1-benzothien-2-ylmethyl) -4-fluorophenyl] -D-glucitol and L-proline compound, and crystalline cellulose
- a solid pharmaceutical composition comprising [2] The solid pharmaceutical composition according to [1], wherein the amount of crystalline cellulose is 5% by weight or more and 90% by weight or less in the pharmaceutical composition, [3] The solid pharmaceutical composition according to [1] or [2], further comprising a disintegrant, [4] The disintegrant is one or more selected from the group consisting of sodium starch glycolate and hydroxypropylcellulose having a hydroxypropoxyl group content of 5 wt% or more and less than 16 wt%, [3] A solid pharmaceutical composition according to claim 1, [5] The solid pharmaceutical composition according to [3] or [4], wherein the amount of the disintegrant is 5% by weight or more and 90% by weight or less in the pharmaceutical composition, [6] In the dissolution
- the method for producing a solid pharmaceutical composition according to [7] or [8], wherein the amount of crystalline cellulose is 5% by weight or more and 90% by weight or less in the pharmaceutical composition [10]
- the method further includes adding a disintegrant during the step (1), between the steps (1) and (2), during the step (2), or after the step (2).
- the production method according to [11] The disintegrant is one or more selected from the group consisting of sodium starch glycolate and hydroxypropyl cellulose having a hydroxypropoxyl group content of 5 wt% or more and less than 16 wt%, [10] A method for producing the solid pharmaceutical composition according to claim 1, [12] The method for producing a solid pharmaceutical composition according to [10] or [11], wherein the amount of the disintegrant is 5% by weight or more and 90% by weight or less in the pharmaceutical composition, [13] (1S) -1,5-Anhydro-1- [3- (1-benzothien-2-ylmethyl) -4-fluorophenyl] -D-glucitol in the dissolution test described in the 15th revision Japanese Pharmacopoeia For producing a solid pharmaceutical composition that elutes 65% or more in 30 minutes, [7] to [12], [14] The method for producing a solid pharmaceutical composition according to [7], wherein the wet granulation is performed at a maximum water
- the features of the present invention are (1) a pharmaceutical preparation containing a co-crystal of known compound A and L-proline exhibits good dissolution properties, and (2) a stable pharmaceutical preparation whose dissolution rate does not change over time. (3) Since it exhibits a good dissolution property, the bioavailability (BA) is improved, and a pharmacologically sufficient therapeutic effect can be obtained.
- BA bioavailability
- 2 is an elution profile of the pharmaceutical composition obtained in Example 1.
- 2 is an elution profile of the pharmaceutical composition obtained in Comparative Example 1.
- 3 is an elution profile of the pharmaceutical composition obtained in Comparative Example 2. It is an elution profile of the pharmaceutical composition obtained in Examples 3, 4, 5, 6, and 9.
- co-crystal of known compound A and L-proline form means a single co-crystal formed from known compound A and L-proline form at a molar ratio of 1: 1.
- Means. Identification of the co-crystal structure is shown by results of differential scanning calorimetry analysis (DSC analysis) and powder X-ray diffraction. For example, powder X-ray diffraction can be distinguished by the diffraction angle (2 ⁇ (°)) of the spectrum and the relative intensity (Tables 1 and 2).
- Powder X-ray diffraction is based on the nature of the data, and the crystal lattice spacing and overall pattern are important in determining the identity of the crystal. The relative intensity depends somewhat on the crystal growth direction, particle size, and measurement conditions. Since it can change, it should not be interpreted strictly. Further, if a peak peculiar to the known compound A structure appears in X-ray diffraction and is negligibly small, it is defined as a co-crystal of the known compound A and L-proline.
- Powder X-ray diffraction was measured under the following conditions.
- Humidity adjustment measurement “MAC Science MXP18TAHF22 with multifunctional humidity temperature converter (VAISALA MHP235)” was used, tube: Cu, tube current: 350 mA, tube voltage: 50 kV, sampling width: 0.020 °, scanning Measurement was performed under conditions of speed: 3 ° / min, wavelength: 1.54056 mm, measurement diffraction angle range (2 ⁇ ): 5 to 40 °.
- “good dissolution” means that the dissolution is equivalent to or equivalent to that of a normal preparation.
- the dissolution rate after 30 minutes is 65% or more, and in another embodiment, the dissolution rate after 30 minutes is 75% or more. It is defined that the elution rate after 30 minutes is 80% or more.
- “elution stability” means that the drug elution rate from the pharmaceutical composition is less changed with time than the start of storage.
- a dissolution test is performed by the dissolution test method described in the 15th revised Japanese Pharmacopoeia, it is defined that the drug dissolution rate is less changed with time than when storage was started.
- the dissolution rate 30 minutes after the start of the dissolution test after 6 months storage at 40 ° C. 75% is within ⁇ 15% compared to the storage start time.
- C-glycoside derivative A The chemical name is (1S) -1,5-anhydro-1- [3- (1-benzothien-2-ylmethyl) -4-fluorophenyl] -D-glucitol (hereinafter referred to as “C-glycoside derivative A”). Or simply “known compound A”).
- the known compound A forms a co-crystal structure with L-proline as shown by the following formula (II).
- the co-crystal has an endothermic peak at 201 to 213 ° C. by DSC analysis and / or 2 ⁇ (°) 4.44, 8.98, 12.4, 16.5, 17.5 by powder X-ray diffraction. , 18.7, 20.5, and 21.5.
- the known compound A and the co-crystal of the known compound A and L-proline can be distinguished by the diffraction angle (2 ⁇ (°)) and relative intensity in the powder X-ray diffraction spectrum.
- the clinical dose (therapeutically effective amount) of a known compound A co-crystal for humans is appropriately determined in consideration of the patient's symptoms, body weight, age and sex, etc.
- the dose is 0.1 to 500 mg, which is administered once or divided into several times. Since the dosage varies depending on various conditions, an amount smaller than the above dosage range may be sufficient.
- the crystalline cellulose used in the present invention is obtained by partially depolymerizing and purifying ⁇ -cellulose obtained as a pulp from a fibrous plant with an acid (15th revised Japanese Pharmacopoeia). As long as it is pharmaceutically acceptable and can maintain the good dissolution property and dissolution stability of the known compound A, the crystalline cellulose can be used with no particular limitation on the bulk density and the average degree of polymerization. .
- the shape of crystalline cellulose is not particularly limited, such as granular or acicular. Needle-shaped ones can also be crushed and used.
- As the crystalline cellulose a commercially available mixture as a mixture with other additives (carrageenan, sodium carboxymethyl cellulose, guar gum, etc.) can be used.
- the average particle size is preferably 20 to 200 ⁇ m when measured by the second method (sieving method) of the powder particle size measurement method described in the Japanese Pharmacopoeia.
- the crystalline cellulose ones having different grades, shapes, average particle sizes and the like can be used alone or in combination of two or more.
- the blending amount of the crystalline cellulose is not particularly limited as long as the known compound A can usually exhibit good dissolution properties. For example, it is 5 to 90% by weight in the pharmaceutical composition of the present invention. % By weight, 20 to 1500% by weight relative to the amount of co-crystal of known compound A and L-proline, and in other embodiments 50 to 1100% by weight or 40 to 350% by weight.
- the disintegrant used in the present invention is not particularly limited as long as generally known compound A exhibits good dissolution properties.
- Examples include low-substituted hydroxypropyl cellulose, sodium starch glycolate corn starch, potato starch, carmellose calcium, carmellose sodium, partially pregelatinized starch, crospovidone, and croscarmellose sodium.
- Other embodiments include low substituted hydroxypropylcellulose and sodium starch glycolate.
- One or two or more disintegrants can be used in appropriate combination.
- the amount of the disintegrant to be added is not particularly limited as long as the known compound A can usually exhibit good dissolution properties. For example, it is 5 to 90% by weight in the pharmaceutical composition of the present invention. % By weight or 5 to 55% by weight, 10 to 1500% by weight relative to the amount of co-crystal of known compound A and L-proline, and in another embodiment 10 to 1100% by weight or 25 to 300% by weight.
- the low-substituted hydroxypropyl cellulose is not particularly limited as long as it is pharmaceutically acceptable.
- hydroxypropyl cellulose having a hydroxypropoxyl group content of 5% by weight or more and less than 16% by weight Specific examples include L-HPC (LH-11, LH-21, LH-22, LH-B1, LH-31, LH-32, LH-B1) (all Shin-Etsu Chemical).
- the shape of the low-substituted hydroxypropyl cellulose is not particularly limited, such as granular or fibrous. Fibrous materials can also be crushed and used.
- the average particle size is preferably 10 to 100 ⁇ m when measured by the second method (sieving method) of the particle size measurement method described in the Japanese Pharmacopoeia. .
- Such low-substituted hydroxypropylcellulose can be used alone or in combination of two or more.
- the compounding amount of the low-substituted hydroxypropyl cellulose is 5 to 90% by weight in the pharmaceutical composition of the present invention, and in another embodiment, 5 to 70% by weight or 5 to 55% by weight. It is 10 to 1500% by weight with respect to the amount of co-crystal, and in other embodiments 10 to 1100% by weight or 25 to 300% by weight.
- Sodium starch glycolate is not particularly limited as long as it is pharmaceutically acceptable.
- Primogel (DMV), Exprotab (Kimura Sangyo) and the like can be mentioned.
- the shape of sodium starch glycolate is not particularly limited, such as granular, needle-like, egg-like, or spherical shape. Needle-shaped ones can also be crushed and used.
- the shape of sodium starch glycolate is granular, for example, the average particle size is preferably 10 to 100 ⁇ m when measured by the second method (sieving method) of the powder particle size measurement method described in the Japanese Pharmacopoeia. .
- Such sodium starch glycolate can be used singly or in combination of two or more different grades.
- the compounding amount of sodium starch glycolate is not particularly limited as long as it is an amount that usually shows a well-dissolved compound A.
- it is 5 to 90% by weight in the pharmaceutical composition of the present invention. 70% by weight, 10-1500% by weight with respect to the amount of co-crystal of known compound A and L-proline, and in another embodiment, 10-1100% by weight.
- various pharmaceutical additives are appropriately used and formulated as desired.
- a pharmaceutical additive is not particularly limited as long as it is pharmaceutically acceptable and pharmacologically acceptable.
- excipients, binders, disintegrants, acidulants, foaming agents, artificial sweeteners, flavors, lubricants, colorants, stabilizers, buffers, antioxidants, surfactants, coating agents, etc. used.
- Excipients include D-mannitol, lactose and the like.
- binder examples include hydroxypropylmethylcellulose, hydroxypropylcellulose, gum arabic and the like.
- sour agent examples include citric acid, tartaric acid, malic acid and the like.
- foaming agents examples include baking soda.
- artificial sweetener examples include saccharin sodium, dipotassium glycyrrhizin, aspartame, stevia and thaumatin.
- fragrances include lemon, lemon lime, orange and menthol.
- lubricant examples include magnesium stearate, calcium stearate, sucrose fatty acid ester, polyethylene glycol, talc, stearic acid and the like.
- Examples of the colorant include yellow ferric oxide, red ferric oxide, edible yellow No. 4, No. 5, edible red No. 3, No. 102, and edible blue No. 3.
- Buffers include citric acid, succinic acid, fumaric acid, tartaric acid, ascorbic acid or salts thereof, glutamic acid, glutamine, glycine, aspartic acid, alanine, arginine or salts thereof, magnesium oxide, zinc oxide, magnesium hydroxide, phosphoric acid Boric acid or a salt thereof.
- antioxidants examples include ascorbic acid, dibutylhydroxytoluene, propyl gallate and the like.
- surfactant examples include polysorbate 80, sodium lauryl sulfate, polyoxyethylene hydrogenated castor oil, and the like.
- Coating agents include talc, polyethylene glycol, hypromellose, titanium oxide and the like.
- an appropriate amount can be appropriately added in combination of one or more kinds.
- the blending amount of the pharmaceutical additive is 0.1 to 70% by weight in the pharmaceutical composition of the present invention.
- the pharmaceutical composition of the present invention can be made into various preparations. For example, tablets, capsules, powders, granules, dry syrups and the like can be mentioned.
- the solid pharmaceutical composition of the present invention is a tablet.
- the pharmaceutical composition of the present invention can be produced by a known method including steps such as pulverization, wet granulation, drying, tableting, and film coating.
- the solid pharmaceutical composition of the present invention in the form of a powder, granule or dry syrup comprises (1) a step of mixing a co-crystal of a known compound A and an L-proline, and crystalline cellulose, and (2 ) It can be produced by a method comprising a step of wet granulating the obtained mixture.
- the pharmaceutical additive may be any one during the step (1), between the steps (1) and (2), and during the step (2). It can be added at this stage.
- disintegrants are used as pharmaceutical additives, and disintegrants are exemplified by sodium starch glycolate and L-HPC.
- the solid pharmaceutical composition of the present invention in the form of a tablet comprises (1) a step of mixing a co-crystal of a known compound A and an L-proline body with crystalline cellulose, and (2) an obtained mixture. It can be manufactured by a method including a step of wet granulation, and (3) a step of compression-molding the granulated product.
- the pharmaceutical additives are used in the step (1), between the steps (1) and (2), in the step (2), and ( It can be added at any stage such as between step 2) and step (3).
- disintegrants are used as pharmaceutical additives, and disintegrants are exemplified by sodium starch glycolate and L-HPC.
- the co-crystal of known compound A and L-proline, crystalline cellulose, and pharmaceutical additives can each be subjected to a pulverization step before the mixing step and adjusted to an arbitrary size.
- the pulverization step is not particularly limited to any device and means as long as the drug and / or pharmaceutical additive can be pharmaceutically pulverized normally.
- the mixing step of each component continuous with the pulverization is not particularly limited to any device or means as long as it is a method that can generally uniformly mix each component pharmaceutically.
- a granulation product is prepared by wet granulation of the mixture using a granulator.
- the granulating apparatus include a fluidized bed granulator, a rolling fluidized bed granulator, and a stirring granulator.
- a binder In wet granulation, a binder is used.
- the binder can be added as an aqueous solution to a mixture containing cocrystal of known compound A and L-proline and crystalline cellulose.
- the rate of addition of the binder varies depending on the granulation method or scale to be produced. For example, when produced on a 1 kg scale by the fluidized bed granulation method, the binder solution is 1 to 30 g / min. It can be added at a rate of 5-20 g / min.
- the wet granulation can be performed in a temperature range of 15 to 35 ° C., for example.
- water can be added, including an aqueous solution of a binder.
- a mode in which a binder is added in advance to a mixture containing a co-crystal of a known compound A and an L-proline compound and crystalline cellulose, and then granulation is performed while adding water can be employed.
- the water can be added so that the maximum water content during granulation is 5 to 40% by weight or 5 to 30% by weight.
- the maximum moisture content during granulation is the maximum moisture content measured for the granulated product during granulation, and is mainly the moisture content of the granulated product at the end of the addition of the binder aqueous solution (or water).
- the moisture in the granulated product can be measured by allowing it to stand at 105 ° C. for 5 minutes using a halogen moisture meter (manufactured by METTLER TRADE).
- the solid pharmaceutical composition of the present invention contains a co-crystal structure of known compound A and L-proline, and the co-crystal structure is maintained during the preparation of the composition (from L-proline to co-crystal). It is necessary that the known compound A does not become a free form by detachment. In order to maintain the co-crystal structure, it is preferable to control the production conditions so as not to give a strong shearing force and excessive moisture to the granulated product. In terms of not giving a strong shearing force, the pharmaceutical composition of the present invention is preferably granulated by fluidized bed granulation.
- the prepared granulated product can be dried by any means.
- drying apparatuses such as a fluidized bed granulator, a multiplex, and a shelf dryer can be used.
- the drying temperature is, for example, 40 to 90 ° C.
- the tableting method is not particularly limited as long as it is a method in which a compression-molded product is usually produced pharmaceutically.
- a method of tableting by mixing a disintegrant and a lubricant into the granulated product may be mentioned.
- the tableting device is not particularly limited as long as it is a method in which a compression-molded product is usually produced pharmaceutically, and examples thereof include a rotary tableting machine and a single tableting machine.
- the tablet hardness is, for example, 40 to 250 N, and in another embodiment, 50 to 200 N.
- the tablet surface may be coated with a film.
- the method is not particularly limited as long as it is a pharmaceutically coating method.
- pan coating and the like can be mentioned.
- the film coating agent is not particularly limited as long as it is a pharmaceutical additive that is usually pharmaceutically coated.
- As the film coating agent one or a combination of two or more can be added as appropriate.
- the coating rate is not particularly limited as long as the tablet surface can be normally coated. For example, it is 1.0 wt% or more and 5.0 wt% or less with respect to the uncoated tablet which is a tablet before coating.
- the method for producing the pharmaceutical composition of the present invention or its pharmaceutical preparation is not particularly limited as long as it is a method for producing a pharmaceutical preparation having the desired effect of the present invention by appropriately combining the above-described methods or methods known per se. .
- the use of the crystalline cellulose of the present invention is the use of crystalline cellulose for producing a solid pharmaceutical composition that maintains the good dissolution property and dissolution stability of the known compound A.
- the co-crystal (1: 1) of known compound A and L-proline was prepared according to the method described in International Publication No. WO2007 / 114475.
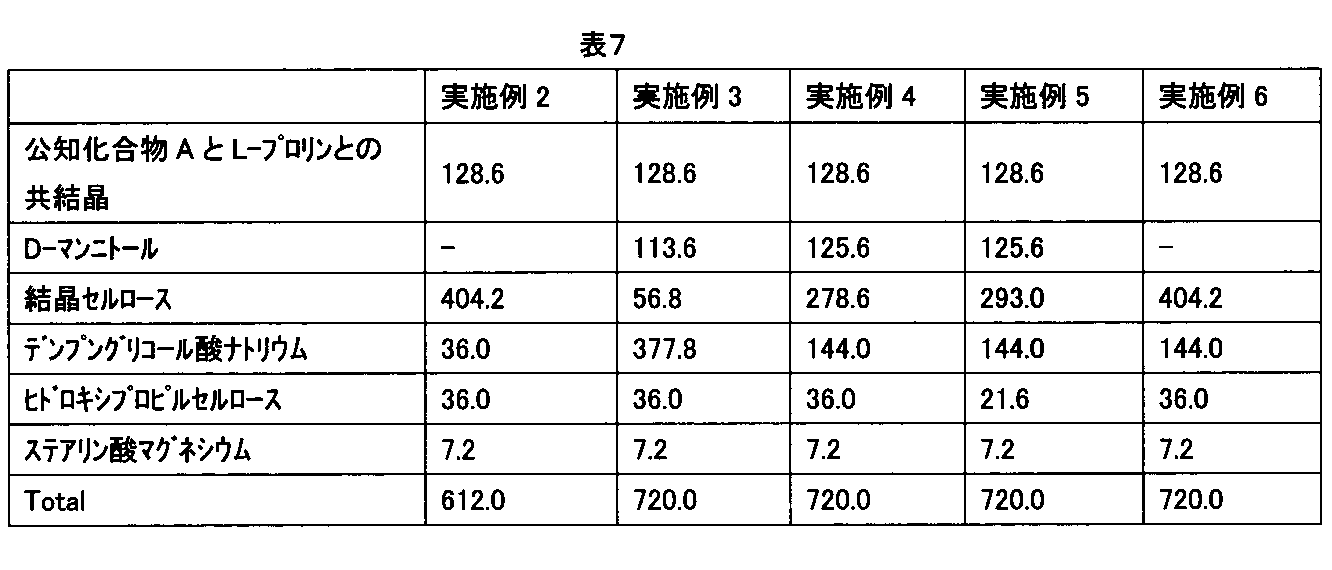
- the compositions of each Example and Comparative Example were prepared based on the formulations shown in Tables 3 to 5, Table 7, and Table 8.
- surface is the weight (g) of each used component.
- Example 1 Tablets were prepared based on the formulation in Table 3 using crystalline cellulose as an excipient.
- Co-crystal of known compound A and L-proline, and crystalline cellulose (product name: Theolas PH101, manufactured by Asahi Kasei, the same shall apply hereinafter), followed by hydroxypropyl cellulose (product name: HPC-L, manufactured by Nippon Soda, the same shall apply hereinafter)
- Aqueous solution (10% by weight) was sprayed as a binding solution (addition rate: 10 g / min), and fluidized bed granulation was performed (granulating device (product name: rolling fluidized bed granulated drying coating device, manufactured by POWREC).
- Granulation temperature 24-25 ° C., granulation time: 48 minutes).
- the maximum moisture value of the granulated product during granulation was 29.2%, and the average particle size of the obtained granulated product was 292 ⁇ m.
- sodium starch glycolate product name: Primogel, manufactured by DMV, the same applies hereinafter
- magnesium stearate product name: Parteck LUB MST, manufactured by Merck, The same applies hereinafter
- tableting was performed to obtain tablets (A-1) ( ⁇ diameter: 9.5 mm ⁇ 11.4R, tableting pressure: 6 kN).
- the moisture content of the granulated product was extracted from the granulator at intervals of 12 minutes from the start to the end of spraying of the binding liquid, and allowed to stand still at 105 ° C for 5 minutes using a halogen moisture meter (manufactured by METTLER TRADE). It was measured by placing. The highest moisture value was observed at the end of spraying.
- Croscarmellose sodium (product name: Ac-Di-Sol, manufactured by FMC Biopolymer, the same applies hereinafter), low-substituted hydroxypropylcellulose having a hydroxypropoxyl group content of 10 to 12.9% (product name: L-HPC,
- tablets A-2 ⁇ were used except that Shin-Etsu Chemical Co., Ltd. (hereinafter the same) or crospovidone (product name: Kollidon CL, BASF Co., Ltd., hereinafter the same) was used instead of sodium starch glycolate.
- A-4 was obtained (A-2: maximum moisture value: 29.2%, heel diameter: 9.5 mm ⁇ 11.4R, tableting pressure: 5 kN; A-3: maximum moisture value: 29.2%, heel diameter: 9.5 mm ⁇ 11.4R, tableting pressure: 5 kN; A-4: maximum moisture value: 29.2%, heel diameter: 9.5 mm ⁇ 11.4R, tableting pressure: 5 kN).
- Example 1 A granulated product (average particle size: average particle diameter: same as in Example 1) except that D-mannitol (product name: PEARITOL 50C, manufactured by Rocket Co., Ltd., hereinafter the same) was used instead of crystalline cellulose as an excipient.
- D-mannitol product name: PEARITOL 50C, manufactured by Rocket Co., Ltd., hereinafter the same
- B-1 maximum moisture value: 3.7%, heel diameter: 9.5 mm ⁇ 11.4R, tableting pressure: 10 kN
- B-2 maximum moisture value: 3.7%, heel diameter: 9.5mm ⁇ 11.4R, tableting pressure: 13kN
- B-3 maximum moisture value: 3.7%, heel diameter: 9.5mm ⁇ 11.4R, tableting pressure: 11kN
- B-4 maximum moisture Value: 3.7%, heel diameter: 9.5 mm ⁇ 11.4R, tableting pressure: 11 kN).
- anhydrous calcium hydrogen phosphate product name: GS Carica, manufactured by Kyowa Chemical Industry Co., Ltd.
- C-1 to C-4 were obtained (C-1: maximum moisture value: 8.1%, heel diameter: 9.5 mm ⁇ 11.4R, tableting pressure: 15 kN; C-2: maximum moisture value: 8.1%, heel diameter: 9.5mm ⁇ 11.4R, tableting pressure: 15kN; C-3: Maximum moisture value: 8.1%, heel diameter: 9.5mm ⁇ 11.4R, tableting pressure: 9kN; C-4: Maximum moisture value: 8.1%, ⁇ Diameter: 9.5 mm ⁇ 11.4R, tableting pressure: 13 kN;).
- Example 1 Each tablet obtained in Example 1, Comparative Example 1 and Comparative Example 2 was subjected to a dissolution test of the composition immediately after production of the preparation (at the start of storage).
- the dissolution test was performed by the paddle method described in the 15th revised Japanese Pharmacopoeia.
- the test solution was 900 mL (0.1N hydrochloric acid aqueous solution) of the first dissolution test solution.
- the rotation speed of the paddle was 50 rpm.
- Table 6 shows the dissolution rate of the known compound A 30 minutes after the start of the test.
- the elution profiles are shown in FIG. 1, FIG. 2, and FIG.
- FIG. 1, FIG. 2, and FIG. 3 the tablets prepared using crystalline cellulose are compared to tablets prepared using D-mannitol or anhydrous calcium hydrogen phosphate. Good elution was exhibited. Moreover, the dissolution property of the tablet prepared using crystalline cellulose was good without depending on the type of disintegrant.
- a solid pharmaceutical composition having a good dissolution property can be provided by combining a co-crystal of known compound A and L-proline with crystalline cellulose.
- Example 2 In the same manner as in Example 1, a co-crystal of known compound A and L-proline and crystalline cellulose were mixed and then sprayed with a hydroxypropylcellulose aqueous solution as a binding liquid to perform fluidized bed granulation (during granulation). Maximum moisture value: 29.2%, average particle size: 144 ⁇ m). After the obtained granulated product was dried, sodium starch glycolate and magnesium stearate were mixed and tableted to obtain a solid pharmaceutical composition of the present invention (inner diameter: 9.5 mm ⁇ 11.4R, tableting pressure). : 7kN).
- Example 3 In the same manner as in Example 1, after mixing a known compound A and L-proline co-crystal, D-mannitol, crystalline cellulose, and sodium starch glycolate, sprayed with an aqueous hydroxypropyl cellulose solution as a binder, Granulation was performed (maximum moisture value during granulation: 15.4%, average particle size: 248 ⁇ m). The obtained granulated product was dried, mixed with magnesium stearate, and tableted to obtain a solid pharmaceutical composition of the present invention ( ⁇ diameter: 9.5 mm ⁇ 11.4R, tableting pressure: 7 kN).
- Example 4 In the same manner as in Example 1, after mixing a co-crystal of a known compound A and L-proline, D-mannitol and crystalline cellulose, and sodium starch glycolate, sprayed with an aqueous hydroxypropyl cellulose solution as a binding liquid, Granulation was performed (maximum moisture value during granulation: 20.1%, average particle size: 164 ⁇ m). The obtained granulated product was dried, mixed with magnesium stearate, and tableted to obtain a solid pharmaceutical composition of the present invention (inner diameter: 10.0 mm ⁇ 15.0R, tableting pressure: 8 kN).
- Example 5 In the same manner as in Example 1, after mixing a co-crystal of a known compound A and L-proline, D-mannitol and crystalline cellulose, and sodium starch glycolate, sprayed with an aqueous hydroxypropyl cellulose solution as a binding liquid, Granulation was performed (maximum moisture value during granulation: 23.2%, average particle size: 115 ⁇ m). After drying the obtained granulated product, magnesium stearate was mixed and tableted to obtain a solid pharmaceutical composition of the present invention (inner diameter: 9.5 mm ⁇ 11.4R, tableting pressure: 8 kN).
- Example 6 In the same manner as in Example 1, a co-crystal of known compound A and L-proline and crystalline cellulose were mixed and then sprayed with a hydroxypropylcellulose aqueous solution as a binding liquid to perform fluidized bed granulation (during granulation). Maximum moisture value: 29.2%, average particle size: 292 ⁇ m). After drying the obtained granulated product, sodium starch glycolate and magnesium stearate were mixed and tableted to obtain a solid pharmaceutical composition of the present invention (inner diameter: 9.5 mm ⁇ 11.4R, tableting) Pressure is 6kN).
- Example 7 In the same manner as in Example 1, a co-crystal of known compound A and L-proline and crystalline cellulose were mixed and then sprayed with a hydroxypropylcellulose aqueous solution as a binding liquid to perform fluidized bed granulation (during granulation). Maximum moisture value: 29.2%, average particle size: 292 ⁇ m). After the obtained granulated product was dried, magnesium stearate was mixed and tableted to obtain a solid pharmaceutical composition of the present invention ( ⁇ diameter: 9.5 mm ⁇ 11.4R, tableting pressure was 5 kN).
- Example 8> In the same manner as in Example 1, after mixing a co-crystal of known compound A and L-proline, D-mannitol, crystalline cellulose and low-substituted hydroxypropylcellulose, sprayed with an aqueous hydroxypropylcellulose solution as a binder, Granulation was performed (maximum moisture value during granulation: 10.4%, average particle size: 231 ⁇ m). After the obtained granulated product was dried, magnesium stearate was mixed and tableted to obtain a solid pharmaceutical composition of the present invention ( ⁇ diameter: 9.5 mm ⁇ 11.4R, tableting pressure: 5 kN).
- Example 9 In the same manner as in Example 1, after co-crystallizing a known compound A and L-proline, D-mannitol, and crystalline cellulose were mixed and then sprayed with a hydroxypropylcellulose aqueous solution as a binding solution to perform fluidized bed granulation ( Maximum moisture value during granulation: 16.6%, average particle size: 197 ⁇ m). The obtained granulated product was dried, mixed with low-substituted hydroxypropylcellulose and magnesium stearate, and tableted to obtain a solid pharmaceutical composition of the present invention (inner diameter: 9.5 mm ⁇ 11.4R, punched) Tablet pressure: 5kN).
- Example 10 In the same manner as in Example 1, after co-crystallizing a known compound A and L-proline, D-mannitol, and crystalline cellulose were mixed and then sprayed with a hydroxypropylcellulose aqueous solution as a binding solution to perform fluidized bed granulation ( Maximum moisture value during granulation: 16.6%, average particle size: 197 ⁇ m). The obtained granulated product was dried, mixed with low-substituted hydroxypropylcellulose and magnesium stearate, and tableted to obtain a solid pharmaceutical composition of the present invention (inner diameter: 9.5 mm ⁇ 11.4R, punched) Tablet pressure: 5kN).
- Example 3 Evaluation of dissolution property and dissolution stability
- the dissolution test was performed by the paddle method described in the 15th revised Japanese Pharmacopoeia.
- the test solution was 900 mL (0.1N hydrochloric acid aqueous solution) of the first dissolution test solution.
- the rotation speed of the paddle was 50 rpm.
- Table 9 shows the dissolution rate of the known compound A 30 minutes after the start of the test. The elution profile is shown in FIG.
- the solid pharmaceutical composition containing the co-crystal of the known compound A and L-proline and the crystalline cellulose maintains the co-crystal structure of the known compound A and L-proline and is good. Showed good elution properties.
- crystalline cellulose By adding crystalline cellulose to the formulation, it is considered that strong cohesiveness, which is a characteristic of the co-crystal of compound A and L-proline, can be alleviated and dispersibility can be improved.
- the present invention relates to a solid pharmaceutical composition containing a co-crystal of a known compound A and L-proline and maintaining good dissolution properties and dissolution stability of the known compound A, a method for producing the same, and the solid composition
- the co-crystal of the known compound A and L-proline exhibits good elution, the bioavailability (BA) is improved, and a pharmacologically sufficient therapeutic effect is obtained. Play.
Abstract
Description
[1](1S)-1,5-アンヒドロ-1-[3-(1-ベンゾチエン-2-イルメチル)-4-フルオロフェニル]-D-グルシトールとL-プロリン体との共結晶、及び結晶セルロースを含有してなる固形医薬組成物、
[2]結晶セルロースの量が、医薬組成物中5重量%以上90重量%以下である、[1]に記載の固形医薬組成物、
[3]更に、崩壊剤を含有する、[1]または[2]に記載の固形医薬組成物、
[4]崩壊剤が、デンプングリコール酸ナトリウム、及びヒドロキシプロポキシル基含量が5重量%以上16重量%未満のヒドロキシプロピルセルロースからなる群より選択される1種または2種以上である、[3]に記載の固形医薬組成物、
[5]崩壊剤の量が、医薬組成物中5重量%以上90重量%以下である、[3]または[4]に記載の固形医薬組成物、
[6]第15改正日本薬局方に記載の溶出試験において、(1S)-1,5-アンヒドロ-1-[3-(1-ベンゾチエン-2-イルメチル)-4-フルオロフェニル]-D-グルシトールが30分で65%以上溶出する[1]~[5]の何れかに記載の固形医薬組成物、
[7](1)(1S)-1,5-アンヒドロ-1-[3-(1-ベンゾチエン-2-イルメチル)-4-フルオロフェニル]-D-グルシトールとL-プロリン体との共結晶と、結晶セルロースとを混合する工程、及び
(2)得られた混合物を、(1S)-1,5-アンヒドロ-1-[3-(1-ベンゾチエン-2-イルメチル)-4-フルオロフェニル]-D-グルシトールとL-プロリン体との共結晶構造を維持した状態で湿式造粒する工程、からなる固形医薬組成物の製造方法、
[8]更に、(3)造粒物を圧縮成形する工程、を含む[7]に記載の固形医薬組成物の製造方法、
[9]結晶セルロースの量が、医薬組成物中5重量%以上90重量%以下である、[7]または[8]に記載の固形医薬組成物の製造方法、
[10](1)の工程中、(1)と(2)の工程の間、(2)の工程中、または(2)の工程の後に、崩壊剤を添加することを更に含む、[7]に記載の製造方法、
[11]崩壊剤が、デンプングリコール酸ナトリウム、及びヒドロキシプロポキシル基含量が5重量%以上16重量%未満のヒドロキシプロピルセルロースからなる群より選択される1種または2種以上である、[10]に記載の固形医薬組成物の製造方法、
[12]崩壊剤の量が、医薬組成物中5重量%以上90重量%以下である、[10]または[11]に記載の固形医薬組成物の製造方法、
[13]第15改正日本薬局方に記載の溶出試験において、(1S)-1,5-アンヒドロ-1-[3-(1-ベンゾチエン-2-イルメチル)-4-フルオロフェニル]-D-グルシトールが30分で65%以上溶出する固形医薬組成物を製造するための、[7]~[12]の何れかに記載の製造方法、
[14]湿式造粒が、造粒中の固形医薬組成物の最大水分値が5~30重量%で行われる、[7]に記載の固形医薬組成物の製造方法、
[15](1S)-1,5-アンヒドロ-1-[3-(1-ベンゾチエン-2-イルメチル)-4-フルオロフェニル]-D-グルシトールとL-プロリン体との共結晶を含有してなる固形医薬組成物を製造するための結晶セルロースの使用、
を提供するものである。


(1)標準測定:「MAC Science MXP18TAHF22」を用い、管球:Cu、管電流:200mA、管電圧:40kV、サンプリング幅:0.020°、走査速度:3°/min、波長:1.54056Å、測定回折角範囲(2θ):3~40°の条件で測定した。
(2)調湿測定:「多機能湿度温度変換器(VAISALA MHP235)付 MAC Science MXP18TAHF22」を用い、管球:Cu、管電流:350mA、管電圧:50kV、サンプリング幅:0.020°、走査速度:3°/min、波長:1.54056Å、測定回折角範囲(2θ):5~40°の条件で測定した。

賦形剤として、結晶セルロースを使用して表3の処方に基づいて錠剤を調製した。
賦形剤として、結晶セルロースに代えてD-マンニトール(製品名:PEARITOL 50C、ロケット社製、以下同じ)を用いたことを除いて、実施例1と同様にして造粒物(平均粒子径:144μm)、及び錠剤B-1~B-4を得た(B-1:最高水分値:3.7%、杵径:9.5mm×11.4R、打錠圧:10kN;B-2:最高水分値:3.7%、杵径:9.5mm×11.4R、打錠圧:13kN;B-3:最高水分値:3.7%、杵径:9.5mm×11.4R、打錠圧:11kN;B-4:最高水分値:3.7%、杵径:9.5mm×11.4R、打錠圧:11kN)。
賦形剤として、結晶セルロースに代えて、無水リン酸水素カルシウム(製品名:GSカリカ、協和化学工業社製)を用いたことを除いて、実施例1と同様にして造粒物および錠剤C-1~C-4を得た(C-1:最高水分値:8.1%、杵径:9.5mm×11.4R、打錠圧:15kN;C-2:最高水分値:8.1%、杵径:9.5mm×11.4R、打錠圧:15kN;C-3:最高水分値:8.1%、杵径:9.5mm×11.4R、打錠圧:9kN;C-4:最高水分値:8.1%、杵径:9.5mm×11.4R、打錠圧:13kN;)。



実施例1、比較例1及び比較例2で得た各錠剤について、製剤の製造直後(保存開始時)における組成物の溶出試験を行った。溶出試験は、第15改正日本薬局方に記載のパドル法により行った。試験液は溶出試験第1液900mL(0.1Nの塩酸水溶液)とした。パドルの回転数は50回転/分とした。試験開始後30分における公知化合物Aの溶出率を表6に示す。また、溶出プロファイルを図1、図2、及び図3に示す。

実施例1と同様にして、公知化合物AとL-プロリンとの共結晶、及び結晶セルロースを混合後、ヒドロキシプロピルセルロース水溶液を結合液として噴霧し、流動層造粒を行った(造粒中の最高水分値:29.2%、平均粒子径:144μm)。得られた造粒物を乾燥した後、デンプングリコール酸ナトリウム及びステアリン酸マグネシウムを混合し、打錠し、本発明の固形医薬組成物を得た(杵径:9.5mm×11.4R、打錠圧:7kN)。
実施例1と同様にして、公知化合物AとL-プロリンとの共結晶、D-マンニトール、結晶セルロース、及びデンプングリコール酸ナトリウムを混合後、ヒドロキシプロピルセルロース水溶液を結合液として噴霧し、流動層造粒を行った(造粒中の最高水分値:15.4%、平均粒子径:248μm)。得られた造粒物を乾燥した後、ステアリン酸マグネシウムを混合し、打錠し、本発明の固形医薬組成物を得た(杵径:9.5mm×11.4R、打錠圧:7kN)。
実施例1と同様にして、公知化合物AとL-プロリンとの共結晶、D-マンニトール及び結晶セルロース、及びデンプングリコール酸ナトリウムを混合後、ヒドロキシプロピルセルロース水溶液を結合液として噴霧し、流動層造粒を行った(造粒中の最高水分値:20.1%、平均粒子径:164μm)。得られた造粒物を乾燥した後、ステアリン酸マグネシウムを混合し、打錠し、本発明の固形医薬組成物を得た(杵径:10.0mm×15.0R、打錠圧:8kN)。
実施例1と同様にして、公知化合物AとL-プロリンとの共結晶、D-マンニトール及び結晶セルロース、及びデンプングリコール酸ナトリウムを混合後、ヒドロキシプロピルセルロース水溶液を結合液として噴霧し、流動層造粒を行った(造粒中の最高水分値:23.2%、平均粒子径:115μm)。得られた造粒物を乾燥した後、ステアリン酸マグネシウムを混合し、打錠し、本発明の固形医薬組成物を得た(杵径:9.5mm×11.4R、打錠圧:8kN)。
実施例1と同様にして、公知化合物AとL-プロリンとの共結晶、及び結晶セルロースを混合後、ヒドロキシプロピルセルロース水溶液を結合液として噴霧し、流動層造粒を行った(造粒中の最高水分値:29.2%、平均粒子径:292μm)。得られた造粒物を乾燥した後、デンプングリコール酸ナトリウム、及びステアリン酸マグネシウムを混合し、打錠し、本発明の固形医薬組成物を得た(杵径:9.5mm×11.4R、打錠圧は6kN)。
実施例1と同様にして、公知化合物AとL-プロリンとの共結晶、及び結晶セルロースを混合後、ヒドロキシプロピルセルロース水溶液を結合液として噴霧し、流動層造粒を行った(造粒中の最高水分値:29.2%、平均粒子径:292μm)。得られた造粒物を乾燥した後、ステアリン酸マグネシウムを混合し、打錠し、本発明の固形医薬組成物を得た(杵径:9.5mm×11.4R、打錠圧は5kN)。
実施例1と同様にして、公知化合物AとL-プロリンとの共結晶、D-マンニトール、結晶セルロース及び低置換度ヒドロキシプロピルセルロースを混合後、ヒドロキシプロピルセルロース水溶液を結合液として噴霧し、流動層造粒を行った(造粒中の最高水分値:10.4%、平均粒子径:231μm)。得られた造粒物を乾燥した後、ステアリン酸マグネシウムを混合し、打錠し、本発明の固形医薬組成物を得た(杵径:9.5mm×11.4R、打錠圧:5kN)。
実施例1と同様にして、公知化合物AとL-プロリンとの共結晶、D-マンニトール、及び結晶セルロースを混合後、ヒドロキシプロピルセルロース水溶液を結合液として噴霧し、流動層造粒を行った(造粒中の最高水分値:16.6%、平均粒子径:197μm)。得られた造粒物を乾燥した後、低置換度ヒドロキシプロピルセルロース及びステアリン酸マグネシウムを混合し、打錠し、本発明の固形医薬組成物を得た(杵径:9.5mm×11.4R、打錠圧:5kN)。
実施例1と同様にして、公知化合物AとL-プロリンとの共結晶、D-マンニトール、及び結晶セルロースを混合後、ヒドロキシプロピルセルロース水溶液を結合液として噴霧し、流動層造粒を行った(造粒中の最高水分値:16.6%、平均粒子径:197μm)。得られた造粒物を乾燥した後、低置換度ヒドロキシプロピルセルロース及びステアリン酸マグネシウムを混合し、打錠し、本発明の固形医薬組成物を得た(杵径:9.5mm×11.4R、打錠圧:5kN)。

実施例2~10について、製剤の製造直後における結晶構造について、粉末X線回折を用いて評価したところ、公知化合物AとL-プロリンの共結晶に相当するピークが観測されたが、公知化合物A単独のものに相当するピークは観測されなかった。これより、製剤後においても公知化合物AとL-プロリンの共結晶構造が維持されていることが判った(表9)。
実施例2~10について、製剤の製造直後(保存開始時)、及び40℃75%相対湿度下で6ヶ月保存後における組成物の溶出試験を行った。溶出試験は、第15改正日本薬局方に記載のパドル法により行った。試験液は溶出試験第1液900mL(0.1Nの塩酸水溶液)とした。パドルの回転数は50回転/分とした。試験開始後30分における公知化合物Aの溶出率を表9に示す。また、溶出プロファイルを図4に示す。

Claims (15)
- (1S)-1,5-アンヒドロ-1-[3-(1-ベンゾチエン-2-イルメチル)-4-フルオロフェニル]-D-グルシトールとL-プロリン体との共結晶、及び結晶セルロースを含有してなる固形医薬組成物。
- 結晶セルロースの量が、医薬組成物中5重量%以上90重量%以下である、請求項1記載の固形医薬組成物。
- 更に、崩壊剤を含有する、請求項1または請求項2に記載の固形医薬組成物。
- 崩壊剤が、デンプングリコール酸ナトリウム、及びヒドロキシプロポキシル基含量が5重量%以上16重量%未満のヒドロキシプロピルセルロースからなる群より選択される1種または2種以上である、請求項3記載の固形医薬組成物。
- 崩壊剤の量が、医薬組成物中5重量%以上90重量%以下である、請求項3または請求項4に記載の固形医薬組成物。
- 第15改正日本薬局方に記載の溶出試験において、(1S)-1,5-アンヒドロ-1-[3-(1-ベンゾチエン-2-イルメチル)-4-フルオロフェニル]-D-グルシトールが30分で65%以上溶出する請求項1乃至5の何れか1項に記載の固形医薬組成物。
- (1)(1S)-1,5-アンヒドロ-1-[3-(1-ベンゾチエン-2-イルメチル)-4-フルオロフェニル]-D-グルシトールとL-プロリン体との共結晶と、結晶セルロースとを混合する工程、及び
(2)得られた混合物を、(1S)-1,5-アンヒドロ-1-[3-(1-ベンゾチエン-2-イルメチル)-4-フルオロフェニル]-D-グルシトールとL-プロリン体との共結晶構造を維持した状態で湿式造粒する工程、からなる固形医薬組成物の製造方法。 - 更に、(3)造粒物を圧縮成形する工程、を含む請求項7に記載の固形医薬組成物の製造方法。
- 結晶セルロースの量が、医薬組成物中5重量%以上90重量%以下である、請求項7または請求項8記載の固形医薬組成物の製造方法。
- (1)の工程中、(1)と(2)の工程の間、(2)の工程中、または(2)の工程の後に、崩壊剤を添加することを更に含む、請求項7に記載の製造方法。
- 崩壊剤が、デンプングリコール酸ナトリウム、及びヒドロキシプロポキシル基含量が5重量%以上16重量%未満のヒドロキシプロピルセルロースからなる群より選択される1種または2種以上である、請求項10記載の固形医薬組成物の製造方法。
- 崩壊剤の量が、医薬組成物中5重量%以上90重量%以下である、請求項10または請求項11に記載の固形医薬組成物の製造方法。
- 第15改正日本薬局方に記載の溶出試験において、(1S)-1,5-アンヒドロ-1-[3-(1-ベンゾチエン-2-イルメチル)-4-フルオロフェニル]-D-グルシトールが30分で65%以上溶出する固形医薬組成物を製造するための、請求項7乃至12の何れか1項に記載の製造方法。
- 湿式造粒が、造粒中の固形医薬組成物の最大水分値が5~30重量%で行われる、請求項7に記載の固形医薬組成物の製造方法。
- (1S)-1,5-アンヒドロ-1-[3-(1-ベンゾチエン-2-イルメチル)-4-フルオロフェニル]-D-グルシトールとL-プロリン体との共結晶を含有してなる固形医薬組成物を製造するための結晶セルロースの使用。
Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2778411A CA2778411A1 (en) | 2009-10-23 | 2010-10-22 | Pharmaceutical composition for oral administration |
| CN201080047947.8A CN102596206B (zh) | 2009-10-23 | 2010-10-22 | 口服用医药组合物 |
| MX2012004782A MX2012004782A (es) | 2009-10-23 | 2010-10-22 | Composicion farmaceutica para administracion oral. |
| US13/502,453 US20120214753A1 (en) | 2009-10-23 | 2010-10-22 | Pharmaceutical composition for oral administration |
| EP10825040A EP2491933A1 (en) | 2009-10-23 | 2010-10-22 | Pharmaceutical composition for oral administration |
| BR112012007364A BR112012007364A2 (pt) | 2009-10-23 | 2010-10-22 | composição farmacêutica para administração oral |
| RU2012121183/15A RU2012121183A (ru) | 2009-10-23 | 2010-10-22 | Фармацевтическая композиция для перорального введения |
| KR1020127010513A KR101476011B1 (ko) | 2009-10-23 | 2010-10-22 | 경구 투여용 의약 조성물 |
| JP2011511934A JP4759102B2 (ja) | 2009-10-23 | 2010-10-22 | 経口投与用医薬組成物 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US25444309P | 2009-10-23 | 2009-10-23 | |
| US61/254443 | 2009-10-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011049191A1 true WO2011049191A1 (ja) | 2011-04-28 |
Family
ID=43900414
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/068660 WO2011049191A1 (ja) | 2009-10-23 | 2010-10-22 | 経口投与用医薬組成物 |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US20120214753A1 (ja) |
| EP (1) | EP2491933A1 (ja) |
| JP (1) | JP4759102B2 (ja) |
| KR (1) | KR101476011B1 (ja) |
| CN (1) | CN102596206B (ja) |
| AR (1) | AR078750A1 (ja) |
| BR (1) | BR112012007364A2 (ja) |
| CA (1) | CA2778411A1 (ja) |
| MX (1) | MX2012004782A (ja) |
| RU (1) | RU2012121183A (ja) |
| WO (1) | WO2011049191A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012144592A1 (ja) * | 2011-04-22 | 2012-10-26 | アステラス製薬株式会社 | 固形医薬組成物 |
| JP2020529450A (ja) * | 2017-08-07 | 2020-10-08 | エナンティア,エセ.エレ. | 2−[(1r,6r)−6−イソプロペニル−3−メチルシクロヘキサ−2−エン−1−イル]−5−ペンチルベンゼン−1,3−ジオールの共結晶 |
| JP2021505674A (ja) * | 2017-12-11 | 2021-02-18 | アーテロ バイオサイエンシズ,インコーポレイテッド | カンナビジオールの新しい固体形態およびその使用 |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106924206A (zh) * | 2015-12-31 | 2017-07-07 | 深圳翰宇药业股份有限公司 | 一种依格列净口服固体制剂及其制备方法 |
| CN113039176A (zh) * | 2018-09-26 | 2021-06-25 | 莱西肯医药有限公司 | N-(1-((2-(二甲基氨基)乙基)氨基)-2-甲基-1-氧代丙-2-基)-4-(4-(2-甲基-5-((2s,3r,4r,5s,6r)-3,4,5-三羟基-6-(甲硫基)四氢-2h-吡喃-2-基)苯甲基)苯基)丁酰胺的结晶形式及其合成方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007114475A1 (ja) * | 2006-04-05 | 2007-10-11 | Astellas Pharma Inc. | C-グリコシド誘導体とl-プロリンとの共結晶 |
| WO2009022010A1 (en) * | 2007-08-16 | 2009-02-19 | Boehringer Ingelheim International Gmbh | Pharmaceutical composition comprising a sglt2 inhibitor in combination with a dpp-iv inhibitor |
| WO2009096455A1 (ja) * | 2008-01-31 | 2009-08-06 | Astellas Pharma Inc. | 脂肪性肝疾患の治療用医薬組成物 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6852336B2 (en) * | 1995-11-15 | 2005-02-08 | J. Rettenmaier & Soehne Gmbh + Co. Kg | Directly compressible high load acetaminophen formulations |
| PT1980560E (pt) * | 2003-03-14 | 2011-06-29 | Astellas Pharma Inc | Derivados de c-glicósido para o tratamento da diabetes |
-
2010
- 2010-10-22 KR KR1020127010513A patent/KR101476011B1/ko active IP Right Review Request
- 2010-10-22 EP EP10825040A patent/EP2491933A1/en not_active Withdrawn
- 2010-10-22 CN CN201080047947.8A patent/CN102596206B/zh not_active Expired - Fee Related
- 2010-10-22 MX MX2012004782A patent/MX2012004782A/es unknown
- 2010-10-22 BR BR112012007364A patent/BR112012007364A2/pt not_active Application Discontinuation
- 2010-10-22 RU RU2012121183/15A patent/RU2012121183A/ru not_active Application Discontinuation
- 2010-10-22 CA CA2778411A patent/CA2778411A1/en not_active Abandoned
- 2010-10-22 US US13/502,453 patent/US20120214753A1/en not_active Abandoned
- 2010-10-22 WO PCT/JP2010/068660 patent/WO2011049191A1/ja active Application Filing
- 2010-10-22 AR ARP100103894A patent/AR078750A1/es unknown
- 2010-10-22 JP JP2011511934A patent/JP4759102B2/ja active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007114475A1 (ja) * | 2006-04-05 | 2007-10-11 | Astellas Pharma Inc. | C-グリコシド誘導体とl-プロリンとの共結晶 |
| WO2009022010A1 (en) * | 2007-08-16 | 2009-02-19 | Boehringer Ingelheim International Gmbh | Pharmaceutical composition comprising a sglt2 inhibitor in combination with a dpp-iv inhibitor |
| WO2009096455A1 (ja) * | 2008-01-31 | 2009-08-06 | Astellas Pharma Inc. | 脂肪性肝疾患の治療用医薬組成物 |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012144592A1 (ja) * | 2011-04-22 | 2012-10-26 | アステラス製薬株式会社 | 固形医薬組成物 |
| KR101841087B1 (ko) | 2011-04-22 | 2018-03-23 | 아스텔라스세이야쿠 가부시키가이샤 | 고형 의약 조성물 |
| JP2020529450A (ja) * | 2017-08-07 | 2020-10-08 | エナンティア,エセ.エレ. | 2−[(1r,6r)−6−イソプロペニル−3−メチルシクロヘキサ−2−エン−1−イル]−5−ペンチルベンゼン−1,3−ジオールの共結晶 |
| JP7186460B2 (ja) | 2017-08-07 | 2022-12-09 | エナンティア,エセ.エレ. | 2-[(1r,6r)-6-イソプロペニル-3-メチルシクロヘキサ-2-エン-1-イル]-5-ペンチルベンゼン-1,3-ジオールの共結晶 |
| JP2021505674A (ja) * | 2017-12-11 | 2021-02-18 | アーテロ バイオサイエンシズ,インコーポレイテッド | カンナビジオールの新しい固体形態およびその使用 |
| JP7429013B2 (ja) | 2017-12-11 | 2024-02-07 | アーテロ バイオサイエンシズ,インコーポレイテッド | カンナビジオールの新しい固体形態およびその使用 |
Also Published As
| Publication number | Publication date |
|---|---|
| AR078750A1 (es) | 2011-11-30 |
| CA2778411A1 (en) | 2011-04-28 |
| JP4759102B2 (ja) | 2011-08-31 |
| CN102596206B (zh) | 2014-06-04 |
| JPWO2011049191A1 (ja) | 2013-03-14 |
| EP2491933A1 (en) | 2012-08-29 |
| US20120214753A1 (en) | 2012-08-23 |
| CN102596206A (zh) | 2012-07-18 |
| BR112012007364A2 (pt) | 2017-06-06 |
| RU2012121183A (ru) | 2013-11-27 |
| MX2012004782A (es) | 2012-09-12 |
| KR20120127387A (ko) | 2012-11-21 |
| KR101476011B1 (ko) | 2014-12-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI750143B (zh) | 含有2-{4-[n-(5,6-二苯基吡-2-基)-n-異丙基胺基]丁基氧基}-n-(甲基磺醯基)乙醯胺之醫藥組合物 | |
| CA3045418C (en) | Orally disintegrating tablet comprising edoxaban | |
| JP6759390B2 (ja) | トホグリフロジンを含有する固形製剤及びその製造方法 | |
| JP4759102B2 (ja) | 経口投与用医薬組成物 | |
| TW201716069A (zh) | 安定的經口投予用醫藥組成物 | |
| JP6063379B2 (ja) | 固形医薬組成物 | |
| JP6455611B2 (ja) | 経口投与用医薬組成物 | |
| EP2263671B1 (en) | Amide derivative-containing pharmaceutical composition | |
| JPWO2020045607A1 (ja) | 経口投与用医薬組成物 | |
| WO2010038689A1 (ja) | 経口投与用医薬組成物 | |
| JP6893687B2 (ja) | 口腔内崩壊錠 | |
| JP3637968B1 (ja) | 胃内崩壊性錠剤 | |
| AU2015287336B2 (en) | Pharmaceutical dosage forms | |
| TW202404585A (zh) | 含有匹密特匹(Pimitespib)之醫藥組合物 | |
| WO2014157603A1 (ja) | 経口投与用医薬組成物 | |
| CN116322653A (zh) | 包括酞嗪酮衍生物的药物组合物 | |
| WO2007113371A1 (en) | Pharmaceutical composition and preparation method thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080047947.8 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011511934 Country of ref document: JP |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10825040 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010825040 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13502453 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2778411 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2012/004782 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 20127010513 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1111/KOLNP/2012 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012121183 Country of ref document: RU |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112012007364 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112012007364 Country of ref document: BR Kind code of ref document: A2 Effective date: 20120330 |