WO2013042695A1 - 金属ポルフィリン錯体、その製造方法及びそれからなる二酸化炭素固定化触媒、並びに、環状炭酸エステルの製造方法 - Google Patents
金属ポルフィリン錯体、その製造方法及びそれからなる二酸化炭素固定化触媒、並びに、環状炭酸エステルの製造方法 Download PDFInfo
- Publication number
- WO2013042695A1 WO2013042695A1 PCT/JP2012/073957 JP2012073957W WO2013042695A1 WO 2013042695 A1 WO2013042695 A1 WO 2013042695A1 JP 2012073957 W JP2012073957 W JP 2012073957W WO 2013042695 A1 WO2013042695 A1 WO 2013042695A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- general formula
- represented
- formula
- group
- following general
- Prior art date
Links
- 0 CC*(CC(C)(C1C=CCC11)C1(C)C(C)=C(C)C)C(C)C Chemical compound CC*(CC(C)(C1C=CCC11)C1(C)C(C)=C(C)C)C(C)C 0.000 description 6
Images

Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/16—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes
- B01J31/22—Organic complexes
- B01J31/2282—Unsaturated compounds used as ligands
- B01J31/2295—Cyclic compounds, e.g. cyclopentadienyls
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/02—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides
- B01J31/0234—Nitrogen-, phosphorus-, arsenic- or antimony-containing compounds
- B01J31/0235—Nitrogen containing compounds
- B01J31/0239—Quaternary ammonium compounds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/02—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides
- B01J31/0234—Nitrogen-, phosphorus-, arsenic- or antimony-containing compounds
- B01J31/0235—Nitrogen containing compounds
- B01J31/0244—Nitrogen containing compounds with nitrogen contained as ring member in aromatic compounds or moieties, e.g. pyridine
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/02—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides
- B01J31/0234—Nitrogen-, phosphorus-, arsenic- or antimony-containing compounds
- B01J31/0255—Phosphorus containing compounds
- B01J31/0267—Phosphines or phosphonium compounds, i.e. phosphorus bonded to at least one carbon atom, including e.g. sp2-hybridised phosphorus compounds such as phosphabenzene, the other atoms bonded to phosphorus being either carbon or hydrogen
- B01J31/0268—Phosphonium compounds, i.e. phosphine with an additional hydrogen or carbon atom bonded to phosphorous so as to result in a formal positive charge on phosphorous
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/16—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes
- B01J31/18—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes containing nitrogen, phosphorus, arsenic or antimony as complexing atoms, e.g. in pyridine ligands, or in resonance therewith, e.g. in isocyanide ligands C=N-R or as complexed central atoms
- B01J31/1805—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes containing nitrogen, phosphorus, arsenic or antimony as complexing atoms, e.g. in pyridine ligands, or in resonance therewith, e.g. in isocyanide ligands C=N-R or as complexed central atoms the ligands containing nitrogen
- B01J31/181—Cyclic ligands, including e.g. non-condensed polycyclic ligands, comprising at least one complexing nitrogen atom as ring member, e.g. pyridine
- B01J31/1815—Cyclic ligands, including e.g. non-condensed polycyclic ligands, comprising at least one complexing nitrogen atom as ring member, e.g. pyridine with more than one complexing nitrogen atom, e.g. bipyridyl, 2-aminopyridine
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/16—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes
- B01J31/18—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes containing nitrogen, phosphorus, arsenic or antimony as complexing atoms, e.g. in pyridine ligands, or in resonance therewith, e.g. in isocyanide ligands C=N-R or as complexed central atoms
- B01J31/1805—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes containing nitrogen, phosphorus, arsenic or antimony as complexing atoms, e.g. in pyridine ligands, or in resonance therewith, e.g. in isocyanide ligands C=N-R or as complexed central atoms the ligands containing nitrogen
- B01J31/181—Cyclic ligands, including e.g. non-condensed polycyclic ligands, comprising at least one complexing nitrogen atom as ring member, e.g. pyridine
- B01J31/1825—Ligands comprising condensed ring systems, e.g. acridine, carbazole
- B01J31/183—Ligands comprising condensed ring systems, e.g. acridine, carbazole with more than one complexing nitrogen atom, e.g. phenanthroline
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/10—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 not condensed with other rings
- C07D317/32—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 not condensed with other rings with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D317/34—Oxygen atoms
- C07D317/36—Alkylene carbonates; Substituted alkylene carbonates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/22—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains four or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/54—Quaternary phosphonium compounds
- C07F9/5442—Aromatic phosphonium compounds (P-C aromatic linkage)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/54—Quaternary phosphonium compounds
- C07F9/5449—Polyphosphonium compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2231/00—Catalytic reactions performed with catalysts classified in B01J31/00
- B01J2231/30—Addition reactions at carbon centres, i.e. to either C-C or C-X multiple bonds
- B01J2231/32—Addition reactions to C=C or C-C triple bonds
- B01J2231/321—Hydroformylation, metalformylation, carbonylation or hydroaminomethylation
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2231/00—Catalytic reactions performed with catalysts classified in B01J31/00
- B01J2231/30—Addition reactions at carbon centres, i.e. to either C-C or C-X multiple bonds
- B01J2231/34—Other additions, e.g. Monsanto-type carbonylations, addition to 1,2-C=X or 1,2-C-X triplebonds, additions to 1,4-C=C-C=X or 1,4-C=-C-X triple bonds with X, e.g. O, S, NH/N
- B01J2231/341—1,2-additions, e.g. aldol or Knoevenagel condensations
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/02—Compositional aspects of complexes used, e.g. polynuclearity
- B01J2531/0238—Complexes comprising multidentate ligands, i.e. more than 2 ionic or coordinative bonds from the central metal to the ligand, the latter having at least two donor atoms, e.g. N, O, S, P
- B01J2531/0241—Rigid ligands, e.g. extended sp2-carbon frameworks or geminal di- or trisubstitution
- B01J2531/025—Ligands with a porphyrin ring system or analogues thereof, e.g. phthalocyanines, corroles
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/10—Complexes comprising metals of Group I (IA or IB) as the central metal
- B01J2531/16—Copper
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/20—Complexes comprising metals of Group II (IIA or IIB) as the central metal
- B01J2531/22—Magnesium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/20—Complexes comprising metals of Group II (IIA or IIB) as the central metal
- B01J2531/26—Zinc
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/40—Complexes comprising metals of Group IV (IVA or IVB) as the central metal
- B01J2531/42—Tin
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/80—Complexes comprising metals of Group VIII as the central metal
- B01J2531/84—Metals of the iron group
- B01J2531/845—Cobalt
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/80—Complexes comprising metals of Group VIII as the central metal
- B01J2531/84—Metals of the iron group
- B01J2531/847—Nickel
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/90—Catalytic systems characterized by the solvent or solvent system used
- B01J2531/98—Phase-transfer catalysis in a mixed solvent system containing at least 2 immiscible solvents or solvent phases
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2540/00—Compositional aspects of coordination complexes or ligands in catalyst systems
- B01J2540/40—Non-coordinating groups comprising nitrogen
- B01J2540/42—Quaternary ammonium groups
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2540/00—Compositional aspects of coordination complexes or ligands in catalyst systems
- B01J2540/50—Non-coordinating groups comprising phosphorus
- B01J2540/54—Quaternary phosphonium groups
Definitions
- the present invention relates to a novel metal porphyrin complex, a production method thereof, and a carbon dioxide-immobilized catalyst comprising the same.
- the carbon dioxide-immobilized catalyst is suitably used for producing a cyclic carbonate.
- the present invention also relates to a method for producing a cyclic carbonate using a catalyst comprising a metal porphyrin complex or metal phthalocyanine complex and a specific cocatalyst.
- C1 carbon sources So far, cyanide ions (or hydrocyanic acid), carbon monoxide, phosgene, and the like have been used as C1 carbon sources. However, since these have very strong toxicity, safe chemical raw materials to replace them are desired. Carbon dioxide is a renewable and safe C1 carbon source. However, since carbon dioxide has low reactivity, its use has been limited.
- cyclic carbonates are widely used as electrolytes for lithium ion secondary batteries, polycarbonate raw materials, aprotic polar solvents, and the like.
- the production of cyclic carbonates has mainly employed a method using 1,2-diol and phosgene as raw materials.
- the use of highly toxic phosgene and the by-product of corrosive hydrogen chloride gas have been problems for many years.
- the synthesis method for obtaining a cyclic carbonate by coupling reaction of carbon dioxide and epoxide is a very clean method without any by-products.
- catalysts that promote this reaction have been reported so far.
- Non-Patent Document 1 uses a porphyrin complex whose central metal is Cr as a catalyst, and N, N-dimethyl-4-aminopyridine (hereinafter sometimes abbreviated as DMAP) or N-methylimidazole as a cocatalyst.
- DMAP N, N-dimethyl-4-aminopyridine
- N-methylimidazole N-methylimidazole
- Non-Patent Document 2 discloses a method for producing a cyclic carbonate using a porphyrin complex whose central metal is Co as a catalyst and using DMAP, pyridine, N-methylimidazole, tricyclohexylphosphine oxide or triphenylphosphine as a cocatalyst. Are listed. However, this method sometimes has a low reaction yield. Moreover, since dichloromethane was used as a solvent, there was an environmental problem.
- Non-Patent Document 3 describes a method for producing a cyclic carbonate using a porphyrin complex or phthalocyanine complex whose central metal is Cu as a catalyst and using DMAP as a cocatalyst.
- this method sometimes has a low reaction yield.
- dichloromethane was used as a solvent, there was an environmental problem.
- Non-Patent Document 4 uses a porphyrin complex whose central metal is Co, Fe, Ru, or Mn as a catalyst, phenyltrimethylammonium tribromide (hereinafter sometimes abbreviated as PTAT), tetrabutylammonium bromide, or DMAP.
- PTAT phenyltrimethylammonium tribromide
- tetrabutylammonium bromide tetrabutylammonium bromide
- DMAP phenyltrimethylammonium tribromide
- a method for producing a cyclic carbonate used as a cocatalyst is described. However, this method sometimes has a low reaction yield.
- Non-Patent Document 5 describes a method for producing a cyclic carbonate using a porphyrin complex whose central metal is Mg as a catalyst and triethylamine as a cocatalyst. However, this method sometimes has a low reaction yield.
- the present invention has been made to solve the above problems, and provides a novel metalloporphyrin complex that exhibits high catalytic activity when used as a carbon dioxide-immobilized catalyst, has a low environmental impact, and can be easily synthesized. It is intended to do. Moreover, it aims at providing the manufacturing method of such a metal porphyrin complex, and the carbon dioxide fixed catalyst consisting thereof. Furthermore, it aims at providing the manufacturing method of cyclic carbonate which is highly efficient and has little environmental impact.
- M is a metal.
- a 1 to A 4 are each independently a substituent represented by the following general formula (2).
- D is a divalent organic group having 1 to 20 carbon atoms.
- E + is a quaternary ammonium group or quaternary phosphonium group having 3 to 60 carbon atoms.
- X is a halogen atom. .
- E + represents the following general formula (3)
- G is a nitrogen atom or a phosphorus atom.
- R 1 to R 3 are each independently a monovalent organic group having 1 to 20 carbon atoms. R 1 to R 3 are bonded to each other.) To form a ring.) It is preferable that it is represented.
- D is an organic group represented by the following general formula (4).
- J represents an oxygen atom, —CO—O—, —O—CO—, sulfur atom, —O—CO—NH—, —NH—CO—O—, —CO—NH—, —NH— CO— or a single bond, a is an integer of 0 or more, and b is an integer of 1 or more.
- the metal porphyrin complex is represented by the following general formula (5).
- the metal porphyrin complex is produced by reacting the metal complex with a tertiary amine or tertiary phosphine to obtain the metal porphyrin complex represented by the general formula (1). It is also solved by providing.
- a carbon dioxide-immobilized catalyst comprising the metal porphyrin complex is a preferred embodiment of the present invention.
- R 4 to R 7 are each independently a hydrogen atom or a monovalent organic group having 1 to 20 carbon atoms. R 4 to R 7 may be bonded to each other to form a ring. Good.) Is reacted with an epoxide represented by the following general formula (9):
- M represents magnesium or zinc
- Ar represents an aromatic ring which may have a substituent.
- R 8 and R 9 are each independently a monovalent organic group having 1 to 20 carbon atoms. R 8 and R 9 may be bonded to each other to form a ring.
- R 4 to R 7 are each independently a hydrogen atom or a monovalent organic group having 1 to 20 carbon atoms. R 4 to R 7 may be bonded to each other to form a ring. Good.
- the quaternary ammonium monohalide and the quaternary phosphonium monohalide are represented by the following general formula (13).
- R 26 to R 29 are each independently a monovalent organic group having 1 to 20 carbon atoms. R 26 R 29 may be bonded to each other to form a ring.
- M is magnesium or zinc.
- R 10 to R 25 are each independently a hydrogen atom, a monovalent hydrocarbon group having 1 to 20 carbon atoms, or a halogen atom.
- R 8 and R 9 are each independently a monovalent organic group having 1 to 20 carbon atoms. R 8 and R 9 may be bonded to each other to form a ring.
- R 4 to R 7 are each independently a hydrogen atom or a monovalent organic group having 1 to 20 carbon atoms. R 4 to R 7 may be bonded to each other to form a ring. Good.
- the quaternary ammonium monohalide and the quaternary phosphonium monohalide are represented by the following general formula (13).
- R 26 to R 29 are each independently a monovalent organic group having 1 to 20 carbon atoms. R 26 R 29 may be bonded to each other to form a ring.
- the metalloporphyrin complex of the present invention exhibits high catalytic activity when used as a carbon dioxide-fixing catalyst, has a low environmental load, and can be easily synthesized. Therefore, it can be suitably used for immobilization of carbon dioxide, particularly for production of cyclic carbonate. Moreover, the production method of the cyclic carbonate of the present invention is highly efficient and has a low environmental load.
- the metal porphyrin complex of the present invention is represented by the following general formula (1).
- the compound is a novel compound.
- M is a metal.
- a 1 to A 4 are each independently a substituent represented by the following general formula (2).
- D is a divalent organic group having 1 to 20 carbon atoms.
- E + is a quaternary ammonium group or quaternary phosphonium group having 3 to 60 carbon atoms.
- X is a halogen atom. .
- a feature of the metal porphyrin complex of the present invention is that it has a quaternary ammonium group or a quaternary phosphonium group represented by E + .
- the substituents are halogen ions X - to form a quaternary ammonium salt or quaternary phosphonium salt.
- Such salts have strong nucleophilicity.
- E + is bonded to the porphyrin ring via the divalent organic group D, E + has an appropriate degree of freedom and the distance between E + and the central metal M is appropriate.
- metal M and X ⁇ having E + as a counter ion can act simultaneously. This significantly accelerates the reaction.
- M is a metal.
- M has Lewis acidity.
- M is not particularly limited as long as it is a metal, but a divalent or trivalent metal that exhibits high catalytic activity when used as a carbon dioxide fixing catalyst is suitable.
- divalent metals examples include magnesium, zinc, copper, nickel, cobalt, and iron.
- M is a combination of a trivalent metal and a monovalent counter anion.
- the trivalent metal in this case include cobalt, iron, manganese, chromium, and aluminum
- the counter anion include a halide anion and an acetate anion.
- M is more preferably a divalent metal. Of these, magnesium and zinc are more preferable, and magnesium is particularly preferable.
- a 1 to A 4 are each independently a substituent represented by the general formula (2).
- E + is a quaternary ammonium group or quaternary phosphonium group having 3 to 60 carbon atoms.
- the number of carbon atoms is preferably 3-30.
- G is a nitrogen atom or a phosphorus atom.
- R 1 to R 3 are each independently a monovalent organic group having 1 to 20 carbon atoms. R 1 to R 3 are bonded to each other.) To form a ring.
- R 30 to R 34 are each independently a hydrogen atom or a monovalent hydrocarbon group having 1 to 20 carbon atoms. R 30 to R 34 are bonded to each other to form a ring. May be good.
- substituents are bulky.
- the substituent E + is bulky, so that X ⁇ having E + as a counter ion functions more efficiently as a nucleophile.
- the catalytic activity is further improved.
- G is a nitrogen atom or a phosphorus atom.
- R 1 to R 3 are each independently a monovalent organic group having 1 to 20 carbon atoms.
- the number of carbon atoms is preferably 2 or more. From the viewpoint of easy synthesis, the number of carbon atoms is preferably 10 or less.
- an alkyl group which may have a substituent an alkenyl group which may have a substituent, an alkynyl group which may have a substituent, an aryl group which may have a substituent, a substituted group
- examples of the substituent include an alkyl group having 1 to 5 carbon atoms and a halogen atom.
- the organic group is preferably an alkyl group which may have a substituent or an aryl group which may have a substituent, and more preferably an alkyl group or an aryl group.
- alkyl group examples include linear alkyl groups such as a methyl group, an ethyl group, an n-propyl group, an n-butyl group, and an n-pentyl group, an isopropyl group, an isobutyl group, a sec-butyl group, and a tert-butyl group.
- branched chain alkyl groups such as isopentyl group, neopentyl group, and tert-pentyl group. Among them, n-butyl group is preferable.
- aryl group examples include a phenyl group, a biphenyl group, a naphthyl group, an anthryl group, and a phenanthryl group, and among them, a phenyl group is preferable.
- R 1 to R 3 may be bonded to each other to form a ring.
- the ring may be a monocycle or a bicyclo ring.
- the total number of carbon atoms forming the ring may be 2 to 40, and 3 to 20 is preferable.
- the total number of carbon atoms forming the bicyclo ring may be 3 to 60, and 5 to 30 is preferable.
- a hetero atom may be contained in a part of atoms forming the ring, and examples thereof include a nitrogen atom and an oxygen atom.
- R 30 to R 34 are each independently a hydrogen atom or a monovalent hydrocarbon group having 1 to 20 carbon atoms.
- the hydrocarbon group preferably has 2 or more carbon atoms. From the viewpoint of easy synthesis, the number of carbon atoms is preferably 10 or less.
- hydrocarbon group examples include alkyl groups, alkenyl groups, alkynyl groups, aryl groups, arylalkyl groups, arylalkenyl groups, arylalkynyl groups, cycloalkyl groups, and the like. Of these, an alkyl group is preferred.
- R 30 to R 34 may combine with each other to form a ring.
- the total number of carbon atoms forming the ring may be 2 to 40, and 3 to 20 is preferable.
- R 30 to R 34 are preferably hydrogen atoms.
- D is a divalent organic group having 1 to 20 carbon atoms.
- D connects the porphyrin ring and E + in the general formula (2).
- E + is bonded to the porphyrin ring via D, the degree of freedom of E + is improved.
- X ⁇ having E + as a counter ion easily undergoes a nucleophilic attack. Thereby, high catalytic activity is obtained.
- D preferably has 2 or more carbon atoms.
- the carbon number of D exceeds 20, the cost increases.
- the carbon number of D is preferably 15 or less.
- J represents an oxygen atom, —CO—O—, —O—CO—, sulfur atom, —O—CO—NH—, —NH—CO—O—, —CO—NH—, —NH— CO— or a single bond, a is an integer of 0 or more, and b is an integer of 1 or more.
- J represents an oxygen atom, —CO—O—, —O—CO—, a sulfur atom, —O—CO—NH—, —NH—CO—O—, —CO—NH—. , -NH-CO- or a single bond.
- J is preferably an oxygen atom, —CO—O— or —O—CO—, and more preferably an oxygen atom.
- a is an integer of 0 or more, and b is an integer of 1 or more. From the viewpoint of easy synthesis, it is preferable that a is 0. That is, it is preferable that J is directly bonded to the porphyrin ring.
- X in the general formula (2) is a halogen atom.
- X becomes an anion (that is, X ⁇ ) to form a salt with E + in the general formula (2).
- X is not particularly limited, but when the metal porphyrin complex of the present invention is used as a catalyst or the like in the synthesis of a cyclic carbonate, X is preferably a bromine atom or an iodine atom from the viewpoint of easy nucleophilic attack. More preferably, it is a bromine atom.
- the bonding positions of the A 1 to A 4 in the general formula (1) to the phenyl group are the same.
- the bonding position of A 1 to A 4 is a meta position or a para position of the phenyl group because a stereoisomer does not occur during synthesis.
- the bonding position of A 1 to A 4 is the meta position or para position of the phenyl group from the viewpoint that steric hindrance does not occur. Is preferred.
- the general formula (1) is represented by the following general formula (5)
- a 1 to A 4 are all the same.
- the method for synthesizing the metal porphyrin complex of the present invention represented by the above general formula (1) is not particularly limited, but a suitable synthesis method is the following general formula (6).
- metal porphyrin complex represented by the general formula (1).
- the metal M is the same as the general formula (1).
- a tertiary amine and a tertiary phosphine become an ammonium salt and a phosphonium salt, respectively, and form E + in the general formula (2).
- a porphyrin and a central metal are made to react at the final stage of a synthesis process.
- the yield of the metal porphyrin complex is increased by reacting the tertiary amine or tertiary phosphine after introducing the metal M into the porphyrin represented by the general formula (6). improves. Moreover, since this method has a short reaction step and uses a very general reaction for each step, the metal porphyrin complex of the present invention can be easily produced. Therefore, it is very advantageous economically.
- the porphyrin represented by the general formula (6) can be synthesized, for example, as follows.
- Acid catalyst of pyrrole and benzaldehyde having halogenated organic group such as trifluoroacetic acid, boron trifluoride diethyl ether complex, etc. (these may be used alone or in combination of two or more)
- DDQ 2,3-dichloro-5,6-dicyano-p-benzoquinone
- dichloromethane can be used as the solvent at this time.
- the reaction product can be purified by ordinary separation means such as column chromatography or recrystallization.
- the method for introducing the central metal into the porphyrin represented by the general formula (6) thus obtained may be appropriately selected depending on the type of the central metal.
- a metal complex by mixing porphyrin and metal salt in a solvent
- water is added to wash the metal salt, and purification is performed by ordinary separation means such as column chromatography or recrystallization. can get.
- it is preferable that 3 to 20 moles of metal salt are mixed and reacted with 1 mole of porphyrin.
- the solvent chloroform, methanol, methylene chloride or the like is used. These may be used alone or in combination of two or more.
- the reaction temperature is usually appropriately selected within the range of 0 to 100 ° C.
- the metal porphyrin complex represented by the general formula (1) is obtained by dissolving the obtained metal complex represented by the general formula (7) and the tertiary amine or tertiary phosphine in the solvent and then stirring the solvent. can get. At this time, it is preferable that 4 to 30 mol of tertiary amine or tertiary phosphine is mixed and reacted with 1 mol of the metal complex.
- the solvent chloroform, acetonitrile or the like is used. These may be used alone or in combination of two or more.
- the reaction temperature is usually appropriately selected within the range of 0 to 100 ° C.
- the reaction product can be purified by ordinary separation means such as column chromatography or recrystallization.
- a carbon dioxide-immobilized catalyst comprising the metal porphyrin complex of the present invention thus obtained is a preferred embodiment of the present invention.
- the metalloporphyrin complex of the present invention significantly accelerates the synthesis reaction using carbon dioxide as the C1 carbon source.
- Carbon dioxide is a renewable and safe C1 carbon source, and the merit of using carbon dioxide as a C1 carbon source is great. It is also preferable from the viewpoint of reducing carbon dioxide emissions.
- the metal porphyrin complex of the present invention significantly accelerates the reaction of synthesizing a cyclic carbonate from carbon dioxide and epoxide.
- R 4 to R 7 are each independently a hydrogen atom or a monovalent organic group having 1 to 20 carbon atoms. R 4 to R 7 may be bonded to each other to form a ring. Good.
- a method for producing a cyclic carbonate to obtain a cyclic carbonate represented by the formula is a more preferred embodiment. Since the method does not use a solvent, no waste liquid is generated and no by-product is generated. Therefore, the environmental load is very small. Furthermore, the carbon dioxide-immobilized catalyst of the present invention has extremely high catalytic activity as compared with the conventional method using a porphyrin complex and a cocatalyst. Therefore, replacement with the existing method using phosgene can be expected.
- the epoxide represented by the general formula (8) and the metal porphyrin complex represented by the general formula (1) are added to a pressure vessel such as an autoclave, and then the vessel is filled with carbon dioxide. Can be performed.
- the amount of the metal porphyrin complex used is not particularly limited, but it is preferable to use 0.0001 to 0.1 mol% with respect to the epoxide (the metal porphyrin complex is 0.000001 to 0.001 mol with respect to 1 mol of the epoxide).
- the initial pressure in the container is preferably 0.1 to 5 MPa.
- the reaction temperature is preferably 10 to 200 ° C. According to the production method of the present invention, a cyclic carbonate can be obtained efficiently even under such mild conditions.
- R 4 to R 7 are each independently a hydrogen atom or a monovalent organic group having 1 to 20 carbon atoms. From the viewpoint of easy synthesis, the organic group preferably has 10 or less carbon atoms.
- Examples of the organic group include an alkyl group that may have a substituent, an alkenyl group that may have a substituent, an alkynyl group that may have a substituent, and an aryl group that may have a substituent.
- a heterocyclic group which may have a group, an alkoxy group, an aryloxy group, an aldehyde group, an optionally protected carboxyl group or a salt thereof, an alkylcarbonyl group, an arylcarbonyl group, an alkyloxycarbonyl group, an aryloxycarbonyl group, Alkylcarbonyloxy group, arylcarbonyloxy group, alkylamino group, arylamino group,
- R 4 to R 7 may be bonded to each other to form a ring.
- the total number of carbon atoms forming the ring may be 2 to 40, and 3 to 20 is preferable.
- a hetero atom may be contained in a part of atoms forming the ring, and examples thereof include a nitrogen atom and an oxygen atom.
- any one of R 4 to R 7 is an organic group and the other is a hydrogen atom.
- M represents magnesium or zinc
- Ar represents an aromatic ring which may have a substituent.
- R 8 and R 9 are each independently a monovalent organic group having 1 to 20 carbon atoms. R 8 and R 9 may be bonded to each other to form a ring.
- this method using a specific cocatalyst may be referred to as a “two-component method using a metal porphyrin complex”.
- the catalytic activity is remarkably improved.
- the use of such metals is a major feature of the two-component method.
- magnesium complexes and zinc complexes were considered to have low catalytic activity, and thus were not used as metal complexes for catalysts. It is surprising that the catalytic activity is remarkably improved by using inexpensive general-purpose metals such as magnesium and zinc.
- a cocatalyst composed of at least one selected from quaternary ammonium monohalide, quaternary phosphonium monohalide and the compound represented by the general formula (11).
- the above reaction is performed by the epoxide represented by the general formula (8), the metal porphyrin complex represented by the general formula (10), the quaternary ammonium monohalide, the quaternary phosphonium monohalide, and the general formula (11).
- the reaction can be carried out by adding at least one cocatalyst selected from the compounds represented to a pressure vessel such as an autoclave and filling the vessel with carbon dioxide.
- the amount of the metal porphyrin complex used is not particularly limited, but it is preferable to use 0.0001 to 1 mol% with respect to the epoxide (0.000001 to 0.01 mol of metal porphyrin complex with respect to 1 mol of epoxide).
- the amount of the cocatalyst used is not particularly limited, but it is preferable to use 0.0001 to 1 mol% with respect to the epoxide (cocatalyst 0.000001 to 0.01 mol with respect to 1 mol of the epoxide).
- the reaction conditions other than the use amount of the metal porphyrin complex and the cocatalyst are the carbon dioxide fixation composed of the metal porphyrin complex having a quaternary ammonium group or quaternary phosphonium group represented by the general formula (1) in the molecule. It can carry out on the same conditions as the manufacturing method (henceforth "bifunctional catalyst method") of the cyclic carbonate using a catalyst.
- M is magnesium or zinc.
- Ar represents an aromatic ring which may have a substituent.
- the aromatic ring include a phenyl group, a biphenyl group, a naphthyl group, an anthryl group, and a phenanthryl group. Of these, a phenyl group is preferred.
- the aromatic ring has a substituent, examples of the substituent include an alkyl group having 1 to 5 carbon atoms and a halogen atom.
- R 26 to R 29 are each independently a monovalent organic group having 1 to 20 carbon atoms.
- R 26 to R 29 are And may be bonded to each other to form a ring, and
- X is the same as in the general formula (2).
- R 26 to R 29 are each independently a monovalent organic group having 1 to 20 carbon atoms.
- R 26 to R 29 are the same as R 1 to R 3 in the general formula (3).
- R 26 to R 29 may be bonded to each other to form a ring.
- the total number of carbon atoms forming the ring may be 2 to 40, and 2 to 20 is preferable.
- a hetero atom may be contained in a part of atoms forming the ring, and examples thereof include a nitrogen atom and an oxygen atom.
- R 8 and R 9 are each independently a monovalent organic group having 1 to 20 carbon atoms. From the viewpoint of easy synthesis, the number of carbon atoms in R 8 and R 9 is preferably 10 or less.
- the organic group an alkyl group which may have a substituent, an alkenyl group which may have a substituent, an alkynyl group which may have a substituent, an aryl group which may have a substituent, a substituted group
- alkyl group examples include a methyl group, ethyl group, n-propyl group, n-butyl group, n-pentyl group, isopropyl group, isobutyl group, sec-butyl group, tert-butyl group, isopentyl group, neopentyl group, Examples thereof include a tert-pentyl group.
- a methyl group that can be obtained at low cost is preferable.
- R 8 and R 9 may be bonded to each other to form a ring.
- the total number of carbon atoms of R 8 and R 9 may be 2 to 40, and is preferably 2 to 20.
- a hetero atom may be contained in a part of atoms forming the ring, and examples thereof include a nitrogen atom and an oxygen atom.
- the epoxide represented by the general formula (8) is the same as the epoxide used in the bifunctional catalyst method.
- the method for producing the metal porphyrin complex represented by the general formula (10) is not particularly limited. For example, after forming the metal complex by mixing the porphyrin and the metal salt in a solvent, excess water is added. The metal salt can be obtained by washing. A commercially available porphyrin can be suitably used. In this case, the cost is further reduced. Moreover, in the manufacturing method of the porphyrin shown by General formula (6) in the above-mentioned bifunctional catalyst method, it can obtain also by using various aromatic aldehydes instead of the benzaldehyde which has a halogenated organic group.
- the cyclic carbonate represented by the general formula (9) is represented by the following general formula (12).
- M is magnesium or zinc.
- R 10 to R 25 are each independently a hydrogen atom, a monovalent hydrocarbon group having 1 to 20 carbon atoms, or a halogen atom.
- This method is a method using phthalocyanine instead of porphyrin in the above-mentioned “two-component method using a metal porphyrin complex”.
- the central metal M is magnesium or zinc, and at least one selected from a quaternary ammonium monohalide, a quaternary phosphonium monohalide and a compound represented by the above general formula (11) as a cocatalyst. Is considered important.
- the reaction can be carried out by adding at least one cocatalyst selected from the above compounds to a pressure vessel such as an autoclave and filling the vessel with carbon dioxide.
- the amount of the metal phthalocyanine complex used is not particularly limited, but it is preferable to use 0.0001 to 1 mol% with respect to the epoxide (0.000001 to 0.01 mol of metal phthalocyanine complex with respect to 1 mol of epoxide).
- the amount of the cocatalyst used is not particularly limited, but it is preferable to use 0.0001 to 1 mol% with respect to the epoxide (cocatalyst 0.000001 to 0.01 mol with respect to 1 mol of the epoxide).
- the reaction conditions other than the amounts of the metal phthalocyanine complex and the cocatalyst used can be carried out under the same conditions as in the bifunctional catalyst method described above.
- R 10 to R 25 are each independently a hydrogen atom, a monovalent hydrocarbon group having 1 to 20 carbon atoms, or a halogen atom.
- hydrocarbon group examples include alkyl groups, alkenyl groups, alkynyl groups, aryl groups, arylalkyl groups, arylalkenyl groups, arylalkynyl groups, cycloalkyl groups, and the like. Of these, an alkyl group is preferred. From the viewpoint of easy synthesis, the hydrocarbon group preferably has 10 or less carbon atoms.
- R 10 to R 25 are preferably a hydrogen atom or a halogen atom.
- the method for producing the metal phthalocyanine complex represented by the general formula (11) is not particularly limited. For example, after forming a metal complex by mixing phthalocyanine and a metal salt in a solvent, excess water is added. The metal salt can be obtained by washing. A commercially available phthalocyanine can be suitably used. This further reduces the cost.
- the measuring equipment used for the measurement is as follows. Melting point measuring device: Mettler Toledo, FP-62 IR measuring device: Shimadzu, FTIR-8900 1 H NMR (600 MHz) measuring apparatus: Varian, Unity Inova AS600 13 C NMR (150 MHz) measuring apparatus: Varian, Unity Inova AS600
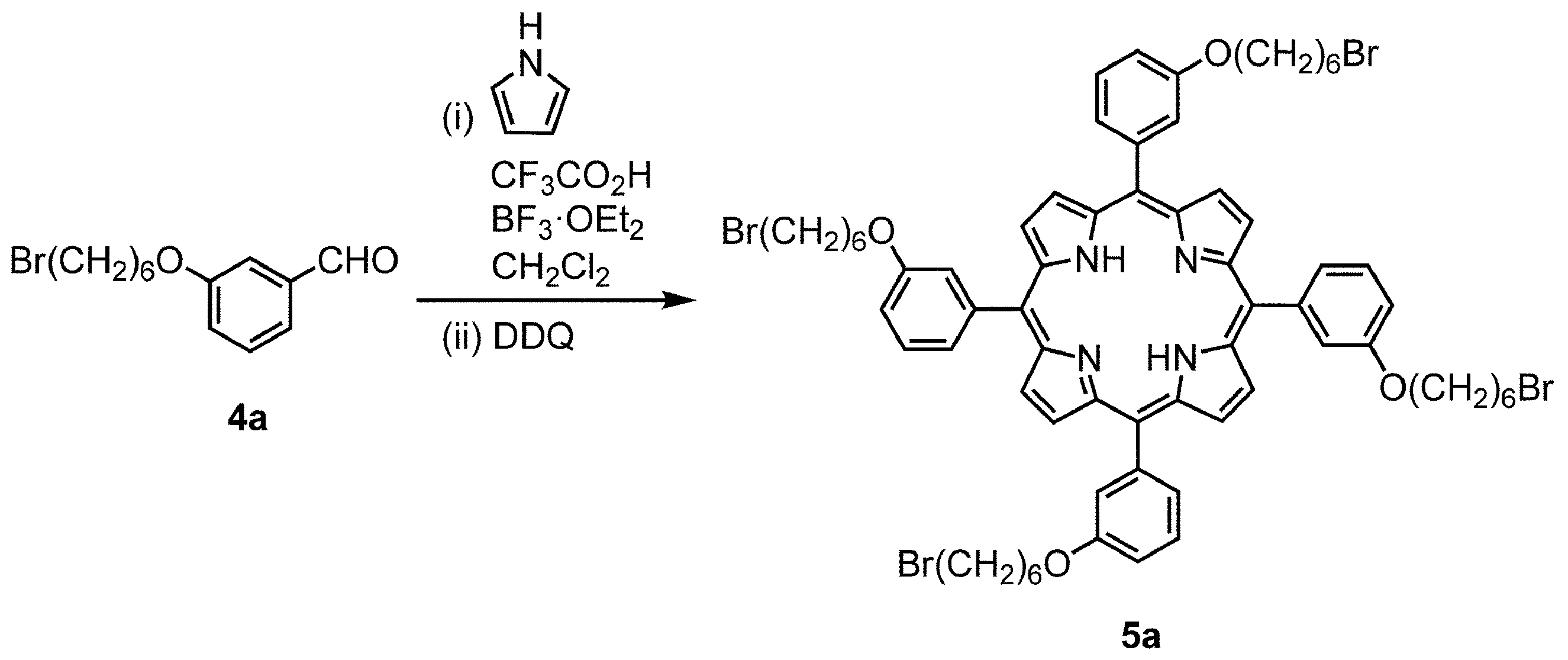
- Triethylamine (1.0 mL, 7.2 mL) was added to the reaction mixture and concentrated. Purification by silica gel column chromatography [chloroform / hexane (2: 1)] gave compound 5a (purple solid) (yield 1.731.7g, yield 52%).
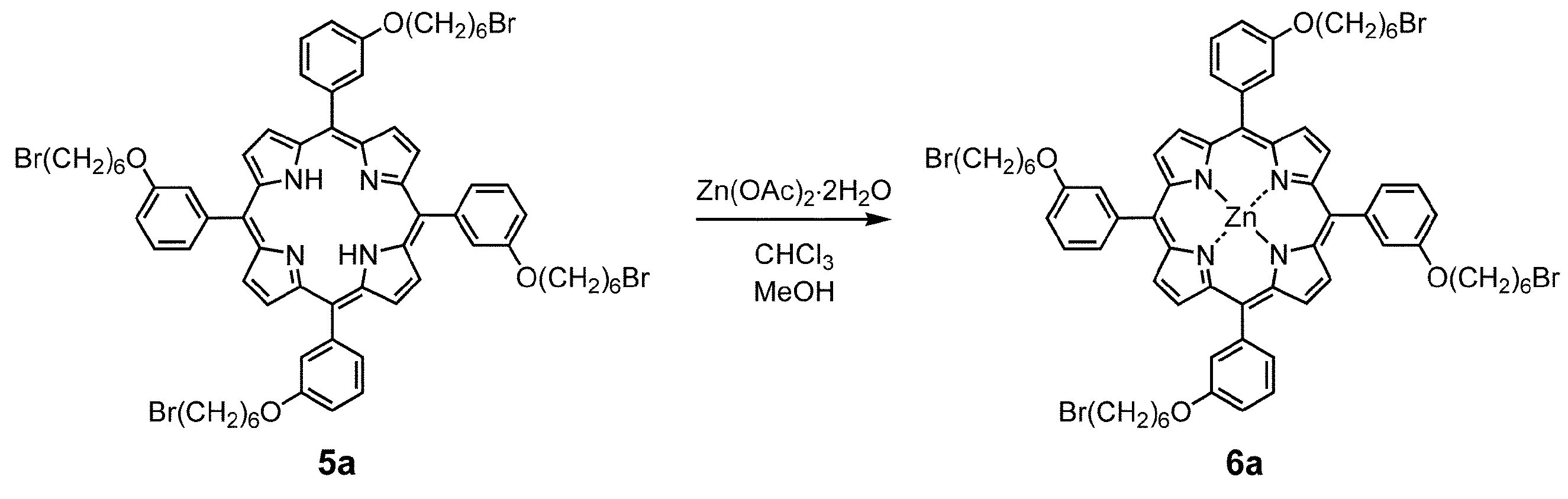
- Porphyrin 5a (266 mg, 0.200 mmol) was dissolved in dehydrated chloroform and stirred at 70 ° C. for 45 minutes under a nitrogen atmosphere.
- Zinc acetate dihydrate (439 mg, 2.00 mmol) was dissolved in dehydrated methanol (4 mL) and added, followed by stirring at 70 ° C. for 4 hours. The mixture was cooled to room temperature, concentrated, and washed with water. The mixture was dried over sodium sulfate and concentrated. Purification by silica gel column chromatography [methylene chloride / hexane (2: 1)] gave compound 6a (red purple solid) (yield 272 mg, yield 98%).
- Example 1 A cyclic carbonate was synthesized using zinc porphyrin complex 1a as a catalyst. The chemical reaction formula at this time is shown below.
- R is an n-butyl group.
- Examples 2 to 15 The synthesis of the cyclic carbonate 3a and the measurement of the NMR yield were carried out in the same manner as in Example 1 except that the type and amount of catalyst used, the pressure after filling with carbon dioxide, and the reaction time were as shown in Table 1. went. The respective NMR yields are shown in Table 1.
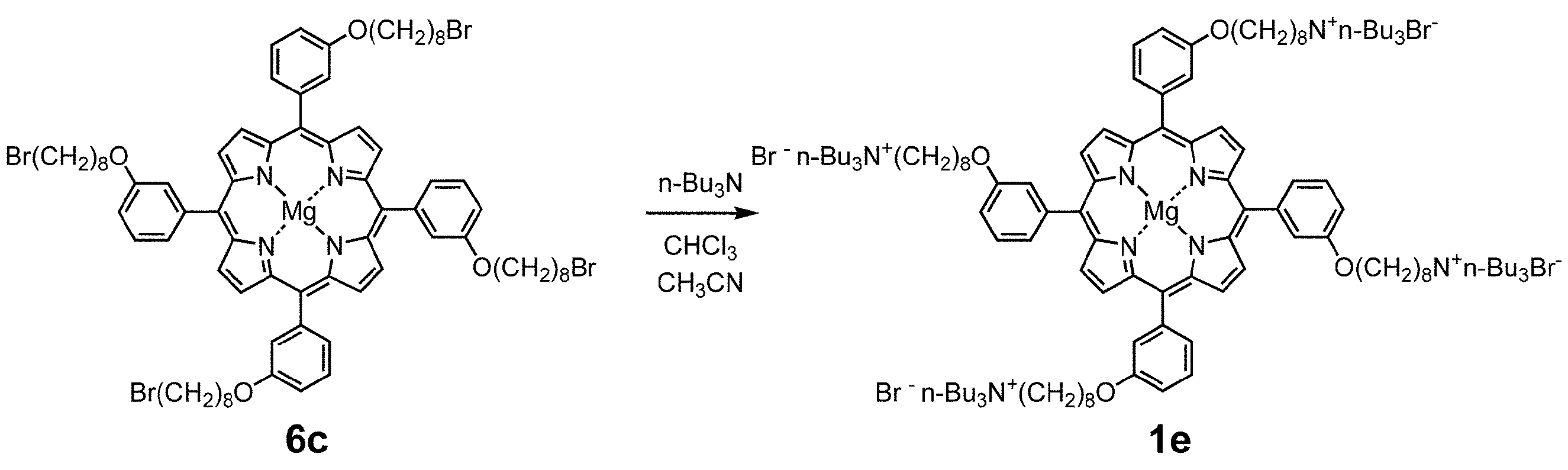
- Examples 16-20 Except that magnesium porphyrin complex 1d (0.005 mol% with respect to epoxide) was used as the catalyst, the pressure after filling with carbon dioxide was 1.5 MPa, and the type and reaction time of epoxide were as shown in Table 2. Cyclic carbonates (3a, 3b, 3c, 3d, 3e) were synthesized in the same manner as in Example 1. Each compound obtained was purified by silica gel column chromatography to determine the isolation yield. The results are shown in Table 2.
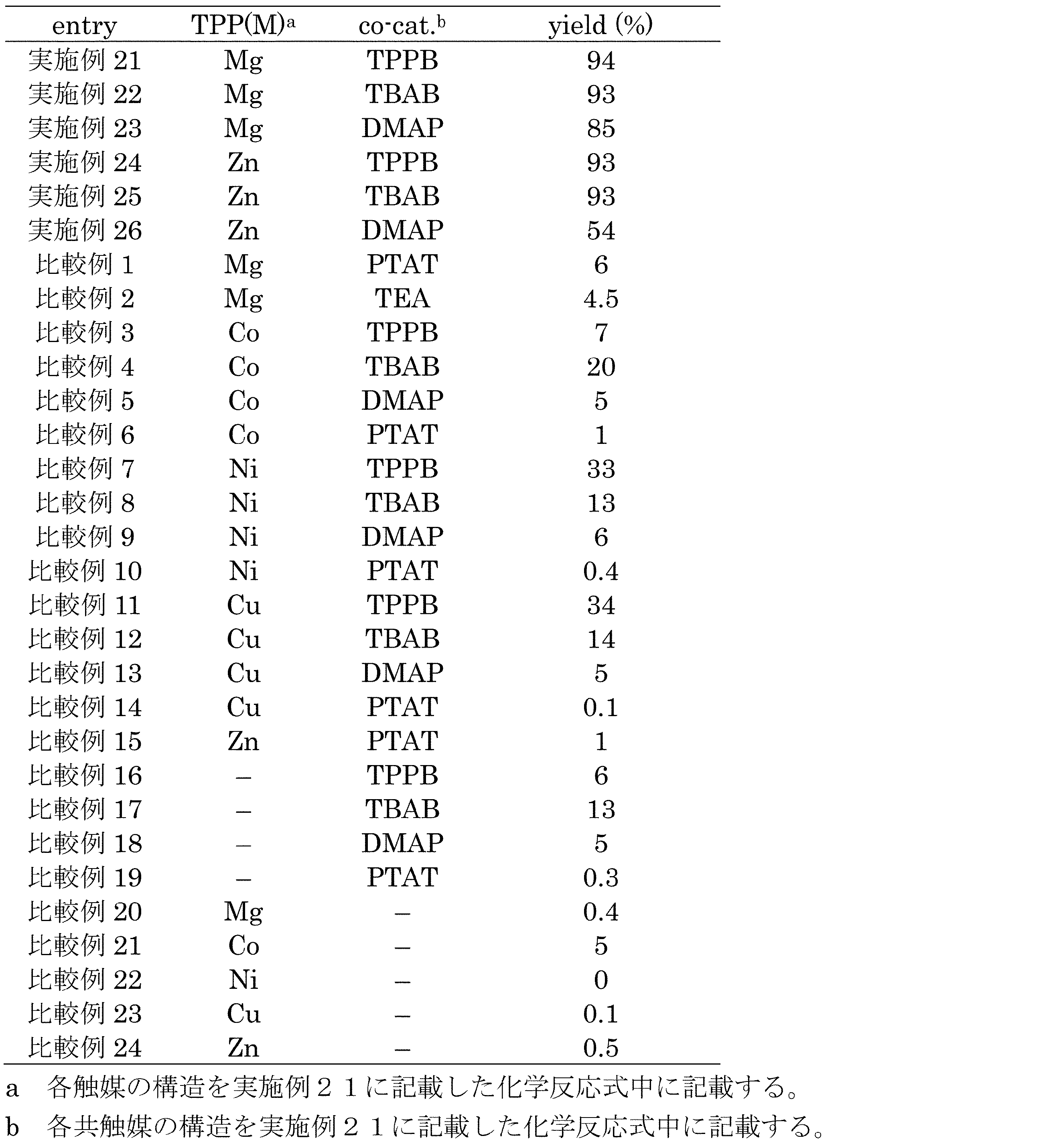
- Example 21 Cyclic carbonates were synthesized using magnesium porphyrin complex TTP (Mg) as a catalyst and tetraphenylphosphonium bromide (TPPB) as a cocatalyst.
- Mg magnesium porphyrin complex
- TPPB tetraphenylphosphonium bromide
- the chemical reaction formula at this time is shown below.
- a 30 mL autoclave was charged with epoxide 2a (10.0 mmol), catalytic magnesium porphyrin complex TPP (Mg) [0.01 mmol (0.1 mol% relative to 2a)], and co-catalyst TPPB ([0.01 mmol (0.1 relative to 2a)). mol%)] and charged with carbon dioxide until the pressure was 1 MPa, the mixture was stirred for 3 hours at 120 ° C.
- Example 22 Comparative Examples 1 to 24
- synthesis of the cyclic carbonate 3a and measurement of the NMR yield were performed in the same manner as in Example 21 except that the types of the catalyst and the cocatalyst were as shown in Table 3.
- the respective NMR yields are shown in Table 2 and FIG.
- Examples 27-29 The catalyst shown below was used as the catalyst, the amount added was as shown in Table 4, TBAB was used as the cocatalyst, and the amount added was as shown in Table 4. Synthesis of carbonic acid ester 3a and measurement of NMR yield were performed. Commercial products were used for the following complexes. Each reaction condition and NMR yield are shown in Table 4.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Inorganic Chemistry (AREA)
- Catalysts (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Polyesters Or Polycarbonates (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description


で表されるものであることが好適である。


で表されるポルフィリンと、金属Mの塩を反応させて、下記一般式(7)

で表される金属錯体を得た後に、当該金属錯体と、3級アミン又は3級ホスフィンを反応させて、上記一般式(1)で表される金属ポルフィリン錯体を得る前記金属ポルフィリン錯体の製造方法を提供することによっても解決される。

で表されるエポキシドを反応させて、下記一般式(9)

で表される環状炭酸エステルを得る環状炭酸エステルの製造方法がより好適な実施態様である。




























融点測定器:Mettler Toledo,FP-62
IR測定装置:Shimadzu,FTIR-8900
1H NMR (600 MHz)測定装置:Varian,Unity Inova AS600
13C NMR (150 MHz)測定装置:Varian,Unity Inova AS600
13C NMR (重クロロホルム, 150 MHz, 50 ℃) 25.4, 28.0, 29.2, 32.7, 33.5, 68.2, 114.3, 120.0, 121.4, 127.5, 127.6, 131.1, 143.6, 146.7, 157.6
IR (臭化カリウム) 3317, 3063, 2932, 2862, 1589, 1466, 1435, 1342, 1281, 1173, 1042, 972, 926, 795, 733, 648 cm-1
Anal. Calcd for C68H74Br4N4O4: C, 61.36; H, 5.60; N, 4.21. Found: C, 60.99; H, 5.76; N, 4.11
MS (FAB) calcd for C68H75 79Br2 81Br2N4O4 1331.2, found 1331.3 (M + H)
13C NMR (重クロロホルム, 150 MHz) 25.3, 27.9, 29.1, 32.6, 33.7, 67.9, 114.0, 120.9, 121.0, 127.3, 127.5, 132.0, 144.0, 150.1, 157.1
IR (臭化カリウム) 3062, 2936, 2858, 1597, 1578, 1477, 1435, 1339, 1285, 1258, 1184, 1049, 999, 937, 799, 721, 702, 648 cm-1
Anal. Calcd for C68H72Br4N4O4Zn: C, 58.57; H, 5.20; N, 4.02. Found: C, 58.65; H, 5.25 N, 3.70
MS (FAB) calcd for C68H73 79Br2 81Br2N4O4 64Zn 1393.2, found 1393.2 (M + H)

13C NMR (重メタノール, 150 MHz) 22.4 (d, JCP = 51.6 Hz), 23.3, 26.3, 29.8, 30.9 (d, JCP = 15.6 Hz), 68.8, 114.2, 119.7 (d, JCP = 85.7 Hz), 121.6, 122.8, 128.4, 128.6, 131.4 (d, JCP = 12.6 Hz), 132.6, 134.6 (d, JCP = 9.7 Hz), 136.0, 145.8, 151.2, 158.5
31P NMR (重メタノール, 243 MHz) 24.2
IR (臭化カリウム) 3055, 2932, 2862, 1582, 1474, 1435, 1327, 1281, 1173, 1111, 1057, 995, 934, 787, 725, 687 cm-1
HRMS (ESI) calcd for C140H132 81Br3N4O4P4 64Zn 2363.5979, found 2363.5986 (M - Br)

13C NMR (重メタノール, 150 MHz) 14.0, 20.5, 22.58, 22.62, 24.6, 26.6, 26.9, 30.1, 59.3, 68.9, 114.3, 121.6, 122.7, 128.4, 128.6, 132.7, 145.9, 151.3, 158.7
IR (臭化カリウム) 3063, 2936, 2870, 1597, 1574, 1474, 1431, 1385, 1335, 1285, 1258, 1180, 1049, 995, 937, 880, 791, 718, 702 cm-1

13C NMR (重クロロホルム, 150 MHz, 50 ℃) 25.3, 27.9, 29.1, 32.7, 33.5, 68.3, 113.9, 121.3, 121.7, 126.9, 128.0, 131.8, 145.1, 149.9, 156.9
IR (臭化カリウム) 3055, 2932, 2862, 1666, 1597, 1474, 1427, 1389, 1335, 1281, 1180, 1049, 995, 941, 880, 795, 725, 640 cm-1
Anal. Calcd for C68H72Br4MgN4O4: C, 60.35; H, 5.36; N, 4.14. Found: C, 60.12; H, 5.48; N, 3.76
MS (FAB) calcd for C68H73 79Br2 81Br2MgN4O4 1353.2, found 1353.2 (M + H)

13C NMR (重メタノール, 150 MHz) 22.4 (d, JCP = 49.9 Hz), 23.3, 26.3, 29.8, 31.0 (d, JCP = 15.9 Hz), 68.8, 114.0, 119.7 (d, JCP = 85.1 Hz), 122.6, 122.9, 128.3, 128.8, 131.4 (d, JCP = 12.3 Hz), 132.7, 134.6 (d, JCP = 9.2 Hz), 136.1, 146.3, 151.1, 158.5
31P NMR (重メタノール, 243 MHz) 28.2
IR (臭化カリウム) 3055, 2939, 2862, 1593, 1516, 1477, 1435, 1331, 1285, 1254, 1180, 1111, 1061, 995, 934, 883, 795, 721, 691 cm-1

13C NMR (重メタノール, 150 MHz) 14.0, 20.6, 22.6, 22.7, 24.7, 26.6, 26.9, 30.1, 59.3, 68.9, 114.2, 122.6, 122.8, 128.3, 128.8, 132.7, 146.4, 151.2, 158.6
IR (臭化カリウム) 3038, 2963, 2876, 1597, 1578, 1474, 1431, 1383, 1333, 1279, 1184, 1165, 997, 937, 880, 799, 727, 710, 694 cm-1
HRMS (ESI) calcd for C116H180 79Br81Br2MgN8O4 2014.1487, found 2014.1243 (M - Br)

13C NMR (重クロロホルム, 150 MHz, 50 ℃) 26.0, 28.1, 28.6, 29.2, 29.4, 32.8, 33.6, 68.3, 114.3, 120.0, 121.4, 127.4, 127.6, 131.1, 143.5, 146.8, 157.6
IR (塩化メチレン) 3317, 3031, 2932, 2862, 1597, 1466, 1435, 1396, 1350, 1281, 1180, 1042, 995, 980, 918, 872, 802, 748, 702, 640 cm-1
MS (FAB) calcd for C76H91 79Br2 81Br2N4O4 1443.4, found 1443.4 (M + H)

13C NMR (重クロロホルム, 150 MHz) 25.7, 25.76, 25.80, 25.83, 28.0, 28.59, 28.60, 28.9, 28.96, 29.04, 29.07, 29.10, 32.7, 33.9, 68.3, 113.76, 113.84, 121.3, 121.4, 126.9, 127.8, 131.9, 144.8, 149.7, 156.5, 156.6 (アトロップ異性体のシグナルが観測された)
IR (塩化メチレン) 3047, 2936, 2858, 1597, 1576, 1518, 1472, 1431, 1391, 1333, 1285, 1265, 1207, 1184, 1165, 1067, 999, 937, 870, 800, 785, 756, 708, 644 cm-1
Anal. Calcd for C76H88Br4MgN4O4: C, 62.29; H, 6.05; N, 3.82. Found: C, 62.24; H, 5.97; N, 3.57
HRMS (FAB) calcd for C76H89 79Br2 81Br2MgN4O4 1465.3, found 1465.4 (M + H)
13C NMR (重メタノール, 150 MHz) 14.0, 20.5, 22.6, 24.6, 27.0, 27.1, 29.9, 30.1, 30.4, 59.2, 59.4, 69.2, 114.2, 122.6, 122.8, 128.3, 128.8, 132.8, 146.4, 151.2, 158.7
IR (臭化カリウム) 3038, 2939, 2876, 1597, 1578, 1474, 1433, 1383, 1333, 1281, 1207, 1184, 1165, 997, 937, 880, 799, 712, 696 cm-1
HRMS (ESI) calcd for C124H196 79Br81Br2MgN8O4 2126.2739, found 2126.2368 (M - Br)
触媒に亜鉛ポルフィリン錯体1aを用いた環状炭酸エステルの合成を行った。このときの化学反応式を以下に示す。

使用した触媒の種類及び使用量、二酸化炭素充填後の圧力、反応時間を表1に示すとおりにしたこと以外は、実施例1と同様にして環状炭酸エステル3aの合成及びNMR収率の測定を行った。それぞれのNMR収率を表1に示す。

触媒としてマグネシウムポルフィリン錯体1d(エポキシドに対して0.005 mol %)を使用し、二酸化炭素充填後の圧力を1.5MPaとし、エポキシドの種類及び反応時間が表2に示すとおりにしたこと以外は、実施例1と同様にして環状炭酸エステル(3a、3b、3c、3d、3e)の合成を行った。得られた各化合物をシリカゲルカラムクロマトグラフィーで精製し単離収率を求めた。その結果を表2に示す。

触媒としてマグネシウムポルフィリン錯体TTP(Mg)、共触媒としてテトラフェニルホスホニウムブロマイド(TPPB)を用いた環状炭酸エステルの合成を行った。このときの化学反応式を以下に示す。

実施例21において、触媒及び共触媒の種類を表3に示すとおりにしたこと以外は、実施例21と同様にして環状炭酸エステル3aの合成及びNMR収率の測定を行った。それぞれのNMR収率を表2及び図1に示す。
触媒として以下に示すものを用い、その添加量を表4に示すとおりにし、共触媒としてTBABを用い、その添加量を表4に示すとおりにしたこと以外は、実施例21と同様にして環状炭酸エステル3aの合成及びNMR収率の測定を行った。下記の錯体は、市販品を用いた。それぞれの反応条件及びNMR収率を表4に示す。


Claims (10)
- 下記一般式(1)で表される金属ポルフィリン錯体。


- 前記一般式(2)において、E+が、下記一般式(3)

で表されるものである請求項1に記載の金属ポルフィリン錯体。 - 前記一般式(2)において、Dが、下記一般式(4)で表される有機基である請求項1又は2に記載の金属ポルフィリン錯体。

- 下記一般式(5)で表される請求項1~3のいずれかに記載の金属ポルフィリン錯体。

- 下記一般式(6)

で表されるポルフィリンと、金属Mの塩を反応させて、下記一般式(7)

で表される金属錯体を得た後に、当該金属錯体と、3級アミン又は3級ホスフィンを反応させて、上記一般式(1)で表される金属ポルフィリン錯体を得ることを特徴とする請求項1~4のいずれかに記載の金属ポルフィリン錯体の製造方法。 - 請求項1~4のいずれかに記載の金属ポルフィリン錯体からなる二酸化炭素固定化触媒。
- 請求項6に記載の二酸化炭素固定化触媒の存在下、二酸化炭素と下記一般式(8)

で表されるエポキシドを反応させて、下記一般式(9)

で表される環状炭酸エステルを得ることを特徴とする環状炭酸エステルの製造方法。 - 下記一般式(10)

で表される金属ポルフィリン錯体からなる触媒、並びに、4級アンモニウムモノハライド、4級ホスホニウムモノハライド及び下記一般式(11)

で表される化合物から選択される少なくとも1種からなる共触媒の存在下、二酸化炭素と下記一般式(8)

で表されるエポキシドを反応させて、下記一般式(9)

で表される環状炭酸エステルを得ることを特徴とする環状炭酸エステルの製造方法。 - 下記一般式(12)

で表される金属フタロシアニン錯体からなる錯体、並びに、4級アンモニウムモノハライド、4級ホスホニウムモノハライド及び下記一般式(11)

で表される化合物から選択される少なくとも1種からなる共触媒の存在下、二酸化炭素と下記一般式(8)

で表されるエポキシドを反応させて、下記一般式(9)

で表される環状炭酸エステルを得ることを特徴とする環状炭酸エステルの製造方法。 - 前記4級アンモニウムモノハライド及び前記4級ホスホニウムモノハライドが下記一般式(13)

で表されるものである請求項8又は9に記載の環状炭酸エステルの製造方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP12834314.2A EP2759546B1 (en) | 2011-09-21 | 2012-09-19 | Metalloporphyrin complex, manufacturing process therefor and its use as carbon dioxide fixation catalyst, as well as process for manufacturing cyclic carbonate |
| JP2013534731A JP6182775B2 (ja) | 2011-09-21 | 2012-09-19 | 金属ポルフィリン錯体、その製造方法及びそれからなる二酸化炭素固定化触媒、並びに、環状炭酸エステルの製造方法 |
| CN201280057181.0A CN103987714B (zh) | 2011-09-21 | 2012-09-19 | 金属卟啉配位化合物、其制造方法及由其构成的二氧化碳固定化催化剂、以及环状碳酸酯的制造方法 |
| US14/345,657 US9211534B2 (en) | 2011-09-21 | 2012-09-19 | Metalloporphyrin complex, manufacturing process therefor and carbon dioxide fixation catalyst therefrom, as well as process for manufacturing cyclic carbonate |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011-206623 | 2011-09-21 | ||
| JP2011206623 | 2011-09-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013042695A1 true WO2013042695A1 (ja) | 2013-03-28 |
Family
ID=47914458
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/073957 WO2013042695A1 (ja) | 2011-09-21 | 2012-09-19 | 金属ポルフィリン錯体、その製造方法及びそれからなる二酸化炭素固定化触媒、並びに、環状炭酸エステルの製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US9211534B2 (ja) |
| EP (1) | EP2759546B1 (ja) |
| JP (1) | JP6182775B2 (ja) |
| CN (1) | CN103987714B (ja) |
| WO (1) | WO2013042695A1 (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9447511B2 (en) | 2013-10-04 | 2016-09-20 | Centre National De La Recherche Scientifique (Cnrs) | Iron-based catalyst for selective electrochemical reduction of CO2 into CO |
| CN107739363A (zh) * | 2017-12-06 | 2018-02-27 | 河南工程学院 | 一种钴基催化剂制备环状碳酸酯的方法 |
| CN109289921A (zh) * | 2018-11-13 | 2019-02-01 | 北京林业大学 | 一种基于植酸的用于合成环状碳酸酯的催化体系 |
| WO2019026883A1 (ja) * | 2017-07-31 | 2019-02-07 | 住友化学株式会社 | 大環状化合物の製造方法 |
| WO2022130796A1 (ja) * | 2020-12-14 | 2022-06-23 | サンアプロ株式会社 | 光酸発生剤及びこれを用いた感光性組成物 |
| KR20230068756A (ko) | 2021-11-11 | 2023-05-18 | 광주과학기술원 | 고활성 및 고선택도의 전기화학적 이산화탄소 전환용 단원자 니켈 촉매 및 이를 포함하는 이산화탄소 전환용 전극 |
Families Citing this family (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104492488B (zh) * | 2014-12-15 | 2017-04-26 | 南京工业大学 | 一种双配体锌配合物催化剂及其应用 |
| JP6498470B2 (ja) * | 2015-02-20 | 2019-04-10 | デクセリアルズ株式会社 | 透明導電膜およびその製造方法 |
| CN105111426B (zh) * | 2015-09-16 | 2017-05-17 | 武汉理工大学 | 一种季铵盐官能化卟啉催化剂及其制备方法 |
| CN105566623A (zh) * | 2016-03-09 | 2016-05-11 | 中国科学院长春应用化学研究所 | 一种聚碳酸酯材料及其制备方法 |
| CN107433205B (zh) * | 2016-05-25 | 2020-02-21 | 中国科学院大连化学物理研究所 | 共价有机框架负载钴催化剂及其制备和应用 |
| TWI601571B (zh) | 2016-12-07 | 2017-10-11 | 財團法人工業技術研究院 | 觸媒及以此觸媒合成環狀碳酸酯的方法 |
| TWI609719B (zh) * | 2016-12-09 | 2018-01-01 | National Taiwan University Of Science And Technology | 用於燃料電池的觸媒及其製造方法 |
| CN106831698A (zh) * | 2016-12-20 | 2017-06-13 | 中山大学 | 一种多相催化合成环状碳酸酯的方法 |
| CN106928238B (zh) * | 2017-03-09 | 2019-04-12 | 中国科学院化学研究所 | 卟啉衍生物及其制备方法和催化剂-底物对和水相体系中断裂酰胺键和/或酯键的方法 |
| JP6909096B2 (ja) * | 2017-08-16 | 2021-07-28 | 公益財団法人微生物化学研究会 | 触媒、アミド結合の形成方法、及びアミド化合物の製造方法 |
| CN107903239A (zh) * | 2017-12-06 | 2018-04-13 | 河南工程学院 | 一种钒基催化剂转化二氧化碳制备环状碳酸酯的方法 |
| CN107954973A (zh) * | 2017-12-06 | 2018-04-24 | 河南工程学院 | 一种锌基催化剂制备环状碳酸酯的方法 |
| CN107827857A (zh) * | 2017-12-06 | 2018-03-23 | 河南工程学院 | 一种铬基催化剂制备环状碳酸酯的方法 |
| CN107827860A (zh) * | 2017-12-06 | 2018-03-23 | 河南工程学院 | 一种锌基催化剂转化二氧化碳制备环状碳酸酯的方法 |
| CN107954972A (zh) * | 2017-12-06 | 2018-04-24 | 河南工程学院 | 一种锰基催化剂制备环状碳酸酯的方法 |
| CN107987050A (zh) * | 2017-12-06 | 2018-05-04 | 河南工程学院 | 一种镁基催化剂制备环状碳酸酯的方法 |
| CN107827858A (zh) * | 2017-12-06 | 2018-03-23 | 河南工程学院 | 一种铝基催化剂制备环状碳酸酯的方法 |
| CN107827859A (zh) * | 2017-12-06 | 2018-03-23 | 河南工程学院 | 一种铝基催化剂转化二氧化碳制备环状碳酸酯的方法 |
| CN107827856A (zh) * | 2017-12-06 | 2018-03-23 | 河南工程学院 | 一种钒基催化剂制备环状碳酸酯的方法 |
| CN107903238A (zh) * | 2017-12-06 | 2018-04-13 | 河南工程学院 | 一种铜基催化剂制备环状碳酸酯的方法 |
| CN107827862A (zh) * | 2017-12-06 | 2018-03-23 | 河南工程学院 | 一种镍基催化剂转化二氧化碳制备环状碳酸酯的方法 |
| CN107827861A (zh) * | 2017-12-06 | 2018-03-23 | 河南工程学院 | 一种铁基催化剂制备环状碳酸酯的方法 |
| CN107973771A (zh) * | 2017-12-06 | 2018-05-01 | 河南工程学院 | 一种钴基催化剂转化二氧化碳制备环状碳酸酯的方法 |
| CN107973772A (zh) * | 2017-12-06 | 2018-05-01 | 河南工程学院 | 一种铁基催化剂转化二氧化碳制备环状碳酸酯的方法 |
| CN107827863A (zh) * | 2017-12-06 | 2018-03-23 | 河南工程学院 | 一种铜基催化剂转化二氧化碳制备环状碳酸酯的方法 |
| CN107954974A (zh) * | 2017-12-06 | 2018-04-24 | 河南工程学院 | 一种锰基催化剂转化二氧化碳制备环状碳酸酯的方法 |
| CN111925400B (zh) * | 2020-08-17 | 2024-03-26 | 中国科学院长春应用化学研究所 | 一种氧化还原响应性金属卟啉配合物、其制备方法和聚乳酸的制备方法 |
| KR20230011080A (ko) * | 2021-07-13 | 2023-01-20 | 삼성에스디아이 주식회사 | 염료, 이를 포함하는 조성물, 필름, 광학 부재 및 디스플레이 장치 |
| CN114669332B (zh) * | 2022-04-24 | 2023-08-01 | 齐齐哈尔大学 | 一种离子型超高交联多孔有机聚合物负载钴催化剂的制备方法 |
| CN115449073B (zh) * | 2022-10-25 | 2024-01-12 | 广东工业大学 | 一种金属卟啉基超交联离子聚合物和制备方法及其应用 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS56127384A (en) * | 1980-03-10 | 1981-10-06 | Hidetoshi Tsuchida | Cation picket fence-type tetraphenylporphyrine and gas adsorbent |
| JPS59101488A (ja) * | 1982-12-01 | 1984-06-12 | Hidetoshi Tsuchida | 水酸基を有する鉄−テトラフェニルポルフィン錯体 |
| JPH11509180A (ja) * | 1995-06-07 | 1999-08-17 | デューク・ユニバーシティ | 酸化体脱除剤 |
| JP2006512301A (ja) * | 2002-10-21 | 2006-04-13 | エル.モルテニ アンド シー.デイ フラテッリ アリッチ ソシエタ’ディ エセルチヅィオ ソシエタ ペル アチオニ | メソ置換されたポルフィリン |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4663467A (en) * | 1984-03-05 | 1987-05-05 | The Dow Chemical Company | Novel porphyrinate and amine composition useful as catalysts in the preparation of alkylene carbonates |
| US5283356A (en) * | 1992-08-03 | 1994-02-01 | Texaco Chemical Company | Process for manufacturing alkylene carbonates using metal phthalocyanine catalysts |
| US5994339A (en) | 1993-10-15 | 1999-11-30 | University Of Alabama At Birmingham Research Foundation | Oxidant scavengers |
| GB2352449B (en) | 1999-04-29 | 2003-06-25 | Secr Defence | Reactions of strained ring heterocyclic monomers in supercritical CO2 |
| SG10201701421PA (en) * | 2008-08-22 | 2017-04-27 | Novomer Inc | Catalysts and methods for polymer synthesis |
| CN101514195B (zh) | 2009-03-11 | 2012-06-06 | 兰州大学 | 一种环碳酸酯的制备方法 |
-
2012
- 2012-09-19 US US14/345,657 patent/US9211534B2/en not_active Expired - Fee Related
- 2012-09-19 JP JP2013534731A patent/JP6182775B2/ja active Active
- 2012-09-19 EP EP12834314.2A patent/EP2759546B1/en not_active Not-in-force
- 2012-09-19 CN CN201280057181.0A patent/CN103987714B/zh not_active Expired - Fee Related
- 2012-09-19 WO PCT/JP2012/073957 patent/WO2013042695A1/ja active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS56127384A (en) * | 1980-03-10 | 1981-10-06 | Hidetoshi Tsuchida | Cation picket fence-type tetraphenylporphyrine and gas adsorbent |
| JPS59101488A (ja) * | 1982-12-01 | 1984-06-12 | Hidetoshi Tsuchida | 水酸基を有する鉄−テトラフェニルポルフィン錯体 |
| JPH11509180A (ja) * | 1995-06-07 | 1999-08-17 | デューク・ユニバーシティ | 酸化体脱除剤 |
| JP2006512301A (ja) * | 2002-10-21 | 2006-04-13 | エル.モルテニ アンド シー.デイ フラテッリ アリッチ ソシエタ’ディ エセルチヅィオ ソシエタ ペル アチオニ | メソ置換されたポルフィリン |
Non-Patent Citations (13)
| Title |
|---|
| AHMADI F. ET AL: "Electron-deficient tin(IV) tetraphenylporphyrin perchlorate: A highly efficient catalyst for chemical fixation of carbon dioxide", POLYHEDRON, vol. 32, no. 1, 2012, pages 68 - 72, XP028356229 * |
| AHMADI F. ET AL: "Highly efficient chemical fixation of carbon dioxide catalyzed by high-valent tetraphenylporphyrinatotin(IV) triflate", INORGANIC CHEMISTRY COMMUNICATIONS, vol. 14, no. 9, 2011, pages 1489 - 1493, XP028244392 * |
| EMA T. ET AL: "A bifunctional catalyst for carbon dioxide fixation: cooperative double activation of epoxides for the synthesis of cyclic carbonates", CHEMICAL COMMUNICATIONS, vol. 48, no. 37, 11 May 2012 (2012-05-11), pages 4489 - 4491, XP003030969 * |
| JIN ET AL., J. MOL. CATAL. A: CHEM., vol. 261, 2007, pages 262 - 266 |
| LIU, Y.: "Functional multiwalled carbon nanotube nanocomposite with iron picket-fence porphyrin and its electrocatalytic behavior", ELECTROCHEMISTRY COMMUNICATIONS, vol. 9, no. 10, 2007, pages 2564 - 2570, XP022259114 * |
| R. L. PADDOCK ET AL., TETRAHEDRON LETT., vol. 45, 2004, pages 2023 - 2026 |
| R. SRIVASTAVA ET AL., J. MOL. CATAL. A: CHEM., vol. 226, 2005, pages 199 - 205 |
| ROBIC N. ET AL: "Synthesis and preliminary DNA- interaction studies of a new cationic porphyrin", TETRAHEDRON LETTERS, vol. 31, no. 33, 1990, pages 4739 - 4742, XP026024995 * |
| See also references of EP2759546A4 |
| W. J. KRUPER ET AL., J. ORG. CHEM., vol. 60, 1995, pages 725 - 727 |
| W. MEI ET AL., CHINESE JOURNAL OF CHEMICAL ENGINEERING, vol. 19, no. 3, 2011, pages 446 - 451 |
| WANG M. ET AL: "Efficient Solvent-free Synthesis of Chloropropene Carbonate from the Coupling Reaction of CO2 and Epichlorohydrin Catalyzed by Magnesium Porphyrins as Chlorophyll-like Catalysts", CHINESE JOURNAL OF CHEMICAL ENGINEERING, vol. 19, no. 3, June 2011 (2011-06-01), pages 446 - 451, XP003030970 * |
| YAMASHITA T. ET AL: "Stabilization of guanine quadruplex DNA by the binding of porphyrins with cationic side arms", BIOORGANIC & MEDICINAL CHEMISTRY, vol. 13, no. 7, 2005, pages 2423 - 2430, XP027637615 * |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9447511B2 (en) | 2013-10-04 | 2016-09-20 | Centre National De La Recherche Scientifique (Cnrs) | Iron-based catalyst for selective electrochemical reduction of CO2 into CO |
| WO2019026883A1 (ja) * | 2017-07-31 | 2019-02-07 | 住友化学株式会社 | 大環状化合物の製造方法 |
| CN110914269A (zh) * | 2017-07-31 | 2020-03-24 | 住友化学株式会社 | 大环化合物的制造方法 |
| JPWO2019026883A1 (ja) * | 2017-07-31 | 2020-05-28 | 住友化学株式会社 | 大環状化合物の製造方法 |
| CN110914269B (zh) * | 2017-07-31 | 2022-07-29 | 住友化学株式会社 | 大环化合物的制造方法 |
| JP7150728B2 (ja) | 2017-07-31 | 2022-10-11 | 住友化学株式会社 | 大環状化合物の製造方法 |
| CN107739363A (zh) * | 2017-12-06 | 2018-02-27 | 河南工程学院 | 一种钴基催化剂制备环状碳酸酯的方法 |
| CN109289921A (zh) * | 2018-11-13 | 2019-02-01 | 北京林业大学 | 一种基于植酸的用于合成环状碳酸酯的催化体系 |
| CN109289921B (zh) * | 2018-11-13 | 2021-08-10 | 北京林业大学 | 一种基于植酸的用于合成环状碳酸酯的催化体系 |
| WO2022130796A1 (ja) * | 2020-12-14 | 2022-06-23 | サンアプロ株式会社 | 光酸発生剤及びこれを用いた感光性組成物 |
| KR20230068756A (ko) | 2021-11-11 | 2023-05-18 | 광주과학기술원 | 고활성 및 고선택도의 전기화학적 이산화탄소 전환용 단원자 니켈 촉매 및 이를 포함하는 이산화탄소 전환용 전극 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2759546A1 (en) | 2014-07-30 |
| JP6182775B2 (ja) | 2017-08-23 |
| CN103987714A (zh) | 2014-08-13 |
| EP2759546B1 (en) | 2018-03-21 |
| US9211534B2 (en) | 2015-12-15 |
| CN103987714B (zh) | 2016-03-16 |
| US20140228561A1 (en) | 2014-08-14 |
| EP2759546A4 (en) | 2016-10-19 |
| JPWO2013042695A1 (ja) | 2015-03-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6182775B2 (ja) | 金属ポルフィリン錯体、その製造方法及びそれからなる二酸化炭素固定化触媒、並びに、環状炭酸エステルの製造方法 | |
| US9850223B2 (en) | Metal catalysts for selective formation of cyclic carbonates, process for preparing cyclic carbonate using the same and use of cyclic carbonate | |
| CN112979400A (zh) | 一种在碱金属氢化物作用下制备2-碘代芳醚的方法 | |
| JP3085722B2 (ja) | アルキレンカーボネートの製造法 | |
| WO2015170688A1 (ja) | 金属担持多孔性配位高分子触媒 | |
| CN113072517B (zh) | 一种五元含氧杂环化合物的合成方法 | |
| WO2011009934A1 (en) | Tris(1,2,3-triazol-4-yl)methane organometallic compounds as catalysts and processes using them. | |
| JP4612219B2 (ja) | 置換芳香族化合物の製造方法 | |
| US6545166B2 (en) | Process for producing spiro acetal derivative | |
| CN112321433B (zh) | 一种3-(羟甲基)环己烷羧酸叔丁酯的合成方法 | |
| KR102376869B1 (ko) | 요오드 함유 규소 화합물의 제조 방법 | |
| KR102100611B1 (ko) | 고리형 카보네이트 제조용 금속 착화합물 촉매 및 이를 이용한 고리형 카보네이트의 제조방법 | |
| KR20240060025A (ko) | N-아릴 아마이드 화합물의 제조방법 | |
| KR102418587B1 (ko) | 이산화탄소 고정화 반응용 촉매 조성물 | |
| WO2024080317A1 (ja) | ヒドロキシビオチン誘導体及びビニルビオチン誘導体の製造方法 | |
| JP6754131B2 (ja) | 脱離基を有する有機化合物と有機ホウ素化合物とのカップリング体の製造方法 | |
| EP1031555B1 (en) | Difluoromethoxybenzene derivatives and their use as intermediates | |
| WO2019151419A1 (ja) | ポリカルボニル化合物、その誘導体及びそれらの製造方法 | |
| JP2022100189A (ja) | キノロンイソインドリン誘導体の製造方法 | |
| US20030060637A1 (en) | Cyclic aniline sulfide and process for preparing the same | |
| JP2022090590A (ja) | イソインドリンボロン酸誘導体の製造方法 | |
| JP3012933B1 (ja) | ジチアナフタレノファン化合物とその製造方法 | |
| US20090048469A1 (en) | Iodine-containing fluoropolyether and process for producing the same | |
| JP3515257B2 (ja) | 2,3,6,7,10,11−ヘキサアセトキシトリフェニレンの製造方法 | |
| CN117069661A (zh) | 一种合成哒嗪衍生物的方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201280057181.0 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12834314 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2013534731 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14345657 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012834314 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |